

Abstract
The pathologic lesions of Alzheimer’s disease (AD) are characterized by accumulation of protein aggregates consisting of intracellular or extracellular misfolded proteins. The amyloid-β (Aβ) protein accumulates extracellularly in senile plaques and cerebral amyloid angiopathy, whereas the hyperphosphorylated tau protein accumulates intracellularly as neurofibrillary tangles. “Professional chaperones”, such as the heat shock protein family, have a function in the prevention of protein misfolding and subsequent aggregation. “Amateur” chaperones, such as apolipoproteins and heparan sulfate proteoglycans, bind amyloidogenic proteins and may affect their aggregation process. Professional and amateur chaperones not only colocalize with the pathological lesions of AD, but may also be involved in conformational changes of Aβ, and in the clearance of Aβ from the brain via phagocytosis or active transport across the blood–brain barrier. Thus, both professional and amateur chaperones may be involved in the aggregation, accumulation, persistence, and clearance of Aβ and tau and in other Aβ-associated reactions such as inflammation associated with AD lesions, and may, therefore, serve as potential targets for therapeutic intervention.
Keywords: Alzheimer’s disease, Amyloid, Heat shock proteins, Heparan sulfate proteoglycans, Apolipoproteins, Amyloid beta-protein, Chaperones
Introduction
Pathological lesions consisting of intra- and/or extracellular accumulations of misfolded proteins are characteristic for neurodegenerative diseases such as Alzheimer’s disease (AD). AD is characterized by three distinct pathological lesions: senile plaques (SPs), neurofibrillary tangles (NFTs), and cerebrovascular amyloid angiopathy (CAA) [1]. Both SPs and CAA are formed by extracellular deposition of aggregated amyloid-beta protein (Aβ), whereas NFTs consist of intracellular aggregates of hyperphosphorylated tau protein in the cytoplasm of neurons [2, 3]. The Aβ protein is a 4-kDa proteolytic cleavage product [2] of the transmembrane amyloid-β precursor protein (APP). The two major forms of Aβ in human brain are Aβ1-40 and Aβ1-42, differing from each other only by two amino acids. Cerebral production of Aβ is balanced by clearance from the brain either via active transport across the blood–brain barrier (BBB) or via uptake and degradation of Aβ by microglial cells and astrocytes [4–6]. Active transport of Aβ is mediated by Aβ receptors that are capable of transporting Aβ, or Aβ in complex with other proteins, across the BBB [7]. In contrast to normal brain, the cerebral Aβ balance is disturbed in AD brains, resulting in accumulation and aggregation of Aβ.
Aβ aggregation includes the formation of Aβ oligomers, protofibrils, and eventually, mature fibrils. Both Aβ oligomers and protofibrils are considered the most toxic forms of Aβ that initiate degeneration of neurons and of cells within the vasculature, such as smooth muscle cells and pericytes [8, 9]. Aβ aggregates do not clear from the brain as efficiently as soluble Aβ, and thus, directly lead to increased levels of Aβ in the brain [10]. Furthermore, deposition of Aβ in SPs is accompanied by attraction and activation of both microglial cells and astrocytes [11–13]. Activation of these cell types results in increased secretion of pro-inflammatory cytokines as part of a neuro-inflammatory reaction.
Chaperones can be defined as proteins that: (1) have a role in the intracellular handling of misfolded proteins, (2) induce conformational changes of proteins, (3) act as transporter of proteins. “Professional” chaperones, such as the heat shock protein family (Hsp), are defined as proteins that have a specific function in facilitating normal folding of proteins and intracellular handling of misfolded proteins. Members of the Hsp family recognize misfolded proteins and transport them to the proteasome for degradation. Therefore, this protein family acts as the first line of defense against toxicity induced by misfolded proteins such as Aβ and tau. In contrast to professional chaperones, “amateur” chaperones can be defined as proteins that bind to other proteins and induce conformational changes or, alternatively, serve as transporter proteins. Examples of putative amateur chaperones are apolipoprotein E (ApoE), heparan sulfate proteoglycans (HSPGs), and complement factors such as C1q. They have, in contrast to the professional chaperones, primarly an extracellular function. In this paper, we will review the role of both amateur and professional chaperones in the pathogenesis of AD.
Aβ-Binding Proteins in Extracellular Interaction with Aβ
Apolipoproteins
The apolipoprotein family consists of proteins that conjugate with lipids to form different classes of lipoprotein particles. In human brain, several members of this protein family are expressed, such as apolipoprotein E (ApoE), apolipoprotein J (ApoJ), and apolipoprotein D (apoD).
ApoE is a major determinant of lipid transport and metabolism and is expressed in brain by astrocytes, microglia, pericytes, and smooth muscle cells [14–18]. In human, three common isoforms are expressed: apoE2, apoE3, and apoE4 which are all products of alleles at a single gene locus [19, 20].
The ɛ4 allele of ApoE is the major genetic risk factor for AD, whereas the ɛ2 allele appears to be protective against AD [21–24]. As ApoE immunoreactivity was found in extracellular amyloid deposits in subjects with AD, it has been suggested that it affects amyloidogenesis [25, 26]. In vitro studies provided evidence for a direct interaction of ApoE with Aβ and the formation of stable complexes [27, 28]. Binding of ApoE to Aβ is, however, ApoE isoform-dependent (ɛ2>ɛ3>>ɛ4) [29, 30] and depends on the degree of lipidation [29]. Lipidation of ApoE also seems a major factor in its effect on Aβ-mediated cellular toxicity [18]. In addition, ApoE4 promotes the conversion of soluble Aβ into β-sheet-rich amyloid more than ApoE3 [31–33].
In contrast to its effect on Aβ in vitro where a consistent accelerating effect of ApoE on Aβ aggregation is observed, the effect of ApoE on Aβ deposition in transgenic (Tg) mice studies is less equivocal. In early studies, both Aβ immunoreactivity and amyloid formation were reduced in ApoE knockout mice [31, 32]. In addition, CAA and associated microhemorrhages were also suppressed in ApoE knockout mice [34]. This effect might be due to the absence of ApoE/Aβ complexes [35]. In contrast to the effects of murine ApoE in the early studies, both human ApoE3 and ApoE4 suppressed Aβ deposition in Tg mice [36]. In addition, when these mice aged, ApoE4 induced a tenfold higher deposition of fibrillar Aβ than ApoE3 [37]. Consistent with these latter results, human ApoE4 accelerated Aβ deposition in APPSwe Tg mice relative to human ApoE3 [38]. In addition, when human ApoE3 or ApoE4 were knocked in in Tg mice, Aβ deposition was reduced compared to mice carrying endogenous ApoE at 9 months, and at 15 months, substantial CAA was observed in mice with human ApoE4, but not with human ApoE3, and, in either case, parenchymal Aβ was sparse [39]. Thus isoform- and species-specific differences in ApoE direct the aggregation or clearance of Aβ. Furthermore, it is suggested that the presence of ApoE facilitates internalization and degradation of Aβ from brain parenchyma by astrocytes [40] and human ApoE may reduce Aβ deposition in mouse brain by facilitating Aβ transport across the BBB [36]. Although these Tg mice studies seem contradictional, ApoE clearly affects conformational changes of Aβ and functions as an Aβ-transporter protein.
Besides colocalization of ApoE with Aβ in AD brains, ApoE is also found within neurons containing NFTs [25] where it is able to interact directly with tau protein [41]. Furthermore, ApoE has an isoform-dependent effect on tau phosphorylation, as ApoE3 binds to tau in vitro, whereas ApoE4 fails to bind tau [42]. In addition, an ApoE4-dependent increase in phosphorylated tau has been observed [43–45].
Neuroinflammation in AD comprises both activation of microglial cells and astrocytes and activation of the complement system. Aβ deposits in brain are associated with activated microglia and astrocytes, but also with elevated levels of complement [5, 6, 46]. ApoE may have an anti-inflammatory effect by suppressing microglial and astrocytic activation [47–50]. ApoE-deficient mice demonstrate increased levels of IL-6 and TNFα after LPS stimulation, suggesting a role of ApoE in inflammatory gene regulation [51]. In addition, ApoE isoform-dependent (ɛ2<ɛ3<ɛ4) differences in nitric oxide (NO) levels have been observed in microglia cells [52]. Transgenic mice expressing the ApoE4 protein isoform show a greater NO production than mice expressing the ApoE3 protein isoform. These data indicate that ApoE4 has a less efficient anti-inflammatory affect, and thus, may accelerate the development of AD.
Apolipoprotein J, also known as clusterin or SP-40/40, is a highly conserved heterodimeric secreted glycoprotein expressed in brain by epithelial and neuronal cells [53]. ApoJ colocalizes with fibrillar Aβ deposits, and it is suggested that it prevents misfolding and aggregation of soluble Aβ [54–56]. ApoD is a glycoprotein associated with high-density lipoproteins in human plasma and also has a high expression level in human brain [57], but neither its physiological role nor its ligand has been identified. ApoD levels are increased in the hippocampus of AD patients and in ApoE-deficient mice [58, 59].
In conclusion, ApoE and ApoJ can be regarded as amateur chaperones that regulate Aβ aggregation in vitro. By accelerating the Aβ aggregation process towards mature fibril formation, (human) ApoE prevents formation of toxic Aβ intermediates such as oligomers and protofibrils, and thus, may have a protective function towards development of AD. Moreover, ApoE protects against the development of AD by suppressing the inflammatory reactions associated with AD lesions. Besides its role in inducing conformational changes in Aβ, ApoE facilitates Aβ clearance from brain by serving as a transporter molecule of Aβ, which will be discussed in paragraph 4.
Heparan Sulfate Proteoglycans
Proteoglycans are members of a large family of macromolecules with a wide variety of functions ranging from simple physical support to various effects on cell adhesion, motility, proliferation, differentiation, and even tissue morphogenesis. They are composed of linear sulfated polysaccharides (glycosaminoglycans, GAGs), consisting of disaccharide units, covalently bound to a core protein. One of the members of this superfamily is the heparan sulfate proteoglycan (HSPG) family characterized by polymers of repeating disaccharides, N-acetylglucosamine and glucuronic acid, which can be subsequently modified by sulfatation [60, 61]. HSPGs can be subdivided into a family of extracellular matrix proteins, including perlecan, agrin, and collagen XVIII, and a family of cell surface proteins, including syndecans and glypicans [60, 62].
Ever since GAGs were demonstrated in amyloid deposits, the proteoglycans became of interest in amyloidogenesis. The presence of HSPGs in SPs, CAA, and NFTs in AD brains was already demonstrated in the late 1980s [63–65]. Only when antibodies became available that could identify the various individual HSPG species was it described that perlecan colocalized with all three lesions characteristic of AD brains [65–67]. However, we were not able to confirm these findings [68, 69]. Furthermore, it was shown that in both diffuse and classic SPs, several other HSPGs were found, such as agrin, glypican 1, and syndecan 1-3, whereas collagen XVIII is only present in classic SPs and CAA [69–72].
These data suggest that HSPGs interact with Aβ, thereby contributing to development or persistence of SPs or CAA. HSPGs isolated from Engelbreth–Holm–Swarm tumor prevented proteolytic breakdown of aggregated Aβ [73]. In addition, both agrin and perlecan directly interacted with Aβ and promoted conversion of non-fibrillar Aβ into fibrillar Aβ [70, 74–76]. Although the interaction between HSPGs and Aβ is likely mediated predominantly by the sulfate moieties of the GAGs, a role for the protein backbone in Aβ aggregation could not be excluded [77, 78]. As sulfated GAGs were also demonstrated in NFTs in AD brains [79], these macromolecules may also play a role in tangle development. Indeed, sulfated GAGs may induce the formation of paired helical filaments by stimulating tau phosphorylation [80].
As heparan sulfates bind to Aβ and interfere with its fibrillogenesis, they are interesting candidates for therapeutic intervention [81]. GAG mimetics are able to inhibit this binding and may block the formation of β-pleated sheets and adherence of Aβ to the cell surface [82]. The use of GAG mimetics has already been explored in mouse models where they reduced progression of inflammation-associated amyloidosis [83]. The efficacy of one of these compounds is currently being tested in a human phase III trial.
As exemplified by ApoE, Aβ-binding proteins may play a role in the inflammatory reactions in AD brains. Recently, it was demonstrated that the semi-synthetic proteoglycan analogue dextran sulfate blocks activation of the complement cascade [84]. In addition, chondroitin sulfate proteoglycans are also known to bind to C1q and prevent the formation of the C1 complex in vitro [85]. By doing so, chondroitin sulfate proteoglycans inhibit normal complement function. Furthermore, heparin has long been regarded as a potential complement inhibitor [86].
In conclusion, HSPGs do not only colocalize with Aβ and tau, but they also contribute to the development of these lesions. The role of HSPGs in Aβ aggregation might even be a protective one. HSPGs prevent the persistence of toxic Aβ forms, e.g., oligomers or protofibrils, and transform them into more harmless aggregates, i.e., the classic senile plaques containing mature Aβ fibrils that are less toxic than the intermediate aggregates. In addition, HSPGs might play a role in the development of AD lesions by inhibiting complement activation. According to the definitions, HSPGs can therefore be regarded as amateur chaperones. Their ability to recognize a variety of proteins may originate from the heterogeneous structure of the heparan sulfate chains. The negatively charged HS chains are structurally heterogeneous and bind a diverse repertoire of proteins, such as amyloid A, protease-resistant prion protein, α-synuclein, and tau, providing HSPGs with the ability to interact with a wide range of intracellular and extracellular amyloidogenic proteins [61].
Complement Factors
The complement system is an ancient host defense mechanism which is involved in boosting antibody activity. The system consists of a group of soluble serum proteins C1–C9 and is activated either by immunoglobulin M or G bound to a foreign particle or directly by microorganisms. Proteins such as Hageman factor, C4 binding protein, CDS46, CD59, and C1 inhibitor regulate the complement system. In AD, the complement system is overexpressed and activated [46]. The Aβ protein itself activates this system, and complement factor concentrations are increased in AD brains [87–89]. Aβ induces C3 and C4 in AD, and elevated levels of the membrane attack complex (MAC) composed of C5–C9 have been observed [90, 91]. In addition, factors such as Hageman factor, C1q, C3, and C5–9 are commonly found in SPs and NFTs [87, 92, 93]. C1q is associated with Aβ deposits and directly binds fibrillar Aβ which activates the complement cascade [94]. In contrast, the complement inhibitor (C1 INH) is downregulated in AD [95, 96]. Thus, an activated complement system is a general feature observed in AD. However, the contribution of complement to the pathogenesis of AD is controversial.
On the one hand, it is suggested that complement activation protects against Aβ-induced toxicity and even contributes to reducing the accumulation of Aβ in SPs [97]. Transgenic mice expressing complement inhibitors develop increased AD-pathology, whereas increased complement C3 production was associated with a reduction of Aβ deposition [97]. Thus, the complement activation in the brain may be beneficial in AD and possibly also other neurodegenerative diseases [98–100].
However, complement activation may lead to accelerated neurodegeneration as well. Activation of complement in an antibody-independent fashion is achieved by binding of aggregated, but not soluble, Aβ to C1q [12, 90, 101, 102]. This latter finding suggests that in AD, aggregated Aβ induces chronic complement activation. Thus, C1 binding to fibrillar Aβ deposits may precede microglial activation. Both Aβ and pro-inflammatory stimuli are able to activate microglia, which results in increased Aβ and cytokine production [103]. Furthermore, cultured human microglial cells show an increase in cytokine production after co-stimulation of Aβ with C1q and serum amyloid P (SAP) [104]. This suggests that microglia may get triggered by both Aβ- and SP-associated factors such as C1q, which results in the secretion of pro-inflammatory cytokines and Aβ, both accelerating neurodegeneration.
Although none of the complement factors directly regulate conformational changes of Aβ, complement activation as a whole plays a role in the Aβ aggregation in vivo. Therefore, complement factors might act as amateur chaperones, although their exact role in Aβ aggregation remains to be elucidated.
Professional Chaperones
Heat shock proteins (Hsp) are professional chaperones. They are highly conserved proteins constitutively expressed in most cells under normal conditions where they play a role in cellular metabolism and help normal folding processes [105]. In addition, during cell stress, they bind unfolded proteins to keep them in their native state [105]. Heat shock proteins can be divided into two different families based on size and function: classic Hsps such as Hsp100, Hsp90, Hsp70, Hsp60, and the small heat shock proteins (sHsps). Hsps with a molecular weight of 60 kD or more possess an ATP-binding site and are actively involved in the process of refolding of misfolded proteins [106]. Small Hsps, with a molecular weight of 40 kD or less, lack this ATP-binding site and assist the Hsps in their refolding function [107]. The role of Hsps in misfolded protein recognition and refolding is illustrated in Fig. 1.
Fig. 1.
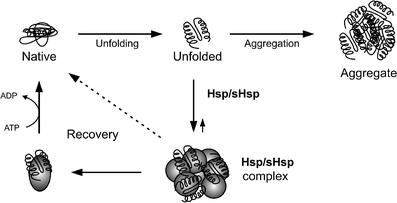
The role of heat shock proteins (Hsp) and small heat shock proteins (sHsps) in recognition and refolding of unfolded and misfolded proteins. Unfolded or misfolded proteins are recognized by Hsps and sHsps. Together with these unfolded or misfolded proteins, Hsps and sHsps form a complex. In addition, Hsps recover unfolded or misfolded proteins back to their native form using ATP. If unfolded or misfolded protein are not recognized by the Hsp/sHsps, these unfolded or misfolded proteins are capable of forming aggregates
Small Heat Shock Proteins
Small Hsps function as molecular chaperones that can prevent proteins from adopting an incorrect conformation [108]. The sHsp family is characterized by the presence of an α-crystallin domain, a stretch of 80–100 amino acids in the C terminal half of the proteins [109]. So far, the sHsp family comprises ten sHsps, including αB-crystallin, Hsp27, Hsp20, HspB8, and HspB2/B3 [110]. Although sHsps are predominantly expressed in muscle cells, several family members are also found in human brain.
In AD, αB-crystallin and Hsp27 are upregulated and expressed by astrocytes surrounding SPs and NFTs [111–114], whereas Hsp20, HspB2, and HspB8 colocalize with Aβ in SPs and CAA [115, 116]. Although αB-crystallin or Hsp27 do not colocalize with Aβ in SPs, direct interaction between Aβ and these sHsps in addition to Hsp20 and HspB8 has been demonstrated [111, 117–119]. In addition, high-affinity binding of αB-crystallin and Aβ has been observed in eye lenses from AD patients [120]. Furthermore, αB-crystallin is able to prevent mature Aβ fibril formation, retaining it in a non-fibrillar, but likely a protofibrillar state, which is highly toxic to neurons [121]. Recently, we demonstrated that αB-crystallin, Hsp20, and HspB8 inhibit Aβ-mediated toxicity towards cerebrovascular cells probably by preventing aggregation of Aβ at the cell surface [116, 117]. Others showed that Hsp27 directly binds to hyperphosphorylated tau, thereby protecting against cell death [122].
Hsps are involved in the formation and persistence of misfolded protein aggregates. They are upregulated in several neurodegenerative diseases, such as AD, Creutzfeldt–Jakob disease, and Parkinson’s disease probably as a reaction to the formation of misfolded proteins [113, 123–126]. However, despite of increased intracellular levels, they are unable to prevent accumulation of Aβ in AD possibly because of decreased chaperone activity. In aged rats, this was illustrated by a significant decrease of Hsp90 function [127], resulting in diminished hepatic chaperone capacity. Furthermore, the increasing amount of damaged or misfolded proteins as a result of defects in protein degradation might lead to a total decrease in chaperone activity in aged cells [128]. Thus, the state of misfolded protein recognition and repair systems, such as the (s)Hsp system, might be of great importance in the development of neurodegenerative diseases.
Miscellaneous Proteins
Apart from the above-described proteins, several other molecules are also associated with the pathological lesions of AD, and some of these can be regarded as amateur chaperones. Acute phase proteins, such as α1-antichymotrypsin (ACT), α2-macroglobulin (α2M), and SAP, are all associated with Aβ deposition [129–132]. ACT is a serine protease inhibitor of the serpin family, and in AD, ACT levels are upregulated, and binding of ACT with Aβ induces Aβ fibrillogenesis [133–135]. Furthermore, when ACT is overexpressed in transgenic mice, an increased plaque load in the brains of these mice and impaired spatial learning is observed [134, 135]. α2M also binds Aβ, although in contrast to ACT, this binding prevents Aβ fibril formation and fibril-associated neurotoxicity [136, 137]. α2M promotes the protease-mediated degradation of α2M/Aβ complexes and contributes to clearance of Aβ from the brain (discussed in paragraph 4) [138, 139]. The glycoprotein SAP belongs to the pentraxin family and is a common component of all known types of amyloid fibrils. SAP is upregulated in AD and protects amyloid fibrils from proteolysis in vitro [140, 141]. SAP not only colocalizes with SPs and interacts with aggregated Aβ; SAP oligomers also bind and activate C1 [142]. Both C1 and SAP may bind to fibrillar Aβ deposits in vivo and induce microglial activation, as cultured human microglial cells show an increase in cytokine production after co-stimulation of Aβ with C1q and SAP [104]. These observations further strengthen the above-noted suggestion that not only Aβ, but also several Aβ-binding proteins, are capable of activating the complement system, and thus, contribute to neuroinflammation in AD. In addition, both α2M and ACT, in contrast to SAP, can be regarded as amateur chaperones, as they regulate conformational changes of Aβ.
Tissue-type plasminogen activator (tPA) regulates activation of plasminogen into plasmin and is expressed in various regions of the brain especially in the hippocampus [143]. Several reports suggested an important role for tPA in AD, as the tPA system is involved in Aβ turnover [144, 145]. Fibrillar forms of Aβ stimulate tPA activity in vitro, whereas in AD patients, a reduction of tPA activity is observed in the affected areas [144, 146]. Although tPA has no effect on conformational changes of Aβ, it might play a role in the clearance of Aβ from the brain (paragraph 4].
The actin-regulatory protein gelsolin is found both intracellularly and in plasma [147, 148]. Plasma gelsolin can be considered an amateur chaperone, as it binds Aβ and not only inhibits its Aβ fibrillization but is also capable of degrading preformed Aβ fibrils [149, 150]. Furthermore, gelsolin inhibits Aβ-mediated neurotoxicity [151].
One of the major gangliosides in the brain is GM1. Soluble Aβ binds GM1 and the formed complexes accelerate Aβ fibrillogenesis by acting as a seed for Aβ [152]. In the presence of GM1, Aβ is more neurotoxic than Aβ alone, and cholesterol-rich membranes demonstrate accelerated Aβ binding due to the formation of GM1 clusters [153, 154]. As GM1 is a major component of lipid rafts and recent studies suggest that Aβ accumulation in these lipid rafts is an early event in AD development, GM1 might play an important role in the early steps in AD pathogenesis [155, 156].
In summary, several proteins are associated with Aβ aggregates in the AD brain and contribute to the aggregation of Aβ and should, therefore, be considered as amateur chaperones. In addition, they might play a role in triggering inflammation.
Aβ-Binding Proteins and Intracellular Interactions with Aβ
Intracellular accumulation of Aβ already starts in the ER or in the Golgi apparatus of the cell [157–159]. Intracellular Aβ is associated with neuronal damage [160, 161], and intraneuronal accumulation of Aβ in transgenic mice was correlated with impairments in synaptic plasticity [162]. Intraneuronal accumulation of Aβ in those brain areas affected earliest in AD suggests a possible relation between intracellular Aβ and development of AD [160].
A few proteins that interact with intracellular Aβ and affect its intracellular fate have been identified. The endoplasmic reticulum amyloid beta-peptide-binding protein binds intracellular Aβ and mediates neurotoxicity in neuronal cells by forming an intracellular target for Aβ [163]. In addition, the mitochondrial enzyme amyloid-β alcohol dehydrogenase also binds Aβ inside neurons, resulting in the production of free radicals [164]. However, whether these intracellular Aβ-binding proteins affect aggregation of Aβ within the cells remains unknown. Therefore, both these proteins cannot, for the time being, be defined as amateur chaperones of Aβ.
The first lines of defense against misfolded and aggregated proteins are the professional chaperones, which counteract these processes and are able to stimulate clearance of misfolded proteins by proteosomal degradation. Newly synthesized proteins are folded by several other proteins, such as immunoglobulin-binding protein (BiP)/glucose-regulated protein (GRP78), and calnexin. GRP78 is a member of the Hsp70 protein family and interacts with intracellular APP. GRP78 regulates APP and Aβ secretion by intervening between APP and β-/γ-secretases within the cell [165].
It is not surprising that in AD, where misfolded protein molecules accumulate, both Hsp90 and Hsp70 synthesis is increased. Several members of the Hsp family directly interact with intracellular Aβ, but only recently, Hsp70 was identified as a protector against intracellular Aβ accumulation [166, 167]. Besides, immunoreactivity of both Hsp90, 70, and Hsp60 is found in SPs [132], which suggests that these professional chaperones may not only interact with misfolded protein in the cell interior [168–171]. In addition, it has also been postulated that up-regulation of Hsp90 and Hsp70 may suppress the formation of NFTs by partitioning tau into a productive folding pathway and thereby preventing its aggregation [172]. Recently, it was demonstrated that the chaperone CHIP–Hsc70 complex conjugates ubiquitin to hyperphosphorylated tau, which enhances cell survival by elimination of soluble hyperphosphorylated tau [173]. These data suggest that the cell increases production of the Hsps to cope with the presence of misfolded proteins such as hyperphosphorylated tau and accumulating Aβ. At some point, this protective mechanism seems to fail, however. In line with this hypothesis, it was shown that the actin and tubulin specific chaperone Hsp60 is decreased in AD, resulting in a decrease of cytoskeletal proteins in AD-affected neurons [174]. Thus, both production and function of Hsps seems to be disturbed in AD, which might result in the accumulation of misfolded proteins. The role of other Hsps in regulating intracellular Aβ or tau folding remains to be investigated (Table 1).
Table 1.
Summary of the expression of chaperones in AD brains and their interaction and effects on Aβ and tau
| SP/CAA | NFT | Direct interaction | Effects on Aβ or tau in general | |
|---|---|---|---|---|
| Apolipoproteins | ||||
| ApoE | + | + | Aβ/tau | ↑ Fibrillar Aβ /↓ hyperph. Tau |
| ApoJ | + | ? | Aβ | ↓ Aβ aggregation |
| HSPGs | ||||
| Perlecan | ± | ± | Aβ | HSPGs: |
| Agrin | + | − | Aβ | ↓ Proteolytic breakdown Aβ |
| Glypican 1 | + | − | ? | ↑ Non-fibrillar → fibrillar Aβ |
| Syndecan 1–3 | + | − | ? | ↑ Phosphorylation tau |
| Collagen XVIII | + | − | ? | |
| GAGs | + | + | Aβ/tau | |
| Complement factors | ||||
| Hageman Factor | + | + | ? | Aβ activates complement in AD |
| C1q | + | + | Aβ | C3 ↓ Aβ deposition |
| C3/C4 | + | + | Aβ | |
| C5-9 | + | + | ? | |
| Heat shock proteins | ||||
| Hsp90 | + | ? | Tau | ↓ Tau aggregation |
| Hsp70 | + | ? | Aβ/tau | ↓ Tau aggregation |
| Small Hsps | ||||
| αB-crystallin | − | − | Aβ | ↓ Aβ fibril formation |
| Hsp27 | − | ± | Aβ/tau | ↓ Aβ fibril formation |
| Hsp20 | + | − | Aβ | ↓ Aβ fibril formation |
| HspB2/B3 | + | + | – | No effect |
| HspB8 | + | − | Aβ | ↓ Aβ fibril formation |
| Acute phase proteins | ||||
| α1-antichymotrypsin | + | − | Aβ | ↑ Aβ fibrillization |
| α2-macroglobulin | + | − | Aβ | |
| serum amyloid P | + | + | Aβ | ↑ Aβ fibrillization |
| Miscellaneous compounds | ||||
| tPA | − | − | Aβ | ↓ Aβ fibril formation |
| Gelsolin | − | − | Aβ | ↑ Aβ fibrillization |
Expression of chaperones in a specific lesion is illustrated as follows: present (+), by conflicting reports (±), absence (−), and unknown (?); ↓ = inhibition or down-regulation, ↑ = induction or up-regulation
SP Senile plaques, CAA cerebral amyloid angiopathy, NFT neurofibrillary tangles, HSPGs heparan sulphate proteoglycans, Aβ amyloid-beta, Hsp heat shock proteins, Apo apolipoproteins, SAP serum amyloid P, tPA tissue-type plasminogen activator, GAGs glycosaminoglycans, LDLR low-density lipoprotein receptor, LRP-1 LDL receptor protein-1, BBB blood–brain barrier
Aβ-Binding Proteins and Aβ Clearance
Aβ-binding proteins, amateur chaperones, play a role in the clearance of Aβ from brain by functioning as a transporter molecule. Two major pathways govern Aβ clearance. By the first pathway, Aβ is removed from brain to blood via active transport across the BBB. This active transport is performed by specialized transporters, so-called “Aβ-receptors”, expressed by endothelial cells. Second, Aβ is removed from brain via phagocytosis by both microglial cells and astrocytes. In both pathways, interaction of Aβ with cell surface Aβ-receptors is crucial; therefore, the expression levels of Aβ-binding proteins might contribute to Aβ clearance by regulating its binding with Aβ receptors.
The low-density lipoprotein receptor-related protein-1 (LRP-1) binds Aβ in a complex with ApoE at the abluminal side of the endothelium and internalizes these ApoE/Aβ complexes followed by degradation in lysosomes or transport into the plasma [4, 175]. However, LRP-1 also mediates transport of free Aβ across the BBB [10]. In contrast to LRP-1, the receptor for advanced glycation end products (RAGE) transports Aβ from the circulation into the central nervous system [176]. Similar to RAGE, the Aβ receptor megalin is also involved in the transport of Aβ from blood to brain, although megalin probably plays only a minor role in Aβ transport. Furthermore, megalin binds Aβ/ApoE complexes rather than free Aβ [177, 178]. Clearance of Aβ/ApoE complexes from brain might be ApoE isoform-dependent. ApoE4 forms less stable complexes with Aβ than ApoE3 or ApoE2; therefore, ApoE4 reduces Aβ transport efficiency across the BBB. Additionally, as described above (paragraph 2), ApoE4 enhances Aβ aggregation more efficiently than ApoE3, which also inhibits clearance. On the other hand, the LDL receptor shows a marked preference for the ApoE3 and ApoE4 isoforms and binds the ApoE2 isoform poorly [179]. Given the similarity between the LDL receptor family, other LDL receptors, such as the LRP-1 receptor, might display similar specificities towards the ApoE isoforms, but this has not been reported yet. Moreover, lipidation of ApoE also affects clearance of ApoE and ApoE/Aβ complexes from brain, as LRP preferentially binds lipid-rich forms of ApoE [179]. These data indicate that Aβ-binding proteins, especially ApoE, and possibly, ApoJ, play an important role in transport of Aβ across the BBB and that both the ApoE isoform and the ApoE lipidation state affect Aβ clearance. In addition to ApoE and ApoJ, the Aβ-binding protein α2M also forms complexes with Aβ. As α2M is a ligand for LRP-1, these α2M/Aβ complexes might undergo LRP-1-mediated endocytosis and degradation or translocation into the plasma [7, 139].
Stimulation of the transport of Aβ across the BBB demonstrated to be an effective therapeutic approach in AD, as several studies demonstrated elevated levels of Aβ in the plasma of mice after passive immunization with anti-Aβ antibodies or Fab fragments [180–182], and decline in cognitive performance was arrested in patients that received vaccination [183]. However, the occurrence of severe meningoencephalitis in human patients after active immunization with Aβ hampered widespread application of this type of therapy. Administration of Aβ-binding proteins that demonstrate similar positive effects, but possibly, without the severe immune reactions associated with antibody therapy, might provide an alternative strategy. An interesting example of such an Aβ-binding protein is gelsolin. This protein has high affinity for Aβ and reduces Aβ levels in a transgenic mouse model of AD [184]. Furthermore, administration of gelsolin or GM1 in PS/APP mice resulted in decreased Aβ aggregation in the brains [184]. Both gelsolin and GM1 act as a “peripheral sink” for Aβ. Although both compounds did not enter the brain, they lowered soluble Aβ concentrations in the blood, shifted the balance between blood and cerebral Aβ concentrations, and accordingly, stimulated Aβ transport over the BBB. Therefore, other Aβ-binding proteins administered in the circulation might also act as “peripheral sinks” [181, 184].
Both activated microglial cells and activated astrocytes are associated with Aβ deposition and may internalize Aβ fragments via phagocytosis [185–187]. Activation of the complement system is, among others, achieved by Hsps such as Hsp60 and Hsp70, which are able to induce phagocytosis by microglia, and thus, clearance of Aβ [132, 188, 189]. In addition, the absence of ApoE reduces internalization and degradation of Aβ by astrocytes in the brain, demonstrating that ApoE is directly involved in the clearance of Aβ from brain via phagocytosis by microglial cells and astrocytes [40]. tPA might also contribute to clearance of Aβ, as it accelerates Aβ clearance from transgenic mouse brains [146]. Thus, as Aβ-chaperones contribute to activation of the complement system or activation of microglial cells and astrocytes, these proteins might contribute to the clearance of Aβ from the brain via phagocytosis.
Concluding Remarks
Professional chaperones, such as the heat shock protein family, and amateur chaperones, such as apolipoproteins and HSPGs and several other proteins, have a role in the intracellular handling of misfolded proteins, induce conformational changes of proteins, or act as transporter of proteins (Fig. 2). This suggests that these chaperones form interesting therapeutic targets in the prevention and treatment of neurodegenerative diseases.
Fig. 2.
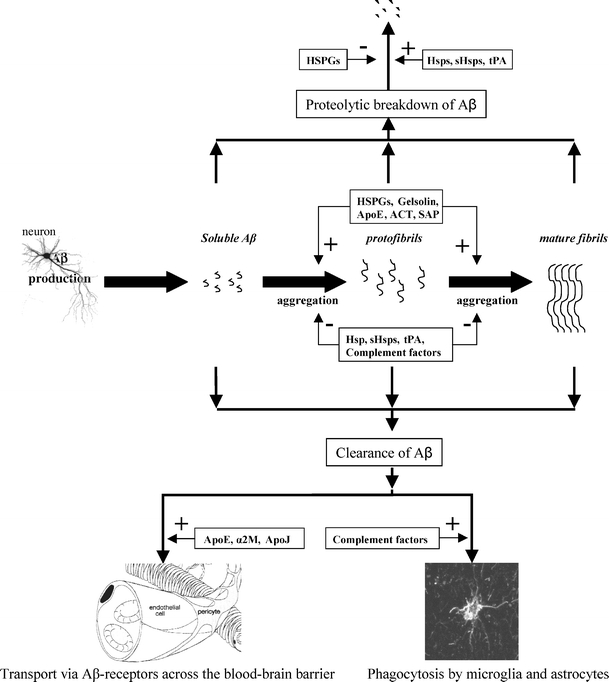
The putative role of chaperones in amyloid-β (Aβ) fibril formation, proteolytic breakdown, and clearance from the brain. In Alzheimer’s disease, soluble Aβ, predominantly produced in neurons, is converted into β-sheet rich protofibrils and eventually forms mature Aβ fibrils. The conversion from soluble Aβ to protofibrils and fibrils, which accumulate in senile plaques and cerebral amyloid angiopathy, is enhanced by chaperones as apolipoprotein E (ApoE), Gelsolin, α1-antichymotrypsin (ACT) and several heparan sulphate proteoglycans (HSPGs), which function as catalysts. In contrast, the heat shock protein family, tissue-type plasminogen activator (tPA) and complement factors prevent the transition of soluble Aβ into protofibrils and mature fibrils. Furthermore, heat shock proteins and tPA stimulate the proteolytic breakdown of (proto)fibrils, whereas HSPGs prevent this breakdown. Finally, the clearance of Aβ from the brain across the blood–brain barrier is stimulated by ApoE, ApoJ, and α2-macroglobulin (α2M), whereas complement factors stimulate phagocytosis-mediated clearance of Aβ by activated microglia and astrocytes
In the process of clearance of Aβ from the brain, Aβ-binding partners might play important roles by acting as Aβ transporter proteins in both the receptor-mediated clearance of Aβ across the BBB but also as a “peripheral sink” for Aβ. Both ApoE isotype and local concentrations in the brain might regulate Aβ transport across the BBB, but as this transport is receptor-mediated, other Aβ-binding proteins might also fulfill such a role. In addition, transport of aggregated Aβ across the BBB is less efficient than soluble Aβ. Thus, by preventing self-aggregation of Aβ, Aβ-binding proteins contribute to the clearance of Aβ from the brain. As a therapeutic strategy, Aβ-binding proteins serving as a “peripheral sink”, such as gelsolin, seem promising [184].
Overexpression of professional chaperones, such as the Hsps, to prevent aggregation of misfolded proteins will have to be evaluated carefully, as they also interact with other chaperones and are dependent on this interaction to fulfill some of their functions. This strategy may therefore result in instability of the cell-stress mechanism, which may cause the system to collapse. A solution may be found in the overexpression of several chaperones, which may be required to achieve an impact on the progression of the disease.
Another pitfall in the use of professional chaperones as therapeutic agents is their ability to bind misfolded proteins and keep them in an intermediate conformation. This type of conformation might even be more toxic than the aggregated state. As an example, co-incubations of αB-cystallin with Aβ are more toxic to neurons than Aβ alone [121]. Furthermore, Hsps are most likely to be involved in early development of neurodegenerative diseases, given their natural function. Yet, the role of this protein family in maturation of the neurodegenerative lesions remains to be elucidated.
In conclusion, studying the role of chaperones, both professional and amateur, in the pathophysiology of AD will provide us with a better understanding of the mechanisms underlying the formation and accumulation of toxic aggregates in AD, which, eventually, will lead to the design of more effective therapeutic strategies.
Acknowledgments
This work was supported by grants from the “Internationale Stichting Alzheimer Onderzoek” (ISAO, grants 01506 and 03517), Zon-MW Innovational Research (number 917.46.331, “Vidi program”) and the “Hersenstichting Nederland” (number 11F03(2)11 and 14F06.18). We would like to thank Wilbert Boelens for helpful discussions.
References
- 1.Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6:487–498 [DOI] [PubMed]
- 2.Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885–890 [DOI] [PubMed]
- 3.Lee VM, Balin BJ, Otvos L, Jr, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251:675–678 [DOI] [PubMed]
- 4.Zlokovic BV (2004) Clearing amyloid through the blood-brain barrier. J Neurochem 89:807–811 [DOI] [PubMed]
- 5.Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY (2003) Astrocytes accumulate A beta 42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res 971:197–209 [DOI] [PubMed]
- 6.Paresce DM, Ghosh RN, Maxfield FR (1996) Microglial cells internalize aggregates of the Alzheimer’s disease amyloid beta-protein via a scavenger receptor. Neuron 17:553–565 [DOI] [PubMed]
- 7.Tanzi RE, Moir RD, Wagner SL (2004) Clearance of Alzheimer’s Abeta peptide: the many roads to perdition. Neuron 43:605–608 [DOI] [PubMed]
- 8.Selkoe DJ (1997) Alzheimer’s disease: genotypes, phenotypes, and treatments. Science 275:630–631 [DOI] [PubMed]
- 9.Selkoe DJ (2000) The origins of Alzheimer disease: a is for amyloid. JAMA 283:1615–1617 [DOI] [PubMed]
- 10.Deane R, Wu Z, Sagare A, Davis J, Du YS, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV (2004) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43:333–344 [DOI] [PubMed]
- 11.Dickson DW (1997) The pathogenesis of senile plaques. J Neuropathol ExpNeurol 56:321–339 [DOI] [PubMed]
- 12.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P (1992) Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA 89:10016–10020 [DOI] [PMC free article] [PubMed]
- 13.Rogers J, Lue LF (2001) Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer’s disease. Neurochem Int 39:333–340 [DOI] [PubMed]
- 14.Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM (1985) Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 76:1501–1513 [DOI] [PMC free article] [PubMed]
- 15.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW (1987) Astrocytes synthesize apolipoprotein E and metabolize apolipoprotein E-containing lipoproteins. Biochim Biophys Acta 917:148–161 [DOI] [PubMed]
- 16.Pitas RE, Boyles JK, Lee SH, Hui D, Weisgraber KH (1987) Lipoproteins and their receptors in the central nervous system. Characterization of the lipoproteins in cerebrospinal fluid and identification of apolipoprotein B,E(LDL) receptors in the brain. J Biol Chem 262:14352–14360 [PubMed]
- 17.Stone DJ, Rozovsky I, Morgan TE, Anderson CP, Hajian H, Finch CE (1997) Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol 143:313–318 [DOI] [PubMed]
- 18.Wilhelmus MM, Otte-Holler I, Davis J, Van Nostrand WE, de Waal RM, Verbeek MM (2005) Apolipoprotein E genotype regulates amyloid-beta cytotoxicity. J Neurosci 25:3621–3627 [DOI] [PMC free article] [PubMed]
- 19.Mahley RW (1988) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science 240:622–630 [DOI] [PubMed]
- 20.Plump AS, Breslow JL (1995) Apolipoprotein E and the apolipoprotein E-deficient mouse. Annu Rev Nutr 15:495–518 [DOI] [PubMed]
- 21.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923 [DOI] [PubMed]
- 22.Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE (1994) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7:180–184 [DOI] [PubMed]
- 23.Mayeux R, Stern Y, Ottman R, Tatemichi TK, Tang MX, Maestre G, Ngai C, Tycko B, Ginsberg H (1993) The apolipoprotein epsilon 4 allele in patients with Alzheimer’s disease. Ann Neurol 34:752–754 [DOI] [PubMed]
- 24.Strittmatter WJ, Roses AD (1996) Apolipoprotein E and Alzheimer’s disease. Annu Rev Neurosci 19:53–77 [DOI] [PubMed]
- 25.Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K (1991) Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt–Jakob disease. Brain Res 541:163–166 [DOI] [PubMed]
- 26.Wisniewski T, Frangione B (1992) Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 135:235–238 [DOI] [PubMed]
- 27.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90:1977–1981 [DOI] [PMC free article] [PubMed]
- 28.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993) Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:8098–8102 [DOI] [PMC free article] [PubMed]
- 29.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-specific binding of apolipoprotein E to beta-amyloid. J Biol Chem 269:23403–23406 [PubMed]
- 30.Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993) Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90:8098–8102 [DOI] [PMC free article] [PubMed]
- 31.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM (1997) Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17:263–264 [DOI] [PubMed]
- 32.Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B., Paul S.M. (1999) Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 96:15233–15238 [DOI] [PMC free article] [PubMed]
- 33.Dolev I, Michaelson DM (2004) A nontransgenic mouse model shows inducible amyloid-beta (Abeta) peptide deposition and elucidates the role of apolipoprotein E in the amyloid cascade. Proc Natl Acad Sci USA 101:13909–13914 [DOI] [PMC free article] [PubMed]
- 34.Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM (2003) Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci 23:7889–7896 [DOI] [PMC free article] [PubMed]
- 35.Sadowski M, Pankiewicz J, Scholtzova H, Ripellino JA, Li Y, Schmidt SD, Mathews PM, Fryer JD, Holtzman DM, Sigurdsson EM, Wisniewski T (2004) A synthetic peptide blocking the apolipoprotein E/beta-amyloid binding mitigates beta-amyloid toxicity and fibril formation in vitro and reduces beta-amyloid plaques in transgenic mice. Am J Pathol 165:937–948 [DOI] [PMC free article] [PubMed]
- 36.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM (1999) Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer’s disease. J Clin Invest 103:R15–R21 [DOI] [PMC free article] [PubMed]
- 37.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 97:2892–2897 [DOI] [PMC free article] [PubMed]
- 38.Carter DB, Dunn E, McKinley DD, Stratman NC, Boyle TP, Kuiper SL, Oostveen JA, Weaver RJ, Boller JA, Gurney ME (2001) Human apolipoprotein E4 accelerates beta-amyloid deposition in APPsw transgenic mouse brain. Ann Neurol 50:468–475 [DOI] [PubMed]
- 39.Fryer JD, DeMattos RB, McCormick LM, O’Dell MA, Spinner ML, Bales KR, Paul SM, Sullivan PM, Parsadanian M, Bu G, Holtzman DM (2005) The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J Biol Chem 280:25754–25759 [DOI] [PubMed]
- 40.Koistinaho M, Lin S, Wu X, Esterman M, Koger D, Hanson J, Higgs R, Liu F, Malkani S, Bales KR, Paul SM (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat Med 10:719–726 [DOI] [PubMed]
- 41.Strittmatter WJ, Weisgraber KH, Goedert M, Saunders AM, Huang D, Corder EH, Dong LM, Jakes R, Alberts MJ, Gilbert JR (1994) Hypothesis: microtubule instability and paired helical filament formation in the Alzheimer disease brain are related to apolipoprotein E genotype. Exp Neurol 125:163–171 [DOI] [PubMed]
- 42.Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong LM, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD (1994) Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA 91:11183–11186 [DOI] [PMC free article] [PubMed]
- 43.Ghebremedhin E, Schultz C, Braak E, Braak H (1998) High frequency of apolipoprotein E epsilon4 allele in young individuals with very mild Alzheimer’s disease-related neurofibrillary changes. Exp Neurol 153:152–155 [DOI] [PubMed]
- 44.Ohm TG, Scharnagl H, Marz W, Bohl J (1999) Apolipoprotein E isoforms and the development of low and high Braak stages of Alzheimer’s disease-related lesions. Acta Neuropathol (Berl) 98:273–280 [DOI] [PubMed]
- 45.Tesseur I, Van Dorpe J, Spittaels K, Van den HC, Moechars D, Van Leuven F (2000) Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol 156:951–964 [DOI] [PMC free article] [PubMed]
- 46.Yasojima K, Schwab C, McGeer EG, McGeer PL (1999) Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol 154:927–936 [DOI] [PMC free article] [PubMed]
- 47.Barger SW, Harmon AD (1997) Microglial activation by Alzheimer amyloid precursor protein and modulation by apolipoprotein E. Nature 388:878–881 [DOI] [PubMed]
- 48.Hu J, LaDu MJ, Van Eldik LJ (1998) Apolipoprotein E attenuates beta-amyloid-induced astrocyte activation. J Neurochem 71:1626–1634 [DOI] [PubMed]
- 49.Laskowitz DT, Goel S, Bennett ER, Matthew WD (1997) Apolipoprotein E suppresses glial cell secretion of TNF alpha. J Neuroimmunol 76:70–74 [DOI] [PubMed]
- 50.Laskowitz DT, Lee DM, Schmechel D, Staats HF (2000) Altered immune responses in apolipoprotein E-deficient mice. J Lipid Res 41:613–620 [PubMed]
- 51.Lynch JR, Morgan D, Mance J, Matthew WD, Laskowitz DT (2001) Apolipoprotein E modulates glial activation and the endogenous central nervous system inflammatory response. J Neuroimmunol 114:107–113 [DOI] [PubMed]
- 52.Colton CA, Brown CM, Czapiga M, Vitek MP (2002) Apolipoprotein-E allele-specific regulation of nitric oxide production. Ann N Y Acad Sci 962:212–225 [DOI] [PubMed]
- 53.Trougakos IP, Gonos ES (2002) Clusterin/apolipoprotein J in human aging and cancer. Int J Biochem Cell Biol 34:1430–1448 [DOI] [PubMed]
- 54.Calero M, Rostagno A, Matsubara E, Zlokovic B, Frangione B, Ghiso J (2000) Apolipoprotein J (clusterin) and Alzheimer’s disease. Microsc Res Tech 50:305–315 [DOI] [PubMed]
- 55.Rosenberg ME, Dvergsten J, Correa-Rotter R (1993) Clusterin: an enigmatic protein recruited by diverse stimuli. J Lab Clin Med 121:205–214 [PubMed]
- 56.Zlokovic BV, Martel CL, Mackic JB, Matsubara E, Wisniewski T, McComb JG, Frangione B, Ghiso J (1994) Brain uptake of circulating apolipoproteins J and E complexed to Alzheimer’s amyloid beta. Biochem Biophys Res Commun 205:1431–1437 [DOI] [PubMed]
- 57.Rassart E, Bedirian A, Do CS, Guinard O, Sirois J, Terrisse L, Milne R (2000) Apolipoprotein D. Biochim Biophys Acta 1482:185–198 [DOI] [PubMed]
- 58.Belloir B, Kovari E, Surini-Demiri M, Savioz A (2001) Altered apolipoprotein D expression in the brain of patients with Alzheimer disease. J Neurosci Res 64:61–69 [DOI] [PubMed]
- 59.Terrisse L, Seguin D, Bertrand P, Poirier J, Milne R, Rassart E (1999) Modulation of apolipoprotein D and apolipoprotein E expression in rat hippocampus after entorhinal cortex lesion. Brain Res Mol Brain Res 70:26–35 [DOI] [PubMed]
- 60.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M (1999) Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 68:729–777 [DOI] [PubMed]
- 61.van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM (2003) Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol 2:482–492 [DOI] [PubMed]
- 62.Iozzo RV (2001) Heparan sulfate proteoglycans: intricate molecules with intriguing functions. J Clin Invest 108:165–167 [DOI] [PMC free article] [PubMed]
- 63.Perry G, Siedlak SL, Richey P, Kawai M, Cras P, Kalaria RN, Galloway PG, Scardina JM, Cordell B, Greenberg BD (1991) Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J Neurosci 11:3679–3683 [DOI] [PMC free article] [PubMed]
- 64.Snow AD, Mar H, Nochlin D, Kimata K, Kato M, Suzuki S, Hassell J, Wight TN (1988) The presence of heparan sulfate proteoglycans in the neuritic plaques and congophilic angiopathy in Alzheimer’s disease. Am J Pathol 133:456–463 [PMC free article] [PubMed]
- 65.Snow AD, Mar H, Nochlin D, Sekiguchi RT, Kimata K, Koike Y, Wight TN (1990) Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am J Pathol 137:1253–1270 [PMC free article] [PubMed]
- 66.Snow AD, Sekiguchi RT, Nochlin D, Kalaria RN, Kimata K (1994) Heparan sulfate proteoglycan in diffuse plaques of hippocampus but not of cerebellum in Alzheimer’s disease brain. Am J Pathol 144:337–347 [PMC free article] [PubMed]
- 67.Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, Kimata K, Wight TN (1995) Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the beta-amyloid protein of Alzheimer’s disease. Arch Biochem Biophys 320:84–95 [DOI] [PubMed]
- 68.van Horssen J, Otte-Holler I, David G, Maat-Schieman ML, van den Heuvel LP, Wesseling P, de Waal RM, Verbeek MM (2001) Heparan sulfate proteoglycan expression in cerebrovascular amyloid beta deposits in Alzheimer’s disease and hereditary cerebral hemorrhage with amyloidosis (Dutch) brains. Acta Neuropathol (Berl) 102:604–614 [DOI] [PubMed]
- 69.Verbeek MM, Otte-Holler I, van den BJ, van den Heuvel LP, David G, Wesseling P, de Waal RM (1999) Agrin is a major heparan sulfate proteoglycan accumulating in Alzheimer’s disease brain. Am J Pathol 155:2115–2125 [DOI] [PMC free article] [PubMed]
- 70.Cotman SL, Halfter W, Cole GJ (2000) Agrin binds to beta-amyloid (Abeta), accelerates abeta fibril formation, and is localized to Abeta deposits in Alzheimer’s disease brain. Mol Cell Neurosci 15:183–198 [DOI] [PubMed]
- 71.Donahue JE, Berzin TM, Rafii MS, Glass DJ, Yancopoulos GD, Fallon JR, Stopa EG (1999) Agrin in Alzheimer’s disease: altered solubility and abnormal distribution within microvasculature and brain parenchyma. Proc Natl Acad Sci USA 96:6468–6472 [DOI] [PMC free article] [PubMed]
- 72.van Horssen J, Wilhelmus MM, Heljasvaara R, Pihlajaniemi T, Wesseling P, de Waal RM, Verbeek MM (2002) Collagen XVIII: a novel heparan sulfate proteoglycan associated with vascular amyloid depositions and senile plaques in Alzheimer’s disease brains. Brain Pathol 12:456–462 [DOI] [PMC free article] [PubMed]
- 73.Gupta-Bansal R, Frederickson RC, Brunden KR (1995) Proteoglycan-mediated inhibition of A beta proteolysis. A potential cause of senile plaque accumulation. J Biol Chem 270:18666–18671 [DOI] [PubMed]
- 74.Castillo GM, Ngo C, Cummings J, Wight TN, Snow AD (1997) Perlecan binds to the beta-amyloid proteins (A beta) of Alzheimer’s disease, accelerates A beta fibril formation, and maintains A beta fibril stability. J Neurochem 69:2452–2465 [DOI] [PubMed]
- 75.Snow AD, Sekiguchi R, Nochlin D, Fraser P, Kimata K, Mizutani A, Arai M, Schreier WA, Morgan DG (1994) An important role of heparan sulfate proteoglycan (Perlecan) in a model system for the deposition and persistence of fibrillar A beta-amyloid in rat brain. Neuron 12:219–234 [DOI] [PubMed]
- 76.Verbeek MM, Eikelenboom P, de Waal RM (1997) Differences between the pathogenesis of senile plaques and congophilic angiopathy in Alzheimer disease. J Neuropathol Exp Neurol 56:751–761 [PubMed]
- 77.Buee L, Ding W, Anderson JP, Narindrasorasak S, Kisilevsky R, Boyle NJ, Robakis NK, Delacourte A, Greenberg B, Fillit HM (1993) Binding of vascular heparan sulfate proteoglycan to Alzheimer’s amyloid precursor protein is mediated in part by the N-terminal region of A4 peptide. Brain Res 627:199–204 [DOI] [PubMed]
- 78.Narindrasorasak S, Lowery D, Gonzalez-DeWhitt P, Poorman RA, Greenberg B, Kisilevsky R (1991) High affinity interactions between the Alzheimer’s beta-amyloid precursor proteins and the basement membrane form of heparan sulfate proteoglycan. J Biol Chem 266:12878–12883 [PubMed]
- 79.Snow AD, Willmer JP, Kisilevsky R (1987) Sulfated glycosaminoglycans in Alzheimer’s disease. Hum Pathol 18:506–510 [DOI] [PubMed]
- 80.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553 [DOI] [PubMed]
- 81.Gervais F, Chalifour R, Garceau D, Kong X, Laurin J, Mclaughlin R, Morissette C, Paquette J (2001) Glycosaminoglycan mimetics: a therapeutic approach to cerebral amyloid angiopathy. Amyloid 8 Suppl 1:28–35 [PubMed]
- 82.Zhu H, Yu J, Kindy MS (2001) Inhibition of amyloidosis using low-molecular-weight heparins. Mol Med 7:517–522 [PMC free article] [PubMed]
- 83.Kisilevsky R, Lemieux LJ, Fraser PE, Kong X, Hultin PG, Szarek WA (1995) Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer’s disease. Nat Med 1:143–148 [DOI] [PubMed]
- 84.Banz Y, Hess OM, Robson SC, Mettler D, Meier P, Haeberli A, Csizmadia E, Korchagina EY, Bovin NV, Rieben R (2005) Locally targeted cytoprotection with dextran sulfate attenuates experimental porcine myocardial ischaemia/reperfusion injury. Eur Heart J 26:2334–2343 [DOI] [PubMed]
- 85.Kirschfink M, Blase L, Engelmann S, Schwartz-Albiez R (1997) Secreted chondroitin sulfate proteoglycan of human B cell lines binds to the complement protein C1q and inhibits complex formation of C1. J Immunol 158:1324–1331 [PubMed]
- 86.Caughman GB, Boackle RJ, Vesely J (1982) A postulated mechanism for heparin’s potentiation of C1 inhibitor function. Mol Immunol 19:287–295 [DOI] [PubMed]
- 87.Eikelenboom P, Stam FC (1982) Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol (Berl) 57:239–242 [DOI] [PubMed]
- 88.Eikelenboom P, Stam FC (1984) An immunohistochemical study on cerebral vascular and senile plaque amyloid in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol 47:17–25 [DOI] [PubMed]
- 89.McGeer PL, Akiyama H, Itagaki S, McGeer EG (1989) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107:341–346 [DOI] [PubMed]
- 90.Webster S, Bradt B, Rogers J, Cooper N (1997) Aggregation state-dependent activation of the classical complement pathway by the amyloid beta peptide. J Neurochem 69:388–398 [DOI] [PubMed]
- 91.Webster S, Lue LF, Brachova L, Tenner AJ, McGeer PL, Terai K, Walker DG, Bradt B, Cooper NR, Rogers J (1997) Molecular and cellular characterization of the membrane attack complex, C5b-9, in Alzheimer’s disease. Neurobiol Aging 18:415–421 [DOI] [PubMed]
- 92.Yasuhara O, Walker DG, McGeer PL (1994) Hageman factor and its binding sites are present in senile plaques of Alzheimer’s disease. Brain Res 654:234–240 [DOI] [PubMed]
- 93.Rozemuller AJ, Eikelenboom P, Theeuwes JW, Jansen Steur EN, de Vos RA (2000) Activated microglial cells and complement factors are unrelated to cortical Lewy bodies. Acta Neuropathol (Berl) 100:701–708 [DOI] [PubMed]
- 94.Chen S, Frederickson RC, Brunden KR (1996) Neuroglial-mediated immunoinflammatory responses in Alzheimer’s disease: complement activation and therapeutic approaches. Neurobiol Aging 17:781–787 [DOI] [PubMed]
- 95.Veerhuis R, Janssen I, Hoozemans JJ, De Groot CJ, Hack CE, Eikelenboom P (1998) Complement C1-inhibitor expression in Alzheimer’s disease. Acta Neuropathol (Berl) 96:287–296 [DOI] [PubMed]
- 96.Walker DG, Yasuhara O, Patston PA, McGeer EG, McGeer PL (1995) Complement C1 inhibitor is produced by brain tissue and is cleaved in Alzheimer disease. Brain Res 675:75–82 [DOI] [PubMed]
- 97.Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E (2002) Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci USA 99:10837–10842 [DOI] [PMC free article] [PubMed]
- 98.Abraham CR (2001) Reactive astrocytes and alpha1-antichymotrypsin in Alzheimer’s disease. Neurobiol Aging 22:931–936 [DOI] [PubMed]
- 99.Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP (2000) Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 49:171–186 [DOI] [PubMed]
- 100.Wyss-Coray T, Mucke L (2002) Inflammation in neurodegenerative disease—a double-edged sword. Neuron 35:419–432 [DOI] [PubMed]
- 101.Jiang H, Burdick D, Glabe CG, Cotman CW, Tenner AJ (1994) Beta-amyloid activates complement by binding to a specific region of the collagen-like domain of the C1q A chain. J Immunol 152:5050–5059 [PubMed]
- 102.Webster S, Bonnell B, Rogers J (1997) Charge-based binding of complement component C1q to the Alzheimer amyloid beta-peptide. Am J Pathol 150:1531–1536 [PMC free article] [PubMed]
- 103.Bitting L, Naidu A, Cordell B, Murphy GM, Jr (1996) Beta-amyloid peptide secretion by a microglial cell line is induced by beta-amyloid-(25-35) and lipopolysaccharide. J Biol Chem 271:16084–16089 [DOI] [PubMed]
- 104.Veerhuis R, Van Breemen MJ, Hoozemans JM, Morbin M, Ouladhadj J, Tagliavini F, Eikelenboom P (2003) Amyloid beta plaque-associated proteins C1q and SAP enhance the Abeta1-42 peptide-induced cytokine secretion by adult human microglia in vitro. Acta Neuropathol (Berl) 105:135–144 [DOI] [PubMed]
- 105.Walter S, Buchner J (2002) Molecular chaperones—cellular machines for protein folding. Angew Chem Int Ed Engl 41:1098–1113 [DOI] [PubMed]
- 106.Gusev NB, Bogatcheva NV, Marston SB (2002) Structure and properties of small heat shock proteins (sHsp) and their interaction with cytoskeleton proteins. Biochemistry (Mosc) 67:511–519 [DOI] [PubMed]
- 107.MacRae TH (2000) Structure and function of small heat shock/alpha-crystallin proteins: established concepts and emerging ideas. Cell Mol Life Sci 57:899–913 [DOI] [PMC free article] [PubMed]
- 108.Buchner J (1996) Supervising the fold: functional principles of molecular chaperones. FASEB J 10:10–19 [PubMed]
- 109.de Jong WW, Caspers GJ, Leunissen JA (1998) Genealogy of the alpha-crystallin–small heat-shock protein superfamily. Int J Biol Macromol 22:151–162 [DOI] [PubMed]
- 110.Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JA, de Jong WW (2003) The human genome encodes 10 alpha-crystallin-related small heat shock proteins: HspB1-10. Cell Stress Chaperones 8:53–61 [DOI] [PMC free article] [PubMed]
- 111.Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman JE (1992) Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am J Pathol 140:345–356 [PMC free article] [PubMed]
- 112.Renkawek K, Voorter CE, Bosman GJ, van Workum FP, de Jong WW (1994) Expression of alpha B-crystallin in Alzheimer’s disease. Acta Neuropathol (Berl) 87:155–160 [DOI] [PubMed]
- 113.Renkawek K, Bosman GJ, de Jong WW (1994) Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol (Berl) 87:511–519 [DOI] [PubMed]
- 114.Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K (1993) Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J Neurol Sci 119:203–208 [DOI] [PubMed]
- 115.Wilhelmus MM, Otte-Holler I, Wesseling P, de Waal RM, Boelens WC, Verbeek MM (2006) Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer’s disease brains. Neuropathol Appl Neurobiol 32:119–130 [DOI] [PubMed]
- 116.Wilhelmus MM, Boelens WC, Otte-Holler I, Kamps B, Kusters B, Maat-Schieman ML, de Waal RM, Verbeek MM (2006) Small heat shock protein HspB8: its distribution in Alzheimer’s disease brains and its inhibition of amyloid-beta protein aggregation and cerebrovascular amyloid-beta toxicity. Acta Neuropathol (Berl) 111:139–149 [DOI] [PubMed]
- 117.Wilhelmus MM, Boelens WC, Otte-Holler I, Kamps B, de Waal RM, Verbeek MM (2006) Small heat shock proteins inhibit amyloid-beta protein aggregation and cerebrovascular amyloid-beta protein toxicity. Brain Res 1089:67–78 [DOI] [PubMed]
- 118.Kudva YC, Hiddinga HJ, Butler PC, Mueske CS, Eberhardt NL (1997) Small heat shock proteins inhibit in vitro A beta(1-42) amyloidogenesis. FEBS Lett 416:117–121 [DOI] [PubMed]
- 119.Liang JJ (2000) Interaction between beta-amyloid and lens alphaB-crystallin. FEBS Lett 484:98–101 [DOI] [PubMed]
- 120.Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X, Mavros C, Coccia JA, Faget KY, Fitch KA, Masters CL, Tanzi RE, Chylack LT, Bush AI (2003) Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer’s disease. Lancet 361:1258–1265 [DOI] [PubMed]
- 121.Stege GJ, Renkawek K, Overkamp PS, Verschuure P, van Rijk AF, Reijnen-Aalbers A, Boelens WC, Bosman GJ, de Jong WW (1999) The molecular chaperone alphaB-crystallin enhances amyloid beta neurotoxicity. Biochem Biophys Res Commun 262:152–156 [DOI] [PubMed]
- 122.Shimura H, Miura-Shimura Y, Kosik KS (2004) Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J Biol Chem 279:17957–17962 [DOI] [PubMed]
- 123.Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM (2002) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295:865–868 [DOI] [PubMed]
- 124.McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem 83:846–854 [DOI] [PubMed]
- 125.Renkawek K, de Jong WW, Merck KB, Frenken CW, van Workum FP, Bosman GJ (1992) alpha B-crystallin is present in reactive glia in Creutzfeldt–Jakob disease. Acta Neuropathol (Berl) 83:324–327 [DOI] [PubMed]
- 126.Renkawek K, Bosman GJ, Gaestel M (1993) Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport 5:14–16 [DOI] [PubMed]
- 127.Nardai G, Csermely P, Soti C (2002) Chaperone function and chaperone overload in the aged. A preliminary analysis. Exp Gerontol 37:1257–1262 [DOI] [PubMed]
- 128.Csermely P (2001) Chaperone overload is a possible contributor to ‘civilization diseases’. Trends Genet 17:701–704 [DOI] [PubMed]
- 129.Abraham CR, Shirahama T, Potter H (1990) Alpha 1-antichymotrypsin is associated solely with amyloid deposits containing the beta-protein. Amyloid and cell localization of alpha 1-antichymotrypsin. Neurobiol Aging 11:123–129 [DOI] [PubMed]
- 130.Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M (1991) Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett 285:111–114 [DOI] [PubMed]
- 131.Holm NE, Nybo M, Junker K, Toftedal HP, Rasmussen IM, Svehag SE (2000) Localization of human serum amyloid P component and heparan sulfate proteoglycan in in vitro-formed Abeta fibrils. Scand J Immunol 52:110–112 [DOI] [PubMed]
- 132.Prohaszka Z, Singh M, Nagy K, Kiss E, Lakos G, Duba J, Fust G (2002) Heat shock protein 70 is a potent activator of the human complement system. Cell Stress Chaperones 7:17–22 [DOI] [PMC free article] [PubMed]
- 133.Licastro F, Mallory M, Hansen LA, Masliah E (1998) Increased levels of alpha-1-antichymotrypsin in brains of patients with Alzheimer’s disease correlate with activated astrocytes and are affected by APOE 4 genotype. J Neuroimmunol 88:105–110 [DOI] [PubMed]
- 134.Eriksson S, Janciauskiene S, Lannfelt L (1995) Alpha 1-antichymotrypsin regulates Alzheimer beta-amyloid peptide fibril formation. Proc Natl Acad Sci USA 92:2313–2317 [DOI] [PMC free article] [PubMed]
- 135.Ma J, Yee A, Brewer HB, Jr, Das S, Potter H (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature 372:92–94 [DOI] [PubMed]
- 136.Du Y, Bales KR, Dodel RC, Liu X, Glinn MA, Horn JW, Little SP, Paul SM (1998) Alpha2-macroglobulin attenuates beta-amyloid peptide 1-40 fibril formation and associated neurotoxicity of cultured fetal rat cortical neurons. J Neurochem 70:1182–1188 [DOI] [PubMed]
- 137.Hughes SR, Khorkova O, Goyal S, Knaeblein J, Heroux J, Riedel NG, Sahasrabudhe S (1998) Alpha2-macroglobulin associates with beta-amyloid peptide and prevents fibril formation. Proc Natl Acad Sci USA 95:3275–3280 [DOI] [PMC free article] [PubMed]
- 138.Lauer D, Reichenbach A, Birkenmeier G (2001) Alpha 2-macroglobulin-mediated degradation of amyloid beta 1–42: a mechanism to enhance amyloid beta catabolism. Exp Neurol 167:385–392 [DOI] [PubMed]
- 139.Narita M, Holtzman DM, Schwartz AL, Bu G (1997) Alpha2-macroglobulin complexes with and mediates the endocytosis of beta-amyloid peptide via cell surface low-density lipoprotein receptor-related protein. J Neurochem 69:1904–1911 [DOI] [PubMed]
- 140.Emsley J, White HE, O’Hara BP, Oliva G, Srinivasan N, Tickle IJ, Blundell TL, Pepys MB, Wood SP (1994) Structure of pentameric human serum amyloid P component. Nature 367:338–345 [DOI] [PubMed]
- 141.McGeer EG, Yasojima K, Schwab C, McGeer PL (2001) The pentraxins: possible role in Alzheimer’s disease and other innate inflammatory diseases. Neurobiol Aging 22:843–848 [DOI] [PubMed]
- 142.Ying SC, Gewurz AT, Jiang H, Gewurz H (1993) Human serum amyloid P component oligomers bind and activate the classical complement pathway via residues 14-26 and 76-92 of the A chain collagen-like region of C1q. J Immunol 150:169–176 [PubMed]
- 143.Sappino AP, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli JD (1993) Extracellular proteolysis in the adult murine brain. J Clin Invest 92:679–685 [DOI] [PMC free article] [PubMed]
- 144.Kingston IB, Castro MJ, Anderson S (1995) In vitro stimulation of tissue-type plasminogen activator by Alzheimer amyloid beta-peptide analogues. Nat Med 1:138–142 [DOI] [PubMed]
- 145.Tucker HM, Kihiko-Ehmann M, Wright S, Rydel RE, Estus S (2000) Tissue plasminogen activator requires plasminogen to modulate amyloid-beta neurotoxicity and deposition. J Neurochem 75:2172–2177 [DOI] [PubMed]
- 146.Melchor JP, Pawlak R, Strickland S (2003) The tissue plasminogen activator-plasminogen proteolytic cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci 23:8867–8871 [DOI] [PMC free article] [PubMed]
- 147.Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL (1986) Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature 323:455–458 [DOI] [PubMed]
- 148.Kwiatkowski DJ, Mehl R, Yin HL (1988) Genomic organization and biosynthesis of secreted and cytoplasmic forms of gelsolin. J Cell Biol 106:375–384 [DOI] [PMC free article] [PubMed]
- 149.Chauhan VP, Ray I, Chauhan A, Wisniewski HM (1999) Binding of gelsolin, a secretory protein, to amyloid beta-protein. Biochem Biophys Res Commun 258:241–246 [DOI] [PubMed]
- 150.Ray I, Chauhan A, Wegiel J, Chauhan VP (2000) Gelsolin inhibits the fibrillization of amyloid beta-protein, and also defibrillizes its preformed fibrils. Brain Res 853:344–351 [DOI] [PubMed]
- 151.Qiao H, Koya RC, Nakagawa K, Tanaka H, Fujita H, Takimoto M, Kuzumaki N (2005) Inhibition of Alzheimer’s amyloid-beta peptide-induced reduction of mitochondrial membrane potential and neurotoxicity by gelsolin. Neurobiol Aging 26:849–855 [DOI] [PubMed]
- 152.Naiki H, Nakakuki K (1996) First-order kinetic model of Alzheimer’s beta-amyloid fibril extension in vitro. Lab Invest 74:374–383 [PubMed]
- 153.Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, Yamamoto N, Michikawa M, Yoshikawa Y, Terao K, Matsuzaki K, Lemere CA, Selkoe DJ, Naiki H, Yanagisawa K (2004) A seed for Alzheimer amyloid in the brain. J Neurosci 24:4894–4902 [DOI] [PMC free article] [PubMed]
- 154.Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K (2001) Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem 276:24985–24990 [DOI] [PubMed]
- 155.Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG (2004) Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 24:3801–3809 [DOI] [PMC free article] [PubMed]
- 156.Parton RG (1994) Ultrastructural localization of gangliosides; GM1 is concentrated in caveolae. J Histochem Cytochem 42:155–166 [DOI] [PubMed]
- 157.Greenfield JP, Tsai J, Gouras GK, Hai B, Thinakaran G, Checler F, Sisodia SS, Greengard P, Xu H (1999) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci USA 96:742–747 [DOI] [PMC free article] [PubMed]
- 158.Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K (1997) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3:1016–1020 [DOI] [PubMed]
- 159.Xu H, Sweeney D, Wang R, Thinakaran G, Lo AC, Sisodia SS, Greengard P, Gandy S (1997) Generation of Alzheimer beta-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc Natl Acad Sci USA 94:3748–3752 [DOI] [PMC free article] [PubMed]
- 160.Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR (2000) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156:15–20 [DOI] [PMC free article] [PubMed]
- 161.LaFerla FM, Troncoso JC, Strickland DK, Kawas CH, Jay G (1997) Neuronal cell death in Alzheimer’s disease correlates with apoE uptake and intracellular Abeta stabilization. J Clin Invest 100:310–320 [DOI] [PMC free article] [PubMed]
- 162.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409–421 [DOI] [PubMed]
- 163.Yan SD, Fu J, Soto C, Chen X, Zhu H, Al Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D (1997) An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer’s disease. Nature 389:689–695 [DOI] [PubMed]
- 164.Yan SD, Stern DM (2005) Mitochondrial dysfunction and Alzheimer’s disease: role of amyloid-beta peptide alcohol dehydrogenase (ABAD). Int J Exp Pathol 86:161–171 [DOI] [PMC free article] [PubMed]
- 165.Yang Y, Turner RS, Gaut JR (1998) The chaperone BiP/GRP78 binds to amyloid precursor protein and decreases Abeta40 and Abeta42 secretion. J Biol Chem 273:25552–25555 [DOI] [PubMed]
- 166.Fonte V, Kapulkin V, Taft A, Fluet A, Friedman D, Link CD (2002) Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci USA 99:9439–9444 [DOI] [PMC free article] [PubMed]
- 167.Magrane J, Smith RC, Walsh K, Querfurth HW (2004) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci 24:1700–1706. [DOI] [PMC free article] [PubMed]
- 168.Perez N, Sugar J, Charya S, Johnson G, Merril C, Bierer L, Perl D, Haroutunian V, Wallace W (1991) Increased synthesis and accumulation of heat shock 70 proteins in Alzheimer’s disease. Brain Res Mol Brain Res 11:249–254 [DOI] [PubMed]
- 169.Hamos JE, Oblas B, Pulaski-Salo D, Welch WJ, Bole DG, Drachman DA (1991) Expression of heat shock proteins in Alzheimer’s disease. Neurology 41:345–350 [DOI] [PubMed]
- 170.Kitamura Y, Nomura Y (2003) Stress proteins and glial functions: possible therapeutic targets for neurodegenerative disorders. Pharmacol Ther 97:35–53 [DOI] [PubMed]
- 171.Anthony SG, Schipper HM, Tavares R, Hovanesian V, Cortez SC, Stopa EG, Johanson CE (2003) Stress protein expression in the Alzheimer-diseased choroid plexus. J Alzheimers Dis 5:171–177 [DOI] [PubMed]
- 172.Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, Gouras GK, Greengard P, Xu H (2003) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci USA 100:721–726 [DOI] [PMC free article] [PubMed]
- 173.Shimura H, Schwartz D, Gygi SP, Kosik KS (2004) CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem 279:4869–4876 [DOI] [PubMed]
- 174.Schuller E, Gulesserian T, Seidl R, Cairns N, Lube G (2001) Brain t-complex polypeptide 1 (TCP-1) related to its natural substrate beta1 tubulin is decreased in Alzheimer’s disease. Life Sci 69:263–270 [DOI] [PubMed]
- 175.Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV (2000) Clearance of Alzheimer’s amyloid-ss(1-40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest 106:1489–1499 [DOI] [PMC free article] [PubMed]
- 176.Deane R, Du YS, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B (2003) RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med 9:907–913 [DOI] [PubMed]
- 177.Mackic JB, Stins M, McComb JG, Calero M, Ghiso J, Kim KS, Yan SD, Stern D, Schmidt AM, Frangione B, Zlokovic BV (1998) Human blood–brain barrier receptors for Alzheimer’s amyloid-beta 1- 40. Asymmetrical binding, endocytosis, and transcytosis at the apical side of brain microvascular endothelial cell monolayer. J Clin Invest 102:734–743 [DOI] [PMC free article] [PubMed]
- 178.Zlokovic BV (2005) Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci 28:202–208 [DOI] [PubMed]
- 179.Ruiz J, Kouiavskaia D, Migliorini M, Robinson S, Saenko EL, Gorlatova N, Li D, Lawrence D, Hyman BT, Weisgraber KH, Strickland DK (2005) The apoE isoform binding properties of the VLDL receptor reveal marked differences from LRP and the LDL receptor. J Lipid Res 46:1721–1731 [DOI] [PubMed]
- 180.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 98:8850–8855 [DOI] [PMC free article] [PubMed]
- 181.DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM (2002) Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295:2264–2267 [DOI] [PubMed]
- 182.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci 22:7873–7878 [DOI] [PMC free article] [PubMed]
- 183.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM (2003) Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38:547–554 [DOI] [PubMed]
- 184.Matsuoka Y, Saito M, LaFrancois J, Saito M, Gaynor K, Olm V, Wang L, Casey E, Lu Y, Shiratori C., Lemere C, Duff K (2003) Novel therapeutic approach for the treatment of Alzheimer’s disease by peripheral administration of agents with an affinity to beta-amyloid. J Neurosci 23:29–33 [DOI] [PMC free article] [PubMed]
- 185.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T (2000) Inflammation and Alzheimer’s disease. Neurobiol Aging 21:383–421 [DOI] [PMC free article] [PubMed]
- 186.DeWitt DA, Perry G, Cohen M, Doller C, Silver J (1998) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Exp Neurol 149:329–340 [DOI] [PubMed]
- 187.McGeer PL, McGeer EG (1995) The inflammatory response system of brain: implications for therapy of Alzheimer and other neurodegenerative diseases. Brain Res Brain Res Rev 21:195–218 [DOI] [PubMed]
- 188.Kakimura J, Kitamura Y, Taniguchi T, Shimohama S, Gebicke-Haerter PJ (2001) Bip/GRP78-induced production of cytokines and uptake of amyloid-beta(1-42) peptide in microglia. Biochem Biophys Res Commun 281:6–10 [DOI] [PubMed]
- 189.Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S (2002) Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J 16:601–603 [DOI] [PubMed]