

Abstract
Lucigenin and paraquat are similar in that each can be taken into Escherichia coli and can then mediate O2⨪ production by cycles of univalent reduction, to the corresponding monocation radical, followed by autoxidation. Thus, both compounds caused induction of enzymes that are regulated by the soxRS regulon. The lucigenin cation radical has the added property of reacting with O2⨪, in a radical–radical addition, to yield an unstable dioxetane, whose decomposition yields light. Superoxide dismutases (SOD), by decreasing [O2⨪], inhibit light production and to the same degree inhibit other O2⨪-dependent reactions in the cell. Lucigenin luminescence was used to show that the levels of SOD in the parental strain provide ≈95% protection of all O2⨪-sensitive targets in E. coli. This degree of protection was so close to the limit of 100% that halving the parental level of [SOD], or increasing it 5-fold, had only marginal effects on the intensity of lucigenin-dependent luminescence.
A previous study (1) has established that lucigenin (Luc++) is readily reduced univalently and that the resultant lucigenin cation radical (Luc ·+) autoxidizes and thus generates O2⨪. Luc ·+ also reacts with O2⨪ in a radical–radical addition reaction to yield an unstable dioxetane, whose decomposition yields the excited state acridone, which emits light in returning to the ground state (2–4). Because it can thus act as both a source of and a detector of O2⨪, Luc++ luminescence is not a reliable measure of [O2⨪]. The inhibition of this luminescence can however be used as a measure of superoxide dismutase (SOD) activity.
Scott and Eaton nevertheless recently reported (5) that a sodA sodB strain of Escherichia coli elicited much more Luc++ luminescence than did the SOD-replete parental strain, and they asserted that they were measuring intracellular [O2⨪]. This was of special interest to us because we had previously attempted to measure [O2⨪] in E. coli based on the balance between the rapid inactivation of aconitase by O2⨪ and its reactivation by Fe(II) plus cellular reductants (6). We have also tried to calculate [O2⨪] based on its measured rate of generation and known rates of removal by SOD and by glutathione (7). We therefore undertook an exploration of Luc++ luminescence elicited by E. coli. The results of these studies that lead to an appreciation of the degree of protection by SOD, afforded to cellular targets of O2⨪, are reported herein.
MATERIALS AND METHODS
Luc++ (bis-N-methylacridinium nitrate) was from Aldrich; CuZnSOD was from Diagnostic Data (Mountain View, CA); catalase was from Boehringer Mannheim; PQ++ (methyl viologen), NADPH, glucose-6-phosphate, succinate, xanthine, and cytochrome c were from Sigma; malate was from ICN; yeast extract and bactotryptone were from Difco; and glucose was from Mallinckrodt (Chesterfield, MO). Xanthine oxidase was prepared by R. Wiley (Duke University Medical Center) from bovine cream as described by Waud et al. (8). SOD activity was assayed by the xanthine oxidase/cytochrome c method (9). MnTMPyP was prepared as described by Pasternack et al. (10)
The strains of E. coli were as follows: AB1157, parental strain; JI132, sodA sodB; JI130; sodA; JI131, sodB; and AB1157 + pHS1–4, FeSOD overproducer. These strains were obtained from J. A. Imlay (11). Bacteria were grown overnight in Luria–Bertani (LB) medium, which in the case of AB1157 + pHS1–4 contained 12.5 μM tetracycline. Aeration was maintained by shaking at 200 rpm at 37°C. In the morning, cells were inoculated into fresh medium as specified in the figure legends and were then grown in all cases without antibiotic. In some cases, paraquat or MnTMPyP was added, and subsequently samples were taken, lucigenin was added, and luminescence was measured. In other cases, cells were collected by centrifugation, washed with 50 mM potassium phosphate (pH 7.3), and then suspended to equal A600 in the same buffer plus carbon source, or in LB medium. Subsequently, lucigenin was added and luminescence measured. When it was desirable to suppress MnSOD biosynthesis, the cells were grown for several hours in anaerobic GAS-Pack containers (BBL, Baltimore). In all cases, Luc++ was the last component added before measurement of luminescence in a Turner Designs Model 20E luminometer as described (1). The reaction volume was 0.8 ml, and the instrument was operated with a 5-sec delay and 10-sec integration time.
When enzyme activity assays were indicated, cells were washed with 50 mM potassium phosphate (pH 7.8) and then lysed with a French Press. Lysates were clarified by centrifugation, and the resultant extracts were assayed for protein (12), fumarase (13), and glucose-6-phosphate dehydrogenase (14). NADPH:Luc++ oxidoreductase was assayed as previously described for NADPH:PQ++ oxidoreductase (15).
RESULTS
Comparison of sodA sodB and SOD-Replete Strains.
Overnight cultures in LB medium were diluted 1:40 with fresh LB and were grown for 90 min. At this time, the sodA sodB strain (JI132) had reached A600 = 0.25, whereas the faster growing SOD-replete strain (AB1157) had achieved A600 = 1.0. The cells were collected by centrifugation, washed once with 50 mM potassium phosphate (pH 7.3), and then diluted to A600 = 0.5 in the specified medium containing 0.1 mM Luc++. Fig. 1 demonstrates that JI132 elicited more Luc++ luminescence than did AB1157, a result in agreement with that of Scott and Eaton (5). This difference was greater when the carbon source was glucose (Fig. 1, bars 1 and 2) than when it was LB medium (bars 3 and 4), and the difference was least when the only carbon source was succinate (bars 5 and 6). This luminescence reflected intracellular events since addition of SOD or catalase, to 10 μg/ml, to the suspending medium did not cause significant inhibition. Strains singly defective in either sodA or sodB, or strains overproducing the FeSOD, did not behave significantly differently than the SOD-replete parental strain (data not shown). This makes sense as will be discussed subsequently. The decreased luminescence seen when glucose was replaced by the amino acids in the LB medium, or by succinate, is also understandable. Thus the supply of electrons, from nutrients dependent for their catabolism on the citric acid cycle, would be limited because of the inactivation of aconitase and of fumarases A and B by O2⨪ (16, 17). A supply of electrons is essential both for the reduction of Luc++ and for the generation of O2⨪.
Figure 1.

Lucigenin luminescence elicited by sodA sodB (JI132) and parental (AB1157) E. coli as a function of carbon source. Overnight cultures were diluted 40-fold with fresh LB and were grown for 90 min, at which point A600 was 0.25 for JI132 and 1.0 for AB1157. Cells were collected, washed in 50 mM sodium phosphate (pH 7.3), and resuspended in this buffer, or in LB medium at pH 7.0, to equal densities. Reaction mixtures contained 0.72 ml of medium, 40 μl of cell suspension, and 40 μl of 2.0 mM Luc++. Luc++ was the last component added. A600 in the reaction mixture was 0.5. Luminescence yield from five sequential 10-sec integration times, separated by 5-sec delay times, were averaged. Bars 1 and 2 represent JI132 and AB1157, respectively, both in 50 mM sodium phosphate (pH 7.3) plus 0.25% glucose. Bars 3 and 4 represent JI132 and AB1157, respectively, both in LB medium. Bars 5 and 6 represent JI132 and AB1157, respectively, both in 50 mM sodium phosphate (pH 7.3) plus 0.25% succinate.
The Luc++ luminescence elicited by the sodA sodB strain was a reflection of intracellular O2⨪ in the presence of Luc++. This was shown by the inhibitory effect of a cell-permeant catalyst (18, 19) of the dismutation of O2⨪. Thus, as shown in Fig. 2, the luminescence seen with the sodA sodB strain was decreased by 0.4 μM MnTMPyP (compare lines 2 and 3). It should be noted that MnTMPyP has been shown to inhibit the uptake of paraquat into E. coli (18) and may also interfere with the uptake of Luc++. The structural resemblances among PQ++, Luc++, and MnTMPyP are made clear in Fig. 5. Once again, the luminescence was much less with the SOD-replete strain (Fig. 2, line 1). It is possible that the apparent decrease in luminescence with incubation time was due to increased masking of emitted light by the higher cell densities achieved with time in these growing cultures.
Figure 2.

Effect of the SOD-mimic MnTMPyP on luminescence. Cells grown overnight in LB were diluted 100-fold into fresh, aerated minimal salts plus 0.2% casamino acids/3 μg/ml thiamine/3 μg/ml pantothenic acid/0.2% glucose. At intervals, 0.78-ml aliquots were taken and mixed with 20 μl of 2 mM Luc++, and the luminescence of four consecutive 10-sec integration times separated by 5-sec delay times were averaged. Lines: 1, AB1157; 2, JI132 with MnTMPyP added to 0.4 μM 5 min before removal of the first sample; 3, JI132.
Figure 5.

Structural formulae of compounds used.
Transition from Anaerobic to Aerobic Conditions.
Little difference was seen in the Luc++ luminescences elicited by the SOD-replete parental and the singly defective sodA or sodB mutants, when aerobically grown cells were used. However, when the cells were grown anaerobically and were then aerated for only ≈2 min before the addition of Luc++, there was a substantial difference between the sodA and the sodB strains, as shown in Fig. 3 (compare bars 2 and 3). This is the expected result because sodA (or MnSOD) is not made by the anaerobic cells and is induced by aeration, whereas sodB (or FeSOD) is constitutive. Thus the sodA strain will contain FeSOD even when grown anaerobically, whereas the B strain, being unable to make FeSOD, will contain almost no SOD when grown anaerobically and will have induced very little MnSOD during the brief interval of aeration.
Figure 3.

Luc++ luminescence elicited by anaerobically grown E. coli. Overnight aerobic cultures in LB medium were diluted 1000-fold into fresh LB plus 0.25% glucose and further incubated for 4.5 hr in gas pack jars. The jars were opened, the cultures were shaken in air at 200 rpm for 2 min, 0.78-ml aliquots were mixed with 20 μl of 2.0 mM Luc++, and luminescence was averaged from three consecutive 10-sec integrate periods separated by 5-sec delays. Bars: 1, AB1157; 2, sodA; 3, sodB; 4, sodA sodB.
O2⨪-Independent Luminescence.
The large luminescence seen with the sodA sodB strain was almost entirely due to the interaction of Luc++ with O2⨪ in the cells. Thus it was ≈95% suppressed by raising intracellular SOD, as shown with the SOD-replete parental strain. Moreover, it was dependent on the simultaneous presence of O2, cells, an energy source, and Luc++. In contrast the much smaller luminescence seen with the SOD-replete parental strain was substantially due to O2⨪-independent processes. Thus it was not significantly diminished by raising SOD, as shown with an overproducing strain (data not shown and ref. 5). Moreover, a substantial fraction of the luminescence of the complete system persisted in the absence of cells, Luc++, or glucose and may in part reflect baseline dark noise of the phototube. We are led to conclude that the difference in O2⨪-dependent Luc++ luminescence between sodA sodB and the parental strain is even greater than those shown in Figs. 1, 2, 3.
Effect of Paraquat.
This viologen has been used to impose oxidative stress (20) and to induce the soxRS regulon (21–24), which it does by cycles of univalent reduction followed by autoxidation. If there were no further complications, one might expect that the presence of PQ++ would increase the Luc++ luminescence elicited by sodA sodB E. coli. However, as shown in Fig. 4, PQ++ did not increase this luminescence when added 30 min (line 1) or 135 min (line 2) before the addition of Luc++. Indeed, at higher concentrations and at longer times of exposure, it diminished the luminescence. There are several factors that account for this result. PQ++ and Luc++ share structural similarities, as shown in Fig. 5, and both are capable of cycles of univalent reduction followed by autoxidation (1). Since PQ++ is actively taken up by E. coli (25), it is reasonable to suspect that Luc++ is also. Thus Luc++ and PQ++ are likely to compete for uptake and for electrons. The competition for reduction has been demonstrated in vitro (1). PQ++ may be expected to limit the electron supply available for reduction of Luc++ both by shunting electrons from NAD(P)H and other cellular reductants to dioxygen and by decreasing regeneration of these reductants due to inhibition of aconitase, fumarases A and B, and other [4Fe-4S]-containing dehydratases by O2⨪.
Figure 4.

Effect of PQ++ on Luc++ luminescence elicited by the sodA sodB strain. An overnight culture of JI132 in LB medium was diluted 200-fold into fresh LB medium and grown to A600 = 0.19, and PQ++ was then added to the concentrations shown. At 30 min (line 1) and at 135 min (line 2) thereafter, 0.78-ml aliquots were mixed with 20 μl of 2.0 mM Luc++, and luminescence was measured as in Fig. 3.
Induction of the Members of the soxRS Regulon by Luc++.
We have previously shown that Luc++ could cause the production of O2⨪, by glucose oxidase plus glucose (1). It was of interest to ascertain whether it could also shunt electrons to O2, with production of O2⨪, within E. coli. This was tested by examining the ability of Luc++ to cause induction of members of the soxRS regulon and to do so to a greater degree in a sodA sodB strain than in the parental strain. Fig. 6 demonstrates that Luc++ induced glucose-6-phosphate dehydrogenase, fumarase C, and an NADPH:Luc++ reductase. In all three cases, induction was greater in the sodA sodB strain than in the parental strain. We have previously shown that NADPH:ferredoxin reductase is a member of the soxRS regulon and that it can reduce PQ++ (26). Probably it can also reduce Luc++. The redox similarities between Luc++ and PQ++ are striking, and since PQ++ is known to induce these enzymes through soxRS (27, 28), it is most likely that Luc++ also induces them by way of soxRS.
Figure 6.

Inductions of members of soxRS regulon by Luc++. Overnight cultures of JI132 and AB1157 in LB medium were diluted 25-fold into fresh LB medium. After 45 min of growth, Luc++ was added to the concentrations shown, and incubation was continued for 75 min, at which time cells were collected and assayed for enzyme activities as described in Materials and Methods. (A) Glucose-6-phosphate dehydrogenase; (B) fumarase C; (C) NADPH: Luc++ oxidoreductase.
DISCUSSION
Luc++ acts both as a source of O2⨪ and as an indicating scavenger of this radical within E. coli. The pertinent reactions are as follows:
NADPH:Luc++ diaphorase activity was present in extracts of E. coli and was induced further by exposure to PQ++. There may be several Luc++ reductases in E. coli. It is in any case clear that Luc++ can be reduced within E. coli (reaction a). Reaction b has been demonstrated in vitro (1) and probably occurs also within E. coli. Reaction c represents the sum total of O2⨪ sources within the cell. Reaction d leads to luminescence, and reaction e represents the dismutation of O2− catalyzed by SOD.
If the dismutation of O2⨪ by SOD is indeed the major sink for O2⨪ in the cell, then the absence of SOD will raise [O2⨪] and hence reaction d, as well as all reactions of O2⨪ with diverse targets, within the cell. Thus the ≈20-fold decrease in Luc++ luminescence, caused by the presence of either sodA or sodB, or of both together, is a reflection of the degree of protection afforded by SOD to all targets of O2⨪ within the cell. This statement is justified because the rate of reaction of any target (T) with O2⨪ will be given by k(T)(O2⨪) and will be diminished to the extent that [O2⨪] is decreased. It does not take account of countervailing processes such as repair or replacement of the damaged target.
The reason why so little difference in Luc++ luminescence was seen between the parental strain and mutants singly defective in either sodA or sodB, or overproducing FeSOD, is made clear in Fig. 7, which is a theoretical curve presenting the degree of protection by SOD of any target, including Luc ·+, which is susceptible to attack by O2⨪. The concentration of SOD is given in biological units, where 1 unit provides 50% protection. If 1 unit of SOD gives 50% protection, then 3 units gives 75% protection, and so on to a limit of 100% protection at ∞ SOD. This curve was constructed using the simple equation % protection = (n/n + 1)100, where n is the concentration of SOD in units per volume. Since Luc++ luminescence was ≈95% inhibited in the parental strain, compared with the sodA sodB strain, the parental strain contained ≈19 units of SOD. It is clear from the curve why no difference will be discernible in Luc++ luminescence between the parental strain and the strains defective in one SOD or overproducing one SOD. This is the case not only because of the small differences in % protection as one approaches the limit of 100% protection but also because the signal-to-noise ratio declines precipitously as the luminescence intensity decreases.
Figure 7.

Calculated inhibition of Luc++ luminescence as a function of [SOD] expressed in terms of biological units. One biological unit is defined as that concentration of SOD within E. coli which will cause 50% inhibition of Luc++ luminescence. The curve was generated using the expression % inhibition = (n/n + 1)100, where n is the number of biological units of SOD. The sodA sodB strain was taken to contain no SOD since the CuZnSOD is present in ≈2% (29) the amount of SOD A plus SOD B and moreover is periplasmic (30). The positions of the other strains of E. coli are indicated on the assumptions that the single mutants contain ≈1/2 concentration of SOD contained in the parental strain and that the SOD B overproducer contains ≈5 times the [SOD] of the parental strain.
These estimates of the degree to which SOD decreases [O2⨪] in E. coli, derived from measurements of Luc++ luminescence, are in reasonable agreement with estimates arrived at by other methods. Thus the steady state [O2⨪] was estimated to be 2 × 10−10 M in SOD-competent E. coli and 5 × 10−7 M in SOD-null cells (7), based on in vitro measurements. This is a 2,500-fold difference, whereas the Luc++ luminescence indicates only a ≈20-fold difference in [O2⨪] between sodA sodB and parental strains. However, the earlier estimate (7) assumed that the reaction of O2⨪ with GSH was the major O2⨪-scavenging reaction in the absence of SOD. We now know that O2⨪ reacts rapidly with the [4Fe-4S] clusters of dehydratases such as aconitase (16), 6-phosphogluconate dehydratase (31), and fumarases A and B (17). The steady state of [O2⨪] in the sodA sodB strain was thus seriously overestimated. However in general terms, the earlier study (7) and the present one agree to the extent that both conclude that SOD causes a >10-fold decrease in [O2⨪] in E. coli and would thus provide >90% protection to all susceptible targets.
The estimates of [O2⨪] based on the balance between inactivation of aconitase by O2⨪ and its subsequent reactivation (6) yielded 1.8 × 10−11 M for the SOD-replete strain and 31.8 × 10−11 M for the sodA sodB strain. It is interesting that the ratio between these estimates for [O2⨪] in the sodA sodB and the parental strains is ≈18, in very good agreement with the ratio obtained from measurements of Luc++ luminescence.
Using PQ++ to enhance [O2⨪] and at the same time using Luc++ luminescence to measure [O2⨪], as was done by Scott and Eaton (5), is problematic for several reasons. (i) Luc++ and PQ++ may be expected to compete for uptake by the cells. (ii) Luc++ and PQ++ will be in competition for reducing equivalents and Luc++ cannot respond to O2⨪ with luminescence unless it is first univalently reduced. (iii) Luc++ and PQ++ can both mediate O2⨪ production. (iv) Raising O2⨪ through the action of PQ++ will inactivate fumarases A and B and aconitases and thus diminish the supply of reducing equivalents coming from the citric acid cycle. An additional minor problem with the Scott and Eaton paper (5) is that they equated urate production by xanthine oxidase and O2⨪ production. In fact, only a fraction of the electrons flowing through xanthine oxidase result in O2⨪ production (32, 33). Finally, it should be pointed out that the rate of reduction of Luc++ to Luc ·+ will probably be different in the xanthine oxidase plus xanthine system than within E. coli, and it is Luc ·+ not Luc++ that reacts with O2⨪ to yield luminescence.
It should be emphasized that, in spite of the complications attending its use, Luc++ luminescence by E. coli provided with a carbon source is a reasonable inverse measure of intracellular SOD activity and that its use indicates that the SOD in SOD-competent E. coli provides ≈95% protection of all O2⨪-sensitive targets in the cell.
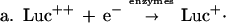
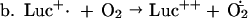
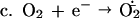
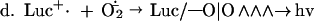
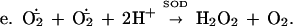
Acknowledgments
This work was supported by grants from the Council for Tobacco Research–U.S.A., Inc. (2871AR2), the U.S. Army Medical Research (Contract DAMD17–95-C-5065), and the National Science Foundation (INT-92–24035).
ABBREVIATIONS
- SOD
superoxide dismutase
- LB
Luria–Bertani
References
- 1.Liochev S I, Fridovich I. Arch Biochem Biophys. 1996;337:115–120. doi: 10.1006/abbi.1997.9766. [DOI] [PubMed] [Google Scholar]
- 2.Greenlee L, Fridovich I, Handler P. Biochemistry. 1962;1:779–783. doi: 10.1021/bi00911a008. [DOI] [PubMed] [Google Scholar]
- 3.Legg K D, Hercules D M. J Am Chem Soc. 1969;91:1902–1907. [Google Scholar]
- 4.Faulkner K, Fridovich I. Free Radical Biol Med. 1993;15:447–451. doi: 10.1016/0891-5849(93)90044-u. [DOI] [PubMed] [Google Scholar]
- 5.Scott M D, Eaton J W. Redox Rep. 1996;2:113–119. doi: 10.1080/13510002.1996.11747037. [DOI] [PubMed] [Google Scholar]
- 6.Gardner P R, Fridovich I. J Biol Chem. 1992;267:8757–8763. [PubMed] [Google Scholar]
- 7.Imlay J A, Fridovich I. J Biol Chem. 1991;266:6957–6965. [PubMed] [Google Scholar]
- 8.Waud W R, Brady F O, Wiley R D, Rajagopalan K V. Arch Biochem Biophys. 1975;169:695–701. doi: 10.1016/0003-9861(75)90214-3. [DOI] [PubMed] [Google Scholar]
- 9.McCord J M, Fridovich I. J Biol Chem. 1969;244:6049–6055. [PubMed] [Google Scholar]
- 10.Pasternack R F, Gibbs E J, Villafranca J J. Biochemistry. 1983;22:2406–2414. doi: 10.1021/bi00279a016. [DOI] [PubMed] [Google Scholar]
- 11.Imlay J A, Linn S. J Bacteriol. 1987;169:2967–2976. doi: 10.1128/jb.169.7.2967-2976.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lowry O H, Rosebrough N T, Farr A L, Randall R J. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 13.Hill R L, Bradshaw R H. Methods Enzymol. 1969;13:91–99. [Google Scholar]
- 14.Kao S M, Hassan H M. J Biol Chem. 1985;260:10478–10481. [PubMed] [Google Scholar]
- 15.Liochev S I, Fridovich I. Free Radical Biol Med. 1994;16:555–559. doi: 10.1016/0891-5849(94)90055-8. [DOI] [PubMed] [Google Scholar]
- 16.Gardner P R, Fridovich I. J Biol Chem. 1991;266:19328–19333. [PubMed] [Google Scholar]
- 17.Liochev S I, Fridovich I. Arch Biochem Biophys. 1993;301:379–384. doi: 10.1006/abbi.1993.1159. [DOI] [PubMed] [Google Scholar]
- 18.Liochev S I, Fridovich I. Arch Biochem Biophys. 1995;321:271–275. doi: 10.1006/abbi.1995.1395. [DOI] [PubMed] [Google Scholar]
- 19.Faulkner K M, Liochev S I, Fridovich I. J Biol Chem. 1994;269:23471–23476. [PubMed] [Google Scholar]
- 20.Hassan H M, Fridovich I. J Biol Chem. 1978;253:8143–8148. [PubMed] [Google Scholar]
- 21.Walkup L K B, Kogoma T. J Bacteriol. 1989;171:1476–1484. doi: 10.1128/jb.171.3.1476-1484.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greenberg J T, Demple B. J Bacteriol. 1989;171:3933–3939. doi: 10.1128/jb.171.7.3933-3939.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsaneva I R, Weiss B. J Bacteriol. 1990;172:4197–4205. doi: 10.1128/jb.172.8.4197-4205.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Demple B. Annu Rev Genet. 1991;25:315–337. doi: 10.1146/annurev.ge.25.120191.001531. [DOI] [PubMed] [Google Scholar]
- 25.Kitzler J W, Minakami H, Fridovich I. J Bacteriol. 1990;172:686–690. doi: 10.1128/jb.172.2.686-690.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liochev S I, Hausladen A, Beyer W F, Jr, Fridovich I. Proc Natl Acad Sci USA. 1994;91:1328–1331. doi: 10.1073/pnas.91.4.1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liochev S I, Fridovich I. Proc Natl Acad Sci USA. 1992;89:5892–5896. doi: 10.1073/pnas.89.13.5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greenberg J T, Monach P, Chou J H, Josephy P O, Demple B. Proc Natl Acad Sci USA. 1990;87:6181–6185. doi: 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benov L, Fridovich I. J Biol Chem. 1994;269:25310–25314. [PubMed] [Google Scholar]
- 30.Benov L, Chang L Y, Day B, Fridovich I. Arch Biochem Biophys. 1995;719:508–511. doi: 10.1006/abbi.1995.1324. [DOI] [PubMed] [Google Scholar]
- 31.Gardner P R, Fridovich I. J Biol Chem. 1991;266:1478–1483. [PubMed] [Google Scholar]
- 32.Fridovich I. J Biol Chem. 1970;245:4053–4057. [PubMed] [Google Scholar]
- 33.Nagano T, Fridovich I. J Free Radical Biol Med. 1985;1:39–42. doi: 10.1016/0748-5514(85)90027-3. [DOI] [PubMed] [Google Scholar]