


Abstract
One of the major challenges in the post-genome era is the correlation between genes and function or phenotype. We have pioneered a strategy for screening of cDNA libraries, which is based on sequential combination of lentiviral and oncoretroviral expression systems and can be used to identify proliferation-modulating genes. Screening of a lentiviral expression library derived from adult human brain cDNA resulted in cloning of the potent proliferation-inducing determinant termed pi1 (proliferation inducer 1). Transduction experiments using GFP-expressing oncoretroviruses to target proliferation-competent cells suggested that overexpression of pi1 initiates proliferation of human umbilical vein endothelial cells (HUVECs). Growth induction of HUVECs as well as Swiss3T3 fibroblasts was confirmed by Brd-uridine incorporation assays, which correlated increased DNA synthesis with expression of pi1. The identified pi1 cDNA is 297 bp long and encodes a 10 kDa polypeptide. Since deregulation of proliferation control accounts for a number of today’s untreatable human diseases such as neurodegenerative disorders and cancer, discovery of novel proliferation-modulating genes is essential for developing new strategies for gene therapy and tissue engineering.
INTRODUCTION
Expression cloning has a track record in discovering novel genes based on their function. Typically, identification of a single gene consists of several screening rounds of a cDNA expression library for a specific trait, such as proliferation, DNA binding or cell surface expression. Successful expression cloning of proliferation-inducing genes requires efficient transfer of cDNA pools into mitotically inactive primary cells. We suggest cDNA libraries engineered into a lentiviral expression setting to be the preferred configuration for functional expression cloning. Retroviruses are lipid-enveloped particles comprising a homodimer of linear, positive-sense, single-stranded RNA genomes that replicate via a DNA intermediate (1). Oncoretroviral and lentiviral vectors exhibit a large cloning capacity and enable long-term expression of desired transgenes following integration into the target chromosomes. Pseudo typing of transgenic retroviral particles extends their tropism and increases their transduction efficiency. The multitropism of VSV-G-pseudotyped lentiviruses facilitates screening of a single lentiviral cDNA expression library in a variety of different cells or tissues. Although very similar in their genetic set-up, oncoretroviruses only infect dividing cells whereas lentiviruses can infect and replicate in non-mitotic cells (2,3). Typically, oncoretroviral/lentiviral transduction rates are a function of the multiplicity of infection (m.o.i.). Depending on the m.o.i., the fraction of transduced cells can vary from 1 to 95%. By increasing the m.o.i., a single cell can be transduced by more than one cDNA-encoding viral particle, resulting in coexpression of a gene pool. This retroviral characteristic enables elucidation of combinatorial effects of several genes coexpressed in a single target cell.
Most of today’s available lentiviral expression vectors have been designed by eliminating pathogenic transcription units from the viral genome while retaining cis-acting elements essential for packaging and reverse transcription. Transgenic non-pathogenic and replication-incompetent lentiviral particles are produced following cotransfection of three separate vectors into a helper cell line: (i) the transfer construct containing viral cis-acting elements as well as the desired transgene, (ii) the packaging construct encoding structural and (iii) envelope proteins (4). In order to increase biosafety and reduce promoter interference, self-inactivating (SIN) lentivectors have become the standard for transduction of proliferation-inert cells. The SIN lentiviruses harbor a deletion in their 3′ LTR that results in inactivation of 5′ LTR-associated promoter/enhancer activities following reverse transcription and integration into the target chromosome (5). Recently, we have engineered a series of self-inactivating lentiviral vectors which contain (i) unmatched polylinkers with up to 29 unique sites for restriction endonucleases, (ii) strong promoters derived from human cytomegalovirus immediate-early or the human elongation factor 1α promoters and/or (iii) tricistronic expression cassettes for coordinated expression of up to three transgenes (6).
Cell proliferation failures are associated with a number of human pathologies including multiple sclerosis, osteoarthritis as well as Parkinson’s and Alzheimer’s disease. Cloning of novel proliferation-inducing genes may have therapeutic implications for tissue regeneration as well as the treatment of tissue-degenerative disorders. We have developed an expression cloning strategy for the discovery of growth-promoting determinants using a combination of retroviral and lentiviral transduction technologies. Screening of a human brain cDNA library expressed in human umbilical vein endothelial cells (HUVEC) resulted in the identification of pi1, a novel proliferation-inducing cDNA.
MATERIALS AND METHODS
Cell culture
HEK293-T (6), Swiss3T3 fibroblasts (provided by Urs Ziegler) and HeLa (ATCC CCL-2) cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal calf serum (FCS) (PAA, Vienna, Austria; lot no. A01129-242), 100 U penicillin and 100 µg/ml streptomycin (Sigma Chemicals, St Louis, MO) in a humidified atmosphere containing 5% CO2 at 37°C. HUVECs were dissected and cultured as previously described (7). HUVECs of the first passages were used. Cells were transfected using either calcium phosphate-based protocols or FuGENE6 transfection reagent (Roche Molecular Biochemicals, Rotkreuz, Switzerland).
FACS analysis
Cells were dissociated from the culture dish by trypsinization, resuspended in PBS containing 1% FCS and the fluorescence was quantified using a Beckman Counter EPICS XL flow cytometer (Beckman Coulter Intl. SA, Nyon, Switzerland).
Genomic DNA isolation
Cells (5 × 105) were washed, collected in PBS and pelleted. The cells were lysed in lysis buffer (10 mM Tris–Cl; 50 mM KCl; 1.5 mM MgCl2; 0.45% Tween 20; 0.5% Triton X-100) and proteinase K (Qiagen, Basel, Switzerland) was added to the lysate for 45 min at 50°C. The DNA was purified by phenol/chloroform extraction followed by ethanol precipitation.
Cell proliferation assay
Cell proliferation assay using Brd-uridine incorporation was performed using the Cell Proliferation ELISA kit (Roche Molecular Biochemicals, Rotkreuz, Switzerland) according to the manufacturer’s instruction. In brief, cells were seeded into 96-well plates at different concentrations, allowed to adhere for 24 h and transfected with either the pPI1-His or isogenic control vectors. Proliferation was quantified 36 h post-transfection.
Western blotting
Bacterial lysates were resolved on SDS–polyacrylamide gels and transferred to Immobilon-P membranes (Millipore Corporation, Bedford, MA) using standard procedures. The membrane was blocked with 4% (w/v) instant non-fat milk powder (Coop, Switzerland) and incubated for 2 h with anti-His antibodies (Novagen, Madison, WI) diluted in PBS containing 4% non-fat milk powder. His-tagged proteins were visualized following incubation with goat anti-mouse horseradish peroxidase-coupled antibody and ECL detection reagents (Amersham Biosciences, UK).
Construction of plasmids and the cDNA library
The lentiviral expression vector pDC32 was constructed by cloning the PhCMV-encoding BamHI/KpnI fragment, PCR-amplified from pcDNA3.1/V5-His-TOPO (Invitrogen, Carlsbad, CA), BamHI/KpnI into pMF388 (6). The following primers were used for amplification of PhCMV: ODC1: 5′-GGGGTACCGCCAGATATACGCGTTGACA-3′ and ODC2 5′-CGGGATCCACGTGAGCCAGTAAGCAGTGGGTTC-3′ (annealing sequences are underlined, extensions containing KpnI and BamHI/PmlI restriction sites are shown in bold). The double-stranded (ds) cDNA library was constructed from human adult brain total RNA (Invitrogen, Carlsbad, CA) using the NotI-containing oligo-d(T) for priming and the Copy Kit cDNA synthesis system (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. The ds cDNA was blunted using DNA polymerase I followed by NotI digestion and gel purification. The NotI-digested ds cDNA was ligated into the NotI–PmlI sites of pDC32 resulting in the lentivector-based cDNA library. The lentivector-encoded cDNA library was converted into replication-incompetent lentiviral particles following recently described protocols (6). The titer of the library was determined using the p24 ELISA assay (NEK-050, Perkin Elmer, Boston, MA).
Transduced cDNA inserts of interest were PCR-amplified from the genomic DNA using primers ODC3: 5′-GAGACAGAGACAGATCCATTCG-3′ (annealing to nucleotides 2372–2393 of pMF388) and ODC4: 5′-CGTGTACGGTGGGAGGTCTA-3′ [annealing to nucleotides 786–805 of pcDNA3.1/V5HisTOPO (corresponding to PhCMV)] and the following PCR program: annealing temperature of the first two cycles was 64°C which was subsequently decreased by 2°C per two cycles until a final annealing temperature of 54°C was reached. At 54°C an additional 20 cycles were run. The products of the first amplification round were diluted 1:20 in H2O and used as template for the second PCR employing identical primers and reaction conditions. The final PCR product was subcloned into pcDNA3.1/V5-His-TOPO (Invitrogen, Carlsbad, CA) to result in pPCR102-2. The novel cDNA sequence contained within pPCR102-2 has been deposited in the GenBank database (accession no. AY255855). All PCR reactions were conducted using DyNAzyme.EXT polymerase (Finnzymes, Oulu, Finland). pND-A2 and pND-A8 represent nested deletions of pPCR102-2 which were performed by restricting pPCR102-2 with KpnI and BamHI followed by Exonuclease III (NEB, Beverly, MA) mediated digestion at 37°C for 1, 2 and 3 min, respectively. The Exonuclease III reaction was stopped in liquid nitrogen and the enzyme was subsequently heat inactivated. The digestion products were blunted using Mung Bean Nuclease (BD Biosciences, Palo Alto, CA) and recircularized followed by bacterial transformation and plasmid DNA purification. The pPI1-His construct was obtained by PCR-mediated amplification of pi1 from pPCR102-2 using primers OD5: 5′-CGGGATCCATGGATCTGTCTCAGTCTCAGTCTC-3′ and OD6: 5′-CCCGATATCAGACAGAGACAATCCATTCGAACAGA-3′, (annealing sequences are underlined and extensions containing BamHI and EcoRV restriction sites are shown in bold) followed by cloning of the BamHI/EcoRV-digested pi1-encoding PCR product into BamHI/EcoRV-restricted pEF6/V5-His-TOPO (Invitrogen, Carlsbad, CA), in-frame with histidine tag and stop codon. The prokaryotic pi1 expression vector pDC106 was constructed by PCR-mediated amplification of pi1 from pPI1-His using OD7: 5′-CACCATGGATCTGTCTCAGTCTCAGTCTC-3′ and OD8: 5′-AACTAGAAGGCACAGTCGAGGCTG-3′ (annealing sequences are underlined). OD8 corresponds to nucleotides 1108–1131 of pcDNA3.1/V5-His-TOPO (Invitrogen, Carlsbad, CA) and OD7 to nucleotides 1–25 of the pi1. The PCR product was purified and cloned into the prokaryotic expression vector pET101-D (Invitrogen, Carlsbad, CA). The oncoretroviral vector pLEGFP-N1 encoding the enhanced green fluorescent protein (GFP) was obtained from BD Biosciences (Palo Alto, CA).
Lentivirus and oncoretrovirus production
Lentiviruses were produced as described previously (6). In brief, equal amounts of the helper construct pCD/NL-BH (8), the envelope construct pLTR-G (9) and the transfer construct containing the desired transgene were transiently transfected into HEK293-T cells and the virus-enriched cell supernatant was collected 3 days later, filtered and frozen in aliquots. pLEGFP-N1-derived GFP-encoding oncoretrovirus was produced using the Retro-X Expression System (BD Biosciences, Palo Alto, CA) according to the manufacturer’s protocols.
RESULTS
Lentiviral expression cloning/screening for the discovery of proliferation-inducing genes
In order to identify novel growth-promoting genes we have developed a cDNA library screening system based on the sequential combination of two expression technologies: transduction of proliferation-inert cells or tissues using a lentiviral cDNA expression library, followed by selection of proliferation-competent cells targeted by a GFP-encoding oncoretrovirus. Figure 1A depicts key steps of the screening strategy. (i) Transduction of proliferation-inert primary target cells by a lentiviral cDNA expression library at specific m.o.i. enables expression of one or several cDNAs per cell. A single cDNA or a combinatorial effect of a particular gene pool of the transduced cDNAs may trigger target cell proliferation. (ii) Selection of mitotically active transgenic target cells using a GFP-encoding reporter oncoretrovirus. Since oncoretroviruses selectively infect dividing cells they can be used to monitor cell-cycle re-entry of primary cells following transduction of a lentiviral cDNA expression library. (iii) FACS-mediated cell sorting enables isolation of transgenic GFP-positive proliferating cells. (iv) Following clonal expansion of GFP- and cDNA-expressing cells, proliferation inducing transgene(s) are recovered from chromosomal prolentiviruses by PCR-mediated amplification using primers complementary to heterologous cDNA-flanking sequences.
Figure 1.
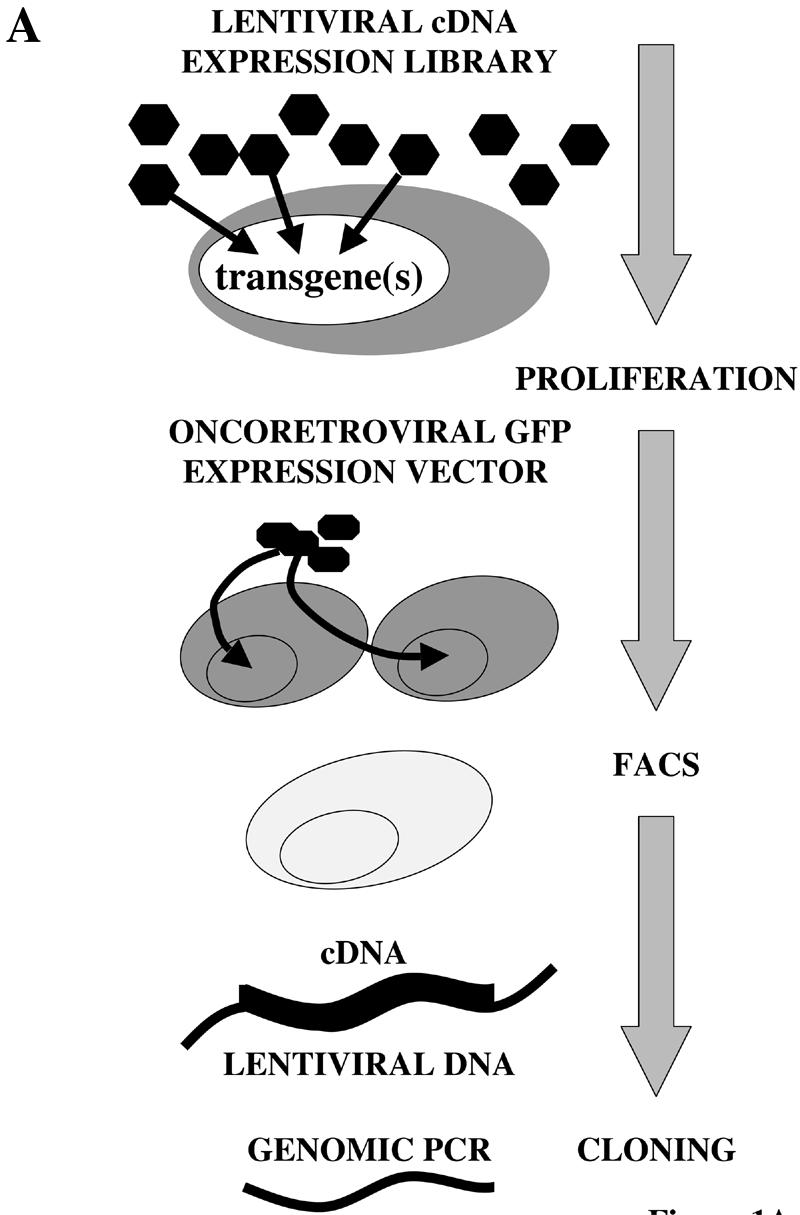

Schematic representation of lentiviral expression cloning for the discovery of proliferation-inducing cDNAs. (A) Proliferation-inert primary cells are transduced by a lentiviral cDNA library at desired m.o.i. Depending on the chosen m.o.i. a single gene or a transgene pool will be expressed in the target cells. Only in the event that the transgene(s) triggers proliferation of a primary cell can it be targeted by a GFP-encoding oncoretrovirus and isolated by FACS-mediated single-cell cloning. Proliferation-inducing cDNAs are recovered from the chromosome by PCR-mediated amplification from flanking lentiviral sequences. (B) Genetic configuration of the self-inactivating (3′ LTRΔU3) lentiviral expression vector pDC32 used for construction of the cDNA library. The cDNA expression cassette is placed antisense to the 5′ LTR in order to prevent interference of the polyadenylation site (pA) with reverse transcription. Following directional cloning of the cDNA inserts into the NotI/PmlI sites, the NotI site survives insertion into pDC32. Black arrows indicate oligonucleotides (ODC3 and ODC4) required for PCR-mediated recovery of integrated cDNAs. The extended lentiviral packaging signal is indicated (Ψ+).
Since RNA synthesis represents a key step in the retroviral life cycle, the desired transgene expression unit integrated into a retroviral transduction configuration should be devoid of any 5′–3′ poly(A) signals. Figure 1B shows a schematic representation of the lentivirus-derived transfer vector pDC32 engineered to support expression of a poly(A) signal-containing cDNA insert. The pMF388 (6) derivative pDC32 is size-optimized and contains a strong constitutive promoter derived from the human cytomegalovirus immediate early promoter (PhCMV) in 3′–5′ orientation. PhCMV is flanked by cis-acting viral elements including the extended packaging signal (Ψ+) as well as 5′ and 3′ long terminal repeats (LTR). Since poly(A) signals are known to function in an orientation-dependent manner they are not expected to interfere with reverse transcription when placed in antisense orientation. In addition, due to a deletion in the 3′ LTR, pCD32-derived lentivectors are self-inactivated, preventing any 5′ LTR-based interference with expression of the cDNA or endogenous genes adjacent to the chromosomal insert. In order to validate the pDC32-based lentiviral expression configuration, a GFP-encoding expression cassette (PhCMV-gfp-pA) was inserted into pDC32 followed by production and transduction of transgenic lentiviral particles into CHO-K1 cells. Based on fluorescence microscopy, close to 50% of the transduced cells scored positive for GFP expression (data not shown), thus confirming that 3′–5′ orientation of the expression cassette is compatible with lentivirus production and enables efficient transgene expression. Next, a ds cDNA library synthesized from adult human brain total RNA was cloned into pDC32. The oligo-d(T) used for priming of first strand synthesis contained a NotI restriction endonuclease site to enable directional cloning into pDC32. Over 4 × 105 independent clones were obtained, pooled and their plasmid DNA isolated. The purified lentivector DNA was used to produce a library of lentiviral particles containing PhCMV-driven adult human brain cDNAs. The virus titer determined using a p24 immunoassay was 127.4 ± 15.1 ng/ml correlating with 2–3 × 107 infectious particles per ml.
Isolation of proliferation-promoting determinants
In order to identify novel proliferation-inducing genes, mitotically quiescent HUVECs were transduced with a lentivirus library transgenic for adult human brain cDNAs at m.o.i. up to 10. Three days post-transduction, the cells were retransduced using a GFP-encoding oncoretrovirus pLEGFP-N1, which exclusively targets HUVECs that underwent cell-cycle re-entry. pLEGFP-N1-transduced cell populations were gated for GFP expression and single cell-sorted by FACS 48 h post pLEGFP-N1 transduction. Single GFP-expressing cells were seeded onto 96-well cell culture plates and allowed to expand. Out of 12 GFP-expressing cell clones, three survived triple passage and expansion indicating that they were transduced by lentiviruses encoding proliferation-inducing genes. Genomic DNA was extracted from the transgenic proliferation-competent HUVEC cell derivatives and a chromosomally integrated library cDNA could be PCR-amplified from a single clone using primers ODC3 and ODC4 (Fig. 1B). The DNA fragment was cloned into the mammalian expression vector pcDNA3.1 to yield pPCR102-2. pPCR102-2 was found to contain a unique novel sequence spanning about 300 nucleotides flanked by the expected lentivector stretches (Fig. 2). The deduced amino acid sequence of the cDNA insert of pPCR102-2 revealed an open reading frame (ORF) of 297 bp coding for a putative protein of 10 kDa. Sequence similarity searches in the human genome database revealed an overall 39% homology of the deduced polypeptide to the hypothetical human protein XP_086050 on chromosome 19 (19p13.11).
Figure 2.

Structure of pPCR102-2 and its derivatives encoding different modules of the proliferation-inducing cDNA. In pPCR102-2, the novel proliferation-inducing cDNA is flanked by viral sequences and contains an ORF. In order to narrow down the proliferation-inducing ORF, we constructed pND-A8 containing 5′ deletions of viral sequences, pND-A2 harboring a deletion of the ATG start codon of ORF and pPI1-His encoding full-length PI1 ORF fused in-frame to a histidine tag. pPCR102-2 derivatives were produced by deleting sequences either upstream or including the putative start ATG of the ORF. All constructs were cloned under control of PhCMV.
Characterization of a novel proliferation-inducing cDNA
In order to demonstrate whether expression of the pPCR102-2-encoded polypeptide can induce proliferation, we quantified GFP-mediated fluorescence of pPCR102-2-transfected HUVECs following transduction with pLEGFP-N1. Since pLEGFP-N1 can integrate only into chromosomes of proliferating cells, GFP fluorescence levels correlate with the number of proliferation-competent cells in transgenic HUVEC populations. Figure 3A shows FACS-assessed GFP-mediated fluorescence levels of HUVECs, transfected either with pPCR102-2 or an isogenic control vector (pcDNA3.1/V5-His-TOPO), 48 h post-transduction with pLEGFP-N1. We observed that pPCR102-2-transfected HUVEC populations showed up to 15-fold higher green fluorescence levels compared to control cells. In order to exclude that proliferation was induced by interference of a non-coding RNA transcribed from the viral DNA encoded on pPCR102-2 clone, we have created several pPCR102-2 derivatives containing truncated/modified cDNA inserts. In pND-A8 viral sequences 5′ of the putative ORF were deleted (Fig. 2). The FACS analysis of HUVECs, transfected with either pND-A8 or isogenic control DNA and subsequently transduced with pLEGFP-N1 revealed nearly 6-fold higher proliferation rate in pND-A8-transfected cells compared to control cell populations (Fig. 3B). Furthermore, pND-A2 lacking viral sequences as well as the putative ATG start codon of the ORF shows no proliferation-modulating effects on HUVECs (Fig. 3C). Finally, we have tagged the 297 bp-long ORF by fusing it N-terminally to a His-tag and a TAA STOP codon (Fig. 2, pPI1-His). FACS analysis of the pLEGFP-N1-transduced HUVECs revealed that cells pretransfected with pPI1-His showed a 4-fold higher GFP-mediated fluorescence compared to HUVEC populations transfected with the control vector (Fig. 3D). The overall reduction in proliferation-inducing potential associated with elimination of viral sequences may result from transcription- and/or translation-enhancing activites contained in cis-acting viral sequences. All results are consistent with the fact that the unique cDNA fragment contained on pPCR102-2 encodes a polypeptide able to induce proliferation in HUVECs, which we call PI1 (proliferation inducer 1). Indeed, conditional IPTG-induced expression of His-tagged piI in Escherichia coli resulted in production of a single 15 kDa (15 kDa, PI1 plus His-tag; 10 kDa PI1 only) protein following 4 h of IPTG induction (Fig. 4).
Figure 3.

Proliferation-inducing capacity of pPCR102-2 and derivatives in HUVECs. HUVECs were transiently transfected with either pPCR102-2 (A), pND-A8 (B), pND-A2 (C) or pPI1-His (D) in order to assess their proliferation-inducing potential. Forty-eight hours post transfection, the cell populations were transduced with a GFP-encoding oncoretroviral vector, which exclusively targets proliferating cells. Forty-eight hours post-transduction, GFP-mediated fluorescence was quantified by FACS. Fluorescence values were calculated by multiplying the number of GFP-expressing cells by the average intensity of GFP expression. The relative fluorescence units were obtained by comparison with GFP-mediated fluorescence control populations (cntrl) transfected with isogenic pcDNA3.1/V5-His-TOPO. Corresponding FACS histograms are also shown. All values are representative of at least three independent experiments. FS, forward scatter; FL, fluoresence.
Figure 4.

Expression of the PI1 protein. Escherichia coli transgenic for pDC106 harboring PI1-His (see Fig. 2) under control of the IPTG-inducible Plac promoter was grown for 4 h in the presence (+) or absence (–) of IPTG. Cell lysates were resolved on SDS–PAGE and analyzed for PI1-His production by His-tag-targeted immunodetection.
PI1-induced cell proliferation
In order to quantify the direct impact of the PI1 on DNA synthesis of mammalian cells we have performed Brd-uridine incorporation-based cell proliferation assays. HUVECs transfected with the pPI1-His showed enhanced Brd-uridine incorporation in comparison with mock-transfected cells which indicates increased proliferation (Fig. 5A). Increases in DNA synthesis ranged from 20 to 80%, depending on the cell density on the day of transfection. Similar results were obtained in Swiss3T3 fibroblasts (Fig. 5B). When PI1 was expressed in confluent cells it was unable to increase proliferation indicating that PI1 is not an oncogene (Fig. 5A and B).
Figure 5.

PI1 induces DNA synthesis in primary (A) HUVECs and (B) Swiss3T3 fibroblasts. The cells were seeded at various densities and transfected with either pPI1-His or control vector followed by 24 h incubation with Brd-Uridine. Incorporation of Brd-Uridine into newly synthesized DNA was quantified by colorimetric ELISA using a peroxidase-conjugated anti-Brd-Uridine antibody. DNA synthesis values correlate with changes in optical densities resulting from Brd-Uridine incorporation.
DISCUSSION
Gene-function analysis is one of the key scientific activities in the post-genomic era. Many genes are only expressed in small quantities, at specific times during development or in a subset of tissues. Based on their (i) high transduction efficiency, (ii) stable integration into the host chromosome, (iii) broad tissue tropism following pseudotyping and (iv) their capacity to target mitotically inactive cells (10–14), we designed a novel lentiviral expression cloning strategy for the detection of proliferation-inducing determinants. As exemplified by cloning of PI1 cDNA the discovery of novel proliferation-inducing determinants using lentiviral expression cloning consists of three consecutive steps. (i) Transduction of a lentiviral cDNA produced from tissues containing proliferation-competent cells into proliferation-inert target cells. (ii) Targeting, isolation and expansion of cDNA expressing proliferating cell clones using a reporter gene-encoding oncoretrovirus which exclusively transduces mitotically active cells. (iii) Cloning of chromosomal cDNA inserts by PCR-mediated amplification from flanking viral sequences.
Our initial decision to screen for proliferation-inducing genes using a human brain cDNA expression library in order to validate lentiviral expression cloning was based on recent findings suggesting that adult human brain cells have the potential to divide and differentiate into neurons (15). There is now abundant evidence that neural stem cells persist in the adult brain and support ongoing neurogenesis in restricted regions of the central nervous system (16). However, genes triggering neurogenesis in the adult brain remain to be identified. Cloning of proliferation-inducing determinants from adult human brain cells is expected to significantly advance the understanding of naturally occurring neurogenesis and will open new perspectives for treatment of neurodegenerative disorders including Parkinson’s and Alzheimer’s diseases. Animal models have substantiated the fact that immature neural precursors can replace lost neurons, restore their function and promote brain self-repair. Ongoing clinical trials for the treatment of Parkinson’s disease are promising and increase hope that pioneering therapeutic strategies established in animal models could be used for the treatment of neurodegenerative diseases (17,18).
An inherent challenge for cloning of proliferation-inducing cDNAs using retroviral systems is their capacity to induce uncontrolled transgene-independent growth as a result of random integration into the target chromosome. In order to minimize unspecific growth-induction it is essential that the target cells are proliferation-inert which is the case for most differentiated primary cells. Our results using HUVECs as a proliferation-inert reporter cell line exemplifies the possibility of isolating growth-promoting genes in a single screening step. PI1 is a 10 kDa protein that induces proliferation in sparse HUVEC and Swiss3T3 fibroblast cultures while it shows no growth-promoting effect on confluent cells. The finding that contact inhibition seems to be dominant over PI1 action suggests that this protein lacks transformation potential. PI1 might therefore be considered for expansion of primary cells in tissue engineering scenarios that require timely growth induction without becoming neoplastic.
The use of lentiviral cDNA expression libraries is not limited to screening for growth-inducing genes. The same library could be used to discover genes involved in any cell phenotype-modulating process as long as there are tools to detect desired cell phenotypes. With the cloning of PI1, lentiviral expression cloning has become a molecular reality and the newest chapter in the success story of expression technology. Taking advantage of pantropic transduction capacity and the ability to deliver transgenes into non-proliferating primary cells, lentiviral expression cloning is expected to enable a new dimension in gene discovery and gene-function analysis in the post genomic era.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Urs Ziegler for providing cell lines, Barbara Mitta for lentivirus production and Katri Pajusola, Oleg Georgiev as well as Josef Jiricny for valuable discussions. This work was supported by the Swiss National Science Foundation (grant no. 631-065946), the Roche Research Foundation (grant no. 118-2001) and the Novartis Foundation (grant no. 01C41).
DDBJ/EMBL/GenBank accession no. AY255855
REFERENCES
- 1.Kay M.A., Glorioso,J.C. and Naldini,L. (2001) Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nature Med., 7, 33–40. [DOI] [PubMed] [Google Scholar]
- 2.Lewis P.F. and Emerman,M. (1994) Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol., 68, 510–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naldini L., Blomer,U., Gallay,P., Ory,D., Mulligan,R., Gage,F.H., Verma,I.M. and Trono,D. (1996) In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science, 272, 263–267. [DOI] [PubMed] [Google Scholar]
- 4.Klimatcheva E., Rosenblatt,J.D. and Planelles,V. (1999) Lentiviral vectors and gene therapy. Front Biosci., 4, D481–D496. [DOI] [PubMed] [Google Scholar]
- 5.Zufferey R., Dull,T., Mandel,R.J., Bukovsky,A., Quiroz,D., Naldini,L. and Trono,D. (1998) Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J. Virol., 72, 9873–9880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitta B., Rimann,M., Ehrengruber,M.U., Ehrbar,M., Djonov,V., Kelm,J. and Fussenegger,M. (2002) Advanced modular self-inactivating lentiviral expression vectors for multigene interventions in mammalian cells and in vivo transduction. Nucleic Acids Res., 30, e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang Z., Ruschitzka,F., Rabelink,T.J., Noll,G., Julmy,F., Joch,H., Gafner,V., Aleksic,I., Althaus,U. and Luscher,T.F. (1997) Different effects of thrombin receptor activation on endothelium and smooth muscle cells of human coronary bypass vessels. Implications for venous bypass graft failure. Circulation, 95, 1870–1876. [DOI] [PubMed] [Google Scholar]
- 8.Mochizuki H., Schwartz,J.P., Tanaka,K., Brady,R.O. and Reiser,J. (1998) High-titer human immunodeficiency virus type 1-based vector systems for gene delivery into nondividing cells. J. Virol., 72, 8873–8883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reiser J., Harmison,G., Kluepfel-Stahl,S., Brady,R.O., Karlsson,S. and Schubert,M. (1996) Transduction of nondividing cells using pseudotyped defective high-titer HIV type 1 particles. Proc. Natl Acad. Sci. USA, 93, 15266–15271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fussenegger M. and Bailey,J.E. (1998) Molecular regulation of cell-cycle progression and apoptosis in mammalian cells: implications for biotechnology. Biotechnol. Prog., 14, 807–833. [DOI] [PubMed] [Google Scholar]
- 11.Fussenegger M., Morris,R.P., Fux,C., Rimann,M., von Stockar,B., Thompson,C.J. and Bailey,J.E. (2000) Streptogramin-based gene regulation systems for mammalian cells. Nat. Biotechnol., 18, 1203–1208. [DOI] [PubMed] [Google Scholar]
- 12.Fussenegger M. (2001) The impact of mammalian gene regulation concepts on functional genomic research, metabolic engineering and advanced gene therapies. Biotechnol. Prog., 17, 1–51. [DOI] [PubMed] [Google Scholar]
- 13.Yancopoulos G.D., Davis,S., Gale,N.W., Rudge,J.S., Wiegand,S.J. and Holash,J. (2000) Vascular-specific growth factors and blood vessel formation. Nature, 407, 242–248. [DOI] [PubMed] [Google Scholar]
- 14.Odelberg S.J., Kollhoff,A. and Keating,M.T. (2000) Dedifferentiation of mammalian myotubes induced by msx1. Cell, 103, 1099–1109. [DOI] [PubMed] [Google Scholar]
- 15.Eriksson P.S., Perfilieva,E., Bjork-Eriksson,T., Alborn,A.M., Nordborg,C., Peterson,D.A. and Gage,F.H. (1998) Neurogenesis in the adult human hippocampus. Nature Med., 4, 1313–1317. [DOI] [PubMed] [Google Scholar]
- 16.Alvarez-Buylla A., Garcia-Verdugo,J.M. and Tramontin,A.D. (2001) A unified hypothesis on the lineage of neural stem cells. Nature Rev. Neurosci., 2, 287–293. [DOI] [PubMed] [Google Scholar]
- 17.Bjorklund A. and Lindvall,O. (2000) Parkinson disease gene therapy moves toward the clinic. Nature Med., 6, 1207–1208. [DOI] [PubMed] [Google Scholar]
- 18.Bjorklund A. and Lindvall,O. (2000) Self-repair in the brain. Nature, 405, 892–893, 895. [DOI] [PubMed] [Google Scholar]