

Abstract
Gal repressor inhibits transcription from the gal promoter (P1) when it binds to the cognate operator (OE). The repression is relieved by the presence of the inducer d-galactose. Compared with its interaction with free repressor, d-galactose binds to the repressor–operator complex with 10-fold reduced affinity as determined by fluorescence enhancement measurements. Thermodynamic analysis and fluorescence anisotropy showed that the stability of the repressor–operator complex is reduced by only 7-fold by the presence of the inducer in the complex. The formation of the inducer–repressor–operator ternary complex has been confirmed by CD spectral analysis. Fluorescence spectroscopy and energy transfer experiments suggest that individual allosteric effects of the two ligands, inducer and operator, on Gal repressor are responsible for the slightly weakened stability of the ternary complex compared with the stability of the inducer–repressor and repressor–operator complexes. In vitro transcription results demonstrated full derepression of transcription of the P1 promoter under conditions in which the concentrations of the inducer–repressor binary complex are severalfold higher than the dissociation constant of the inducer–repressor–operator ternary complex into inducer–repressor and free DNA. These results strongly suggest that the inducer binding to the repressor–operator complex does not lead to dissociation of the repressor from the operator during transcription induction. Because Gal repressor inhibits transcription by modulating the α subunit of the P1-bound RNA polymerase, we conclude that the inducer binding to the operator-bound repressor only allosterically relieves the inhibitory effect of repressor on RNA polymerase without dissociating the repressor from DNA.
Keywords: tryptophan fluorescence, derepression, galactose, circular dichroism
In the classical negative control of gene expression, a repressor protein binds to cognate operator element(s) in DNA at or near the promoter and inhibits transcription initiation (1). Induction of transcription can occur in the presence of an inducer molecule that interacts with and causes an allosteric change in the repressor, which then loses its inhibitory power on transcription. It was originally proposed and commonly believed that repressor sterically prevents RNA polymerase from binding to the promoter site. According to this idea, because of inducer-generated allosteric change(s), the repressor loses its affinity for the operator and dissociates from DNA, thus allowing RNA polymerase to bind. We investigated the dissociation of repressor from the operator DNA by inducer under conditions of transcription derepression in the gal operon of Escherichia coli and found that this long-held view of the mechanism of induction does not apply to the P1 promoter of the operon.
The gal operon is transcribed by two promoters, P1 and P2 (2). Repression of the two gal promoters involves Gal repressor (GalR) binding to two operators, OE and OI (3–6). Derepression occurs by the interaction of GalR with inducer d-galactose or d-fucose (7). Whereas full repression of both P1 and P2 requires the histone-like protein HU (8), interaction of repressor with OE alone represses P1, but not P2. GalR·OE-mediated repression of P1 observed in the absence of HU also is relieved by the presence of d-galactose or d-fucose (6). We report here the results of an analysis of (i) formation and stability of various binary and ternary complexes of operator, repressor, and inducer by fluorescence and CD spectroscopy, (ii) the antagonistic effect of inducer or operator on the binding of the other to repressor, and (iii) DNA-induced conformational changes in GalR. These results lead us to propose that inducer derepresses transcription in this system without dissociating the repressor from the operator.
MATERIALS AND METHODS
Materials.
d-Galactose, d-fucose, adenosine 5′-[γ-thio]triphosphate (ATP[γ-S]), and acrylamide were obtained from Sigma. Fluorescein isothiocyanate and 5-({[(2-iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid (IAEDANS) were purchased from Molecular Probes. Oligonucleotides purified by HPLC were from Midland Certified Reagent (Midland, TX). The sequence of the 20-mer wild-type OE was 5′-TTGTGTAAACGATTCCACTA-3′, whereas that of the mutant OE was 5′-TTGTGTAAATAATTCCACTA-3′. The sequence of the 29-mer wild-type OE was 5′-xACTTCTTGTGTAAACGATTCCACTAATTT-3′, x being a hexylamino group (2). Polynucleotide kinase was obtained from BRL. GalR was purified from a plasmid clone in which the galR gene was expressed from a late promoter of bacteriophage T7 as described previously (9). The concentration of GalR dimer was obtained from its extinction coefficient at 280 nm of 39,600 M−1·cm−1 (10) and is expressed in terms of monomer. Plasmid pSA509 used as the source of DNA in transcription assays has been described before (6).
IAEDANS Labeling of OE.
Twenty nanomoles of OE DNA duplex, 250 μmol of ATP[γ-S], and 10 units of T4 polynucleotide kinase were incubated at 37°C for 2 h in 0.1 M potassium phosphate buffer, pH 8.0. The mixture was extracted with an equal volume of phenol/chloroform/isoamyl alcohol (25:24:1). The aqueous layer was loaded onto a Sephadex G-25 column (1.5 × 12.5 cm), equilibrated with 0.1 M potassium phosphate (pH 8.0) and eluted. Appropriate fractions were pooled, and IAEDANS was added at a final concentration of 0.5 mM. The mixture was incubated for 90 min in the dark and extensively dialyzed. The calculated incorporation ratio obtained from A260 and A340 was approximately 1.2 mol per mol of oligonucleotide duplex.
Synthesis and Purification of Fluorescein-Labeled OE.
The 29-mer complementary oligonucleotides with a hexylamino group (x) attached at their 5′ ends were separately synthesized in an Applied Biosystems 381A DNA synthesizer using amino link supplied by Applied Biosystems. The oligonucleotides were deprotected by incubating them at 55°C for 24 h in 30% ammonium hydroxide, precipitated by 1-butanol, then dried under vacuum and dissolved in water.
Ten milligrams of fluorescein 5-isothiocyanate was dissolved in 200 μl of 1.0 M NaHCO3/Na2CO3 buffer (pH 9.0)/N,N′-dimethylformamide/water in the ratio of 5:2:3. The dye solution, the oligonucleotide solution, and the carbonate buffer were mixed in the ratio of 2:5:3, vortexed briefly, and incubated at 25°C for 20 h. The fluorescein-labeled oligonucleotides were further purified by C18 reverse-phase HPLC using a linear gradient of 0–60% acetonitrile in 0.1 M triethylammonium acetate buffer (pH 7.0). The appropriate fractions of each labeled oligonucleotide were mixed in equimolar proportion, and then annealed.
Fluorescence.
Fluorescence spectra were recorded either in a Hitachi F3010 or SLM-Aminco (Urbana, IL) spectrofluorometer equipped with computers for spectra addition and subtraction facility. Experiments were conducted in a water-circulated thermostat chamber maintained at 25°C. Excitation band pass was 1.5 nm, and emission band pass was 20 nm. Samples were incubated at 25°C for 30 min before conducting the experiment. This incubation was necessary, because initial dilution of protein from stock solution caused the appearance of slight turbidity, which disappeared within several minutes upon standing at room temperature. The excitation wavelength was 295 nm; the emission wavelength was 360 nm. In preliminary experiments, the tryptophan residue in GalR was found to be somewhat photosensitive. The excitation slit was opened only during the fluorescence measurements and only at a single point to reduce exposure time. Repeated measurements under such conditions yielded constant fluorescence values. The experiments were performed in 12.5 mM Tris·HCl buffer (pH 8.0) containing 300 mM KCl and 0.5 mM EDTA. d-Galactose titrations of GalR were done at 2 × 10−6 M GalR at 25°C. For titration of GalR-OE complex by d-galactose, GalR, and OE concentrations were 2.4 × 10−6 M and 1.2 × 10−6 M, respectively. OE titrations were performed at 2 × 10−6 M GalR. Increasing concentrations of DNA were added to GalR before measuring the emission spectra. The normalized integrated intensity was plotted as a function of OE concentration. The fluorescence values were corrected for dilution and inner filter effect. The excitation and emission band passes were 2 nm each.
The direct binding of OE to GalR and to d-galactose·GalR complex was measured fluorometrically by using fluorescein-labeled operator DNA in the buffer described above. The excitation and emission wavelengths were 490 nm and 525 nm, respectively. Excitation and emission band passes were 10 nm.
Fluorescence anisotropy was measured after each addition of GalR as described in Results.
Fluorescence energy transfer efficiency (E) was calculated from excitation spectra using the following equation (11):
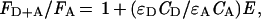 |
where FD+A is the fluorescence intensity of the donor–acceptor pair (dansylated OE and GalR); FA is the fluorescence of the acceptor (dansylated OE) at the same wavelengths; ɛD and ɛA are the molar extinction coefficients of the donor and acceptor, respectively, at the excitation wavelength, and CD and CA are the concentrations of the donor and acceptor, respectively. The distance between the dansyl group and the tryptophan in GalR was determined by the following equation:
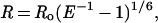 |
where R is the distance between the donor and the acceptor, Ro is the distance for which the energy transfer efficiency is 50%, and E is the experimentally observed energy transfer efficiency.
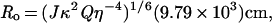 |
where J is the overlap integral (a measure of the spectral overlap), Q is the quantum yield, η is the refraction index, and κ is the orientation factor. The overlap integral was calculated according to the method of Wu and Stryer (12). The value of κ2 was taken as 2/3, which is completely spatially averaged. Such complete spatial averaging is justified here because of aliphatic linkers separating the 5-({[(acetyl)amino]- ethyl}amino)naphthalene-1-sulfonic acid (AEDANS) molecule from the DNA as well as the experimentally obtained low polarization value. The energy transfer experiments were done in the buffer described above at 25°C. The excitation and emission band passes were 4 nm and 16 nm, respectively. The emission wavelength was set at 500 nm.
Circular Dichroism.
CD spectra were obtained using a Jasco J-600 spectropolarimeter at 25°C. DNA concentration was 2.0 μM. Ten scans were averaged to produce a CD spectum. The bandwidth was 1 nm, and the time constant was 1 s. The experiments were conducted in the buffer described above. The buffer spectrum was subtracted from the OE spectrum, whereas the buffer-plus-GalR spectrum was subtracted from OE-plus-GalR spectrum. The protein did not contribute significantly to the intensities at the wavelengths used.
Data Processing.
For all titration experiments, control experiments were carried out in which identical volumes of buffer were added to the protein solution without the ligand. Values from the blank titration were used to correct the volumes for ligand titrations. The corrected fluorescence values then were fitted to an equation of a single class of ligand binding by the nonlinear least-squares method for determining dissociation constants. The three variable parameters chosen (initial fluorescence value, fluorescence value at infinite ligand concentration, and the dissociation constant) were systematically varied within a given range. The values that gave the lowest χ2 were chosen as the best-fit parameters. A similar least-squares fit procedure was used to obtain operator–repressor dissociation constants from anisotropy data.
In Vitro Transcription.
In vitro transcription assays were done as described previously (9) with slight modifications. The 348-bp EcoRI–BamHI fragment of pSA509 containing the gal promoter region was used as the template. The transcription reaction mixture containing 12.5 mM Tris·HCl (pH 8.0), 200 mM potassium chloride, 5 mM magnesium chloride, 2 nM DNA, 20 nM RNA polymerase, 160 or 800 nM GalR, and different concentrations of d-galactose as indicated in Fig. 7 was preincubated for 5 min at 37°C. The reaction was started by the addition of 0.2 mM ATP, GTP, CTP, and 0.02 mM 32P-labeled UTP. After incubation for 15 min at 37°C, the reaction was stopped by the addition of 0.2 vol of 0.2 M EDTA in 40% glycerol with dye. d-Galactose concentrations were 2.0 and 30 mM. The RNA was quantified by PhosphorImager 425 (Molecular Dynamics).
Figure 7.

The effect of d-galactose on galP1 transcription in the presence of GalR. In vitro transcription assays were carried out as described in the text. GalR concentrations were 160 (A) and 800 nM (B).
RESULTS
Inducer Binding to GalR and GalR·OE Complex.
Binding of ligands to proteins often causes quenching or enhancement of fluorescence of tryptophan, depending on the location of the amino acid(s) in the protein. Such enhancement or quenching can be used to derive binding isotherms. GalR is a dimer containing only one tryptophan residue per subunit. The intrinsic fluorescence of this tryptophan was used to monitor ligand binding. Fig. 1 shows the results of d-galactose, d-fucose, and d-glucose titration of GalR at 1.0 × 10−6 M as monitored by the intrinsic tryptophan fluorescence. Binding of d-galactose resulted in a 23% enhancement of tryptophan fluorescence, which quickly saturated. No significant fluorescence increase occurred at d-galactose concentrations greater than 2.0 × 10−4 M. This enhancement also was accompanied by a small (≈2 nm) red shift of the emission maximum and a change in acrylamide quenching pattern (data not shown). The data were used to fit a binding equation for a single class of binding site using a nonlinear least squares fit procedure. It yielded a dissociation constant (KD1) of 2.2 ± 0.3 × 10−5 M. A similar dissociation constant for the interaction of d-galactose and GalR has been reported previously (13). d-Fucose is a nonmetabolizable analog of d-galactose and is a slightly weaker inducer (14). Binding of d-fucose also led to an enhancement of tryptophan fluorescence, although the extent of enhancement (12%) was less, resulting in a dissociation constant of 6.0 ± 0.3 × 10−5 M. d-Glucose, which is not an inducer, caused relatively minor changes in tryptophan fluorescence.
Figure 1.

Relative tryptophan fluorescence change (f/fo) of GalR as a function of hexose concentration. d-Galactose (○), d-fucose (•), d-glucose (▵), and d-galactose plus 20-mer OE-DNA (□). The GalR concentration was 2 × 10−6 M in all of the experiments, except repressor concentration was 2.4 × 10−6 M and OE DNA was 1.2 × 10−6 M in the titration of GalR·OE complex.
The binding of d-galactose to operator–repressor complex was studied by monitoring tryptophan fluorescence changes that occurred upon d-galactose binding. The concentration of GalR was 2.4 × 10−6 M, and that of OE DNA was 1.2 × 10−6 M. As will be shown later, Gal repressor bound to OE DNA with a dissociation constant (KD4) of 4 × 10−9 M under these conditions. Thus, GalR and OE should exist as a 1:1 complex under these conditions. A significant increase of tryptophan fluorescence was achieved by the addition of increasing concentrations of d-galactose, with little enhancement occurring beyond 1.0 × 10−3 M (Fig. 1). At the high d-galactose concentration, tryptophan fluorescence increased by 23% of the initial value. The degree of fluorescence enhancement was very similar to that observed on binding of d-galactose to free GalR. However, d-galactose bound to the GalR·OE complex with a dissociation constant (KD3) of 2.2 × 10−4 M, which is approximately 10-fold higher than KD1, the dissociation constant of d-galactose binding to free GalR. This suggests that although binding of repressor to the operator reduced the affinity of inducer to repressor, because of the sufficient residual affinity of the inducer-bound repressor toward the operator, the inducer bound to the operator-bound GalR without dissociating the complex.
The inability of d-galactose to dissociate repressor from the DNA·protein complex was further tested by fluorescence anisotropy experiments. The OE DNA was thiophosphorylated at the 5′ end by ATP γ S and T4 polynucleotide kinase and subsequently allowed to react with IAEDANS as described in Materials and Methods. The AEDANS-labeled operator at 0.5 × 10−6 M gave an anisotropy value of 0.036 (data not shown), indicating significant internal motion of the IAEDANS probe. The presence of 1.0 × 10−6 M GalR monomer increased the anisotropy value to 0.064. This value is still low compared with that expected for a fully rigid probe, again indicating significant internal motion. Such low anisotropy values are not uncommon in 5′-end-labeled DNAs because of flexibility of the linker arm and lack of steric constraints at the end of the oligonucleotides (15). Further addition of 1.0 × 10−3 M d-galactose resulted in a very small (less than 5%) decrease in the anisotropy value obtained for GalR plus DNA. The value in the presence of d-galactose was still much higher than that of the free operator, indicating that binding of d-galactose to the repressor–operator complex does not lead to significant dissociation of the repressor from the operator.
Determination of the affinity of d-galactose to free repressor and repressor–operator complex allowed an estimation of the relative affinities of OE for the free GalR and the d-galactose–GalR complex based on the binding cycle described in Fig. 2. Two pathways lead from the repressor to the end product, the inducer–repressor–operator ternary complex. Thus,
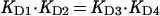 |
or
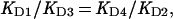 |
where the KDs are the dissociation constants of the respective binding equations. Thus, the predicted ratio of KD4/KD2 is 0.1 as determined from KD1 and KD3 values of 2.2 × 10−5 M and 2.2 × 10−4 M, respectively.
Figure 2.

The binding cycle of GalR (R) to ligands d-galactose (I) and operator (O).
GalR Binding to OE in the Absence and Presence of Inducer.
To determine values of KD4 and KD2 experimentally, the affinity of the OE for GalR and d-galactose–GalR complex was determined by fluorescence anisotropy measurements. A 29-bp OE DNA with a hexylamino group at the 5′ end was labeled with fluorescein isothiocyanate. Fluorescein has a high molar extinction coefficient and quantum yield. The purified duplex was titrated with GalR and anisotropy was measured at each point. A plot of fluorescence anisotropy of the 2.0 × 10−9 M OE DNA at different GalR concentrations is shown in the absence (Fig. 3A) and in the presence (Fig. 3B) of 1 × 10−3 M d-galactose. In both cases, fluorescence anisotropy increased by about 40–50%. The observed values of KD4 and KD2 were 4.2 × 10−9 M and 27.8 × 10−9 M, respectively. The measured dissociation constant (KD4) of the operator–repressor complex under the present conditions was at least an order of magnitude higher than those obtained by gel electrophoresis and DNase footprinting assays (9, 16). This was expected, as the latter measurements were done in the presence of potassium glutamate instead of KCl and at a lower salt concentration, conditions that increase the affinity of DNA-binding proteins for the target sequences (17). Nevertheless, the binding of d-galactose to GalR reduced the affinity of repressor for the operator by approximately 7-fold. The KD4/KD2 ratio of 0.15, measured by fluorescence anisotropy, is very similar to the value (0.1) obtained from the indirect binding cycle analysis.
Figure 3.

Titration of fluorescein-labeled OE DNA with GalR in the absence (A) and in the presence (B) of 1.0 × 10−3 M d-galactose. Fluorescein-labeled oligonucleotides (29-mer) were prepared as described in the text. The OE concentration was 2 × 10−9 M. Each reading was time-averaged for 100 sec. The solution conditions are described in Materials and Methods.
The binding of repressor to the operator was further investigated in the presence and absence of inducer by CD spectroscopy. Binding of the GalR to OE leads to a significant change of CD spectra of the operator (10). Fig. 4A shows the CD spectra of the operator, the repressor–operator complex, and the repressor–operator complex in the presence of d-galactose. Binding of GalR to OE at micromolar concentrations, as expected, resulted in a change of the operator CD spectrum. The direction and magnitude of the spectral change was the same as that observed previously (10). The CD spectral change indicated a DNA distortion that very likely is the DNA bending caused by the binding of GalR to OE (18). The presence of 2 × 10−3 M d-galactose did not cause any significant change in the CD spectrum of the repressor–operator complex. Because the d-galactose concentration was 10-fold higher than the apparent dissociation constant of the d-galactose from the operator-repressor complex (Fig. 1), it was evident that the binding of d-galactose to the operator-repressor complex at micromolar concentrations did not lead to the dissociation of repressor from the operator.
Figure 4.

(A) CD spectrum of the operator OE (20-mer) at 2 × 10−6 M (– – – –), in the presence of GalR at 4.0 × 10−6 M (····), and in the presence of GalR at 4.0 × 10−6 M and d-galactose at 2.0 × 10−3 M (——). (B) CD spectrum of the mutant OEC operator (20 mer) at 2.0 × 10−6 M (– – – –), in the presence of GalR at 4.0 × 10−6 M (——) and in the presence of GalR at 4.0 × 10−6 M and d-galactose at 2.0 × 10−3 M (····). The bandwidth was 1.0 nm, and the scan speed was 200 nm/min. Ten spectra were averaged to improve signal-to-noise ratio.
A control experiment focused on a DNA duplex of the same size that included a mutant OEC sequence (2). Because of a change of 2 bp, the nature of the spectra of the OEC DNA is slightly different from that of O+ DNA. Fig. 4B shows the CD spectra of the mutant operator, the mutant operator in the presence of repressor, and the mutant operator in the presence of repressor and d-galactose. The presence of GalR led to only a very small change in the CD spectrum. The addition of d-galactose did not cause any detectable CD change, indicating that the change in the CD spectrum of OE is a consequence of specific binding of GalR. Furthermore, binding of d-galactose to this complex preserved the nature of the complex.
OE-Induced Conformational Change in GalR.
Similar to the reduction in the affinity of inducer for the operator–repressor complex compared with that for free repressor, the affinity of the operator for the inducer–repressor complex is lower than for free repressor. We tested the possibility that DNA binding induces an allosteric change in repressor conformation by measuring the tryptophan fluorescence of GalR at 2.0 × 10−6 M concentration in the presence of increasing concentrations of OE DNA. As shown in Fig. 5, binding of OE reduced the fluorescence by more than 11%.
Figure 5.

Effect of OE DNA binding on the tryptophan fluorescence (f/fo) of GalR. GalR at a concentration of 2.0 × 10−6 M was titrated with increasing concentrations of the operator (20-mer). The total emission spectra were integrated.
Measurements were taken of the distance of the tryptophan residue in GalR from the AEDANS placed at one end of the operator DNA. AEDANS has a large overlap in its excitation spectra with the tryptophan emission spectra. Fig. 6 shows the excitation spectra of the AEDANS-labeled OE and of a stoichiometric complex of AEDANS-OE and GalR. At 295 nm, the fluorescence of the complex was slightly higher than that of the free repressor, suggesting a small, but significant, energy transfer. The calculated energy transfer efficiency was 0.09, which corresponds to a distance of 38.6 Å between the tryptophan and the AEDANS label. Mutational and modeling studies also have placed the tryptophan residue in the C domain and the DNA-binding motif in the N domain of GalR (19, 20). Because the DNA-binding site and the tryptophan residue in GalR are far from each other and in different domains, quenching of tryptophan fluorescence in GalR by operator binding strongly suggested that a DNA-induced conformational change is transmitted from the N domain to the C domain in GalR. A similar change has been documented in λ repressor (21).
Figure 6.

Fluorescence energy transfer between AEDANS-OE and tryptophan of the GalR. The excitation spectra of 20-mer AEDANS-labeled OE (0.5 × 10−6 M) (– – – –), and that of the same DNA complexed with GalR (1.0 × 10−6 M) (——-) are shown.
In Vitro Transcription.
Because the fluorescence anisotropy and CD results indicated that GalR was bound to the operator in the presence of the inducer, we studied derepression of transcription of the gal P1 promoter in vitro as a function of d-galactose concentration under conditions used for ligand binding studies, except the KCl concentration was 200 mM. The slightly lower salt concentration was chosen to avoid a possible decrease in transcription. The lower salt concentration is expected to further tighten the binding of GalR to the operator. The results are shown in Fig. 7. In the absence of inducer, GalR at 1.6 × 10−7 M transcription initiation from the P1 promoter repressed by about 70% (Fig. 7A, lane 1 vs. 2). In the presence of 2 × 10−3 M d-galactose, transcription was derepressed 78%, whereas in the presence of a saturating concentration of the inducer (3 × 10−2 M) the P1 promoter was derepressed to 99% (lanes 2 and 3). When transcription was studied at 8 × 10−7 M GalR, the presence of 3 × 10−2 M d-galactose derepressed transcription by 38% (Fig. 7B). This result is significant considering the fact that at this high GalR concentration, the repressor also inhibited transcription from nonspecific promoters, such inhibition not being lifted by d-galactose. Given the determined values of the KDs, we calculated the fractional occupancy, Y, of operator by repressor from the equation
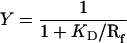 |
1 |
at 1.6 × 10−7 M and 8.0 × 10−7 M GalR. At 2 × 10−9 M DNA and such high repressor concentrations, the free repressor concentration, Rf, is practically the same as the total repressor concentration. At 1.6 × 10−7 M and 8.0 × 10−7 M GalR and saturating inducer concentration, transcription is 99% and 38%, respectively, of the unrepressed levels and the calculated occupancy of operator by repressor is 86% and 94%, respectively.
DISCUSSION
In negative control, binding of repressor to a cognate operator inhibits RNA polymerase action at the promoter, and an allosteric modification of repressor by a small-molecule inducer inhibits repressor action. Likely mechanisms by which the inducer can inhibit repressor action include:
(i) Inducer binds only to free repressor, allosterically inactivating its operator binding site. In this model, induction follows the rate of spontaneous dissociation of operator from repressor.
(ii) Inducer forms an inducer–repressor–operator ternary complex which, because of allosteric change, dissociates into inducer-repressor complex and free operator more readily than into repressor–operator complex and free inducer. In this case, induction follows the rate of dissociation of repressor–inducer complex from free operator.
(iii) Inducer forms an inducer–repressor–operator ternary complex which, because of allosteric change, allows RNA polymerase to act without repressor being physically dissociated from operator.
GalR binds to operator OE and inhibits transcription from the P1 promoter of the gal operon (6, 22). We showed that d-galactose and the OE operator antagonize the binding of each other to GalR. Consistently, both d-galactose and OE DNA binding to GalR exhibit allosteric changes in the protein (ref. 13; results reported in this paper). Despite such an antagonistic effect, inducer binding to the operator-bound repressor was found to be only 10-fold less than the extent of binding to free repressor under conditions of repression and induction of gal transcription. Similarly, the affinity of OE to the inducer–repressor complex is reduced by 7-fold compared with the binding of OE to the free repressor. The results of GalR binding to fluorescein-labeled DNA in the presence of d-galactose also showed the existence of stoichiometric amounts of inducer–repressor–operator ternary complex. The existence of stable ternary complex under physiological conditions also has been confirmed by comparing CD spectra of the free operator, operator bound to repressor, and operator bound to repressor in the presence of inducer. Comparable reduction in the affinity of inducer for repressor when complexed with operator was found in the lac system (23, 24).
In vitro transcription results presented here clearly demonstrated that full derepression of gal P1 occurs under conditions of binding in which (i) the repressor is still bound to the DNA, i.e., the concentrations of inducer–repressor binary complex are severalfold higher than the dissociation constant of the inducer–repressor–operator ternary complex into the inducer–repressor complex and free DNA; (ii) the RNA polymerase concentration was significantly less than the dissociation constant of the RNA polymerase-promoter complex (KB−1) (25, 26), and (iii) nonspecific DNA concentration was negligible. These results strongly suggest that previous dissociation of repressor is not obligatory for full derepression of transcription from the P1 promoter of the gal operon. Whether gal derepression occurs by a similar mechanism in vivo remains to be investigated.
It is unclear how repressor inhibits transcription. Operator-bound repressor either occludes RNA polymerase binding to the promoter or inhibits RNA polymerase activity at one or more of the postbinding steps (27). We previously demonstrated that GalR and RNA polymerase coexist on the DNA, and GalR inhibits the P1 promoter by freezing the RNA polymerase at an intermediate step between closed and open complex formation (22). GalR does so by modulating the activity of the α subunit of the DNA-bound RNA polymerase. As discussed above, dissociation of repressor from the operator is not obligatory for transcription from the P1 promoter. We propose that because a direct contact between DNA-bound GalR and RNA polymerase brings about repression of the P1 promoter, then binding of inducer to the operator–bound repressor neutralizes the inhibitory contact without dissociating the repressor from DNA. A role of repressor at a post-RNA polymerase binding level also has been suggested in the E. coli lac promoter and Bacillus subtilis phage φ 29 A2c promoter (28–30). As in the gal P1 promoter, MerR repressor and RNA polymerase coexist at the repressed promoter in the mer operon (31, 32). Binding of inducer Hg2+ to MerR derepresses transcription without Hg2+–MerR being dissociated from the promoter. But unlike the repressor gal P1 complex, the protein-DNA linkage is obligatory for mer transcription, because the Hg2+–MerR complex is an activator of the mer promoter.
Acknowledgments
We thank D. Jin and G. Gussin for many critical suggestions and D. Jin also for the gift of plasmid DNA and acknowledge the Council of Scientific and Industrial Research for a fellowship to S.C.
ABBREVIATIONS
- IAEDANS
5-({[(2-iodoacetyl)amino]ethyl}amino)- naphthalene-1-sulfonic acid
- AEDANS
5-({[(acetyl)amino]ethyl}- amino)naphthalene-1-sulfonic acid
- ATP[γ-S]
adenosine 5′-[γ-thio]triphosphate
Note added in proof
Since this manuscript was submitted, we noticed that the BetI repressor of E. coli binds to each operator both in the absence and presence of inducer (Røkenes, T. P., Lamark, T., Strøm, A. R. (1996) J. Bacteriol. 178, 1663–1670).
References
- 1.Jacob F, Monod J. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- 2.Adhya S, Miller W. Nature (London) 1979;279:492–494. doi: 10.1038/279492a0. [DOI] [PubMed] [Google Scholar]
- 3.Irani M, Orosz L, Adhya S. Cell. 1983;32:783–788. doi: 10.1016/0092-8674(83)90064-8. [DOI] [PubMed] [Google Scholar]
- 4.Fritz H-J, Bicknase H, Gleumes B, Heibach C, Roshl S, Ehring R. EMBO J. 1983;2:2129–2135. doi: 10.1002/j.1460-2075.1983.tb01713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Majumdar A, Adhya S. Proc Natl Acad Sci USA. 1984;81:6100–6104. doi: 10.1073/pnas.81.19.6100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choy H E, Adhya S. Proc Natl Acad Sci USA. 1992;89:11264–11268. doi: 10.1073/pnas.89.23.11264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Buttin G. J Mol Biol. 1963;7:164–182. doi: 10.1016/s0022-2836(63)80044-3. [DOI] [PubMed] [Google Scholar]
- 8.Aki T, Choy H E, Adhya S. Genes Cells. 1996;1:179–188. doi: 10.1046/j.1365-2443.1996.d01-236.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Y N, Chatterjee S, Roy S, Adhya S. J Mol Biol. 1995;253:414–425. doi: 10.1006/jmbi.1995.0563. [DOI] [PubMed] [Google Scholar]
- 10.Wartell R, Adhya S. Nucleic Acids Res. 1988;16:11531–11541. doi: 10.1093/nar/16.24.11531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantor C, Schimmel P. Biophysical Chemistry. Vol. 2. San Francisco: Freeman; 1981. [Google Scholar]
- 12.Wu C W, Stryer L. Proc Natl Acad Sci USA. 1972;69:1104–1108. doi: 10.1073/pnas.69.5.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown M, Shaikh N, Brenowitz M, Brand L. J Biol Chem. 1994;269:12600–12605. [PubMed] [Google Scholar]
- 14.Nakanishi S, Adhya S, Gottesman S, Pastan I. Proc Natl Acad Sci USA. 1973;70:334–336. doi: 10.1073/pnas.70.2.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Letilly V, Royer C A. Biochemistry. 1993;33:7753–7758. doi: 10.1021/bi00081a021. [DOI] [PubMed] [Google Scholar]
- 16.Brenowitz M, Jamison E, Majumdar A, Adhya S. Biochemistry. 1990;29:3374–3383. doi: 10.1021/bi00465a033. [DOI] [PubMed] [Google Scholar]
- 17.Ha J H, Capp M W, Hohenwalter M D, Baskerville M, Record M T., Jr J Mol Biol. 1992;228:252–264. doi: 10.1016/0022-2836(92)90504-d. [DOI] [PubMed] [Google Scholar]
- 18.Zwieb C, Kim J, Adhya S. Genes Dev. 1989;3:606–611. doi: 10.1101/gad.3.5.606. [DOI] [PubMed] [Google Scholar]
- 19.Lehming N, Sartorius J, Kister-Woike B, von Wilcken-Bergmann B, Muller-Hill B. EMBO J. 1990;9:615–621. doi: 10.1002/j.1460-2075.1990.tb08153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hsieh M, Hensley P, Brenowitz M, Fetro J S. J Biol Chem. 1994;269:13825–13835. [PubMed] [Google Scholar]
- 21.Saha R, Banik U, Bandopadhyay S, Mandal N C, Bhattacharyya B, Roy S. J Biol Chem. 1992;267:5862–5867. [PubMed] [Google Scholar]
- 22.Choy H E, Park S-W, Parrack P, Fujita N, Ishihama A, Adhya S. EMBO J. 1995;14:4523–4529. doi: 10.1002/j.1460-2075.1995.tb00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen J, Alberti S, Matthews K S. J Biol Chem. 1994;269:12482–12487. [PubMed] [Google Scholar]
- 24.Daly T J, Matthews K S. Biochemistry. 1986;25:5479–5484. doi: 10.1021/bi00367a020. [DOI] [PubMed] [Google Scholar]
- 25.Goodrich J A, McClure W R. J Mol Biol. 1992;224:15–29. doi: 10.1016/0022-2836(92)90573-3. [DOI] [PubMed] [Google Scholar]
- 26.Herbert M, Kolb A, Buc H. Proc Natl Acad Sci USA. 1986;83:2807–2811. doi: 10.1073/pnas.83.9.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adhya S. Annu Rev Genet. 1989;23:227–250. doi: 10.1146/annurev.ge.23.120189.001303. [DOI] [PubMed] [Google Scholar]
- 28.Straney S B, Crothers D M. Cell. 1987;51:699–707. doi: 10.1016/0092-8674(87)90093-6. [DOI] [PubMed] [Google Scholar]
- 29.Lee J, Goldfarb A. Cell. 1991;66:793–798. doi: 10.1016/0092-8674(91)90122-f. [DOI] [PubMed] [Google Scholar]
- 30.Monsalve M, Menica M, Salas M, Rojo F. Proc Natl Acad Sci USA. 1996;93:8913–8918. doi: 10.1073/pnas.93.17.8913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Summers A O. J Bacteriol. 1992;174:3097–3101. doi: 10.1128/jb.174.10.3097-3101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ansari A Z, Bradner J E, O’Halloran T V. Nature (London) 1995;374:371–375. doi: 10.1038/374370a0. [DOI] [PubMed] [Google Scholar]