

Abstract
We report the 1.8 Å structure of yeast poly(A) polymerase (PAP) trapped in complex with ATP and a five residue poly(A) by mutation of the catalytically-required aspartic acid 154 to alanine. The enzyme has undergone significant domain movement and reveals a closed conformation with extensive interactions between the substrates and all three polymerase domains. Both substrates and 31 buried water molecules are enclosed within a central cavity that is open at both ends. Four PAP mutants were subjected to detailed kinetic analysis, and studies of the adenylyltransfer (forward), pyrophosphorolysis (reverse), and nucleotidyltransfer reaction utilizing CTP for the mutants are presented. The results support a model in which binding of both poly(A) and the correct nucleotide, MgATP, induces a conformational change, resulting in formation of a stable, closed enzyme state. Thermodynamic considerations of the data are discussed as they pertain to domain closure, substrate specificity, and catalytic strategies utilized by PAP.
Keywords: polyadenylate polymerase, substrate recognition, mRNA processing, nucleotidyltransferase, induced fit mechanism
Introduction
Polyadenylation is an essential step in mRNA maturation, and the poly(A) tails at the 3′ end of mRNA facilitate mRNA transport from the nucleus, enhance translational efficiency, and increase mRNA longevity in the cytoplasm (Edmonds, 2002; Gilmartin, 2005; Zhao et al., 1999). In eukaryotes, the apparatus responsible for polyadenylation is physically coupled to that of mRNA cleavage, and in yeast these processes involve a complex of over a dozen individual subunits (Zhao et al., 1999). These subunits are recruited to the phosphorylated C-terminal tail of RNA polymerase II during transcription, and they recognize polyadenylation signal sequences and enhancer elements on the pre-mRNA transcripts. In mammals the pre-mRNA is likely to be cleaved by CPSF-73 (Mandel et al., 2006) (Ysh1 in yeast), and in all eukaryotes it is then polyadenylated by PAP, the poly(A) polymerase.
PAP is a template-independent polymerase that belongs to the DNA polymerase beta (polβ) family of enzymes. Though it functions as part of the cleavage/polyadenylation complex, the isolated enzyme retains catalytic activity and nucleotide specificity (Edmonds, 1990). Crystal structures of yeast and bovine PAP have shown that PAP is composed of three globular domains which surround a central, substrate-binding cleft (Bard et al., 2000; Martin et al., 2000). The N-terminal domain (residues 40–190) bears homology to the “palm domains” of pol β nucleotidyltransferases (Aravind and Koonin, 1999). The middle domain (residues 1–39 and 190–353) is functionally, though not structurally, analogous to the fingers domains of template-directed polymerases. The C-terminal domain (residues 354–530) is responsible for binding the yeast cleavage/polyadenylation subunit Fip1 (Helmling et al., 2001; Preker et al., 1995). The three PAP domains are connected by a set of hinges which allow them to move as essentially rigid bodies with respect to one another. Domain motion is certainly a component of the induced fit mechanism exhibited by PAP (Balbo et al., 2005). However, the structural basis of the effect of domain movement on catalysis and on substrate binding has not been understood in the absence of a crystal structure of a PAP-MgATP-RNA ternary complex.
Here we present the crystal structure of a single-site mutant (D154A) of yeast poly(A) polymerase in complex with MgATP and a 5-mer oligoadenylate RNA molecule. The structure is in a closed state, and the enzyme makes extensive interactions with four RNA nucleotides and the MgATP which are bound within the central cleft. Previous crystallographic studies (Balbo et al., 2007; Bard et al., 2000; Martin et al., 2000) have shown that the domains can assume various intermediate states of closure, and fluorescence quenching studies (Balbo et al., 2007) have indicated that maximal solvent protection of the active site requires the presence of both ATP and poly(A) substrates, suggesting the presence of these substrates induces the domains to close. In addition to detailing the structural determinants for substrate binding, we present a complete steady state characterization of four site directed mutants. The results support a model in which binding of both substrates (poly(A) and MgATP) induces the conformational change, resulting in stabilization of the closed enzyme state and enabling catalysis.
Results and Discussion
Structure of the PAP ternary complex
PAP(D154A) was crystallized with MgATP and a 5-mer poly(A) oligonucleotide. The mutation of Asp154, a catalytically essential active site residue, renders the enzyme inactive and was introduced to trap the closed, substrate-bound complex. This mutation resulted in only one of the catalytically-required metals being bound at the active site, but the mutation does not appear to significantly perturb the expected positions of the bound substrates. To our knowledge, this is the first crystal structure of a polymerase wherein one of the three catalytically-required aspartic acids has been mutated. There are, however, many instances where structures have been determined with only metal B at the active site, and in many of these the missing metal appears to cause a relatively small change in the nucleotide conformation. Our structure suggests that the strategy of mutating one of the catalytic aspartic acids may be generally useful, particularly in cases where, as in PAP, using unnatural substrates to trap the enzyme-substrate complex proves problematic. The structure was refined to 1.8 Å resolution (Table I), and clear electron density was observed for both MgATP and RNA (Supplemental Figure 1). In the ternary complex, the structure of individual domains are similar to those seen earlier, but these domains have undergone significant movement about both of the previously defined hinge regions; one between the N- and middle domains (residues around 40 and around 190) and the other between the middle and C-terminal domains (residues around 353) (Balbo et al., 2007). As shown in Figure 1A, when compared to the most open structure of yeast PAP (molecule A of PDB code 1FA0) the N-terminal domain is rotated 23.4° towards the middle domain. The trajectory of this rotation is similar to that seen earlier, but this structure is 14.7° more closed than the most closed state reported previously (PDB code 2HHP). The C-terminal domain is in an intermediately closed state, near the average position of this domain observed in the various crystal structures. The translational components associated with movements about both hinges are very small.
Table I.
Crystallographic statistics
| Space group | P212121 |
| Wavelength | 1.0809 Å |
| Cell parameters (Å) | 67.633, 85.907, 107.469 |
| Resolution | 1.8 Å |
| Data source | NSLS beamline X29A |
| Rsym (last shell) | 7.2% (27.5%) |
| Completeness (last shell) | 95.7% (76.0%) |
| I/σI (last shell) | 52.0 (4.6) |
| Redundancy (last shell) | 12.0 (7.5) |
| R-factor (all data) | 19.08% |
| R-free (5.1%) | 22.54% |
| Mean B-factors (Å2) | |
| Protein | 27.79 |
| ATP | 22.38 |
| RNA | 35.34 |
| solvent | 38.29 |
| RMSD from ideal geometry | |
| Bonds | 0.010 Å |
| Angles | 1.24° |
| Ramachandran outliers | 1 (0.2%) |
Figure 1.
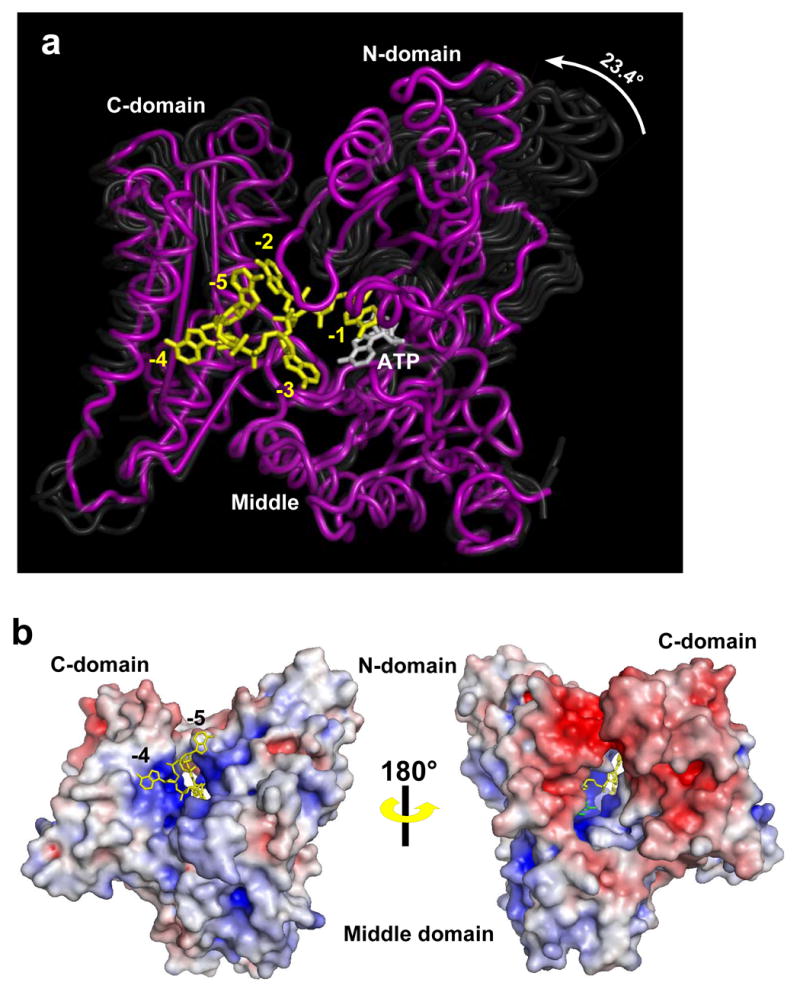
Structure of PAP in complex with ATP and RNA. (A) The structure of the closed, ternary complex of PAP is shown in purple, and ATP and RNA are shown in white and yellow, respectively. The middle domains of the earlier PAP structures have all been superimposed with that of the ternary complex, and the earlier structures are shown as transparent ghosts to highlight the domain movements. (B). Two views of the surface representations of the PAP/ATP/RNA complex are shown. The color reflects the surface electrostatics where the color gradient from red to white to blue corresponds to −8KT to 0 KT to +8KT. The top view is rotated ~45 degrees relative to that shown in 1A. The -5 and -4, RNA nucleotides (yellow) are seen exiting a cleft formed by the three domains. The bottom view has been rotated ~180 about the vertical axis relative to the top, so that the ATP-binding side of the active site cleft is visible. Part of the ATP base (green) and the -2 nucleotide (yellow) are seen. The nucleotide at the 3′ end is hidden within the active site cavity.
In earlier PAP crystal structures, a large cleft (~30 Å long and ~35 Å deep) was observed between the N- and C-terminal domains (Balbo et al., 2007; Bard et al., 2000; Martin and Keller, 2004; Martin et al., 2000). In these structures, the active site is located at the bottom of this cleft, near the interface of the N- and middle domains. In the closed state, the N- and C-terminal domains interact, closing off the top of the cleft, but leaving openings at both ends near the bottom (Figure 1B). The closed structure reveals extensive contacts involving both substrates and residues within the central cavity. In fact, the substrates themselves largely mediate contact across the domains in the closed state. The enzyme-substrate interface buries ~2470 Å2 (1380 Å2 on the RNA and 1090 Å2 on the protein); interactions between the N- and C- terminal domains bury an additional ~360 Å2 of accessible surface area. The latter interface has good shape complementarity and includes a salt bridge between Arg125 in the N-terminal domain and Glu373 in the C-terminal domain, located near the top of the cleft.
The PAP-bound RNA substrate adopts an extended conformation, and there are no contacts between the RNA bases. In this respect, the PAP-RNA complex is quite different from the nucleic acid-containing complexes of two other template-independent polymerases, CCA adding enzyme and terminal deoxynucleotidyltransferase. All three enzymes belong to the polymerase beta family, but contain no homology outside of the catalytic palm domain. In both other structures, the active site cleft into which the nucleic acid substrate binds is wider to accommodate the single-stranded oligonucleotide chains, which are much more solvent exposed and generally form base-stacking interactions (Delarue et al., 2002; Tomita et al., 2004). This is in stark contrast to the extensive protein-RNA interactions observed in PAP ternary complex. The trajectory of the RNA leaving the active site also differs when PAP is compared to these other polymerases. PAP is also distinct from other ssRNA-binding proteins. Though the C-terminal domain of PAP contains an RNA recognition motif (RRM), the way in which this domain interacts with the RNA is completely dissimilar to that seen in other RRM proteins. RNA is generally bound to a particular face of RRM domains (Conte et al., 2000), but in PAP, this RNA-binding surface is largely occluded by C-terminal domain residues 419 through 439. Furthermore, while ssRNA does interact with the C-terminal domain, it does so through residues which are on the opposite side of the RRM’s central beta sheet relative to the RNA-binding parts of the other structures.
Other than those of bovine PAP, the structures most similar to yeast PAP are those of the trypanosomal terminal uridylyltransferases, TbRET2 and TbTUT4 (Deng et al., 2005; Stagno et al., 2007). These proteins add a uradine residue to the 3′ end of a single-stranded RNA primer in a template-independent reaction, and they are homologous to PAP throughout the PAP’s N-terminal and middle domains. TUTases lack PAP’s C-terminal domain, but, like PAP, they are capable of significant domain closure. There is no crystal structure of a TUTase with the 3′ end position occupied. As discussed below, superimposition of PAP with TUTases reveals a very similar incoming nucleotide orientation, and as there is space in the TUTase structures for a nucleotide similar to that at position -1 in PAP, it would appear that the PAP structure is a good model for the TUTase ternary complex (Supplemental Figure 2).
There are 24 protein-RNA interactions that are mediated by an extensive network of highly ordered water molecules, most of which are buried (as defined in Methods) within the interior of the complex (Figures 2 and 3). Overall, 58 water molecules are completely buried, and 31 of these are within the central, substrate-binding cavity. The degree to which the individual water molecules are buried is indicated in Figure 2, and all of the solvent molecules are shown in Supplementary Figure 3. These ordered waters are mostly hydrogen bonded to the polar atoms of the adenine bases as well as to the phosphodiester backbone. Consequently, the poly(A) substrate remains largely solvated in the enzyme ternary complex. Recognition of the 2′ and 3′ hydroxyl groups of the ribose of ATP is also mediated by bridging waters, which seems to be a strategy employed in many structurally unrelated ribonucleotide binding proteins (Babor et al., 2002). As discussed below, some of the waters, particularly those that interact with the base of ATP, could help ensure substrate specificity.
Figure 2.
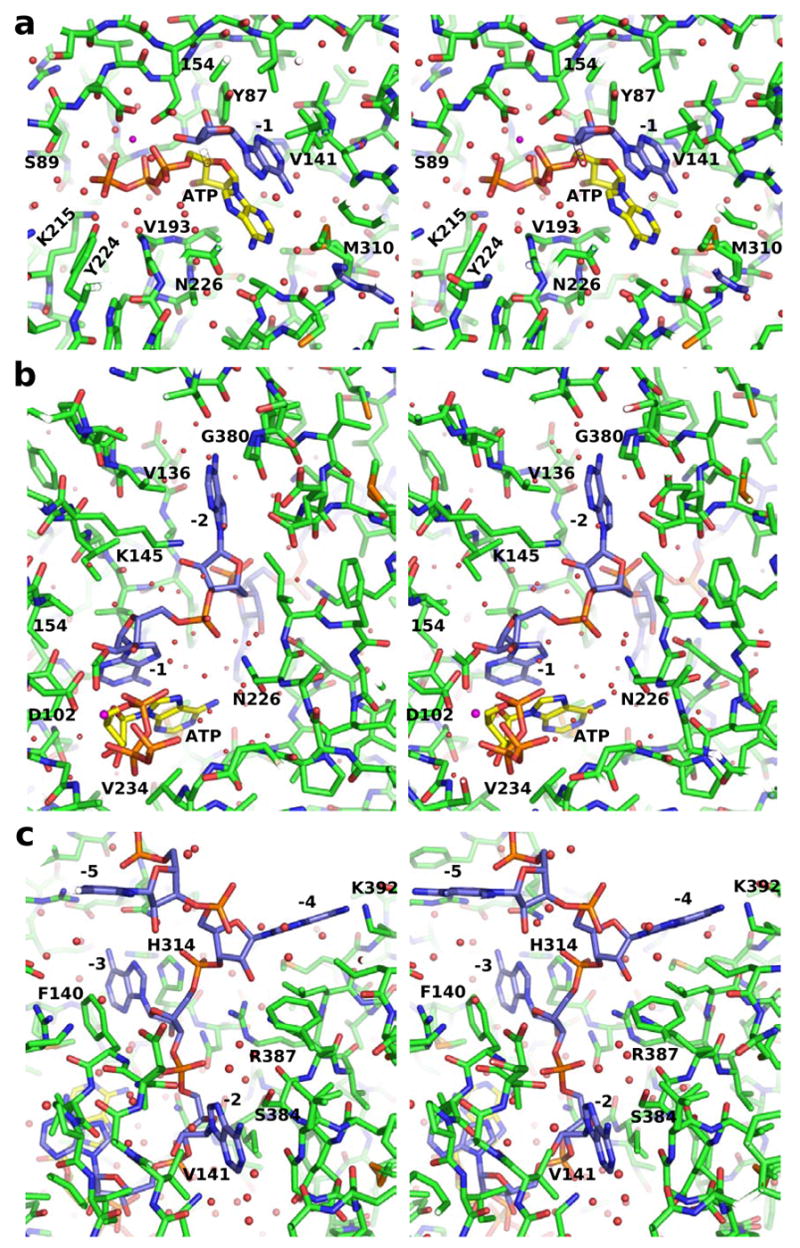
Determinants of ATP and RNA binding. Interacting residues from the N-terminal, middle, and C-terminal domains are colored yellow, green, and cyan, respectively. Residues within cloud-shaped bubbles on either side of the bases indicate hydrophobic/van der Waals interactions. Water molecules (round circles) are color-coded based on the degree to which they are buried within the interior of the protein; those colored dark blue are completely buried (see Methods). Those colored grey-blue are at the protein surface. Others are colored intermediate shades of blue depending on how many shells of water needed to be removed in order for the water atoms to become exposed. Asp154 and the second Mg2+ ion are shown in grey. Eight additional water molecules interact with the triphosphate moiety of the ATP. These are not shown for clarity, and because the water structure in this region may be altered due to the D154 A mutation. The base at position -5 interacts with a neighboring PAP molecule and is not shown.
Figure 3.

Stereo views of the substrate binding sites of PAP. (A–C) Detailed substrate interactions formed in closed, ternary complex. ATP (yellow carbons) and the 3′ end (blue carbons) are shown along with PAP with the surrounding amino acids (green carbons) and water molecules (red spheres).
Substrate binding determinants
In the sections below, the individual bases will be referred to by their position on the chain, with -1 being the 3′ end of the RNA and -5 denoting the 5′ end. The nucleotide at position -5 leaves the protein and interacts with F511 and K478 in a neighboring molecule. We do not believe this interaction is physiologically important. Poly(A) residue -4 is located at the surface of the enzyme, partially exposed to the solvent. The adenine bases at positions -1, -2, and -3 are completely buried within the closed structure, and PAP interacts with the 2′-OH groups of each of these nucleotides. These observations are consistent with earlier studies indicating that DNA primers are poor substrates for the enzyme (Edmonds, 1990) and that efficient polyadenylation requires a primer of at least three residues (Zhelkovsky et al., 1998).
PAP does not exhibit great specificity with respect to the sequence of its RNA substrate (Butler and Platt, 1988; Butler et al., 1990). This is consistent with the requirement for initiating polyadenylation at non-ploy(A) sequences that can occur in freshly cleaved pre-mRNA substrates (Zhao et al., 1999). There are, however, three base-specific interactions with the RNA observed in the crystal structure. Two of these are between the protein and the RNA base at position -4, where the side chains of Lys 392 and Glu487 make hydrogen bonds with the N1 and N6 atoms of the base, respectively. The -4 nucleotide lies on the surface of the C-terminal domain and exhibits relatively high B-factors, suggesting that it may not be as well anchored to the protein as the bases at positions -1, -2 and -3. In addition to the two interactions mentioned above, the hydrophobic face of the -4 base sits atop C-terminal domain residues Leu388 and Leu 491, and the 2′ hydroxyl makes a hydrogen bond with the side chain of His314. As the base-specific interactions we observe at position -4 are at the protein surface, there is likely little energetic penalty associated with their loss when other nucleotides occupy this position because hydrogen bonds to the base can easily be replaced by those to water.
The nucleotide at position -3 interacts with all three domains of the polymerase. The side chain of Gln294 makes a hydrogen bond with the N6 position of the adenine ring, potentially providing base specificity at this position; however, this residue is not conserved. The -3 base is sandwiched between highly conserved residue Phe140, from the N-terminal domain, and His314, from the middle domain. The side chains of both of these residues were exposed to solvent in earlier PAP structures and appeared highly flexible as indicated by their B-factors and in some cases the absence of side chain electron density. Conserved residue Arg387 from the C-terminal domain also interacts with the -3 phosphate moiety, as does His314. The latter residue is replaced with tyrosine in higher eukaryotes, thus presumably retaining both its base stacking and hydrogen bonding functions. Finally, the 2′ hydroxyl makes a hydrogen bond with the side chain of Asn315. As is typical for the remaining nucleotides in this structure, this nucleoside makes six water-mediated interactions. Many of these interactions, as well as the protein atoms to which these water molecules are coordinated, are detailed in Figures 2 and 3.
The base of the -2 nucleotide is sandwiched between the N- and C-terminal domains. One side of its hydrophobic surface interacts with Val136; the other contacts Gly380 and Ser384 from helix N. The 2′ hydroxyl of this nucleotide makes a hydrogen bond with Lys145, and an additional hydrogen bond is made between the phosphate moiety and the main chain amide of Phe140. All of these residues are very highly conserved, and again there are well ordered, buried water molecules which make additional interactions with the nucleotide.
The base of the 3′-terminal residue (-1 position) of the poly(A) substrate is sandwiched between the side chain of Val141 and the base of the incoming ATP. In addition, hydrogen bonds are formed between the 2′ hydroxyl and the side chain hydroxyl of Tyr87 and between the backbone phosphate oxygen and the δN of Asn226. In the two-metal ion dependent nucleotidyltransferases, the 3′-terminal residue is also bound through coordination of the 3′ hydroxyl group to a second, so-called catalytic Mg2+ ion (Batra et al., 2006; Sawaya et al., 1994; Sawaya et al., 1997). The mutation of Asp154 abrogates binding of this metal, and this interaction is not observed in the present structure. Nonetheless, the positions of the MgATP and the poly(A) terminus observed in the structure generally conform to this mechanism. The position of the -1 nucleotide coincides well with the nucleotide (3′-dAMP) bound at the poly(A) subsite in the earlier structure of yeast PAP (Bard et al., 2000) (Figure 4A) and with the nucleotides observed in structures of DNA pol β (Sawaya et al., 1997) (Figure 4B). Furthermore, despite the absence of the second Mg2+, the 3′ hydroxyl is located 3.2 Å from the α-phosphorus atom of ATP, as if poised for nucleophilic attack. This all supports the overall correctness of the nucleotide conformation in this structure.
Figure 4.
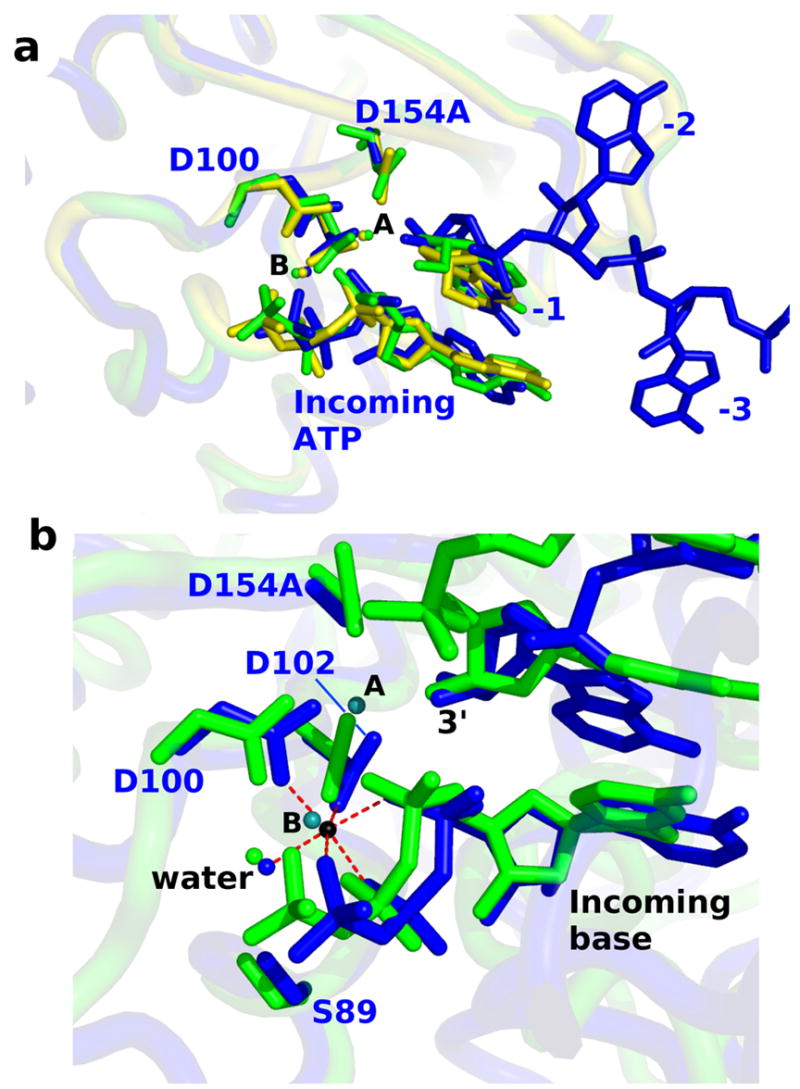
Comparison of the active site with other structures. (A) The alpha carbons of the N-terminal domain of the RNA-bound PAP molecule were superimposed with those of the two crystallographically-independent molecules from PDB code 1FA0. In the earlier structure each of the two molecules contained two molecules of 3′-dATP at the active site. The structure presented in this paper is shown in blue. The earlier structures are shown in green (molecule A) and yellow (molecule B). The middle and C-terminal domains are not shown because they occlude the nucleotides. (B) Superposition of the PAP active site with that of DNA polymerase beta (PDB code 2FMS) demonstrates very similar structure and geometry with respect to the ligand environment about metal B. PAP is shown in blue with Mg2+ ion in black. Polymerase beta is shown in green with its two metal ions in teal. As in polymerase beta, a water molecule (also present in the earlier yeast PAP structures) completes the octahedral geometry of metal ion B. Distances between the Mg2+ in the ternary PAP complex and its six ligands range from 2.02 – 2.16 Angstroms.
The triphosphate moiety of ATP exhibits tridentate coordination to metal B, and this Mg2+ exhibits octahedral geometry involving coordination to the side chain carboxylates of Asp100 and Asp102 and an ordered water, in addition to the triphosphate (Figure 4). This structural arrangement is an essential characteristic of this class of enzymes (Sawaya et al., 1997). Additionally, the γ-phosphate of ATP makes direct contact with the side chains of Ser89, Lys215, and Tyr224. The latter two interactions are formed only in the closed enzyme state. A backbone amide from Ser89 forms an additional hydrogen bond to the non-bridging oxygen of the β-phosphate. The non-bridging α-phosphate oxygen is exposed to solvent molecules and does not interact with active site residues, a result consistent with previous studies (Balbo et al., 2007). The coordination environment about this oxygen is important because a partial negative charge develops here in the transition state (Burgers and Eckstein, 1979; Steitz and Steitz, 1993). Exposure of this atom to solvent and the absence of an appropriately positioned acidic residue is evidence that water-derived protons function in charge stabilization during catalysis.
Structural basis for nucleotide base recognition
A central question regarding the mechanism of PAP concerns the structural determinants for ATP specificity. The present structure confirms that domain closure results in the formation of new interactions, both direct and water-mediated, between the enzyme and the nucleotide substrate. It also suggests that there are multiple factors determining nucleotide base recognition. Overall, the nucleotide binding site exhibits good surface complementarity to the MgATP, but there are no direct hydrogen bonds observed between the enzyme and the adenine base. The surface of the binding site of the adenine-ribose moiety in PAP is defined primarily by two features: (1) non-polar interactions above and below the plane of the adenine base and to the sugar, and (2) water mediated interactions to polar groups in the plane of the base and to the sugar hydroxyl groups. These results seem to conform to general principles utilized in many, structurally unrelated adenylate-binding proteins (Moodie et al., 1996). There is also suggestive evidence that N226 may participate in adenine recognition via an interaction with the N6 group.
As shown in Figure 3, a base stacking interaction occurs between the ATP and the 3′-terminus of the poly(A) substrate. The side chain of Val234 makes van der Waals contact with the ribose-base moiety of ATP, on the opposite face as the base-stacking interaction. Nucleotide bases form stacking interactions both in solution and in nucleic acids (Broom et al., 1967; Ts’o et al., 1969; Ts’o et al., 1962), with the order of stacking preference: purine-purine > purine-pyrimidine > pyrimidine-pyrimidine. The theoretical free energy change realized upon purine-purine base stacking is 2–6 kcal/mol, and A-A stacking is more favorable than A–C stacking by 0.5 – 2 kcal/mol (Friedman and Honig, 1995; Norberg and Nilsson, 1995). As discussed below, these energetic values are consistent with the lower catalytic efficiency exhibited by PAP for nucleotidyltransfer reactions utilizing the pyrimidine substrate, CTP (Balbo et al., 2005). In vivo, the terminal nucleotidyl residue formed by cleavage at the poly(A) site is often, but not always, adenylate (Zhao et al., 1999). Previously, we studied the role of base stacking in substrate binding and the effect of alternative 3′-terminal bases by measuring the kinetics of the reaction: A17-C + MgATP ⇌ A17-CA + MgPPi (Balbo et al., 2005). The catalytic efficiency for this reaction was 10-fold lower than the reaction with A18 and MgATP, supporting the assertion that base stacking is significant.
Base stacking might provide a substantial amount of substrate binding free energy, but it does not explain specificity between ATP and GTP. PAP exhibits 800-fold lower catalytic efficiency when GTP is utilized as a substrate instead of ATP (this work), strongly suggesting that other factors are involved in base recognition. Discrimination between adenine and guanine is largely based on differential electrostatic properties and shape (Moodie et al., 1996; Nobeli et al., 2001). In the present structure, three water molecules, which are completely buried within the interior of the complex, coordinate the N3, N6, and N7 positions of the incoming nucleotides base. Substitution of the N6 of adenine with the O6 of guanine replaces a H-bond donor with a H-bond acceptor, and results in protonation of the N1 atom, altering its H-bonding properties as well. These perturbations would be expected to have significant consequences on base recognition even if molecular recognition at any of these positions is mediated by a buried water. Also, the adenine near the C2 position is packed in a small pocket formed by the side chains of Thr304, Met310, and Ala312. Therefore, steric exclusion might contribute to negative selection against the 2-amino group of GTP.
Finally, the proximity of Asn226 to the adenine moiety of ATP has led to previous speculation of its involvement in base recognition (Balbo et al., 2007; Bard et al., 2000). In the present structure, the δO of Asn226 is only 3.57 Å from the N6 position of the ATP. A small change in the nucleotide or side chain conformation, perhaps by restoration of the Asp154 side chain and the second Mg2+, could align these groups and promote an interaction. Indeed, no other active site residue is close enough to the nucleotide to make a base-specific contact without a large structural rearrangement of the active site. To investigate the role of Ans226 in nucleotide specificity, a kinetic analysis of GTP utilization by PAP (w.t.) and the N226A mutant was performed (Table II). As is the case for CTP (Balbo et al., 2005), substrate selectivity against GTP is manifested in Vmax, rather than Km. Figure 5A reports the apparent ΔΔGcat contributed by each mutated residue as measured for a given reaction. The small discrepancy between the ΔΔGcat for the reactions utilizing ATP and GTP (−1.29 − (−0.74) = −0.54 kcal/mol) is consistent with a small degree of antagonism between N226 and the guanine base, as would be expected if this residue is involved in the negative selection of GTP. However, due to the limitations in interpreting kinetic studies of this nature, such speculation should be made cautiously. Presently, stronger evidence indicating an ATP-Asn226 interaction is unavailable.
Table II.
Steady State Kinetic Parameters for PAP and Mutants
| Adenylyltransfer: | An + ATP ⇌ A(n+1) + PPi | ||||
|---|---|---|---|---|---|
| PAP Δ10 w.t.1 |
N189A
|
N226A
|
K215A
|
Y224F
|
|
| V1 (min−1) | 846 (± 127) | 665 (± 111) | 402 (± 81) | 191 (± 54) | 904 (± 511) |
| Kia (μM) | 93.2 (± 28.9) | 70.2 (± 16.1) | 54.0 (± 7.6) | 123.2 (± 17.9) | 92.8 (± 15.8) |
| Ka (μM) | 46.8 (± 12.4) | 106.0 (± 26.3) | 367.2 (± 87.0) | 195.0 (± 75.8) | 711.6 (± 448.3) |
| Kib (μM) | 71.5 (± 22.2) | 53.0 (± 9.8) | 36.6 (± 4.0) | 256.8 (± 45.7) | 121.1 (± 20.5) |
| Kb (μM) | 35.9 (± 12.6) | 80.0 (± 21.4) | 249.0 (± 62.3) | 406. (± 145.9) | 928.8 (± 583.3) |
|
| |||||
| Pyrophosphorolysis: | An + PPi⇌A(n−1) + ATP | ||||
|
| |||||
| V2 (min−1) | 189 (± 296) | 54.6 (± 53.4) | 21.9 (± 26.1) | n.d.2 | 3.8 (± 1.5) |
| K′a (μM) | 2270 (± 3600) | 1170 (± 1213) | 1678 (± 2089) | n.d | 121 (± 54) |
| Kip3(μM) | 28 (± 46) | 43 (± 42) | 18 (± 21) | 465 (± 62) | 285 (± 28) |
| Kp (μM) | 671 (± 1100) | 721 (± 726) | 563.0 (± 689) | n.d | 375 (± 167) |
|
| |||||
| Keq4 | 85 | 97 | 42 | n.d. | 95 |
|
| |||||
| Cytidylyltransfer: | An + CTP ⇌ An-C + PPi | ||||
|
| |||||
| V1 (min−1) | 8.6 (± 1.9) | 8.9 (± 1.8) | 1.04 (± 0.17) | 0.51 (± 0.24) | 4.7 (± 20.9) |
| Kia (μM) | 71.0 (± 18.1) | 62.4 (± 12.9) | 64.9 (± 11.9) | 216.7 (± 58.0) | 72.2 (± 17.0) |
| Ka (μM) | 64.2(± 22.5) | 145.6 (± 41.9) | 131.8 (± 32.8) | 129.9 (± 105.1) | 3.5 mM (± 15.9) |
| Kib (μM) | 114.6 (± 29.2) | 63.6 (± 12.8) | 69.2 (± 12.4) | 614.7 (± 265.8) | 96.3 (± 22.5) |
| Kb (μM) | 103.6 (± 36.2) | 148.3 (± 40.1) | 140.6 (± 33.1) | 368.5 (± 234.6) | 4.7 mM (± 21.3) |
|
| |||||
| Guanylyltransfer: | An + GTP ⇌ An-G + PPi | ||||
|
| |||||
| V1 (min−1) | 2.1 (± 0.6) | 0.64 (± 0.17) | |||
| Kia (μM) | 101.7 (± 34.1) | 119.4 (± 39.7) | |||
| Ka (μM) | 259.2 (± 90.0) | 246.6 (± 82.5) | |||
| Kib (μM) | 24.2 (± 4.8) | 26.7 (± 4.9) | |||
| Kb (μM) | 61.7 (± 27.8) | 54.9 (± 24.7) | |||
PAPΔ10 w.t. reactions (except GTP utilization) from reference (Balbo et al., 2005)
not determined
Kip values for K215, Y224F were determined from product inhibition experiments, these values were treated as fixed parameters in the analysis of the pyrophosphorolysis data using Equation 1, where the term K′aKip, which is equivalent to KiaKp, was used.
calculated from the Haldane equation (Gold et al., 1970), Keq = V1Kp/V2Kb
Figure 5.
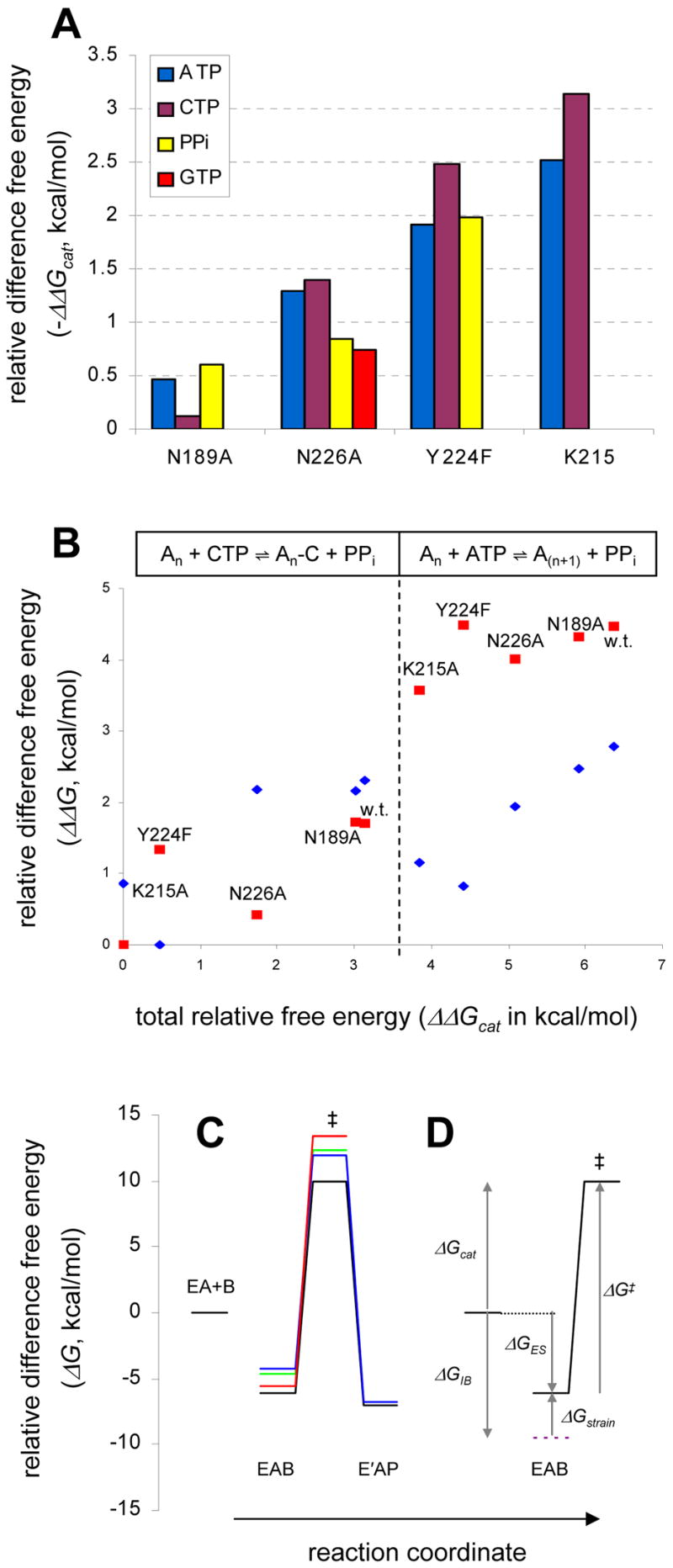
Kinetics and mutangenesis of PAP. (A) Total apparent free energy contributions to catalysis (Δ Gcat) from mutated residues. Values are derived from nucleotidyltransfer (utilizing ATP, CTP or GTP) or pyrophosphorolysis (PPi) reactions. (B) Relative difference free energy changes ΔΔG‡ and ΔΔGES, against the total difference free energy changes for catalysis of nucleotidyltransfer, ΔΔGcat. The ΔΔGcat is the total difference free energy change of a particular enzyme in either reaction utilizing ATP (right) or CTP (left) relative to the cytidylyltransfer reaction, An + MgCTP ⇌ An-C + MgPPi, catalyzed by K215A. The data points for ΔΔG‡ (red) and ΔΔGES (blue) are relative to the appropriate term for the cytidylyltransfer reaction of the K215A and Y224F mutant (the lowest value in each case), respectively. Large values on the y-axis indicate either a faster rate, V1 (ΔΔG‡), or greater free energy release upon formation of the Michaelis complex (ΔΔGES) relative to the reference reaction. (C and D) Thermodynamics of PAP substrate specificity and catalysis. The free energy change upon E+A+B ⇌ EAB is a function of the product of KiaKib (or KaKib). Here, the relative difference free energies were calculated using Kb as the substrate binding term, rather than KiaKb., thus ΔGES here refers to EA+B ⇌ EAB. This comparison is valid since the Kia term for the mutants were determined to be essentially invariant. In each case, the magnitude of the energy barrier from EAB to the transition state was calculated using V1; the barrier from the transition state to EAP for wild type and Y224F was calculated using V2 (Table II). (C) Results for the adenylyltransfer reactions catalyzed by wild type PAP (black), Y224F (blue), K215A (green), and for the cytidylyltransfer reaction of w.t. (red). (D) The wild type adenylyltransfer reaction with labels, illustrating ΔG terms discussed in the text. The magnitude of ΔGIB was estimated from the CTP reaction according to: ΔGIB = ΔGcatATP −ΔGcatCTP, and represents the total free energy difference realized upon molecular recognition of the adenine base of ATP (relative to CTP).
Kinetics of PAP mutants
Kinetic studies were undertaken to investigate the relationship between substrate binding, domain movement, and catalysis. We describe below a model wherein the enzyme converts between open and closed conformations during the catalytic cycle residues in the central cavity function to stabilize the closed conformation of the enzyme-substrate ternary complex. Full active site assembly occurs only upon formation of the closed state, thus proper substrate recognition, which induces formation of the closed state, will promote catalysis. This results in nucleotide specificity appearing in the Vmax term, rather than Km. Previously, yeast and bovine PAP have both been the subject of mutagenesis/activity studies (Martin et al., 1999; Martin et al., 2004; Zhelkovsky et al., 2004), however these studies did not consider such a model. Rather than attempt to reinterpret the existing data, which is complicated by the use of different reaction conditions, different methods of assaying activity, and intrinsic differences between the yeast and mammalian enzymes, we have chosen to perform a highly detailed analysis of a subset of mutations.
Here, the mutants N189A, K215A, Y224F, and N226A were characterized; these residues are all located in the active site cleft and are or have been implicated in substrate binding and recognition. Asn189 was previously suggested to be important in ATP specificity (Martin and Keller, 2004). It is located at the hinge between the N-terminal and middle-domains, and while it does not interact with either substrate, it does make a tertiary contact to the carbonyl oxygen of Tyr307. This bridging interaction between the N- and middle-domains occurs only in the closed state. Lys215 and Tyr224 bind the γ-phosphate of ATP in the closed state and were observed to interact with the product, MgPPi, in the open state (Bard et al., 2000). Asn226 is in the middle domain. It does not interact with substrates in the open state, but in the closed state, it contacts a backbone phosphate of the poly(A) and possibly also the ATP base (see discussion above).
The steady state kinetic mechanism of PAP is rapid equilibrium random, and the overall rate is limited by the chemical step (Balbo et al., 2005). The steady state kinetic parameters (Eqs. 1 and 2) were evaluated for these mutant enzymes in both the forward (polyadenylation) and reverse (pyrophosphorolysis) directions and for the cytidylyltransfer reaction utilizing the alternative nucleotide substrate, CTP. The results are reported in Table II alongside the published results (Balbo et al., 2005) for wild type PAP. For each mutant, difference free energy changes relative to wild-type PAP were calculated for each of the reactions performed (Figure 5A) including nucleotidyltransfer utilizing ATP, CTP or GTP and pyrophosphorolysis. These are defined as ΔΔGcat = −RTln[(V/KmKi)w.t./(V/KmKi)]. These values measure the apparent, total free energy contribution of the residue to both substrate binding and catalysis. Agreement among the values for a given mutation reflects consistency between the different sets of experiments. Additionally, for each experiment involving the nucleotidyltransfer reactions with ATP or CTP, the values, ΔΔGES and ΔΔG‡ (see Methods), were calculated and plotted against the total difference in free energy change (ΔΔGcat) relative to a reference reaction (the cytidylyltransfer reaction catalyzed by K215A) (Figure 5B). This treatment of the data illustrates the relationship between the total free energy change for catalysis, ΔΔGcat, and the individual energy contributions to this term from substrate binding (ΔΔGES) and the chemical (rate determining) step (ΔΔG‡).
Certain generalizations can be made upon inspection of the data in Table II and Figure 5. With regard to adenylyltransfer activity, all mutations primarily exert their effect on the “Km” terms, Ka and Kb. For the rapid equilibrium random mechanism employed by PAP, these terms reflect the equilibrium dissociation constants of the poly(A) (Ka) and ATP (Kb) substrates from the enzyme-substrate ternary complex (see (Balbo et al., 2005) for a thorough discussion). In contrast, the inhibition constants, Kia and Kib, reflect dissociation of poly(A) and ATP, respectively, from the binary complex. Importantly, the observation that a given mutation (see N226A, Y224F) has an essentially equal effect on both Ka and Kb, but essentially no effect on the Ki terms, indicates that these mutations do something other than simply disrupt substrate “binding.” We argue below that these mutations affect the equilibrium between the open- and closed-domain forms of the enzyme. Even N189A, a residue that does not directly contact substrates, but bridges the N- and middle domains in the closed state, exhibits a Km effect. Furthermore, these mutations have a comparatively small effect on V1, suggesting that the catalytic machinery was uncompromised. This result is consistent with the location of these residues, which are at the active site, but away from the chemically reactive parts of the substrate(s).
In contrast, for the pyrophosphorolysis reaction, (Table II) the mutations primarily affect the maximal velocity (V2). For N189A and N226A, the Ki and Km terms were very similar to those of wild type. Interestingly, the Y224F (and K215A) mutant(s) showed lower affinity for MgPPi in the binary complex (Kip), but the total free energy change (calculated from the product K′aKip) upon enzyme-product ternary complex formation by Y224F is similar to that of wild type (ΔΔG = 0.4 kcal/mol) due to a compensatory increase in affinity for An in the ternary complex. Attempts to fit the pyrophosphorolysis data for K215A were unsuccessful; they did suggest, however, that the Km values were much greater than 10 mM (not shown).
The cytidylyltransfer data exhibit more scatter than those for the reactions involving ATP. Even so, the total free energy change upon mutation calculated from these data is mostly consistent with those from the other experiments (Figure 5A). The largest discrepancy (0.5–0.6 kcal/mol) between these and the other data sets involves K215A and Y224F and suggests that the compound effect of mutating either K215 or Y224 and utilization of the CTP is not additive. This could possibly reflect the binding of CTP in an alternative (i.e., non-ATP-like) binding mode. Aside from this complication, the important generalization from this data set, particularly the experiments with wild type, N189A and N226A, is that the ΔΔGcat is primarily accounted for in the maximal velocity rather than substrate binding (Figure 5B), a qualitatively different result from the adenylyltransfer data set. As Figure 5B also illustrates, when CTP is utilized by w.t. PAP, the apparent “binding” energy is similar to that when ATP is utilized, and the main difference is in the reaction velocity. This was previously noted (Balbo et al., 2005) and indicates differences in the mechanisms for utilization of ATP and CTP. As mentioned above, the N226A mutant also exhibits a velocity effect for the guanylyltransfer reaction, mirroring the result for the CTP reaction. A general mechanism accounting for this phenomenon is discussed in detail below.
Mechanism of poly(A) polymerase
The kinetic results can be rationalized by the mechanism described in Scheme 1, where A = poly(A), B = MgATP, P = MgPPi, and E = PAP; the designation, E′, refers to the enzyme when the poly(A) substrate is bound in the product-binding mode. This mechanism amends our previous model (Balbo et al., 2005) by introducing conformational change steps (domain reorientation) on either side of the chemical step. The equilibrium constants describing the open-to-closed conformational change for the enzyme-substrate and enzyme-product ternary complexes are Kc1 and Kc2, respectively, where each Kc is the ratio of closed enzyme to open enzyme. The central step in which the chemical transformation of substrates occurs involves closed complexes and is denoted by the dashed box. In the sections below, we describe the predicted kinetic behavior of the system in Scheme 1 and explain how the mutagenesis results are consistent with such a model.
Because the steady state kinetic mechanism of PAP is rapid equilibrium random (Balbo et al., 2005), relatively simple relationships exist between the kinetic parameters and the thermodynamic equilibrium constants in Scheme 1. It follows from both the previous kinetic study and the present work that domain movement is also rapid. The kinetic effect of internal conformational change steps such as those proposed here was previously described in the Tetrahymena ribozyme system, and these references (Narlikar and Herschlag, 1998; Narlikar et al., 1997) should be consulted for a more detailed description of the kinetic features of this type of system. Briefly, the conformational change steps (Kc1 and Kc2 in Scheme 1) will affect the “Km” terms for each substrate or product. In each case, the Km term equals the equilibrium constant (or function of constants) describing dissociation of the substrate or product from the appropriate enzyme ternary complex in the open state divided by the term, (1 + Kc). This termcauses a reduction in Km when Kc is significantly greater than 1 (closed conformation is favored), and has a negligible effect when Kc is significantly less than 1 (open conformation is favored). Definitions for all kinetic parameters are given in Table III.
Table III.
Steady State Kinetic Parameters of PAP
| Polyadenylation Direction
|
Pyrophosphorolysis Direction
|
|---|---|
| Ka = K4/(1 + Kc1) | Ka′ = K6/(1 + Kc2) |
| Kp = K5· (1 + K7/K1)/(1 + Kc2) | |
| Kb = K3· (1 + K1/K7)/(1 + Kc1) | Kip = K8 |
| Kib = K2 |
The adenylyltransfer reaction catalyzed by wild type PAP can be described by the free energy diagram in Figure 6A. For reactions having a single, rate limiting step, the Michaelis complex corresponds to the lowest-energy ground state enzyme substrate complex that exists prior to the highest-energy transition state barrier (i.e. the rate limiting step). In this model, the closed enzyme conformation is of lower energy than the open conformation, and the Michaelis complex corresponds to EAB(closed). This idea is supported by previous fluorescence quenching results suggesting that MgATP and poly(A), specifically, induce domain closure (Balbo et al., 2007). The free energy required to stabilize the closed, active state is derived from the numerous and extensive interactions formed in the closed state (tertiary interactions) as a result of binding both ATP and poly(A). Residues that contribute tertiary interactions will lower the free energy of both the closed ground state and the transition state by exactly the same amount, since these interactions occur in both states. This phenomenon has been called “uniform binding” (Albery and Knowles, 1976). As shown in Figure 6B, the effect of mutating residues that donate tertiary interactions is to destabilize the closed state, and, hence, lower Kc1 and increase Km. In this situation, the lowest energy ground state remains EAB(closed) and the height of the energy barrier from EAB(closed) to the transition state is unchanged, thus Vmax is unaffected by the mutation. The mutagenesis results for the adenylyltransfer reaction (Table II) are consistent with this mechanism. Furthermore, as is diagramed in Figure 6B, the mutation does not affect the free energy of the enzyme product ternary complex (E′AP), thus the free energy barrier from E′AP to the transition state increases and the mutants exhibit a velocity effect in the reverse direction. Thus, the pyrophosphorolysis results are also consistent with this mechanism. One interpretation of these results is that the Michaelis complex for the reverse reaction corresponds to the open enzyme conformation, E′AP(open). The situation diagramed in Figure 6C represents a different mechanism in which the lowest-energy ground state (the Michaelis complex) is EAB(open). This occurs when there is insufficient free energy available from tertiary interactions in the closed state (i.e., Kc1 < 1). The data for the reactions utilizing either CTP or GTP are consistent with the mechanism of Figure 6C. Substitution of the nucleotide base from adenine to cytidine results in a loss of 3.4 kcal/mol (ΔΔGcat) of available free energy (Balbo et al., 2005); and from adenine to guanine, 4.0 kcal/mol (this work). As a result, the open conformation is favored significantly over the closed conformation. Because the Michaelis complex for this reaction corresponds to the open conformation, the mutants, particularly N226A and K215A, which affect the energy of the closed state, exhibit a velocity effect, as described by the model in Figure 6C.
Figure 6.
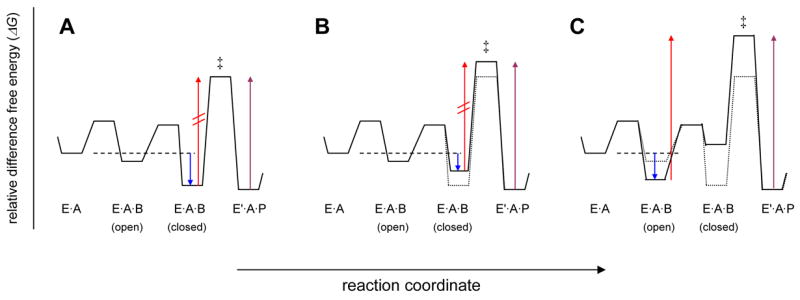
Free energy consideration of PAP. Conceptual free energy diagrams for the reaction segment, (EA + B⇌EAB(open) ⇌EAB(closed) ⇌E′AP), including a rate-limiting conversion of enzyme-bound substrates to products, are depicted. The arrows in each panel refer to free energy differences that correspond to important kinetic parameters: blue corresponds to Km (Kb); red, V1 (maximal rate in the forward direction); violet, V2 (maximal rate in reverse direction). Panel (A) describes the reaction catalyzed by wild type PAP in which the closed conformation of the enzyme substrate ternary complex is strongly favored over the open conformation. Panel (B) depicts the energetic effects of the mutation of residues involved in tertiary interactions that stabilize EAB(closed). The effect of such a mutation is to increase the energy of the EAB(closed) (ground state) and EAB‡ (transition state) by an equal amount, i.e. a uniform binding mutation. Panel (C) describes an alternative situation where the lowest-energy ground state is EAB(open), as is evidently the case for the reaction utilizing the alternative nucleotide substrate, MgCTP. The red arrow in (C) is related to the apparent maximal velocity, Vobs, where Vobs = Fc×V and Fc is the fraction of enzyme substrate complexes that are in the active, closed state.
The main catalytic features of PAP as determined from kinetic experiments, including the site directed mutagenesis (this work) and previous work with alternative substrates (Balbo et al., 2005), are summarized as free energy diagrams in Figure 5C, D. In Figure 5C, the results for three “mutations” in the forward reaction are compared to the adenylyltransfer reaction catalyzed by wild type PAP: Y224F, K215A, and cytidylyltransfer catalyzed by wild type PAP (effectively, a “mutation” of the adenine base of ATP to cytidine). Figure 5D shows only the diagram for wild type adenylyltransfer reaction with the relevant free energy terms indicated. The terms ΔGcat, ΔGES, and ΔG‡ are related to the steady state kinetic parameters V/KiaKb, 1/KiaKb, and V, respectively (see Methods). Also indicated are terms relating to the intrinsic binding energy, ΔGIB, and strain energy, ΔGstrain, where strain represents the energetic penalties incurred upon formation of the ground state. The relationship between intrinsic binding energy and strain has been widely recognized (Fersht, 1974; Herschlag, 1988; Jencks, 1975), and can be represented by: ΔGES = ΔGIB + ΔGstrain. For poly(A) polymerase, ΔGstrain represents the free energy utilized in the ground state destabilization mechanism. We have demonstrated that specificity for ATP (versus CTP or GTP) is manifested primarily in the Vmax term (102–103-fold effect), whereas only a small (3-fold) effect on Km is observed. We proposed an induced fit mechanism for nucleotide specificity that was described as a composite of two effects: uniform binding and ground state destabilization (Balbo et al., 2005). The model of Scheme 1 offers an elaboration of this mechanism in structural terms, specifically that ground state destabilization occurs in the closed conformation. In contrast, the cytidylyl- and guanylyltransfer reactions do not exhibit ground state destabilization (Balbo et al., 2005); this is explained in part by perturbation of Kc1 so that the open complex is favored in the ground state, thus the energetic penalty from ground state destabilization is not incurred in the formation of the Michaelis complex when alternative NTPs are utilized. The polyadenylation results for N189A, N226A, and Y224F, suggest these are classic uniform binding mutations (Albery and Knowles, 1976). The ΔΔGcat for these mutants are ≤ 2 kcal/mol, thus leaving sufficient free energy available to induce the full ground state destabilization effect. In contrast, the K215A mutant, though generally similar to Y224F (Figure 5C), exhibits a measurable effect on ΔG‡ (Figure 5B). We speculate this is due to weaker induction of strain in the Michaelis complex, and could suggest that Lys215 plays a catalytic role, possible by donation of a positive charge in the ground state.
Summary
The mechanism for poly(A) polymerase must account for a number of experimental observations including the previously demonstrated rapid equilibrium random mechanism, domain motion, nucleotide specificity, and the kinetic effects of PAP mutation. According to the induced fit mechanism of PAP, the binding of both MgATP and poly(A) induce a conformational change to form the closed enzyme state. Analogous domain movements have been demonstrated in other polymerases (Doublie et al., 1999), and indeed, rigid body motion such as domain reorientation or flap closure is a common feature in enzymes. In many cases, domain movement is associated with the full assembly of the active site and conversion of the enzyme to a catalytically active state (Hammes, 2002). Numerous tertiary interactions between residues in the central cleft and substrates, as revealed in the present structure, provide the free energy necessary to stabilize the enzyme in the closed state. Furthermore, a substantial portion of the intrinsic binding energy derived from ATP base recognition is utilized to promote the reaction velocity via a ground state destabilization mechanism. This is the basis for manifestation of substrate specificity in the velocity term (Vmax), rather than the Km. Upon substrate binding and formation of the E·A·B ternary complex, domain closure is induced and adenylyltransfer is then catalyzed in the closed enzyme state. Because incorrect nucleotide substrates do not induce the conformational change, their utilization is inefficient, and substrate dissociation (of the incorrect nucleotide) is likely to occur. The details of how catalysis is promoted in the closed, fully assembled active site, including the chemical nature of the proposed ground state destabilization mechanism, remains an important question in the enzymology of PAP and all polymerase beta family enzymes.
PAP functions in vivo as part of a multi-protein complex which includes a number of RNA-binding proteins (Colgan and Manley, 1997; Zhao et al., 1999). These proteins tether the elongating poly(A) substrate to PAP, the effect of which is to increase the local concentration of RNA at the site of polyadenylation. PAP is distributive (Lingner et al., 1991; Wahle, 1991), but appears processive in the presence of other polyadenylation factors (Preker et al., 1997). The kinetic mechanism provides an explanation for how the intrinsic properties of the polymerase allow for efficient catalysis in the context of RNA tethering in vivo. Efficient polyadenylation is governed, in large part, by the thermodynamics of substrate/product binding and domain movement. Poly(A) can bind in either of two, mutually exclusive binding modes (as either a substrate or product), and it is essential that the product poly(A) is released efficiently after each round of catalysis. The high values for K′a and Kp indicate that product dissociation readily occurs even under the cellular condition of locally high poly(A) concentration. One new insight provided by this study (particularly the kinetics of pyrophosphorolysis by the mutants) is that the open conformation of the enzyme-product ternary complex (E′·A·P) is more stable than the closed conformation. This suggests that upon conversion of the enzyme-bound substrates to products, domain opening is induced, facilitating dissociation of products from the enzyme after each round of catalysis. Furthermore, we speculate that the large number of buried waters in the closed complex serves to lower the kinetic barrier to domain opening and closing, which must occur rapidly during successive rounds of adenylyltransfer, by minimizing the extent of substrate desolvation upon Michaelis complex formation. These factors together prevent unproductive reaction in the reverse direction and ensure efficient catalysis and forward flux under cellular conditions.
Materials and methods
Chemicals
All chemicals and buffers were of analytical quality. Luciferin and luciferase were from Promega (Madison, WI). Oligo RNAs (2′-deprotected, and either PAGE- or HPLC-purified) were purchased from Dharmacon (Lafayette, CO). Nucleotide and RNA concentrations were determined by UV absorbance. [2,8-3H]-ATP and [5-3H]-CTP were from PerkinElmer (Boston, MA) as 50% ethanol solutions. Prior to their use, the ethanol was removed by applying a steady stream of air over the surface of the solution, and the amount of [3H]-nucleotide was re-determined by liquid scintillation counting using the specific activity provided by the supplier. The [α-32P]-GTP was from MP Biomedicals (Irvine, CA) in a 5mM Tris solution, pH 7.5.
Site directed mutagenesis and enzyme purification
All experiments utilized the PAPΔ10 enzyme that contains a 32 amino acid C-terminal truncation and a C-terminal His6 tag, as described previously (Balbo et al., 2005) or single point PAP mutants. Mutants were prepared by Quikchange (Stratagene) PCR mutagenesis (Supplemental text) and confirmed by DNA sequencing (Tufts University Core Facility). All PAP used in this study was purified by nickel affinity and ion exchange chromatography, as described previously (Balbo et al., 2005). Protein concentrations were determined by absorbance at 280 nm (ε2800.1% = 0.99 L·g−1·cm−1). Prior to enzymological characterization of the mutants, structural stability was assessed in a thermal denaturation experiment monitored by CD spectroscopy. The buffer was 40 mM KxPO4, pH 7.0, and the enzyme concentration was 10 μg/mL. N189A, N226A, and K215A had a nearly identical Tm as w.t. PAP (~48°C, defined as the temperature at which thermal denaturation is complete); Y224F had altered thermal stability, with a Tm of 44°C. All enzymes were stable at the temperature of the kinetics experiments (30°C).
Crystallization and structure determination
Crystallization of the wild type enzyme substrate complex of PAP was complicated due to catalytic turnover and RNA degradation in the crystallization drops (data not shown). The protein solution used for crystallization included 0.23 mM PAP(D154A), 0.45 mM MgATP and 0.27 mM A5, 20 mM NaCl, 10 mM 2-mercaptoethanol, 5 mM KxHPO4, pH 7.0, and 3% glycerol. Crystals of the PAP/MgATP/A5 complex were grown by mixing the protein solution with an equal volume of crystallization buffer (0.1 M bis-Tris propane, pH 6.4, 0.2 M Li-acetate, and 16% PEG 3350) using the hanging drop method. Crystals appeared in 1 day, and were harvested for data collection after a 3–5 day period of growth. Prior to data collection, crystals were washed in 30% ethylene glycol, 70% crystallization solution then cooled in liquid nitrogen. Drops containing sub-stochiometric quantities of RNA yielded a different crystal form, one in which PAP does not adopt its fully-closed conformation and did not exhibit full occupancy of the substrate binding sites (data not shown), suggesting that the presence of both substrates is required to induce the closed state.
Data to 1.8 Å resolution were collected via “mail-in crystallography” at NSLS beamline X29 and processed using the HKL2000 suite of programs (Otwinowski and Minor, 1997). Data processing statistics are presented in Table I. The structure was solved by molecular replacement, using the program MOLREP. An earlier structure of PAP (PDB code 2HHP) was used as the search model, and the three domains were treated as independent units. The structure was fit to the crystallographic data though multiple rounds of refinement wherein REFMAC (Murshudov et al., 1997) was used for the reciprocal space refinement, and COOT (Emsley and Cowtan, 2004) was used to manually build and modify parts of the model. COOT was also used extensively for real-space fitting of the atomic model to the electron density. The final model contains 519 amino acids, a five nucleotide RNA molecule, a molecule of ATP, one Mg2+ ion, 509 water molecules and 17 molecules of ethylene glycol cryoprotectant. There is only one Ramachandran outlier, Ala225. This residue, which exhibits very good electron density, sits between two very important residues in PAP, both of which were mutated in our analysis, and adopts a similar conformation in earlier structures of the open states of yeast and bovine PAP.
Structure analysis
Accessible surface area calculations were performed within the CCP4 program suite (1994), using the program AREAIMOL (Lee and Richards, 1971) and a probe radius of 1.4 Å. Electrostatics were calculated using PDB2PQR (Dolinsky et al., 2004) and APBS (Baker et al., 2001). Molecular graphics figures were generated using PyMOL (DeLano, 2002). To characterize the surface-exposure of water molecules, the accessible surface area for each individual water molecule was calculated, and those with greater than 3 Å2 exposed to the exterior of the protein/RNA/ATP/Mg2+/water complex were removed. (The surface area of an isolated water molecule is 113 Å2. Thus, in our calculation all water molecules exposing at least 2.6% of their surface to the exterior are considered “exposed.”) This procedure was repeated four times, each cycle removing a new shell of water from the structure. The ethylene glycol molecules were treated in the same manner. On the 5th cycle, no waters were flagged as being exposed, but 58 water molecules remained. These we describe as being “completely buried.”
Kinetics assays
Initial rates of polyadenylation from the reaction of tritium labeled MgATP and A18 were determined at pH 7.0 and 30°C using a discontinuous assay measuring the incorporation of 3H-adenylate into the acid-insoluble fraction, as previously described (Balbo et al., 2005). The initial rates of cytidylyltransfer were measured from the reaction of tritium labeled MgCTP and A18 in an analogous manner. The term, Kip, was independently determined in product inhibition experiments in which the initial rates of polyadenylation were measured at a single [A18] as a function of both MgATP and MgPPi. The initial rates of pyrophosphorolysis from the reaction of MgPPi and A18 at pH 7.0 and 30°C were determined by the previously described method (Balbo et al., 2005) which measures ATP formed by the luciferin/luciferase assay (Ford and Leach, 1998).
Kinetic model and data analysis
The initial rate data were fitted to the steady state rate equation describing a bireactant system, Equation 1, by nonlinear regression, using the program, gnuplot. For competitive (product) inhibition experiments with MgPPi, data were fitted to Equation 2, where v is the measured initial velocity, V is the maximal velocity, V1 (forward) or V2 (reverse), and K is an apparent constant. Throughout the manuscript, reactant concentrations are represented as follows: A = An, B = MgATP2− (or MgNTP2−), P = MgPPi2−. When pyrophosphorolysis was studied, Equation 1 was modified by substituting B with P, Ka with Ka′, and Kb with Kp.
| Equation 1 |
| Equation 2 |
Free energy calculations
The difference free energy (ΔG) terms associated with the steady state kinetic mechanism of PAP as defined here and used in the preparation of Figure 5C, D were calculated as follows: ΔG‡ = −RTln[ηkcat/κT]; Δ GES = RTln[KaKib]; ΔGcat = ΔG‡ + ΔGES, where R is the gas constant (1.9872 cal/mol·K), η is Planck’s constant (6.626 × 10−34 J/s), κ is Boltzman’s constant (1.3807 × 10−23 J/K), T is temperature (K), kcat is the experimentally determined maximal velocity (sec−1) of either the nucleotidyltransfer (V1) or pyrophosphorolysis (V2) reactions, and KaKib is the product of the so-called inhibition and Michaelis constants in units, M2. For Figure 5A, B, the difference difference free energy terms were calculated as follows: ΔΔGcat = −RTln[(V/KmKi)ref/(V/KmKi)], ΔΔGES = RTln[(KmKi)ref/(KmKi)], ΔΔG‡ = −RTln(Vref/V), where the superscript, “ref,” indicates the kinetic parameter from the reference reaction. In these calculations, each concentration dependent term was multiplied by 1 M, therefore the (ΔG) terms here represent standard state values.
Supplementary Material
Acknowledgments
We thank Howard Robinson at NSLS for collecting the high resolution synchrotron data used for this refinement, Viviana Oroleo for preparing one of the mutants discussed, Gretchen Meinke for helping with the electrostatics calculation, and Claire Moore for helpful discussions. Coordinates have been deposited to the Protein Data Bank (2Q66). This work was funded by grant GM065972 to AB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Albery W, Knowles J. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry. 1976;15:5631–5640. doi: 10.1021/bi00670a032. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin E. DNA polymerase β-like nucleotidyltransferase superfamily: Identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999;27:1609–1618. doi: 10.1093/nar/27.7.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babor M, Sobolev V, Edelman M. Conserved positions for ribose recognition: importance of water bridging interactions among ATP, ADP and FAD-protein complexes. J Mol Biol. 2002;323:523–532. doi: 10.1016/s0022-2836(02)00975-0. [DOI] [PubMed] [Google Scholar]
- Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbo PB, Meinke G, Bohm A. Kinetic studies of yeast polyA polymerase indicate an induced fit mechanism for nucleotide specificity. Biochemistry. 2005;44:7777–7786. doi: 10.1021/bi050089r. [DOI] [PubMed] [Google Scholar]
- Balbo PB, Toth J, Bohm A. X-ray Crystallographic and Steady State Fluorescence Characterization of the Protein Dynamics of Yeast Polyadenylate Polymerase. J Mol Biol. 2007;366:1401–1415. doi: 10.1016/j.jmb.2006.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard J, Zhelkovsky AM, Helmling S, Earnest TN, Moore CL, Bohm A. Structure of yeast poly(A) polymerase alone and in complex with 3′-dATP. Science. 2000;289:1346–1349. doi: 10.1126/science.289.5483.1346. [DOI] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium-induced assembly of a complete DNA polymerase catalytic complex. Structure. 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom AD, Schweizer MP, Ts’o POP. Interaction and association of bases and nucleosides in solution. V. Studies of the association of purine nucleosides by vapor pressure osmometry and by proton magnetic resonance. J Am Chem Soc. 1967;89:3612–3622. [Google Scholar]
- Burgers P, Eckstein F. A study of the mechanism of DNA polymerase I from Escherichia coli with diasteromeric phosphorothioate analogs of deoxyadenosine triphosphate. J Biol Chem. 1979;254:6889–6893. [PubMed] [Google Scholar]
- Butler JS, Platt T. RNA processing generates the mature 3′ end of yeast CYC1 messenger RNA in vitro. Science. 1988;242:1270–1274. doi: 10.1126/science.2848317. [DOI] [PubMed] [Google Scholar]
- Butler JS, Sadhale PP, Platt T. RNA processing in vitro produces mature 3′ ends of a variety of Saccharomyces cerevisiae mRNAs. Mol Cell Biol. 1990;10:2599–2605. doi: 10.1128/mcb.10.6.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colgan D, Manley J. Mechanism and regulation of mRNA polyadenylation. Genes Dev. 1997;11:2755–2766. doi: 10.1101/gad.11.21.2755. [DOI] [PubMed] [Google Scholar]
- Conte MR, Grune T, Ghuman J, Kelly G, Ladas A, Matthews S, Curry S. Structure of tandem RNA recognition motifs from polypyrimidine tract binding protein reveals novel features of the RRM fold. Embo J. 2000;19:3132–3141. doi: 10.1093/emboj/19.12.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL User’s Manual. San Carlos, CA: 2002. [Google Scholar]
- Delarue M, Boule JB, Lescar J, Expert-Bezancon N, Jourdan N, Sukumar N, Rougeon F, Papanicolaou C. Crystal structures of a template-independent DNA polymerase: murine terminal deoxynucleotidyltransferase. Embo J. 2002;21:427–439. doi: 10.1093/emboj/21.3.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Ernst NL, Turley S, Stuart KD, Hol WG. Structural basis for UTP specificity of RNA editing TUTases from Trypanosoma brucei. Embo J. 2005;24:4007–4017. doi: 10.1038/sj.emboj.7600861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doublie S, Sawaya MR, Ellenberger T. An open and closed case for all polymerases. Structure. 1999;7:R31–35. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- Edmonds M. Polyadenylate polymerase. Methods Enzymol. 1990;181:161–170. doi: 10.1016/0076-6879(90)81118-e. [DOI] [PubMed] [Google Scholar]
- Edmonds M. A history of poly A sequences: from formation to factors to function. Prog Nucleic Acid Res Mol Biol. 2002;71:285–389. doi: 10.1016/s0079-6603(02)71046-5. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Fersht A. Catalysis, binding and enzyme-substrate complimentarity. Proc R Soc London, Ser B. 1974;187:397–407. doi: 10.1098/rspb.1974.0084. [DOI] [PubMed] [Google Scholar]
- Ford SR, Leach FR. Improvements in the application of firefly luciferase assays. Methods Mol Biol. 1998;102:3–20. doi: 10.1385/0-89603-520-4:3. [DOI] [PubMed] [Google Scholar]
- Friedman RA, Honig B. A free energy analysis of nucleic acid base stacking in aqueous solution. Biophys J. 1995;69:1528–1535. doi: 10.1016/S0006-3495(95)80023-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin GM. Eukaryotic mRNA 3′ processing: a common means to different ends. Genes Dev. 2005;19:2517–2521. doi: 10.1101/gad.1378105. [DOI] [PubMed] [Google Scholar]
- Gold M, Johnson R, Tseng J. Kinetic mechanism of rabbit muscle glycogen phosphorylase a. J Biol Chem. 1970;245:2564–2572. [PubMed] [Google Scholar]
- Hammes GG. Multiple conformational changes in enzyme catalysis. Biochemistry. 2002;41:8221–8228. doi: 10.1021/bi0260839. [DOI] [PubMed] [Google Scholar]
- Helmling S, Zhelkovsky A, Moore CL. Fip1 regulates the activity of Poly(A) polymerase through multiple interactions. Mol Cell Biol. 2001;21:2026–2037. doi: 10.1128/MCB.21.6.2026-2037.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D. The role of induced fit and conformational changes of enzymes in specificity and catalysis. Bioorg Chem. 1988;16:62–96. [Google Scholar]
- Jencks WP. Binding energy, specificity, and enzymic catalysis: the circe effect. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- Lingner J, Radtke I, Wahle E, Keller W. Purification and characterization of poly(A) polymerase from Saccharomyces cerevisiae. J Biol Chem. 1991;266:8741–8746. [PubMed] [Google Scholar]
- Mandel CR, Kaneko S, Zhang H, Gebauer D, Vethantham V, Manley JL, Tong L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature. 2006;444:953–956. doi: 10.1038/nature05363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Jeno P, Keller W. Mapping of ATP binding regions in poly(A) polymerases by photoaffinity labeling and by mutational analysis identifies a domain conserved in many nucleotidyltransferases. Protein Sci. 1999;8:2380–2391. doi: 10.1110/ps.8.11.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W. Sequence motifs that distinguish ATP(CTP):tRNA nucleotidyl transferases from eubacterial poly(A) polymerases. Rna. 2004;10:899–906. doi: 10.1261/rna.5242304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Keller W, Doublie S. Crystal structure of mammalian poly(A) polymerase in complex with an analog of ATP. Embo J. 2000;19:4193–4203. doi: 10.1093/emboj/19.16.4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Moglich A, Keller W, Doublie S. Biochemical and structural insights into substrate binding and catalytic mechanism of mammalian poly(A) polymerase. J Mol Biol. 2004;341:911–925. doi: 10.1016/j.jmb.2004.06.047. [DOI] [PubMed] [Google Scholar]
- Moodie SL, Mitchell JB, Thornton JM. Protein recognition of adenylate: an example of a fuzzy recognition template. J Mol Biol. 1996;263:486–500. doi: 10.1006/jmbi.1996.0591. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Herschlag D. Direct demonstration of the catalytic role of binding interactions in an enzymatic reaction. Biochemistry. 1998;37:9902–9911. doi: 10.1021/bi980495t. [DOI] [PubMed] [Google Scholar]
- Narlikar GJ, Khosla M, Usman N, Herschlag D. Quantitating tertiary binding energies of 2′ OH groups on the P1 duplex of the Tetrahymena ribozyme: intrinsic binding energy in an RNA enzyme. Biochemistry. 1997;36:2465–2477. doi: 10.1021/bi9610820. [DOI] [PubMed] [Google Scholar]
- Nobeli I, Laskowski RA, Valdar WS, Thornton JM. On the molecular discrimination between adenine and guanine by proteins. Nucleic Acids Res. 2001;29:4294–4309. doi: 10.1093/nar/29.21.4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norberg J, Nilsson L. Stacking free energy profiles for all 16 natural ribodinucleoside monophosphates in aqueous solution. J Am Chem Soc. 1995;117:10832–10840. [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Preker PJ, Lingner J, Minvielle-Sebastia L, Keller W. The FIP1 gene encodes a component of a yeast pre-mRNA polyadenylation factor that directly interacts with poly(A) polymerase. Cell. 1995;81:379–389. doi: 10.1016/0092-8674(95)90391-7. [DOI] [PubMed] [Google Scholar]
- Preker PJ, Ohnacker M, Minvielle-Sebastia L, Keller W. A multisubunit 3′ end processing factor from yeast containing poly(A) polymerase and homologues of the subunits of mammalian cleavage and polyadenylation specificity factor. Embo J. 1997;16:4727–4737. doi: 10.1093/emboj/16.15.4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya MR, Pelletier H, Kumar A, Wilson SH, Kraut J. Crystal structure of rat DNA polymerase beta: evidence for a common polymerase mechanism. Science. 1994;264:1930–1935. doi: 10.1126/science.7516581. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Prasad R, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- Stagno J, Aphasizheva I, Rosengarth A, Luecke H, Aphasizhev R. UTP-bound and Apo structures of a minimal RNA uridylyltransferase. J Mol Biol. 2007;366:882–899. doi: 10.1016/j.jmb.2006.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci U S A. 1993;90:6498–6502. doi: 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Fukai S, Ishitani R, Ueda T, Takeuchi N, Vassylyev DG, Nureki O. Structural basis for template-independent RNA polymerization. Nature. 2004;430:700–704. doi: 10.1038/nature02712. [DOI] [PubMed] [Google Scholar]
- Ts’o PO, Kondo NS, Schweizer MP, Hollis DP. Studies of the conformation and interaction in dinucleoside mono- and diphosphates by proton magnetic resonance. Biochemistry. 1969;8:997–1029. doi: 10.1021/bi00831a033. [DOI] [PubMed] [Google Scholar]
- Ts’o POP, Melvin IS, Olson AC. Interaction and association of bases in aqueous solutions. J Am Chem Soc. 1962;85:1289–1296. [Google Scholar]
- Wahle E. Purification and characterization of a mammalian polyadenylate polymerase involved in the 3′ end processing of messenger RNA precursors. J Biol Chem. 1991;266:3131–3139. [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelkovsky A, Helmling S, Bohm A, Moore C. Mutations in the middle domain of yeast poly(A) polymerase affect interactions with RNA but not ATP. Rna. 2004;10:558–564. doi: 10.1261/rna.5238704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhelkovsky A, Helmling S, Moore C. Processivity of the Saccharomyces cerevisiae poly(A) polymerase requires interactions at the carboxyl-terminal RNA binding domain. Mol Cell Biol. 1998;18:5942–5951. doi: 10.1128/mcb.18.10.5942. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.