

Abstract
Evolutionary genomics has benefited from methods that allow identifying evolutionarily important genomic regions on a genomewide scale, including genome scans and QTL mapping. Recently, genomewide scanning by means of microarrays has permitted assessing gene transcription differences among species or populations. However, the identification of differentially transcribed genes does not in itself suffice to measure the role of selection in driving evolutionary changes in gene transcription. Here, we propose and apply a “transcriptome scan” approach to investigating the role of selection in shaping differential profiles of gene transcription among populations. We compared the genomewide transcription levels between two Atlantic salmon subpopulations that have been diverging for only six generations. Following assessment of normality and unimodality on a gene-per-gene basis, the additive genetic basis of gene transcription was estimated using the animal model. Gene transcription h2 estimates were significant for 1044 (16%) of all detected cDNA clones. In an approach analogous to that of genome scans, we used the distribution of the QST values estimated from intra- and intersubpopulation additive genetic components of the transcription profiles to identify 16 outlier genes (average QST estimate = 0.11) whose transcription levels are likely to have evolved under the influence of directional selection within six generations only. Overall, this study contributes both empirically and methodologically to the quantitative genetic exploration of gene transcription data.
ACCORDING to the theory of adaptive radiation, colonization of new environments may promote rapid population divergence as a by-product of local adaptation to differential selective regimes (Schluter 2000). Elucidating the genetic changes underlying phenotypic evolution resulting from this process is a central objective of evolutionary biology. Reaching this goal has recently greatly benefited from the development of various experimental strategies, including genome scans, QTL analysis, and gene expression QTL (eQTL) mapping (Vasemagi and Primmer 2005). Genomewide scanning for gene transcription differences among species or populations using microarrays is increasingly being performed on a wide range of organisms (e.g., Jin et al. 2001; Brem et al. 2002; Oleksiak et al. 2002; Bochdanovits et al. 2003; Townsend et al. 2003; Derome et al. 2006; Giger et al. 2006; Roberge et al. 2006). However, the identification of genes showing significant differences in transcription levels among groups does not in itself suffice to conclude that such changes were driven by divergent selection, even when environmental conditions were controlled. Thus, the role of selection in shaping patterns of gene transcription is still contentious. For instance, Khaitovich et al. (2005) suggested that most interspecific differences in transcript levels evolve neutrally, whereas others argued that strong stabilizing selection dominates the evolution of transcriptional change (Denver et al. 2005; Rifkin et al. 2005). Theory predicts that both the magnitude and the speed of phenotypic change (including change in gene transcription level) in response to selection depend on heritability of a trait (Falconer and Mackay 1996). Yet, only a handful of studies have formally determined the heritability estimates (h2) of gene transcription (reviewed in Stamatoyannopoulos 2004; Gibson and Weir 2005).
Because transcription profiles can be considered as reflecting variation at both phenotypic and genomic levels (Whitehead and Crawford 2006), the synergy of methods that can detect the role of selection in shaping differences among populations at the phenotypic and genetic levels may represent an efficient means of investigating evolutionary changes in gene transcription profiles. On the one hand, comparing the extent of quantitative genetic differences of phenotypic traits among populations (QST) (Spitze 1993) with that observed at neutral marker loci (FST) has been widely used to assess the relative contributions of selection and drift to phenotypic divergence among populations (Merila and Crnokrak 2001; Koskinen et al. 2003). QST is defined as:
 |
(1) |
where  and
and  represent the among- and average within-population components of the genetic variance for quantitative traits, respectively. There is no theoretical constraint for applying the QST framework to gene transcription profiles (Gibson and Weir 2005). Yet, this has never been done to our knowledge. On the other hand, genome scans, which consist in analyzing many (several hundreds to several thousands) molecular markers to reveal patterns of genetic differentiation at the genome scale, have become a popular means for identifying genes evolving under the effect of selection between populations. The underlying principle of this approach is that gene flow will affect all loci across the genome, whereas selection will act locally on specific genes (Luikart et al. 2003). As a result, only a few loci will display an atypical pattern of variation caused by the influence of locus-specific forces (Storz 2005). It is then possible to reveal these “outlier” loci by comparison with the rest of the genome. Namely, outlier genes showing the highest levels of differentiation between populations will represent those that are most likely to evolve under directional selection whereas those with the lowest level of differentiation are more likely to be under the effect of stabilizing selection (Beaumont 2005).
represent the among- and average within-population components of the genetic variance for quantitative traits, respectively. There is no theoretical constraint for applying the QST framework to gene transcription profiles (Gibson and Weir 2005). Yet, this has never been done to our knowledge. On the other hand, genome scans, which consist in analyzing many (several hundreds to several thousands) molecular markers to reveal patterns of genetic differentiation at the genome scale, have become a popular means for identifying genes evolving under the effect of selection between populations. The underlying principle of this approach is that gene flow will affect all loci across the genome, whereas selection will act locally on specific genes (Luikart et al. 2003). As a result, only a few loci will display an atypical pattern of variation caused by the influence of locus-specific forces (Storz 2005). It is then possible to reveal these “outlier” loci by comparison with the rest of the genome. Namely, outlier genes showing the highest levels of differentiation between populations will represent those that are most likely to evolve under directional selection whereas those with the lowest level of differentiation are more likely to be under the effect of stabilizing selection (Beaumont 2005).
In this study, we propose and apply a “transcriptome scan” approach combining both the QST and genome scan frameworks to investigate the role of selection in shaping differential profiles of gene transcription between populations. Recently diverging (six generations) subpopulations of Atlantic salmon (Salmo salar), which, respectively, reproduce in an upstream and downstream stretch of the Sainte-Marguerite River, Canada, were used as a model system. More precisely, a 16,006-gene cDNA microarray was used for comparing the genomewide gene transcription data of the progeny from several half-sib families from these two subpopulations reared in a controlled environment. Using a quantitative genetic approach based on the animal model (Kruuk 2004), we estimated both gene transcription heritability and QST for each gene represented and detected on the microarray. We then performed a “transcriptome scan” by which the distribution of individual QST estimates for all genes with a significant transcription profile h2 was obtained and genes in the upper tail of the distribution identified as potentially under directional selection. Genes with high-transcription profile QST estimates might not be genetically linked to the causal genetic sequence differences. Nevertheless, high-transcription QST estimates are expected to pinpoint the most likely genes for which transcription profiles evolved as a result of selection and thus provide valuable information on the actual functions that have been targeted by selection.
MATERIALS AND METHODS
Habitat characteristics, sampling, and crossing design:
The Sainte-Marguerite River (48°20′ N, 70°00′ W) is located 250 km northeast of Quebec City, Canada, and is subdivided into two main branches. The northeast branch is 85 km long, but access to salmon is limited to the lower 35 km by waterfalls and was limited to the lower 6.5 km until a fish ladder was installed at the level of a waterfall in 1981. In 1997, highly significant genetic differentiation at microsatellite loci (FST = 0.028 and 0.036 (two upstream sites), P < 0.0006) was revealed between salmon subpopulations from downstream and upstream of the fish ladder (Garant et al. 2000), indicating partial reproductive isolation between them. Moreover, Aubin-Horth et al. (2006) also reported a significantly higher proportion of precocious male parr (early maturing males) and a smaller size threshold for male parr to mature upstream rather than downstream from the fish ladder. Environmental conditions also differ between river sections. The downstream stretch is surrounded by bare and unstable clay cliffs. Heavy rainfall leaches the clay from these cliffs into the river, resulting in different water coloration and chemistry relative to the upper reaches (Centre Inter-universitaire de Recherche sur le Saumon Atlantique, unpublished data). Overall, evidence for restricted gene flow as well as differences in habitat characteristics and life histories suggest that upstream and downstream salmon subpopulations may be in the course of becoming genetically adapted to their respective environments. Eight male and two female adult salmon from each of the upstream and downstream populations were captured in the summer of 2003 and 2004, and factorial crosses within subpopulations were made at the provincial fish hatchery in Tadoussac (15 km from the Sainte-Marguerite River) during the fall of those years. Crosses should have yielded a total of 64 half-sib families, but two families were lost in the first year and seven in the second, for a total of 55 families. Fertilized eggs were incubated in controlled conditions (identical for all families) and alevins (immature fish still living on food reserves of their yolk sacs; here resources from the yolk sacs were completely or almost completely depleted) were sampled at the yolk-sac resorption stage. Four individuals from each family were collected and immediately frozen in liquid nitrogen.
RNA extraction, labeling, and cDNA hybridization:
Whole frozen alevins were homogenized individually in 1 ml TRIZOL@Reagent (Invitrogen, San Diego) using a Diax 100 homogenizer (Heidolph Instruments), and total RNA was extracted as previously described (Roberge et al. 2006). Briefly, 200 μl chloroform (Sigma, St. Louis) was added to each ml of fish homogenate in Trizol. After mixing and centrifuging (12,000 × g, 0°, 15 min), the aqueous layers were transferred into new tubes where 1 ml isopropanol (Sigma) was added. Samples were then stored overnight at −80°. On the following day, they were centrifuged for 1 hr (12,000 × g, 0°) and the isopropanol was discarded. The pellets were washed with 1 ml 70% ethanol, dried for 15 min at room temperature, resuspended in 40 μl non-DEPC and treated nuclease-free water (Ambion), and spiked with 1 μl RNAse inhibitor (Ambion). For each sample, 15 μg of the pooled RNA from the four separate extractions was retro-transcribed and labeled using a Genisphere 3DNA array 50 kit, Invitrogen's Superscript II retro-transcriptase, and Cy3 and Alexa 647 dyes (Genisphere). The detailed protocol of the retro-transcription, labeling, and hybridization procedures can be found at http://web.uvic.ca/cbr/grasp/ (Genisphere Array 50 Protocol). Briefly, 15 μg total RNA was reverse transcribed using special oligo(dT) primers with 5′ unique sequence overhangs for the labeling reactions. Microarrays were prepared for hybridization by washing twice for 5 min in 0.1% SDS, five times for 1 min in MilliQ H2O, immersing 3 min in 95° MilliQ H2O, and drying by centrifugation (5 min at 800 × g in 50-ml conical tubes). The cDNA was hybridized to the microarrays in a formamide-based buffer (25% formamide, 4× SSC, 0.5% SDS, 2× Denhardt's solution) with competitor DNA [LNA dT bloker, (Genisphere), human COT-1 DNA (Sigma)] for 16 hr at 51° in a humidified hybridization oven. The arrays were washed once for 5 min at 45° (2× SSC, 0.1% SDS), twice for 3 min in 2× SSC, 0.1% SDS at room temperature (RT), twice for 3 min in 1× SSC at RT, twice again for 3 min in 0.1× SSC at RT, and finally dried by centrifugation. The Cy3 and Alexa 647 fluorescent dye attached to DNA dendrimer probes (3DNA capture reagent, Genisphere) were then hybridized to the bound cDNA on the microarray, using the same hybridization solution as earlier: the 3DNA capture reagents bound to their complementary cDNA capture sequences on the oligo(dT) primers. This second hybridization was carried out over 2 hr at 51° in a humidified hybridization oven. The arrays were then washed and dried as mentioned before.
The design for the experimental microarray hybridizations differed for each of the 2 years. In 2003, we were interested mainly in assessing the functional genomic basis of sexual precocity (C. Roberge, H. Guderley and L. Bernatchez, unpublished data), such that the progeny of one anadromous male and one precocious parr from the same subpopulation and sharing the same mother were always coupled on the same microarray, for a total of 24 microarrays and 48 offspring analyzed. For 2004, one offspring each from the upstream and downstream subpopulation was always coupled on the same microarray, for a total of 21 microarrays and 42 offspring analyzed. Thus, transcription profiles could be obtained for a total of 90 offspring representing 50 half-sib families.
Signal detection, data preparation, and ANOVA:
Hybridization signals were detected using a ScanArray scanner (Packard BioScience). Spots were located and quantified with the QuantArray 3.0 software, using the histogram quantification method and keeping the mean value of intensity for each spot. Local background and the data from bad spots were removed. Missing data were then imputed using the K-nearest neighbors imputer in SAM (Tusher et al. 2001) (15 neighbors). Data were normalized by dividing by the channel mean. Genes with mean intensity smaller than the mean intensity of control empty spots plus twice its standard deviation in both channels were discarded, leaving data from a total of 6484 detected clones to be analyzed. Prepared data were corrected for intensity-linked distortion using a regional LOWESS algorithm in the R/MAANOVA package (http://www.jax.org/staff/churchill/labsite/software/Rmaanova). The significance of the observed differences in transcription level between subpopulations was assessed using the R/MAANOVA package (Kerr et al. 2000, 2002). The ANOVA model included the “site” (above or below the ladder), “sire type” (anadromous or precocious), and “year” and “dye” terms as fixed terms. A permutation-based F-test (FS, with 1000 sample ID permutations) was then performed, and restricted maximum likelihood was used to solve the mixed-model equations. Best linear unbiased estimates of the “dye” effect were subtracted from the normalized data for each gene and dye; the obtained data were used to assess normality before to be fitted into an animal model for heritability and QST estimation (see below).
Normality and uni/multimodality assessment:
Normality is generally assumed in microarray data but rarely verified because of the small sample sizes typical in such experiments (Draghici 2003). Here, we used an R script to obtain, for each of the 6484 detected cDNA clones, Pearson's correlation test P-values and R2 between the observed Q-Q plot of the 90 gene transcription data points and that expected for a normal distribution, using the R program (version 2.3.0). A similar strategy was used by Giles and Kipling (2003) as an alternative to the Shapiro–Wilk normality test, which they considered overstringent (in the sense that, even with a small sample size, it has sufficient power to detect as significant very slight departures from normality). We also performed the Shapiro–Wilk normality test on the gene transcription data of each detected cDNA clone in R. Unimodality of gene transcription data was tested for each of the 6484 detected cDNA clones using an R script and the Diptest package (version 0.25-1). The Diptest package (Maecher and Ringach 2004) computes Hartigan's dip statistic for an empirical distribution, which is the maximum difference between this empirical distribution function and the unimodal distribution function that minimizes that maximum difference; this is consistent for testing any unimodal against any multimodal distribution (Hartigan and Hartigan 1985). Interpolating from the table of quantiles from a large simulation for Hartigan's dip test (qDiptab) in the Diptest package for R, we found that a dip statistic of 0.054 corresponds to a tail probalility of 5% (for n = 90). Hence, we interpreted transcription profiles of genes with dip > 0.054 (P < 0.05) as showing significant departure from unimodality.
Heritability and QST estimation:
Components of the genetic variance in the gene transcription level for each detected gene were estimated from an animal model (Kruuk 2004) using the Parameter Estimation Software 3.0 (PEST; Groeneveld et al. 1990) and Variance Component Estimator 5.1 (VCE; Kovac et al. 2002) programs. PEST was used for recoding phenotypic and pedigree information into a form usable by VCE (Perry et al. 2005). VCE was then used to estimate genetic and phenotypic variance within and between subpopulations using restricted maximum likelihood to solve a linear model of the form:
 |
(2) |
where y is a vector of the transcription levels for a given gene (n = 90 individuals), X is the design matrix relating the appropriate fixed effect (β) to each individual, Z1 refers to design matrices relating the appropriate random genetic effects (a, vector of the additive genetic effect; b, vector of subpopulation effect; m, vector of maternal effect; t, vector of sampling year effect) to each individual, and e is the error term. A Perl script was used to run the programs sequentially and collect the information for all genes into a single output file (available upon request). Heritability estimates and their standard errors were collected directly from VCE output files, whereas QST values were estimated from Equation 1, using the animal genetic variance in gene transcription to represent  and the variance from the parental subpopulation (upstream or downstream) as
and the variance from the parental subpopulation (upstream or downstream) as 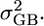 Significance of heritability and
Significance of heritability and  estimates for each gene was assessed by performing the analysis on 500 random permutations of the data, generating neutral distributions for these two quantities. Significance of
estimates for each gene was assessed by performing the analysis on 500 random permutations of the data, generating neutral distributions for these two quantities. Significance of  estimates was used to evaluate the significance of QST for genes with significant heritability estimates. Indeed, permutation tests on QST were much less accurate, generating hundreds of false positives with nonsignificant
estimates was used to evaluate the significance of QST for genes with significant heritability estimates. Indeed, permutation tests on QST were much less accurate, generating hundreds of false positives with nonsignificant  and/or
and/or  but very high and significant QST estimates.
but very high and significant QST estimates.
Functional classification:
Functional classification and assessment of significant differential representation of functional classes between subgroups of cDNA clones and all clones analyzed were performed in the DAVID/EASE environment (http://david.niaid.nih.gov/david/). The DAVID 2.1 (beta version) gene accession conversion tool was first used to convert gene ontology-linked identifications of various types gathered in the GRASP 16,006-gene microarray gene identification file to UNIGEN clusters. Assessment of significant differential representation of functional classes between subgroups of cDNA clones representing genes with significant transcription profile heritability or genes showing significant differences between subpopulations and the group of all analyzed cDNA clones was performed with EASE 2.0.
Transcriptome scan and neutrality test:
The “transcriptome scan” was performed by obtaining the distribution of individual transcription-level QST values for all detected cDNA clones corresponding to genes with significant transcriptional heritability and by identifying those in the upper (1.5%) tail of the distribution as those for which transcription was potentially under directional selection. Additionally, we performed Lande's neutrality test (Lande 1976, 1977; Koskinen et al. 2003) on gene transcription data for all detected genes with significant transcriptional heritability. The null hypothesis of evolution by random drift was tested as follows: F = (Ne  The numerator and denominator degrees of freedom are the number of populations compared −1 and ∞, respectively. Ne was estimated from previously published microsatellite data (Garant et al. 2000) using the NeEstimator software and the linkage disequilibrium method (Peel et al. 2004).
The numerator and denominator degrees of freedom are the number of populations compared −1 and ∞, respectively. Ne was estimated from previously published microsatellite data (Garant et al. 2000) using the NeEstimator software and the linkage disequilibrium method (Peel et al. 2004).
For both the QST scan and neutrality test, we obtained more realistic results when considering only genes with significant heritability estimates. This criterion makes sense from a theoretical point of view since genes with nonsignificant transcriptional h2 or intrapopulational genetic variance of transcription levels are not theoretically expected to respond rapidly to selection and evolve different expression levels (in a few generations only). We considered the complete exhaustion of genetic variance for gene expression by selection unlikely in six generations. Some genes with nonsignificant transcriptional h2 may have high transcriptional QST estimates as a result of leftover environmental variance (although this would come mainly from environmental maternal effects; we included a term for maternal effects in the animal model we used); however, we were interested here mainly in identifying genes that transcription evolved as a result of directional selection. Perhaps even more importantly, using only genes with significant transcriptional h2 allowed us to correct for artifacts resulting from very small  estimates. Hence, both the QST and Lande's neutrality test equations contain a h2 and/or
estimates. Hence, both the QST and Lande's neutrality test equations contain a h2 and/or  term in their denominator. As shown in Figure 2, h2 estimates (and hence
term in their denominator. As shown in Figure 2, h2 estimates (and hence  ) are virtually null for a majority of the 6484 detected genes. For QST, considering the genes with nearly null transcriptional heritability artifactually generates very high estimates, even in cases where
) are virtually null for a majority of the 6484 detected genes. For QST, considering the genes with nearly null transcriptional heritability artifactually generates very high estimates, even in cases where  is not significant. For Lande's test, this generates F-statistics several orders of magnitude above the threshold F-value for significance for a majority of the detected genes where there should in fact be an indeterminate form (
is not significant. For Lande's test, this generates F-statistics several orders of magnitude above the threshold F-value for significance for a majority of the detected genes where there should in fact be an indeterminate form ( ). Finally, there is an increasing number of reports about the predominance of nonadditive interactions at the transcriptome levels (Gibson et al. 2004; Auger et al. 2005; Roberge et al. 2008). Estimation of QST for genes under nonadditive control would not be valid since the QST framework is based on the premise of additivity. Such genes are also more likely to have low or nonsignificant heritability values. Hence, preselecting genes with significant transcriptional h2 minimized the impacts of several problems and possible artifacts on our results and interpretations.
). Finally, there is an increasing number of reports about the predominance of nonadditive interactions at the transcriptome levels (Gibson et al. 2004; Auger et al. 2005; Roberge et al. 2008). Estimation of QST for genes under nonadditive control would not be valid since the QST framework is based on the premise of additivity. Such genes are also more likely to have low or nonsignificant heritability values. Hence, preselecting genes with significant transcriptional h2 minimized the impacts of several problems and possible artifacts on our results and interpretations.
Figure 2.—

Gene transcription heritability. (A) Distribution of heritability estimates of the normalized transcription profiles for the 6484 cDNA clones corresponding to genes for which expression was detected. (B) Relationship between h2 and its standard error for each of the 6484 detected cDNA clones.
RESULTS
Hybridization design:
The design for the experimental microarray hybridization differed for the two sampling years. In 2003, we were interested mainly in assessing the functional genomic basis of sexual precocity (C. Roberge, H. Guderley and L. Bernatchez, unpublished data), such that the progeny of one anadromous male and one precocious parr from the same subpopulation and sharing the same mother were always coupled on a same microarray, for a total of 24 microarrays and 48 offspring analyzed. For 2004, one offspring from each the upstream and downstream subpopulations was always coupled on the same microarray, for a total of 21 microarrays and 42 offspring analyzed. Thus, transcription profiles were obtained for a total of 90 offspring representing 50 half-sib families.
Normality and uni/multimodality assessment:
Pearson's correlation tests between normalized gene transcription data and a normal distribution revealed a highly significant correlation to a normal distribution for all detected genes (the highest observed P-value being 1.57 × 10−8), with Pearson's determination coefficient generally very close to one (average 0.97; Figure 1). Results from the Shapiro–Wilk test were less categorical: 10% of the genes showed a significant departure from normality, even when using a Bonferroni-corrected significance threshold of 7.7 × 10−6. The distributions of the transcription profiles for the three genes that showed the most significant departures from normality according to the Shapiro–Wilk test all included outlier data points (supplemental Figure S1 at http://www.genetics.org/supplemental/), suggesting that the apparent departure from normality was the result of the test's high sensitivity to the presence of outliers. Figure 1A shows the relationship between the log of the P-value from the Shapiro–Wilk test and Pearson's determination coefficients for all detected genes. While normality does not have to be assumed for performing the permutation-based ANOVA, it is assumed in the procedure that we used for assessing genetic parameters. However, restricted maximum-likelihood estimators are robust to deviations from the assumption of normality (Kruuk 2004), and therefore we believe that the observed departure from normality for a minority of genes (in the Shapiro–Wilk test only) should not significantly impact on our main conclusions. The distribution of the dip statistic from Hartigan's dip test for the 6484 detected genes is presented in Figure 1B. While the transcripts from the right tail of the distribution in Figure 1B show expression patterns across all samples that suggest multimodality (four examples are presented in Figure 1C), only 175 transcripts (including those presented in Figure 1C) showed statistical evidence of multimodality (P < 0.05, dip > 0.054), while 324 (0.05 × 6484) could have been expected by chance alone. Thus, there was no strong evidence for the occurrence of bi- or multimodality of transcription profiles in our data set.
Figure 1.—

Assessment of normality and unimodality. (A) Relationship between Pearson's determination coefficient between a normal distribution and the observed distribution of normalized gene transcription data, on the one hand, and the inverse of the log of the P-value from the Shapiro–Wilk normality test of gene transcription data, on the other hand, for each of the 6484 cDNA clones corresponding to genes for which expression was detected. The horizontal line indicates the position of the Bonferroni-corrected significance threshold for the Shapiro–Wilk test. (B) Density curve of Hartigan's dip statistic testing for unimodality of the gene transcription data for all detected genes. Gene transcription data from genes at the right of the vertical line (dip > 0.054, P < 0.05) show significant departure from unimodality. (C) Density curves of the normalized gene transcription data of the 90 sampled individuals for the four genes with the highest dip statistic. The corresponding gene products are (i) an unknown gene product (GenBank accession number of the cDNA sequence: CA059553), (ii) ribosomal protein L35, (iii) ubiquitin, and (iv) α-actin 2.
Heritability estimates, QST estimation, and transcriptome scan:
Figure 2 presents the distribution of heritability estimates of the normalized transcription profiles for the genes corresponding to the 6484 cDNA clones analyzed and their associated standard error. Significant (P ≤ 0.05) transcriptional h2 estimates were obtained for 1044 genes (16% of all detected genes). Transcriptional h2 estimates for these varied between 0.121 and 0.996 and averaged 0.409. A test conducted in EASE 2.0 revealed significant overrepresentation of genes in several gene ontology (GO) categories (9 categories with an EASE score <1 × 10−3) within the genes with significant transcriptional h2 when compared to all genes on the microarray. Most of these categories were related to oxidative phosphorylation, including, for instance, primary active transporter activity (GO molecular function, EASE score = 1.6 × 10−4) and hydrogen ion transporter activity (GO molecular function, EASE score = 7.3 × 10−4).
Transcriptional QST were estimated only for the 1044 cDNA clones corresponding to genes with significant transcriptional h2 for reasons explained previously. Most of those genes had low transcriptional QST estimates that are unlikely to differ from 0 (Figure 3). The 1.5% upper outliers (16 cDNA clones) of this distribution (Table 1) are genes for which transcription levels are the most likely to have diverged between the two subpopulations as a result of directional selection. Transcriptional QST estimates for these outliers ranged from 0.07 (tartrate-resistant acid phosphatase type 5) to 0.19 (“unknown” gene) and averaged 0.11. The identity of 10 of these outliers was unknown, meaning that they did not generate any BLAST hits with e-values <1 × 10−15 and an informative name (for a detailed description of the array annotation process, see http://web.uvic.ca/cbr/grasp). The remaining six outlier cDNA clones represented genes with various functions (Table 1). No functional group appeared significantly overrepresented within these genes when compared to all genes represented on the microarray. It is noteworthy that all of these outliers were overtranscribed in the upstream subpopulation (average = 29% overexpression relative to the downstream subpopulation). The overtranscription ranged from 16% (unknown gene CB513854) to 46% (nucleolar RNA helicase II). Gene transcription h2 among these genes ranged from 0.14 (transmembrane protein 14C) to 0.44 (pterin-4-α-carbinolamine dehydratase) and averaged 0.28. The interpopulation component of additive genetic variance ( ) was significant for all 16 outliers (P < 0.05). We used significance of the
) was significant for all 16 outliers (P < 0.05). We used significance of the  estimates to test for significance of QST estimates since the permutation test on QST estimates was prone to false positives (see materials and methods).
estimates to test for significance of QST estimates since the permutation test on QST estimates was prone to false positives (see materials and methods).
Figure 3.—

Gene transcription QST. Distribution of QST estimates of the normalized transcription profiles of the 1044 cDNA clones representing genes with significant gene transcription heritability (P ≤ 0.05)
TABLE 1.
Results from the transcriptome scan
| Gene product | GenBank accession no. | h2 | QST | P-value h2 |
P-value 
|
Fold difference | F neutrality test | Functional information |
|---|---|---|---|---|---|---|---|---|
| Transmembrane protein 14C | CA042090 | 0.14 | 0.11 | 3.8 × 10−2 | 1.0 × 10−2 | 1.21 | 18.94* | Unknown function |
| Selenoprotein P, 1b | CB509685 | 0.21 | 0.11 | 4.8 × 10−2 | 8.0 × 10−3 | 1.31 | 12.28* | Oxidant defense, thyroid hormone metabolism, defense against viral infections |
| Nucleolar RNA helicase II | CB505664 | 0.32 | 0.08 | 2.0 × 10−2 | 0 | 1.46 | 6.03* | Ribosomal RNA processing, transcription regulation |
| Pterin-4-α- carbinolamine dehydratase | CB497855 | 0.44 | 0.07 | 8.0 × 10−3 | 2.0 × 10−3 | 1.19 | 3.80 (NS) | Tetrahydrobiopterin recycling and biosynthesis, HNF-1-α regulation |
| Lysozyme type II | CA037907 | 0.28 | 0.07 | 2.6 × 10−2 | 1.8 × 10−2 | 1.29 | 5.76* | Bacteriolytic function |
| Tartrate-resistant acid phosphatase type 5 | CA045149 | 0.37 | 0.07 | 1.2 × 10−2 | 2.0 × 10−3 | 1.39 | 4.22* | Cytochemical marker of macrophages, osteoclasts, and dendritic cells; unknown biological function |
| Unknown | CB496434 | 0.22 | 0.19 | 3.8 × 10−2 | 0 | 1.40 | 22.40* | |
| Unknown | CA060749 | 0.23 | 0.16 | 5.0 × 10−2 | 0 | 1.31 | 17.34* | |
| Unknown | CB514770 | 0.38 | 0.14 | 1.0 × 10−2 | 0 | 1.18 | 9.30* | |
| Unknown | CB514561 | 0.27 | 0.13 | 2.4 × 10−2 | 0 | 1.25 | 12.39* | |
| Unknown | CB513854 | 0.20 | 0.13 | 2.6 × 10−2 | 6.0 × 10−3 | 1.16 | 16.42* | |
| Unknown | CA060904 | 0.21 | 0.13 | 4.6 × 10−2 | 6.0 × 10−3 | 1.26 | 15.59* | |
| Unknown | CB517440 | 0.44 | 0.11 | 1.8 × 10−2 | 0 | 1.35 | 6.13* | |
| Unknown | CA056630 | 0.23 | 0.11 | 3.2 × 10−2 | 4.0 × 10−3 | 1.30 | 11.03* | |
| Unknown | CA041889 | 0.31 | 0.07 | 3.2 × 10−2 | 1.6 × 10−2 | 1.25 | 5.18* | |
| Unknown | CA056760 | 0.23 | 0.10 | 3.8 × 10−2 | 6.0 × 10−3 | 1.37 | 9.88* |
cDNA clones corresponding to the 16 genes for which transcription-level QST estimates were in the upper 1.5% of the QST distribution for 1044 cDNA clones representing genes with significant transcription-level heritability. Corresponding QST and heritability estimates (with their P-values from the permutation test), F-statistic from Lande's neutrality test (asterisks indicate significance at the P < 0.05 threshold), fold difference between subpopulations (upstream/downstream), GenBank accession number, and functional information from the literature are presented.
Lande's test, which we also performed on all cDNA clones corresponding to the 1044 genes with significant transcriptional h2, provided evidence of non-neutral evolution for the gene transcription levels of 24 of these genes only, which nevertheless included 15 of the 16 genes identified by the QST scan (Table 1). The 9 remaining genes were also all overtranscribed in the upstream subpopulation. Five of these had no known function, while the remaining 4 were a salmonid toxin-like gene (toxin-1, CB493361), P0-like glycoprotein, myc-associated factor X, and glutamyl-prolyl-tRNA synthetase.
ANOVA results on gene expression divergence between subpopulations:
In parallel to our quantitative genetic approach, we also tested the significance of gene transcription differences between subpopulations in a simpler linear model where the pedigree information was not considered, as usually done in other studies. The simulation-based ANOVA revealed highly significant (P < 0.0001) differences in gene transcription between the progeny of fish from the two subpopulations for 12 cDNA clones (Table 2). The magnitude of the observed significant changes varied between a 30% undertranscription (thioredoxin-like protein p19-coding gene) to a 66% overtranscription (creatine kinase-coding gene) in the upstream subpopulation (average fold difference of 33%). Genes that differed significantly in expression between the two subpopulations were involved in several molecular and biological functions, including energy metabolism and transcription (Table 2). Moreover, a test conducted in EASE 2.0 revealed a highly significant (EASE score = 8.14 × 10−3) overrepresentation of genes of the immune response gene ontology category (biological process) among the genes significantly differentially transcribed at P < 0.005 when compared to all genes represented on the microarray. Notably, four different clones representing major histocompatibility class II-associated invariant chains showed consistent undertranscription (average 17%) in the progeny of the upstream subpopulation. A nuclease-sensitive element-binding protein 1 (Y-box-binding protein 1) coding gene was also significantly undertranscribed in the upstream subpopulation. It is noteworthy that this gene was originally identified as coding for a DNA-binding protein that regulates major histocompatibility complex (MHC) class II genes (Didier et al. 1988).
TABLE 2.
Results from the simulation-based ANOVA
| Gene product | P-value | Fold difference | GenBank accession no. | Functional information |
|---|---|---|---|---|
| Creatine kinase (EC 2.7.3.2) | 9.87 × 10−6 | 1.66 | CK990405 | Energy metabolism |
| Thioredoxin-like protein p19 | 9.87 × 10−6 | 0.70 | CB498161 | Redox regulation, protein refolding, transcription factors regulation |
| α-Globin and β-globin | 8.00 × 10−5 | 1.48 | CA051720 | Oxygen transport |
| Phosphoglycerate mutase 2 (EC 5.4.2.1) | 2.16 × 10−4 | 1.47 | CB497792 | Regulation of glycolysis, cell proliferation |
| Invariant chain INVX | 2.65 × 10−4 | 0.85 | CB503772 | MHC class II-associated invariant chain |
| Invariant chain INVX | 2.91 × 10−4 | 0.87 | CK990275 | MHC class II-associated invariant chain |
| NADH-ubiquinone oxidoreductase chain 2 (EC 1.6.5.3) | 3.27 × 10−4 | 1.34 | CN442556 | Energy metabolism |
| Nuclease-sensitive element-binding protein 1 | 3.28 × 10−4 | 0.82 | CB511419 | Transcription factor |
| Invariant chain S25-7 | 4.16 × 10−4 | 0.86 | CB513162 | MHC class II-associated invariant chain |
| Invariant chain S25-7 | 5.04 × 10−4 | 0.75 | CB502487 | MHC class II-associated invariant chain |
| Nucleolar RNA helicase II | 5.64 × 10−4 | 1.46 | CB505664 | Transcription |
| Acidic mammalian chitinase (EC 3.2.1.14) | 7.32 × 10−4 | 1.44 | CB505509 | Chitin catabolism |
cDNA clones representing genes with significant (P < 0.0001) differences in transcription level between the upstream and downstream subpopulations are presented. The corresponding gene identities, P-values, average fold difference (upstream/downstream), and GenBank accessions are provided for each significant cDNA clone, along with brief functional annotations from the literature.
DISCUSSION
Here, we applied a “transcriptome scan” approach combining both the QST and genome scan frameworks to investigate the role of selection in shaping differential profiles of gene transcription between recently diverging (six generations) subpopulations of Atlantic salmon. The additive genetic basis of gene transcription was estimated for genes corresponding to all detected cDNA clones (6484) and gene transcription heritability estimates were significant for 1044 (16%) of these. The scan of QST values estimated from intra- and interpopulation additive genetic components of the transcription profiles identified 16 outlier cDNA clones (average QST estimate = 0.11) representing the most likely genes for which transcription levels have evolved under the influence of directional selection within only approximately six generations. Additional evidence of non-neutral evolution of the transcription levels for 15 of those 16 genes was obtained by performing Lande's neutrality test on gene transcription data. Overall, this study shows that the transcriptome scan approach can be used for identifying genes of which the transcription profiles are likely to have evolved as a result of directional selection, even at small temporal and spatial scales.
Gene transcription normality and multimodality:
When assessing the genetic parameters of gene transcription profiles taken as phenotypic traits, the question of the normality of the data becomes more than a mere “to-use-or-not-to-use-parametric-statistics” concern: it may reflect either the qualitative or the quantitative nature of the traits. Hence, Gibson and Weir (2005) noted that, in expression QTL mapping studies, eQTL accounting for 25–50% of transcriptional variation prevails. They suggested that since major-effect eQTL are common, gene transcription data should often depart from a normal distribution and exhibit bi- or multimodal distributions, which would make the genetic structure of gene transcription profiles more akin to qualitative than to quantitative phenotypic traits. Yet, very few studies have tested the normality of gene transcription data on a gene-per-gene basis (but see Giles and Kipling 2003) and, to our knowledge, none have examined the modality of such data. Here, we observed strong and highly significant correlations between gene transcription data and the normal distribution and that for all detected genes (cDNA clones with low-intensity signal were filtered prior to this analysis). However, results from the Shapiro–Wilk test suggest that the transcription data of 10% of the detected genes departed significantly from normality. The Shapiro–Wilk test appears very sensitive to outliers (supplemental Figure S1 at http://www.genetics.org/supplemental/), which might increase the number of false positives. Moreover, Giles and Kipling (2003) argued that, even with small sample sizes, the Shapiro–Wilk normality test has the power to detect very slight departures from normality and score them as significant. We also tested the evidence of uni- vs. bi- or multimodality in the gene transcription data distributions of each of the 6484 detected cDNA clones. We found little evidence for bi- or multimodality since only 175 clones showed significant departure from unimodality while 324 were expected by chance alone. Yet, we cannot rule out the possibility that some of these 175 clones represent true positives (genes with bi- or multimodal transcription-level distribution; Figure 1C). In summary, there was little evidence of significant departure from normality in transcription profiles, with the vast majority of the genes showing unimodal distribution. This suggests that most transcription profiles behaved as quantitative rather than qualitative traits in this system. Hence, the high prevalence of major-effect eQTL in eQTL mapping studies (Gibson and Weir 2005) may correspond more to a detection bias toward major-effect eQTL than to a biological fact. However, of the minority of transcripts showing significant departure from either normality or unimodality (Figure 1C), some might yet correspond to genes for which transcription depends on major-effect eQTL.
Gene transcription heritability:
Only a handful of studies have formally estimated the genomewide heritability of gene transcription profiles, and these concerned only model species (mice: Schadt et al. 2003, Chesler et al. 2005, Cui et al. 2006; humans: Monks et al. 2004; yeast: Brem et al. 2002). Results were quite diverse as the median h2 among genes with significantly heritable transcription profiles ranged from 0.11 (Chesler et al. 2005) to 0.84 (Brem et al. 2002), whereas the average h2 among genes with significantly heritable transcription profiles in this study was 0.41. When indicated, the proportion of genes with significant gene transcription h2 in previous studies was ∼30% (Schadt et al. 2003; Monks et al. 2004), while it was 16% here. This proportion varies with the statistical power to detect truly significant h2 and is therefore not formally comparable between studies. Neither are the average or median gene transcription h2 among genes with significant transcriptional h2, since they are likely to be inflated when the power to detect small h2 estimates as significant is low. Heritability estimation is also highly model dependent. To illustrate this point, we reanalyzed the same data with a different animal model that did not take into account the sampling year and maternal effects (supplemental Figure S2 at http://www.genetics.org/supplemental/). The proportion of h2 estimates detected as significant rose to 39% (2543), which shows the importance of choosing an appropriate model (accounting, notably, for maternal effects) to reduce biases in estimating h2 (Kruuk 2004).
Here, the statistical power to detect significant transcriptional h2 was relatively low, given the low genetic variance for gene expression in our system. Hence, the crosses that we used were performed among genitors of the same subpopulation (one of two small subpopulations). Clearly, then, the additive genetic and phenotypic variances cannot be expected to be as high here as in systems where crosses were performed between genitors of different strains or populations. This low power likely caused the average h2 estimates among genes with significant transcription heritability to be higher than in some previous studies assessing gene transcription h2 (Monks et al. 2004; Chesler et al. 2005). Higher average h2 estimates may also reflect genuine differences between species (salmon, mouse, and human) or result from the different statistical models used. Although our sample size was small compared to those commonly used when quantifying heritability for “classical” phenotypic traits, it still ranks among the highest used to date in any study that has investigated transcriptional heritability.
While the 1044 genes with significant gene transcription h2 estimates do not necessarily represent genes for which directional selection was evidenced in this study, they likely represent genes for which levels of transcription have a high potential for responding to selection. Functional analysis showed significant overrepresentation of gene categories related to oxidative phosphorylation among this group of genes when compared to all genes represented on the chip. These categories were not significantly overrepresented in the list of genes that showed significant differences in gene transcription between subpopulations (on the basis of ANOVA) or among the transcriptional QST distribution upper outliers. This might suggest that genes from this basic pathway, although their transcription profiles often appear heritable, were not particularly involved in the recent phenotypic divergence between the salmon subpopulations that we studied.
QST estimates and transcriptome scan:
As illustrated from the distribution of transcription profile QST (Figure 3), transcriptional QST estimates were virtually null for the great majority of the genes tested. While QST estimates were expected to be low, given that divergence between the two subpopulations under study has been possible only since 1981 (about six generations), transcription-level QST estimates for 97% of the genes with significant transcriptional h2 were under (in some cases by orders of magnitude) the FST values (average of 3.2%) estimated in a previous study based on neutral markers (Garant et al. 2000). This observation is consistent with the results of recent studies (Denver et al. 2005; Rifkin et al. 2005) in which the authors concluded that transcription profiles of a large proportion of all genes appear to be under the effect of stabilizing selection in natural systems. However, Beaumont and Balding (2004) recently used a simulation-based approach to show that current statistical methods could not accurately identify loci under balancing or stabilizing selection in a genome scan framework, especially when populations are weakly differentiated. Yet, results from Lande's neutrality test suggest that, for 1020 (97.7%) of the genes tested, neutral evolution of gene transcription profiles cannot be rejected. An alternative explanation for the high number of very small QST estimates in this study could be the lack of genetic variance for gene transcription profiles (Merila and Crnokrak 2001), as discussed above.
Conversely, by targeting genes in the upper outliers of the transcriptional QST distribution, the “transcriptome scan” approach identified 16 cDNA clones (Table 1) representing the most likely genes for which transcription levels may have diverged between the two subpopulations as a result of directional selection in two contrasting habitats. Additional evidence for the role of selection in driving transcriptional divergence for 15 of those 16 genes (plus 9 others) comes from the results of Lande's neutrality test (Table 1). Transcriptional QST estimates for these genes were high (0.07–0.19, mean = 0.11), given the short time frame in which divergence is likely to have occurred between the two subpopulations. Transcriptional QST estimates for these genes were also all above the FST estimated between them using neutral markers. It is noteworthy that 62% of the genes identified as outliers have no known function, which suggests that selection acted on unpredictable targets. The other candidate genes identified with the transcriptome scan represented various functional groups (Table 1). Moreover, the differential transcription of some of these genes could potentially affect gene expression of other genes at different levels. Hence, the nucleolar RNA helicase II is a multifunctional protein that is notably implicated in rRNA biosynthesis and in the regulation of c-Jun-mediated gene expression (Yang et al. 2005). Pterin-4-α-carbinolamine dehydratase, apart from its role in tetrahydrobiopterin biosynthesis, is also known as a dimerization factor of the hepatocyte nuclear factor (HNF) 1 α, one of the master regulators of hepatocyte and pancreatic islet transcription (Resibois et al. 1999).
Between-subpopulations ANOVA and comparison with the results of the transcriptome scan and gene transcription heritability:
The between-subpopulations ANOVA identified 12 cDNA clones corresponding to genes for which the transcription level diverged between subpopulations, albeit apparently not necessarily through the effect of selection (Table 2). Except for a nucleolar RNA helicase II-coding gene, none of these genes overlapped with outliers identified by the transcriptome scan approach, and the null hypothesis of neutral evolution could not be rejected for any of them by Lande's test. Namely, these differences could be the result of six generations of phenotypic divergence in different parts of the Sainte-Marguerite River caused by genetic drift. Yet they still represent genes that potentially differentially affect several physiological functions between the two subpopulations. Of particular interest is the gene coding for nucleolar RNA helicase II, which both was significantly differently transcribed under a conservative significance threshold and exhibited substantial genetic variance in gene transcription between relative to within subpopulation (Tables 1 and 2).
The fact that, except for one gene, there was no overlap between the results of the transcriptome scan and those of the ANOVA may seem paradoxical and therefore deserves explanations. First, the absence of most of the transcriptome scan candidate genes from the list of genes showing significant transcription differences between subpopulations (Table 2) is largely explained by the stringency of the significance threshold chosen for the ANOVA. Had we raised this threshold to 0.05, differences in transcription for 12 (75%) genes in Table 1 would have been significant in the ANOVA analysis. Moreover, heritability estimates for eight of the genes harboring the most significant differences in gene transcription between subpopulations were not significant. Hence, these were not considered in the QST analysis and could therefore not be detected as outliers in the transcriptome scan. With the exception of nucleolar RNA helicase II, the remaining genes in Table 2 (Y-box transcription factor, invariant chain INVX, and invariant chain S25-7) had significant gene transcription h2 and showed highly significantly different expression profiles between subpopulations. Yet, they had small transcriptional QST and hence were not identified as outliers in the transcriptome scan. We propose two interpretations for this result: Either (1) the transcription profiles of these genes evolved neutrally between subpopulations (e.g., by genetic drift, which might be supported by the results of Lande's test) or (2) they evolved as a consequence of selection but their transcription levels have a nonadditive genetic basis. Indeed, recent studies have provided clear evidence that a nonadditive genetic basis for gene transcription is common (Gibson et al. 2004; Auger et al. 2005; Roberge et al. 2008). Here, a possible example of the latter case may be given by genes related to the MHC, three of which had significant transcription levels h2 but very low estimated transcriptional QST. Interestingly, Landry and Bernatchez (2001) showed that directional selection has been acting at the antigen presentation sites of the MHC class II B locus on the subpopulations of this same river. The coding sequence of MHC class II B coding genes could have evolved under the effect of selection while the transcription levels of other MHC-related genes evolved neutrally. Alternatively, both could have evolved through selection on traits harboring nonadditive genetic variance. Another general observation on the comparison of the results of the ANOVA and transcriptome scan is that all of the upper outlier genes of the transcriptional QST distribution were overtranscribed in the upstream vs. downstream population (Table 1; average fold change: 29%). This was, however, not observed for the genes that showed significant transcription differences between the two subpopulations in the ANOVA (Table 2), in which a similar number of genes were over- and undertranscribed for a given subpopulation. This raises the hypothesis that selection may have favored enhanced general metabolism and gene transcription in the upstream subpopulation, which is also characterized by a higher growth rate and smaller size threshold at precocious sexual maturation, resulting in a higher proportion of sexually precocious parr relative to the downstream subpopulation (Aubin-Horth et al. 2006). Gene overexpression associated with higher metabolic demands has been observed in other salmonids. For instance, in a study on the transcriptomics of population divergence involving dwarf and normal whitefish species pairs (Coregonus sp.), Derome et al. (2006) observed that higher swimming activity in the dwarf whitefish was consistently associated with overexpression of genes involved in muscle contraction as well as energy metabolism.
No correlation was observed between the P-values from the ANOVA comparing the subpopulations, the estimated gene transcription h2, and the transcriptional QST estimate for the 1044 genes with significant gene transcription h2 (Figure 4), or for all detected genes (not shown). However, the relationship between transcriptional QST and h2 estimates was marked by a mutual exclusion: few genes had both high-transcriptional QST and h2. This may be at least partly explained by the fact that the animal genetic variance, which we used to estimate  is the numerator of the h2 formula and also contributes to the denominator of the QST formula. High within-population genetic variance naturally imposes a superior limit to values that QST can reach (see Hedrick 2005 for analogous arguments regarding FST estimation). Inversely, very small between-subpopulation genetic variance can result in very high QST estimates for genes with very low within-population genetic variance, which could represent a technical artifact inflating QST estimates for a number of genes. Here, this potential problem was controlled for by considering only genes with significant h2 estimates for the transcriptome scan. Yet, one could argue that genes with high gene transcription QST estimates and very low genetic variance estimates illustrate the erosion of the within-subpopulation genetic variance as a result of the action of divergent selection. Although this possibility cannot be ruled out, we do not deem that this is very likely considering the short time frame (six generations) involved in the divergence of the salmon subpopulations studied here.
is the numerator of the h2 formula and also contributes to the denominator of the QST formula. High within-population genetic variance naturally imposes a superior limit to values that QST can reach (see Hedrick 2005 for analogous arguments regarding FST estimation). Inversely, very small between-subpopulation genetic variance can result in very high QST estimates for genes with very low within-population genetic variance, which could represent a technical artifact inflating QST estimates for a number of genes. Here, this potential problem was controlled for by considering only genes with significant h2 estimates for the transcriptome scan. Yet, one could argue that genes with high gene transcription QST estimates and very low genetic variance estimates illustrate the erosion of the within-subpopulation genetic variance as a result of the action of divergent selection. Although this possibility cannot be ruled out, we do not deem that this is very likely considering the short time frame (six generations) involved in the divergence of the salmon subpopulations studied here.
Figure 4.—

Relationships between gene transcription QST, heritability, and the significance from the ANOVA for the 1044 genes with significant heritability estimates. Scatterplots illustrating the relationships between (A) gene transcription QST estimates and the inverse of the log of the P-values from an ANOVA testing for significant gene transcription-level differences between the progeny of fish from the two subpopulations, (B) gene transcription QST and heritability estimates, and (C) gene transcription heritability estimates and the inverse of the log of the P-values from the above-mentioned ANOVA.
To conclude, this study represents, to our knowledge, the first attempt to translate differences in gene transcription into QST estimates to identify genes for which transcription levels are potentially under directional selection and one of the few studies to evaluate transcription-level h2 on a genomewide scale. It thus contributes both empirically and methodologically to the evolutionary quantitative genetic exploration of transcription data by showing that a combination of QST and genome scan framework can efficiently identify genes for which transcription may have evolved under the effect of directional selection, even at the scale of very recently diverging subpopulations.
Acknowledgments
We thank François Martin, David Páez, Orlane Rossignol, Serge Higgins, and the staff at the Laboratoire de Recherche en Sciences Aquatiques, Université Laval, Québec, for their assistance in fish rearing and sampling. We are also grateful to Stéphane Larose for Perl scripting, Aurélie Labbe for help with R scripting, and Guy Perry for help with PEST/VCE. We also acknowledge the constructive comments of G. Perry, M. Veuille, and three anonymous reviewers on a previous version of the manuscript. Finally, we thank the staff at the provincial fish hatchery in Tadoussac for fish crossing and rearing. This work was supported by a research grant from Natural Science and Engineering Research Council (NSERC strategic program) to L.B., H.G., and J.J. Dodson, and a scholarship from the Fonds Québécois de la Recherche sur la Nature et les Technologies to C.R. This study is a contribution to the research program of the Centre Inter-universitaire de Recherche sur le Saumon Atlantique.
References
- Aubin-Horth, N., J. F. Bourque, G. Daigle, R. Hedger and J. J. Dodson, 2006. Longitudinal gradients in threshold sizes for alternative male life history tactics in a population of Atlantic salmon (Salmo salar). Can. J. Fish. Aquat. Sci. 63: 2067–2075. [Google Scholar]
- Auger, D. L., A. D. Gray, T. S. Ream, A. Kato, E. H. Coe et al., 2005. Nonadditive gene expression in diploid and triploid hybrids of maize. Genetics 169: 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A., 2005. Adaptation and speciation: What can F-st tell us? Trends Ecol. Evol. 20: 435–440. [DOI] [PubMed] [Google Scholar]
- Beaumont, M. A., and D. J. Balding, 2004. Identifying adaptive genetic divergence among populations from genome scans. Mol. Ecol. 13: 969–980. [DOI] [PubMed] [Google Scholar]
- Bochdanovits, Z., H. van der Klis and G. de Jong, 2003. Covariation of larval gene expression and adult body size in natural populations of Drosophila melanogaster. Mol. Biol. Evol. 20: 1760–1766. [DOI] [PubMed] [Google Scholar]
- Brem, R. B., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296: 752–755. [DOI] [PubMed] [Google Scholar]
- Chesler, E. J., L. Lu, S. M. Shou, Y. H. Qu, J. Gu et al., 2005. Complex trait analysis of gene expression uncovers polygenic and pleiotropic networks that modulate nervous system function. Nat. Genet. 37: 233–242. [DOI] [PubMed] [Google Scholar]
- Cui, X. Q., J. Affourtit, K. R. Shockley, Y. Woo and G. A. Churchill, 2006. Inheritance patterns of transcript levels in F1 hybrid mice. Genetics 174: 627–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver, D. R., K. Morris, J. T. Streelman, S. K. Kim, M. Lynch et al., 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat. Genet. 37: 544–548. [DOI] [PubMed] [Google Scholar]
- Derome, N., P. Duchesne and L. Bernatchez, 2006. Parallelism in gene transcription among sympatric lake whitefish (Coregonus clupeaformis Mitchill) ecotypes. Mol. Ecol. 15: 1239–1249. [DOI] [PubMed] [Google Scholar]
- Didier, D. K., J. Schiffenbauer, S. L. Woulfe, M. Zacheis and B. D. Schwartz, 1988. Characterization of the cDNA-encoding a protein-binding to the major histocompatibility complex class II Y box. Proc. Natl. Acad. Sci. USA 85: 7322–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici, S., 2003. Data Analysis Tools for DNA Microarrays. CRC Press, London.
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, New York.
- Garant, D., J. J. Dodson and L. Bernatchez, 2000. Ecological determinants and temporal stability of the within-river population structure in Atlantic salmon (Salmo salar L.). Mol. Ecol. 9: 615–628. [DOI] [PubMed] [Google Scholar]
- Gibson, G., and B. Weir, 2005. The quantitative genetics of transcription. Trends Genet. 21: 616–623. [DOI] [PubMed] [Google Scholar]
- Gibson, G., R. Riley-Berger, L. Harshman, A. Kopp, S. Vacha et al., 2004. Extensive sex-specific nonadditivity of gene expression in Drosophila melanogaster. Genetics 167: 1791–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger, T., L. Excoffier, P. J. R. Day, A. Champigneulle, M. M. Hansen et al., 2006. Life history shapes gene expression in salmonids. Curr. Biol. 16: R281–R282. [DOI] [PubMed] [Google Scholar]
- Giles, P. J., and D. Kipling, 2003. Normality of oligonucleotide microarray data and implications for parametric statistical analyses. Bioinformatics 19: 2254–2262. [DOI] [PubMed] [Google Scholar]
- Groeneveld, E., M. Kovac and T. Wang, 1990. PEST, a general purpose BLUP package for multivariate prediction and estimation. Proceedings of the 4th World Congress on Genetics Applied to Livestock Production, Edinburgh, pp. 488–491.
- Hartigan, J. A., and P. M. Hartigan, 1985. The dip test of unimodality. Ann. Stat. 13: 70–84. [Google Scholar]
- Hedrick, P. W., 2005. A standardized genetic differentiation measure. Evolution 59: 1633–1638. [PubMed] [Google Scholar]
- Jin, W., R. M. Riley, R. D. Wolfinger, K. P. White, G. Passador-Gurgel et al., 2001. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat. Genet. 29: 389–395. [DOI] [PubMed] [Google Scholar]
- Kerr, M. K., M. Martin and G. A. Churchill, 2000. Analysis of variance for gene expression microarray data. J. Comput. Biol. 7: 819–837. [DOI] [PubMed] [Google Scholar]
- Kerr, M. K., C. A. Afshari, L. Bennett, P. Bushel, J. Martinez et al., 2002. Statistical analysis of a gene expression microarray experiment with replication. Statistica Sinica 12: 203–217. [Google Scholar]
- Khaitovich, P., S. Paabo and G. Weiss, 2005. Toward a neutral evolutionary model of gene expression. Genetics 170: 929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koskinen, M. T., T. O. Haugen and C. R. Primmer, 2003. Contemporary fisherian life-history evolution in small salmonid populations. Nature 421: 656. [DOI] [PubMed] [Google Scholar]
- Kovac, M., E. Groeneveld and L. A. Garcia-Cortés, 2002. VCE-5: a package for the optimization of dispersion parameters. Seventh World Congress on Genetics Applied to Livestock Production, Montpellier, France.
- Kruuk, L. E. B., 2004. Estimating genetic parameters in natural populations using the ‘animal model’. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359: 873–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande, R., 1976. Natural selection and random genetic drift in phenotypic evolution. Evolution 30: 314–334. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1977. Statistical tests for natural selection on quantitative characters. Evolution 31: 442–444. [DOI] [PubMed] [Google Scholar]
- Landry, C., and L. Bernatchez, 2001. Comparative analysis of population structure across environments and geographical scales at major histocompatibility complex and microsatellite loci in Atlantic salmon (Salmo salar). Mol. Ecol. 10: 2525–2539. [DOI] [PubMed] [Google Scholar]
- Luikart, G., P. R. England, D. Tallmon, S. Jordan and P. Taberlet, 2003. The power and promise of population genomics: from genotyping to genome typing. Nat. Rev. Genet. 4: 981–994. [DOI] [PubMed] [Google Scholar]
- Maecher, M., and D. Ringach, 2004. The Diptest package. R program documentation.
- Merila, J., and P. Crnokrak, 2001. Comparison of genetic differentiation at marker loci and quantitative traits. J. Evol. Biol. 14: 892–903. [Google Scholar]
- Monks, S. A., A. Leonardson, H. Zhu, P. Cundiff, P. Pietrusiak et al., 2004. Genetic inheritance of gene expression in human cell lines. Am. J. Hum. Genet. 75: 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiak, M. F., G. A. Churchill and D. L. Crawford, 2002. Variation in gene expression within and among natural populations. Nat. Genet. 32: 261–266. [DOI] [PubMed] [Google Scholar]
- Peel, D., J. R. Ovenden and S. L. Peel, 2004. NeEstimator: Software for Estimating Effective Population Size, Version 1.3. Queensland Government, Department of Primary Industries and Fisheries, Brisbane, Queensland, Australia.
- Perry, G. M. L., C. Audet and L. Bernatchez, 2005. Maternal genetic effects on adaptive divergence between anadromous and resident brook charr during early life history. J. Evol. Biol. 18: 1348–1361. [DOI] [PubMed] [Google Scholar]
- Resibois, A., L. Cuvelier, M. Svoboda, C. W. Heizmann and B. Thony, 1999. Immunohistochemical localisation of pterin-4 alpha-carbinolamine dehydratase in rat peripheral organs. Histochem. Cell Biol. 111: 381–390. [DOI] [PubMed] [Google Scholar]
- Rifkin, S. A., D. Houle, J. Kim and K. P. White, 2005. A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 438: 220–223. [DOI] [PubMed] [Google Scholar]
- Roberge, C., S. Einum, H. Guderley and L. Bernatchez, 2006. Rapid parallel evolutionary changes of gene transcription profiles in farmed Atlantic salmon. Mol. Ecol. 15: 9–20. [DOI] [PubMed] [Google Scholar]
- Roberge, C., É. Normandeau, S. Einum, H. Guderley and L. Bernatchez, 2008. Genetic consequences of interbreeding between farmed and wild Atlantic salmon: insights from the transcriptome. Mol. Ecol. (in press). [DOI] [PubMed]
- Schadt, E. E., S. A. Monks, T. A. Drake, A. J. Lusis, N. Che et al., 2003. Genetics of gene expression surveyed in maize, mouse and man. Nature 422: 297–302. [DOI] [PubMed] [Google Scholar]
- Schluter, D., 2000. Ecological character displacement in adaptive radiation. Am. Nat. 156: S4–S16. [Google Scholar]
- Spitze, K., 1993. Population structure in Daphnia obtusa: quantitative genetic and allozymic variation. Genetics 135: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatoyannopoulos, J. A., 2004. The genomics of gene expression. Genomics 84: 449–457. [DOI] [PubMed] [Google Scholar]
- Storz, J. F., 2005. Using genome scans of DNA polymorphism to infer adaptive population divergence. Mol. Ecol. 14: 671–688. [DOI] [PubMed] [Google Scholar]
- Townsend, J. P., D. Cavalieri and D. L. Hartl, 2003. Population genetic variation in genomewide gene expression. Mol. Biol. Evol. 20: 955–963. [DOI] [PubMed] [Google Scholar]
- Tusher, V. G., R. Tibshirani and G. Chu, 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasemagi, A., and C. R. Primmer, 2005. Challenges for identifying functionally important genetic variation: the promise of combining complementary research strategies. Mol. Ecol. 14: 3623–3642. [DOI] [PubMed] [Google Scholar]
- Whitehead, A., and D. L. Crawford, 2006. Variation within and among species in gene expression: raw material for evolution. Mol. Ecol. 15: 1197–1211. [DOI] [PubMed] [Google Scholar]
- Yang, H. S., D. Henning and B. C. Valdez, 2005. Functional interaction between RNA helicase II Gu alpha and ribosomal protein L4. FEBS J. 272: 3788–3802. [DOI] [PubMed] [Google Scholar]