

Summary
Site-specific recombinases of the λ-integrase family recognize and cleave their cognate DNA sites through cooperative binding to opposite sides of the DNA substrate by a C-terminal catalytic domain and a flexibly linked ‘core-binding’ domain; regulation of this cleavage is achieved via the formation of higher-order complexes. We report that the core-binding domain of λ-integrase is able to stimulate the activity of the catalytic domain even when the two domains are not linked. This trans stimulation is accomplished without significantly increasing the affinity of the catalytic domain for its DNA substrate. Moreover, we show that mutations in the DNA substrate can abrogate this effect while retaining specificity determinants for cleavage. Since the domains do not significantly interact directly, this finding implies that trans activation is achieved via the DNA substrate in a manner that may be mechanistically important in this and similar DNA binding and cleaving enzymes.
Keywords: lambda integrase, DNA recombinase, tyrosine recombinase, covalent intermediate, suicide substrate, substrate binding, DNA binding, NMR, single turnover kinetics, fluorescence anisotropy
Introduction
Bacteriophage λ integrase (λ-Int) 1 is the prototypical member of a family of enzymes that mediate site-specific DNA recombination via the formation of a covalent 3’ phosphotyrosyl linkage at the site of DNA cleavage in a manner similar to that of type I eukaryotic and viral topoisomerases 2. DNA cleavage and recombination by λ-Int at its cognate “core” sites proceeds upon assembly of four protomers of the enzyme at a pair of T-rich semi-symmetrical inverted sequences on the phage and bacterial genomic DNA 3. Although the enzyme functions as a tetramer during recombination, the enzyme can also bind and cleave DNA without tetramer assembly 4, as experiments in vitro show it exhibits topoisomerase activity 5 and is capable of cleaving small DNA substrates consisting of core half-sites 6.
The 356-residue λ-Int enzyme consists of three structurally independent domains connected by extended peptide linkers: the amino-terminal “arm binding” domain (residues 1–64), the central “core-binding” domain (residues 65–169), and the C-terminal catalytic domain (residues 170–356) 7; 8; 9. Although the catalytic domain is capable of sequence-specific cleavage of core DNA sites on its own, maximal cleavage activity requires a construct containing both the catalytic and “core-binding” domains of the protein; this construct is as active as the full-length protein in cleaving DNA 8; 9. The “arm binding” domain and other protein factors are not directly involved in core site recognition and cleavage, but are required for efficient recombination 9; 10; 11; 12; 13; 14; 15.
Crystallographic 16 and solution 17 structural studies of the isolated catalytic domain (residues 170–356) revealed that in the absence of DNA the tyrosine nucleophile (Y342) resides on a flexible C-terminal segment that adopts an average conformation far from that required for substrate cleavage. This observation led to the proposal that a concerted structural rearrangement plays a regulatory role and is required for generation of an “active” conformation with the tyrosine residue suitably positioned for DNA cleavage 16; 18. Intriguingly, although stoichiometric DNA binding by the catalytic domain was found to stabilize the protein structure 19, this interaction was insufficient to significantly populate the active conformation in solution 17.
On the other hand, the crystal structures of a λ-Int construct containing the core-binding and catalytic domains of the enzyme, residues 75–356, trapped in covalent complexes with half-site 20 or full core site 15 DNA substrates show the tyrosine residue properly positioned for DNA cleavage. In a fashion similar to that of two other members of the family, Cre and Flp recombinases 21; 22, the structures show that the two domains bind to opposite sides of the substrate DNA and are connected by an extended linker (Figure 1)15; 20. In the tetrameric complexes with full sites 15, modest protein-protein interactions are observed between the core binding domains of adjacent protomers, while more extensive interactions between the catalytic domains may be important for regulating activity. In particular, inter-protomer interactions mediated by the C-terminal segment of the protein are believed to have an important regulatory role 15; 20; 23; 24. In each of these structures, the DNA is distorted from canonical B-form, with a widening of the major groove at the sites of helix insertion and a narrowing of the minor groove at the site of cleavage. The arrangement of the core-binding domain on the opposite side of the DNA as the site of cleavage is consistent with a role for this domain in increasing the affinity of the enzyme for its substrate by anchoring the catalytic domain on the DNA.
Figure 1.

Cartoon diagram of the tetrameric complex observed in the crystal structure of residues 75–356 of λ-Int covalently bound to a suicide DNA substrate (1Z19.pdb) 15. (A) The core-binding domain (top) binds on the opposite side of the DNA substrate as the catalytic domain (bottom). An extended linker connects the domains in the intact enzyme. DNA is shown in molecular surface representation, with each continuous strand colored differently; the bright orange strand is covalently attached to the green protomer. (B) Close-up of the protein-DNA and inter-protomer interfaces observed in the crystal structure. The core-binding domain binds the DNA opposite the site of cleavage (indicated by Y342, in red). The only contacts between the two domains are the linker (indicated in the green protomer), and a contact between the sidechain of K93 (indicated in the blue protomer) and the backbone carbonyl of S234 in a hairpin loop. The C-terminus of the green protomer, containing W350, makes contacts to the blue protomer.
To reconcile the observed properties of the C-terminal segment of the protein in the isolated catalytic domain (IntCat)16; 17 with those observed in a construct also containing the core-binding domain (IntCB) 15; 20, we investigated the effect of isolated IntCB on the catalytic and DNA binding properties of IntCat. Here we report the surprising finding that IntCB and IntCat act cooperatively even when they are not covalently linked. Since crystallographic 15; 20 and solution data (this work) show that the two domains do not significantly interact directly, this cooperativity must be accomplished in an indirect manner. The mechanistic implications for such an indirect means of cooperation between catalytic and accessory DNA binding domains of enzymes are discussed.
Results
The core-binding domain activates catalysis in trans
Proteolytic mapping experiments previously identified the three structural domains of λ-Int 9; 25; 26, and crystallographic and NMR studies established the structural independence of the catalytic 16 and arm- binding domains 27. We expressed and purified four different protein constructs comprising fragments of the enzyme involved in core site binding and cleavage: (1) IntCB, a protein construct containing the core- binding domain of λ-Int, (2) IntCB+Cat, containing both the central and catalytic domains, (3) IntCat, the isolated catalytic domain, and (4) IntCatΔ349C197S, a fragment of the catalytic domain bearing a Cys-Ser substitution at position 197 to improve sample behavior and lacking the last seven residues from the C-terminus; these residues have been implicated in mediating inter-protomer interactions important for coordinated cleavage 23; 24; 28; 29. All constructs containing the catalytic domain, (i.e., IntCB+Cat, IntCat and IntCatΔ349C197S) were competent for site-specific cleavage of DNA half-site substrates 6; 26, with the IntCatΔ349C197S construct exhibiting slightly increased activity 24 and improved stability compared to IntCat.
Incubation of IntCat with a suicide core half-site DNA substrate 6; 17; 30 results in site-specific DNA cleavage and generates a covalent complex with IntCat attached to the DNA via a 3’-phosphotyrosine linkage that is electrophoretically stable under denaturing conditions (Figure 2, lane 1). As previously noted 8, the cleavage activity of the catalytic domain alone (IntCat) is several fold lower than that of a construct containing both the catalytic and core-binding domains (IntCB+Cat; Figure 2, lane 3). Surprisingly, we found that IntCB stimulates the activity of IntCat in trans, i.e., even when the two domains are not covalently linked (Figure 2, lanes 1 and 2). This result indicates that IntCB plays a role in DNA cleavage beyond enhancing the affinity of the catalytic domain for its substrate DNA.
Figure 2.

Trans activation of IntCat by IntCB. DNA cleavage activity monitored by denaturing SDS-PAGE analysis of the formation of a covalent complex between the protein and a 5’-radiolabeled DNA suicide substrate. Lane 1, the catalytic domain alone (IntCat); lane 2, the catalytic domain in the presence of the (separately added) core-binding domain (IntCat + IntCB); lane 3, the linked core and catalytic domains in the context of IntCB+Cat (lane 3). The concentration of the DNA substrate was 0.1 μM, while the IntCat, IntCB and IntCB+Cat concentrations were 4 μM. Addition of IntCB results in a large increase in complex formation, while linking the two domains provides an additional enhancement.
Because of the low activity and relatively low affinity of IntCat for DNA 8, several controls were performed to establish whether the observed enhancement was due to nonspecific or chaperone-like effects 31. Bovine serum albumin was used in place of IntCB to show that the increased activity was not due to solution stabilization 32. The core-binding domain of Cre recombinase (CreCB; residues 1–130) 33 was used in place of IntCB to show that nonspecific DNA binding cannot account for the effect. Excess salmon sperm DNA was used as a nonspecific competitor to show that trans complementation could not be eliminated by dilution of weakly binding proteins. The bottom strand was 32P-labeled to show that the observed DNA cleavage activity by IntCat is not due to nonspecific topoisomerase-like activity,5 which would be expected to result in cleavage of either strand. In each of these controls, catalytic enhancement of IntCat was only observed when IntCB was added, nonspecific competitor DNA had no effect, and only site-specific cleavage was observed 31. The results of these control experiments effectively rule out nonspecific effects as being responsible for the observed trans complementarity beween IntCB and IntCat.
IntCB and IntCat do not interact in the absence of DNA
Comparison of the two-dimensional 1H-15N correlation NMR spectrum of the larger IntCB+Cat with that of IntCat (Figure 3) reveals the positions of resonances from the catalytic domain in IntCB+Cat are nearly identical to those in spectra of catalytic domain alone 17. This finding indicates that in the absence of DNA, except for the linker connecting them the two domains do not significantly interact. Biophysical and biochemical analysis of IntCB alone showed that the isolated domain, a helical bundle 20, is monomeric but not well folded when free in solution, becoming more structured upon binding to its cognate DNA 38. The NMR spectra of IntCB+Cat indicate that the core-binding domain is also not well structured in the context of the larger construct, as most of the resonances not attributable to IntCat are clustered in the random coil region of the spectrum, with amide proton shifts of 8–8.5 ppm (Figure 3).
Figure 3.

In the absence of DNA, catalytic and core binding domains of λ-Int are independent. Left, overlay of two-dimensional 1H/15N-HSQC spectra of IntCB+Cat (black) and IntCat (red). Near-perfect superposition of signals from the catalytic domain in isolation (IntCat) and when linked in tandem with the core binding domain indicate the domains do not significantly interact. Right, ribbon model of IntCB+Cat from the crystal structure of its complex with a half-site DNA substrate 20.
Trans activation is DNA sequence dependent
Since the catalytic and core binding domains do not significantly interact in the absence of DNA, nor do they exhibit significant inter-domain contacts in the presence of DNA, we examined whether the observed trans-activating effect might be mediated via the DNA substrate. This hypothesis was tested by examining the effect of alterations of the DNA substrate on the DNA cleavage activity of IntCat in the absence and presence of IntCB. Examination of the crystal structure of IntCB+Cat (residues 75–356) covalently bound to DNA (Figure 4A) reveals that the only bases contacted by both domains are two adenines at positions 5 and 6 downstream from the center of the overlap region 17, and one nucleotide from the site of cleavage; these residues are two of three invariant adenines in the consensus λ-Int recognition sites. Inspired by studies of cooperativity between the homeodomain and POU-specific domain of the Oct-1 transcription factor 34, we reasoned that if cooperativity between the two domains were mediated by the DNA substrate, alteration of these two nucleotides would interfere with trans activation.
Figure 4.
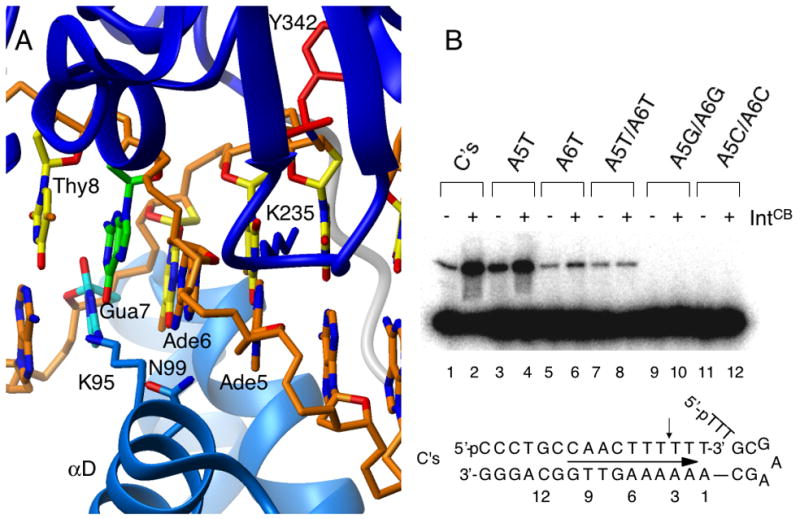
Trans complementarity between IntCB and IntCat is DNA sequence dependent. (A) Close-up of the closest contact between IntCat (blue) and IntCB (cyan) in the crystal structure (1P7D) 20 (B) Cleavage of DNA substrates by IntCat in the absence (odd-numbered lanes) and presence (even lanes) of IntCB. The oligonucleotide substrates tested are the “wild-type” C’s and variants with adenine nucleotides at positions +5 and +6 mutated to T, G or C in pairs and individually. Trans activation is lost when both adenines are mutated to T, while variants with those positions mutated to G or C are no longer recognized and cleaved by IntCat.
As shown in Figure 4B, a substrate with the AT basepair at position +5 altered to TA was recognized and cleaved by IntCat, and addition of IntCB resulted in a strong enhancement in cleavage activity (lanes 3, 4). Alteration of the AT basepair at position +6 to TA resulted in decreased cleavage by IntCat, but addition of IntCB nevertheless enhanced cleavage (lanes 5, 6). However, cleavage of a substrate variant in which both AT basepairs are changed to TA is no longer enhanced by addition of IntCB although it is still cleaved specifically by the catalytic domain (lanes 7, 8). As an additional control, altering both AT basepairs to either GC or CG (lanes 9–12), completely abrogated recognition and cleavage by IntCat. These results suggest that contacts between IntCB and these nucleotides are important for the observed trans activation and are consistent with a DNA-mediated interaction between the domains.
IntCB has a large effect on kcat
Trans activation of an enzyme can be accomplished by increasing the affinity of the enzyme for its substrate (i.e., lowering KM) and/or by promoting catalytic efficiency (i.e., increasing kcat). Low solubility and a tendency to aggregate complicate detailed kinetic and thermodynamic analyses of DNA binding by λ-Int, and IntCat in particular. Nevertheless, we endeavored to characterize this trans effect by measuring the effect of IntCB on the kinetics of DNA cleavage/covalent complex formation.
The efficiency of IntCat for DNA cleavage is strongly dependent on solution conditions 31. Furthermore, since the reaction catalyzed by the domain is slow, degradation (i.e., denaturation, precipitation) of the protein during the reaction can be a complicating factor. To minimize these complications, we sampled a large array of reaction conditions and arrived at a set that enabled collection of interpretable data (see Materials and Methods). Most notably, measurements of initial rates of product formation were performed at 4 °C under the same solution conditions we routinely use for long-term storage (2–3 months), which we have found both from kinetic and spectroscopic analyses to preserve the integrity of IntCat over timescales relevant to the kinetic measurements (hours). Representative time course data are shown in Figure 5.
Figure 5.
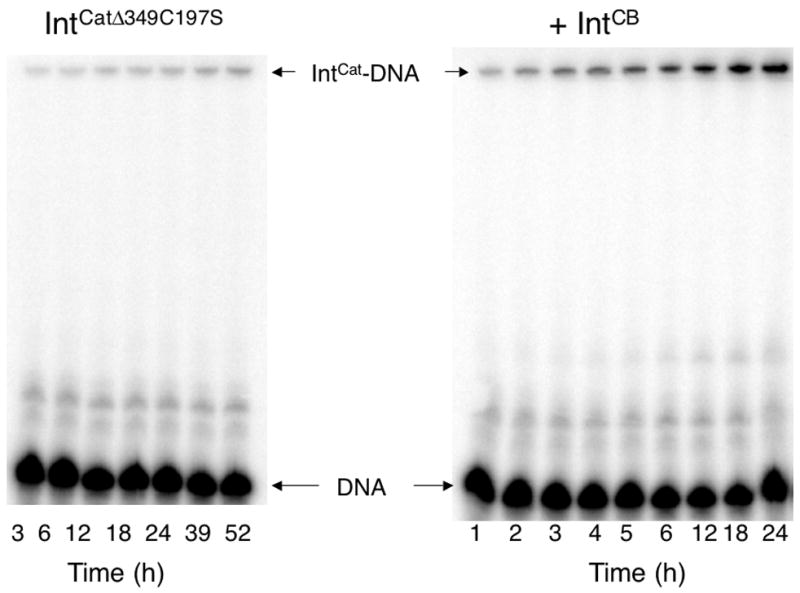
Time course for DNA cleavage by IntCat in the absence (left) and presence (right) of IntCB. Bands in the denaturing SDS-PAGE gel indicate the migration position of uncleaved 32P-labeled substrate DNA and of IntCat in covalent complex with the trapped suicide substrate. IntCatΔ349C197S was used to obtain the kinetic data because the mutant is more active and is less prone to precipitation than wild-type IntCat; trans activation by IntCB was observed for both.
Single turnover DNA cleavage kinetics were monitored by measuring the time-dependent formation of covalent IntCat-DNA complexes as a function of enzyme concentration while using a double-stranded oligonucleotide suicide substrate radiolabeled on the 5’ end of the cleaved strand 17. Although the reactions were quite slow under the conditions of the experiments (i.e., 4 °C), timepoints recorded in triplicate showed the data to be reasonably reproducible (Figure 5), allowing determination by nonlinear regression (Figure 6) of the initial rates of DNA cleavage by IntCat, in both the absence and presence of IntCB. Although auto-inhibition at higher enzyme concentrations increased uncertainty in the extracted parameters, analysis of the initial rates allowed determination of the corresponding kcat and KM values. IntCat was found to have a of 25 ± 23 μM in the absence of IntCB, and 5 ± 4 μM in the presence, while kcat increased from 0.08 ± 0.04 s−1 to 3 ± 1 s−1. Although the uncertainty in the kinetic parameters is large, the data indicate that addition of IntCB strongly enhances the turnover rate of the enzyme (kcat) by a factor of ~40, while there appears to be a less significant effect on KM (Figure 6). Overall, the efficiency of DNA cleavage ( kcat/KM) is enhanced by ~200 fold in the presence of IntCB.
Figure 6.

Pre-steady state kinetics analysis of DNA cleavage by IntCat indicates trans activation by IntCB is due to improved efficiency, not tighter substrate binding. Left, initial rates of product formation were monitored as a function of IntCat concentration in the absence (top) and presence (bottom) of IntCB. Right, concentration-dependence of the initial DNA cleavage rates, in the absence (top) and presence of IntCB (bottom).
IntCB does not enhance DNA binding by IntCat
Because of the uncertainties in the extracted KM values, the affinity of IntCB for its substrate was determined independently. Both IntCat and IntCB demonstrate relatively weak affinities for DNA, with neither forming electrophoretically stable (non-covalent) complexes in non-denaturing electrophoresis experiments 31. Because the specific activity of the enzyme is quite low at 4 °C, by performing binding assays quickly (relative to the enzyme’s activity), equilibrium affinity for substrate could be measured directly without significant contribution from covalent product formation.
To measure DNA binding affinity by the various λ-Int constructs, a duplex DNA substrate matching the recognition sequence used for the kinetic experiments (see Methods) was labeled at the 3’-end of one strand with 6-carboxy-Fluorescein, and protein binding was monitored by changes in fluorescence anisotropy (Figure 7). These measurements yielded equilibrium dissociation constants ( KD) for this substrate of 17 ± 2 μM for IntCB, 11 ± 1 μM for IntCat and 34 ± 3 nM for IntCB+Cat. Notably, if the two domains were perfectly cooperative in binding to DNA, one would expect the affinity of IntCB+Cat to correspond to the product of the dissociation constants of the individual domains, i.e., 0.19 nM. The ratio of the affinities of the separate and linked constructs, (KD,CBKD,Cat/KD,CB+Cat) yields the effective concentration of one linked domain in the presence of the other 35, which in this case is 5.5 mM; this result is similar to that obtained for the cooperating domains of Oct-1 34. Importantly, the affinity of IntCat for the DNA was not significantly enhanced by the presence of near-saturating IntCB. Although the binding curve could be fit to a KD of 60 ± 10 μM, the small amplitude change in anisotropy renders this result qualitative, not quantitative: that is, the effect on binding is well below the effect on catalysis (i.e., much less than the ~200 fold enhancement in kcat/KM). This finding indicates that the strong trans activating effect of IntCB is accomplished by enhancing catalytic efficiency, not by promoting substrate binding by IntCat.
Figure 7.

IntCB has a negligible effect of DNA binding affinity by IntCat. (A) Binding to a fluorescently labeled DNA substrate by IntCB (“+” marks), IntCat alone (open circles) and IntCat in the presence of near-saturating (70%) IntCB (filled circles) was monitored by fluorescence anisotropy. Data were fit using nonlinear regression to a one-site binding model; contributions from non-specific binding were determined from global fitting of all three datasets. (B) Binding of IntCB+Cat to the same fluorescently labeled DNA substrate as in A.
Discussion
Although the isolated catalytic domain is capable of specific recognition and cleavage of half-core DNA substrates, maximal cleavage activity requires the core binding domain. The poor activity of isolated IntCat is attributable in part to a reduced binding affinity for DNA: in the absence of a covalent bond to the DNA it does not produce electrophoretically stable complexes in the presence of nonspecific competitor DNA 8; 9; 31. The difference in activity between IntCB+Cat and IntCat could be potentially be interpreted in a straightforward manner by invoking the “chelate effect” 35: IntCB, in the context of IntCB+Cat serves as an anchor to increase the effective concentration of the catalytic domain in the vicinity of the DNA cleavage site. However, the NMR data indicate there is little or no interaction between the two domains in the absence of DNA (Figure 3), consistent with the observed absence of interdomain contacts in the IntCB+Cat-DNA crystal structures (Figure 1). Thus, although the chelate effect clearly contributes to the DNA cleavage activity of the bipartite protein (IntCB+Cat), the observed trans complementation indicates there’s more to the story.
In this work we report three critical observations: 1) IntCB activates IntCat even when the two domains are not linked, 2) the trans activation can be abrogated through mutations in the DNA substrate, and 3) the effect is manifested through increased catalytic efficiency ( kcat), as opposed to substrate binding (KM). These observations invite intriguing mechanistic hypotheses.
One plausible explanation for the observed activation derives from the native functional form of the enzyme, in a tetrameric “intasome” 36 structure, implicating IntCB-promoted tetramerization. The crystal structure of IntCB+Cat in a tetrameric complex with two core recognition full-sites (Figure 1) 15 reveals protein-protein interactions that must be important for coordinated cleavage of two strands at a time. Close inspection of those structures reveals that packing of IntCB domains appears to be somewhat loose, with only a few interprotomer contacts (only ~ 600 Å2 buried) involving residues Ala125, Thr168 and Arg169 on one protomer and Ala156, Arg152, Asp149 and Glu153 on the other. On the catalytic domain, residues at the C-terminus of the protein (347–356) rearrange relative to their observed position 16 in the isolated IntCat structure and pack against strands β1– β3 on the adjacent protomer. Indeed, it has been proposed that these interactions between adjacent catalytic domains is a dominant contributor to stabilization of the “active” conformation of the enzyme 16; 18; 23. Additional close IntCat contacts were observed between Lys334 and Ser335 on one protomer, and Asp344 and Ser340 on the Tyr342-bearing helix (αM) on the other. Consequently, if trans addition of IntCB were to promote the formation of higher-order complexes that bring together IntCat domains, this protein-protein interaction could lead to an increase in kcat without affecting DNA binding.
Although attractive in some respects, this model is not well supported by other data. First, the DNA substrate used for these studies, a “half-core site” is not long enough to support assembly of two protomers on the same DNA strand in a geometry that would favor stimulatory protein-protein interactions. Second, full-length λ-integrase, although prone to aggregation, has been shown to be monomeric in the absence of DNA 37, indicating that it is not a strong promoter of higher-order complexes. The monomeric nature of IntCat and IntCB are also well supported by experimental evidence: size exclusion chromatography and NMR resonance linewidths (IntCat)17; 31, and NMR and analytical ultracentrifugation data (IntCB) 38, which show both constructs to be monomeric under a wide range of solution conditions. Importantly, the construct of the catalytic domain used to obtain the kinetic parameters, IntCatΔ349C197S, lacks most of the C-terminal residues, including Trp350 and Ile353, which are attributed with making stimulatory protein-protein interactions in the recombining intasome 23; 24. The fact that deletion of these residues does not diminish trans activation by IntCB argues against multimerization playing an important role in the process. Although it is difficult to rule out the possibility that transient formation of protein-protein contacts are formed that could be stimulatory, it is not clear how isolated IntCB could promote, in a DNA sequence-dependent fashion, productive interactions between IntCat protomers.
An more compelling explanation for these observations is that the trans activating effect of IntCB on IntCat is mediated through the DNA substrate, by inducing a conformational change in the substrate so that it adopts a structure that is more readily acted upon by the catalytic domain. Indeed, the fact that sequence alterations at the IntCB binding site can specifically abrogate trans activation without affecting cleavage specificity strongly argues for a DNA substrate-mediated effect. In each of the available structures of Int-DNA complexes the DNA duplex is distorted away from its B-form structure with a widening of the major groove at the site of IntCB binding and a narrowing of the minor groove at the site of cleavage 15; 20; this is also a feature of the DNA cleavage sites of the related recombinases, Cre 22; 39; 40 and Flp 21. Although the structure of the IntCB-DNA complex has not been determined, it seems likely that the domain is responsible for at least some of the observed distortions in the cognate DNA structure.
The notion that IntCB pre-forms the DNA substrate for efficient cleavage in an “induced fit” manner 41; 42 is in line with biochemical 43 and single molecule force 44 studies arguing for an active role by DNA substrates in cleavage by restriction enzymes. This finding may also help explain important determinants of DNA site recognition and cleavage efficiency: the protein makes many contacts to the DNA phosphate backbone but very few contacts to the bases 15; 20; a clear and perhaps critical contact is that between Asn99 and Ade+6 (Figure 4a). Nevertheless, the enzyme exhibits a high degree of specificity for its cognate sites and sufficient versatility to recognize both halves of the asymmetric phage and bacterial core sites 3; 45. The enzyme’s versatility and fidelity might be explained by the fact that both core half-site sequences are AT-rich, suggesting that shape and/or deformability are important determinants for DNA binding and cleavage by λ-Int. Given the existence of integrase homologs that possess only the catalytic domain (i.e., the E. coli FimB, FimE inversion elements 46; 47), the findings reported here invite some tantalizing speculation about the evolution and mechanism of recruitment of the core binding domain by a functional catalytic domain.
Conclusion
The activity of λ-Int is regulated through intricately intertwined intra- and inter-molecular couplings; the findings reported here reveal yet a new level of complexity with potentially broad implications. The most interesting observation of this work is that IntCB, an auxiliary domain of an enzyme, serves a novel role in DNA substrate recognition and cleavage, not via protein-protein interactions with the catalytic domain, but via important interactions with the substrate. In addition, considering the prevalence of bipartite structures in DNA cleaving enzymes (e.g., topoisomerases, restriction enzymes, base excision repair enzymes, ligases; see 48; 49 for reviews), it may be worth investigating whether the present means of domain cooperation may be a general feature of such enzymes. These observations yield new insights into the complexities of sequence recognition, induced fit and catalysis.
Methods
Protein samples
The IntCB+Cat, IntCat and IntCB protein expression constructs used in this study 8; 9 (generously provided by A. Landy, Brown University) encompass residues 65–356, 170–356 and 62–176 of the wild type λ-Int, respectively. A truncated form of the catalytic domain, IntCatΔ349C197S, lacking the seven C-terminal residues and a cysteine to serine mutation at residue 197, was generated by introducing an extra stop codon using the “Quik-Change” procedure (Stratagene, Inc) 31. Protein expression and purification were performed as described previously 17; 19, except that for the DNA cleavage assays the final gel filtration column was omitted; sample purity was judged to be > 90% by SDS-PAGE analysis. After purification, the proteins were exchanged by gel filtration (Sephadex G-25) into cleavage buffer: 25 mM Tris (pH 9.1), 100 mM NaCl, 5 mM DTT, 5 mM EDTA, 10% DMSO, 0.5 mM 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (Pefabloc; Roche). Uniformly 15N-labeled IntCat and IntCB+Cat were obtained by culturing transformed bacterial cells in vitamin-enriched M9 minimal media with 15N-ammonium chloride as the sole nitrogen source 17; NMR spectra were obtained in 25 mM Tris (pH 7.2), 400 mM NaCl, 1 mM EDTA, 2 mM DTT. The core-binding domain of Cre recombinase (CreCB; residues 1–130) was obtained by site-directed mutagenesis from a plasmid encoding the full-length protein (obtained from G. van Duyne, University of Pennsylvania). The protein was expressed in E. coli using an approach similar to that used for IntCB and transferred into cleavage buffer (C. Amero and M. Foster, unpublished).
DNA samples
DNA oligonucleotides were obtained from MWG Biotech, Inc. or IDT, Inc. For DNA cleavage experiments, a hybrid C/C’ hairpin “flap” substrate was used (Figure 7)17; 18; 20, with a top strand (5’-*pCCC TGC CAA CTT T↓TT T-3’) radiolabeled with 32P at the 5’-end 50. The bottom strand (5’-pTTT GCG AAG CAA AAA AGT TGG CAG GG-3’) was chemically phosphorylated at the 5’ end (or enzymatically labeled with 32P as a control). The site of cleavage of the top strand between the −3 and −4 nucleotides is indicated by “↓”, and the +5 and +6 adenines and their corresponding (−6, −5) thymines are bold. Nucleotides incorporated for increased stability are italicized, and the three-nucleotide hairpin in the bottom strand is underlined. Sequence specificity of the trans activating effect was tested with single or double alterations of both strands of the substrate, as indicated in Figure 7.
Duplex DNA oligonucleotides for fluorescence anisotropy measurements corresponded to an 18-bp consensus C'/C half-site 17, with a top-strand sequence of 5'-CCC TGC CAA CTT T↓TT TGC-3'-6-FAM, where 6-FAM is 6-carboxyfluorescein; italicized nucleotides were added to enhance duplex stability. DNA duplexes were assembled by mixing equimolar concentrations of the DNA strands dissolved in buffer, heating to 95 °C for 5 min and cooling on ice for 15 min.
Single Turnover DNA Cleavage Experiments
Covalent complex formation was monitored upon incubating the enzyme with 50 nM 5’ radiolabeled suicide DNA substrate in cleavage buffer at 4 °C. Single time-point and control experiments were performed with 0.1 μM DNA, 4 μM IntCat, and when present, 4 μM IntCB, 4 μM CreCB, 0.1% or 1% (w/v) BSA, 0.1 or 1 mg/ml salmon sperm DNA31. Formation of the suicide complex was monitored after 18 hours by quenching with SDS Laemmli buffer, separating by electrophoretic mobility shift using 12% SDS-PAGE gels, and visualizing by autoradiography.
For kinetics experiments, IntCat concentrations were as indicated in the legend to Figure 6; IntCB was provided at a concentration of 134 μM (~80% saturated, based on measured KD, below). Time points for kinetic studies were obtained by quenching aliquots of the reaction with an equal volume of SDS Laemmli buffer, subjected to electrophoretic separation, visualized by autoradiography and quantitated by phosphorimager analysis (ImageQuant, GE Healthcare). Experiments were performed in triplicate and band intensities were used to calculate the mean and standard deviation of the reaction progress. Initial velocities were obtained by fitting the mean fractional progress to the integrated first order rate expression for product formation:
where P and S are the intensities of the product and substrate bands and v0 is the initial velocity. The concentration dependence of the initial velocities were used to determine kcat and KM values from a nonlinear weighted least-squares fit of the initial velocity data to:
where [E]T and [S]T correspond to the total, enzyme and substrate concentrations (see appendix for details). Reported uncertainties in kcat and KM values correspond to the 95% confidence interval obtained from the Student’s T test. Nonlinear regression analysis was performed with Igor Pro v. 5.05 (Wavemetrics, Inc)
Spectroscopy
Two-dimensional FHSQC51 NMR spectra of IntCat and IntCB+Cat on a 800 MHz NMR spectrometer at 25 ºC, and processed with NMRPipe 52. Fluorescence anisotropy data were recorded on a Fluorolog spectrophotometer (Jobin-Yvon, Inc.) with a 2 s integration time, 5 nm bandwidth, excitation at 495 nm and detection at 520 nm. Initial DNA concentrations were 0.015 μM (25 mM Tris pH 9.13, 100 mM NaCl, 5 mM EDTA, 5 mM DTT, 4º C).
Binding affinity
Protein-DNA dissociation constants, KD, were determined by measuring the change in fluorescence anisotropy of 6-Fam-labeled DNA as a function of protein concentration. KD values were obtained for IntCB alone, IntCat alone, IntCB+Cat and IntCat in the presence of IntCB (112 μM initial concentration), by fitting to a standard bimolecular binding quadratic53:
where A is the measured anisotropy, AFree and ABound are the anisotropy values for the free and protein-bound DNA, DT and PT are the total titrated protein and DNA concentrations, KD is the dissociation constant and m is the nonspecific effect of protein concentration on the measured anisotropy. Titrations were performed at 4 °C, with initial DNA concentrations of 15 nM in 3 mL, and stock protein concentrations were 1.37, 1.8 and 0.036 mM for IntCat, IntCB and IntCB+Cat, respectively. Data were fit by nonlinear least squares regression (Igor Pro 5.05; Wavemetrics, Inc); for the IntCB and IntCat titrations, the contribution from nonspecific effects subject to global fitting for all three titrations. Reported uncertainties correspond to the standard deviation of the fitted values. Because titration experiments were performed under conditions (4 °C) where the cleavage rate is much slower than the measurement time (~ 2 h), the effect of trace amounts of DNA cleavage on the measured anisotropy change was ignored.
Acknowledgments
We thank A. Landy (Brown U.), S. Nunes-Duby (Brown U.), G. van Duyne (U. Penn), V. Gopalan (OSU), D. Pulukkunat (OSU), C. Amero (OSU) and P. Mehta (OSU) for reagents, protocols, guidance and suggestions, V. Gopalan, Z. Suo, and M. Ibba (OSU) for access to resources and instrumentation, the OSU CCIC, and Departments of Biochemistry and Microbiology for instrumentation support. This material is based upon work supported by the National Science Foundation under Grant No. MCB-0092962.
Appendix
Single turnover kinetics analysis
The analytical treatment for single turnover kinetics with enzyme in excess has a similar form to the Michaelis-Menten kinetics for steady state, multiple turnover kinetics with substrate in excess. For the simple two-step enzymatic mechanism:
the rate of EP formation is given by:
where [ES] is the concentration of the enzyme-substrate complex. Since, for IntCat, kcat is slow relative to k1 and k−1, over short time periods (i.e., little change in [S]) we can make the pre-existing equilibrium approximation for formation of the ES complex:
where [E] and [S] refer to the free concentrations of the enzyme and substrate, respectively. Since the enzyme is present in great excess over substrate (i.e., [E]T ? [S]T, [ES]), we use conservation of mass to relate the free concentrations to the total concentrations of E and S:
Thus, the rate expression becomes:
Solving for [ES]:
The rate of product formation is then:
where Vmax is limited by [S]T at high [E]T, i.e., Vmax = kcat[S]T
Equilibrium binding quadratic
The change in fluorescence polarization (anisotropy) of the fluorescently labeled DNA due to reduced molecular tumbling upon complex formation was used to quantitate the affinity of IntCB and IntCat for DNA. For a binding-dependent spectroscopic parameter, the experimentally measured parameter (here anisotropy) is a sum of the contributions from the free and bound species as 53:
where [D] and [PD] are concentrations of free and protein-bound DNA, DT is the total DNA concentration and Ax is the anisotropy of species x. Normalizing to the total DNA concentration:
The concentrations of the free and bound species are related to the equilibrium dissociation constant KD and total protein and DNA concentrations as:
where [P] is the free protein concentration and PT is the total concentration (free plus bound). Solving this equation for [PD] and dividing by DT yields the quadratic:
Substituting this into the expression for the measured anisotropy (A), and adding a linear term for the effect of weak non-specific protein-DNA interactions the equation used to obtain the binding constants for IntCB and IntCat:
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Craig NL, Nash HA. The mechanism of phage lambda site-specific recombination: site-specific breakage of DNA by Int topoisomerase. Cell. 1983;35:795–803. doi: 10.1016/0092-8674(83)90112-5. [DOI] [PubMed] [Google Scholar]
- 2.Cheng C, Kussie P, Pavletich N, Shuman S. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell. 1998;92:841–50. doi: 10.1016/s0092-8674(00)81411-7. [DOI] [PubMed] [Google Scholar]
- 3.Ross W, Landy A. Patterns of lambda Int recognition in the regions of strand exchange. Cell. 1983;33:261–72. doi: 10.1016/0092-8674(83)90355-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee SY, Aihara H, Ellenberger T, Landy A. Two structural features of {lambda} integrase that are critical for DNA cleavage by multimers but not by monomers. Proc Natl Acad Sci U S A. 2004;101:2770–2775. doi: 10.1073/pnas.0400135101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kikuchi Y, Nash HA. Nicking-closing activity associated with bacteriophage lambda int gene product. Proc Natl Acad Sci U S A. 1979;76:3760–4. doi: 10.1073/pnas.76.8.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nunes-Duby SE, Matsumoto L, Landy A. Half-att site substrates reveal the homology independence and minimal protein requirements for productive synapsis in lambda excisive recombination. Cell. 1989;59:197–206. doi: 10.1016/0092-8674(89)90881-7. [DOI] [PubMed] [Google Scholar]
- 7.Moitoso de Vargas L, Pargellis CA, Hasan NM, Bushman EW, Landy A. Autonomous DNA binding domains of lambda integrase recognize two different sequence families. Cell. 1988;54:923–9. doi: 10.1016/0092-8674(88)90107-9. [DOI] [PubMed] [Google Scholar]
- 8.Tirumalai RS, Healey E, Landy A. The catalytic domain of lambda site-specific recombinase. Proc Natl Acad Sci U S A. 1997;94:6104–9. doi: 10.1073/pnas.94.12.6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tirumalai RS, Kwon HJ, Cardente EH, Ellenberger T, Landy A. Recognition of core-type DNA sites by lambda integrase. J Mol Biol. 1998;279:513–27. doi: 10.1006/jmbi.1998.1786. [DOI] [PubMed] [Google Scholar]
- 10.Landy A. Dynamic, structural, and regulatory aspects of lambda site-specific recombination. Annu Rev Biochem. 1989;58:913–49. doi: 10.1146/annurev.bi.58.070189.004405. [DOI] [PubMed] [Google Scholar]
- 11.Azaro MA, Landy A. Lambda Integrase and the Lambda Int Family. In: Lambowitz AM, editor. Mobile DNA II. ASM Press; Washington, D.C: 2002. [Google Scholar]
- 12.Sarkar D, Radman-Livaja M, Landy A. The small DNA binding domain of lambda integrase is a context-sensitive modulator of recombinase functions. Embo J. 2001;20:1203–12. doi: 10.1093/emboj/20.5.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sarkar D, Azaro MA, Aihara H, Papagiannis CV, Tirumalai R, Nunes-Duby SE, Johnson RC, Ellenberger T, Landy A. Differential affinity and cooperativity functions of the amino-terminal 70 residues of lambda integrase. J Mol Biol. 2002;324:775–89. doi: 10.1016/s0022-2836(02)01199-3. [DOI] [PubMed] [Google Scholar]
- 14.Radman-Livaja M, Shaw C, Azaro M, Biswas T, Ellenberger T, Landy A. Arm sequences contribute to the architecture and catalytic function of a lambda integrase-Holliday junction complex. Mol Cell. 2003;11:783–94. doi: 10.1016/s1097-2765(03)00111-4. [DOI] [PubMed] [Google Scholar]
- 15.Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T. A structural basis for allosteric control of DNA recombination by lambda integrase. Nature. 2005;435:1059–66. doi: 10.1038/nature03657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–31. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramaniam S, Tewari AK, Nunes-Duby SE, Foster MP. Dynamics and DNA substrate recognition by the catalytic domain of lambda integrase. J Mol Biol. 2003;329:423–39. doi: 10.1016/s0022-2836(03)00469-8. [DOI] [PubMed] [Google Scholar]
- 18.Tekle M, Warren DJ, Biswas T, Ellenberger T, Landy A, Nunes-Duby SE. Attenuating functions of the C-terminus of lambda Integrase. J Mol Biol. 2002;324:649–65. doi: 10.1016/s0022-2836(02)01108-7. [DOI] [PubMed] [Google Scholar]
- 19.Kamadurai HB, Subramaniam S, Jones RB, Green-Church KB, Foster MP. Protein folding coupled to DNA binding in the catalytic domain of bacteriophage lambda integrase detected by mass spectrometry. Protein Sci. 2003;12:620–6. doi: 10.1110/ps.0234303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aihara H, Kwon HJ, Nunes-Duby SE, Landy A, Ellenberger T. A conformational switch controls the DNA cleavage activity of lambda integrase. Mol Cell. 2003;12:187–98. doi: 10.1016/s1097-2765(03)00268-5. [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Narendra U, Iype LE, Cox MM, Rice PA. Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell. 2000;6:885–97. [PubMed] [Google Scholar]
- 22.Guo F, Gopaul DN, van Duyne GD. Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature. 1997;389:40–6. doi: 10.1038/37925. [DOI] [PubMed] [Google Scholar]
- 23.Kazmierczak RA, Swalla BM, Burgin AB, Gumport RI, Gardner JF. Regulation of site-specific recombination by the C-terminus of lambda integrase. Nucleic Acids Res. 2002;30:5193–204. doi: 10.1093/nar/gkf652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tekle M, Warren DJ, Biswas T, Ellenberger T, Landy A, Nunes-Duby SE. Attenuating Functions of the C Terminus of lambda Integrase. J Mol Biol. 2002;324:649–65. doi: 10.1016/s0022-2836(02)01108-7. [DOI] [PubMed] [Google Scholar]
- 25.Han YW, Gumport RI, Gardner JF. Mapping the functional domains of bacteriophage lambda integrase protein. J Mol Biol. 1994;235:908–25. doi: 10.1006/jmbi.1994.1048. [DOI] [PubMed] [Google Scholar]
- 26.Tirumalai RS, Healey E, Landy A. The catalytic domain of lambda site-specific recombinase. Proceedings of the National Academy of Sciences of the United States America. 1997;94:6104–9. doi: 10.1073/pnas.94.12.6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wojciak JM, Sarkar D, Landy A, Clubb RT. Arm-site binding by lambda -integrase: solution structure and functional characterization of its amino-terminal domain. Proc Natl Acad Sci U S A. 2002;99:3434–9. doi: 10.1073/pnas.052017999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Swalla BM, Gumport RI, Gardner JF. Conservation of structure and function among tyrosine recombinases: homology-based modeling of the lambda integrase core-binding domain. Nucleic Acids Res. 2003;31:805–18. doi: 10.1093/nar/gkg142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hazelbaker D, Radman-Livaja M, Landy A. Receipt of the C-terminal Tail from a Neighboring lambda Int Protomer Allosterically Stimulates Holliday Junction Resolution. J Mol Biol. 2005;351:948–55. doi: 10.1016/j.jmb.2005.06.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nunes-Duby SE, Matsumoto L, Landy A. Site-specific recombination intermediates trapped with suicide substrates. Cell. 1987;50:779–88. doi: 10.1016/0092-8674(87)90336-9. [DOI] [PubMed] [Google Scholar]
- 31.Subramaniam S. PhD. The Ohio State University; 2005. Studies of Conformational Changes and Dynamics Accompanying Substrate Recognition, Allostery and Catalysis in Bacteriophage Lambda Integrase. [Google Scholar]
- 32.Jaenicke R, Rudolph R. Folding proteins. In: Creighton TE, editor. Protein structure: a practical approach. IRL Press; Oxford: 1989. pp. 191–223. [Google Scholar]
- 33.Hoess R, Abremski K, Irwin S, Kendall M, Mack A. DNA specificity of the Cre recombinase resides in the 25 kDa carboxyl domain of the protein. J Mol Biol. 1990;216:873–82. doi: 10.1016/S0022-2836(99)80007-2. [DOI] [PubMed] [Google Scholar]
- 34.Klemm JD, Pabo CO. Oct-1 POU domain-DNA interactions: cooperative binding of isolated subdomains and effects of covalent linkage. Genes Dev. 1996;10:27–36. doi: 10.1101/gad.10.1.27. [DOI] [PubMed] [Google Scholar]
- 35.Zhou HX. The affinity-enhancing roles of flexible linkers in two-domain DNA-binding proteins. Biochemistry. 2001;40:15069–73. doi: 10.1021/bi015795g. [DOI] [PubMed] [Google Scholar]
- 36.Richet E, Abcarian P, Nash HA. The interaction of recombination proteins with supercoiled DNA: defining the role of supercoiling in lambda integrative recombination. Cell. 1986;46:1011–21. doi: 10.1016/0092-8674(86)90700-2. [DOI] [PubMed] [Google Scholar]
- 37.Kikuchi Y, Nash HA. The bacteriophage lambda int gene product. A filter assay for genetic recombination, purification of int, and specific binding to DNA. J Biol Chem. 1978;253:7149–57. [PubMed] [Google Scholar]
- 38.Kamadurai HB. Mechanistic Insights into Catalysis and Allosteric Enzyme Activation in Bacteriophage Lambda Integrase. The Ohio State University; 2007. [Google Scholar]
- 39.Gopaul DN, Guo F, Van Duyne GD. Structure of the Holliday junction intermediate in Cre-loxP site- specific recombination. Embo J. 1998;17:4175–87. doi: 10.1093/emboj/17.14.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guo F, Gopaul DN, Van Duyne GD. Asymmetric DNA bending in the Cre-loxP site-specific recombination synapse. Proc Natl Acad Sci U S A. 1999;96:7143–8. doi: 10.1073/pnas.96.13.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Post CB, Ray WJ., Jr Reexamination of induced fit as a determinant of substrate specificity in enzymatic reactions. Biochemistry. 1995;34:15881–5. doi: 10.1021/bi00049a001. [DOI] [PubMed] [Google Scholar]
- 43.Jeltsch A, Pleckaityte M, Selent U, Wolfes H, Siksnys V, Pingoud A. Evidence for substrate-assisted catalysis in the DNA cleavage of several restriction endonucleases. Gene. 1995;157:157–62. doi: 10.1016/0378-1119(94)00617-2. [DOI] [PubMed] [Google Scholar]
- 44.Gemmen GJ, Millin R, Smith DE. Tension-dependent DNA cleavage by restriction endonucleases: two-site enzymes are "switched off" at low force. Proc Natl Acad Sci U S A. 2006;103:11555–60. doi: 10.1073/pnas.0604463103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dorgai L, Sloan S, Weisberg RA. Recognition of core binding sites by bacteriophage integrases. J Mol Biol. 1998;277:1059–70. doi: 10.1006/jmbi.1998.1642. [DOI] [PubMed] [Google Scholar]
- 46.Klemm P. Two regulatory fim genes, fimB and fimE, control the phase variation of type 1 fimbriae in Escherichia coli. Embo J. 1986;5:1389–93. doi: 10.1002/j.1460-2075.1986.tb04372.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A. Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res. 1998;26:391–406. doi: 10.1093/nar/26.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lilley DMJ. Frontiers in molecular biology. IRL Press at Oxford University Press; Oxford ; New York: 1995. DNA-protein : structural interactions; p. 7. [Google Scholar]
- 49.Garvie CW, Wolberger C. Recognition of specific DNA sequences. Mol Cell. 2001;8:937–46. doi: 10.1016/s1097-2765(01)00392-6. [DOI] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch EF, Maniatis T. Molecular cloning : a laboratory manual. 2. Cold Spring Harbor, NY: 1989. [Google Scholar]
- 51.Mori S, Abeygunawardana C, Johnson MO, van Zijl PC. Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J Magn Reson B. 1995;108:94–8. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 52.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–93. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 53.Fersht A. Structure and mechanism in protein science: a guide to enzyme catalysis and protein folding. W.H. Freeman; New York: 1999. [Google Scholar]