

Abstract
S-1 [3-(4-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide] is one member of a series of potent selective androgen receptor modulators (SARMs) that are being explored and developed for androgen-dependent diseases. Recent studies showed that S-1 holds great promise as a novel therapeutic agent for benign hyperplasia [W. Gao, J. D. Kearbey, V. A. Nair, K. Chung, A. F. Parlow, D. D. Miller, and J. T. Dalton (2004) Endocrinology 145:5420–5428]. We examined the pharmacokinetics and metabolism of S-1 in rats as a component of our preclinical development of this compound and continued interest in structure-activation relationships for SARM action. Forty male Sprague-Dawley rats were randomly assigned to treatment groups and received either an i.v. or a p.o. dose of S-1 at a dose level of 0.1, 1, 10, or 30 mg/kg. S-1 demonstrated a low clearance (range, 3.6–5.2 ml/min/kg), a moderate volume of distribution (range, 1460–1560 ml/kg), and a terminal half-life ranging from 3.6 to 5.2 h after i.v. doses. The oral bioavailability of S-1 ranged from 55% to 60%. Forty phase I and phase II metabolites of S-1 were identified in the urine and feces of male Sprague-Dawley rats dosed at 50 mg/kg via the i.v. route. The two major urinary metabolites of S-1 were a carboxylic acid and a sulfate-conjugate of 4-nitro-3-trifluoromethylphenylamine. Phase I metabolites arising from A-ring nitro reduction to an aromatic amine and B-ring hydroxylation were also identified in the urinary and fecal samples of rats. Furthermore, a variety of phase II metabolites through sulfation, glucuronidation, and methylation were also found. These studies demonstrate that S-1 is rapidly absorbed, slowly cleared, moderately distributed, and extensively metabolized in rats.
Androgens play important roles in male phenotype development and maintenance of male and female physiology and reproduction (Mooradian et al., 1987). Testosterone and 5α-dihydrotestosterone are the two main endogenous steroidal androgens involved in reproductive and nonreproductive tissue. Synthesized steroidal androgens have been used as therapeutic agents to treat a broad spectrum of disorders due to androgen deficiency. However, androgen preparations available in the market exhibit severe limitations. Testosterone shows low oral bioavailability, whereas testosterone esters are normally administered via intramuscular injection in oily vehicles, resulting in variable testosterone levels (Handelsman et al., 1990; Wilson, 1996). Although 17α-alkylated testosterones are orally available, they exhibit a high incidence of hepatotoxicity and less efficacy (Ishak and Zimmerman, 1987). Thus, they are not recommended for long-term androgen therapy. In addition, cross-reactivity occurs between steroidal androgens and/or their in vivo metabolites with steroid receptors other than the androgen receptor, further limiting their clinical use (Wilson et al., 1980; Bhasin and Bremner, 1997). Therefore, there is an urgent need to develop an orally bioavailable drug for patients suffering from a variety of androgen disorders.
Nonsteroidal selective androgen receptor modulators (SARMs) possess several advantages over synthesized steroidal androgens, including better receptor selectivity and greater flexibility in structural modification (Zhi and Martinborough, 2001; Yin et al., 2003b,c; Brown, 2004). Our laboratory recently reported the discovery of a series of novel SARMs, with propanamide as a template structure, having potent and tissue-selective in vivo pharmacologic activity (Dalton et al., 1998; Yin et al., 2003a,b,c; Gao et al., 2004; Marhefka et al., 2004; Chen et al., 2005). Among the SARMs discovered in our laboratory, 3-(4-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide (denoted hereafter as S-1; Fig. 1) demonstrated tissue-selective pharmacologic activity in both castrated and intact male rats (Yin et al., 2003a; Gao et al., 2004).
Fig. 1.
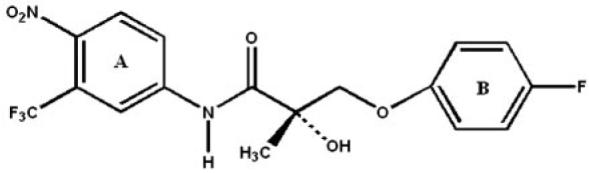
Chemical structure of 3-(4-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide, denoted as S-1.
In vivo pharmacologic studies in castrated rats showed that S-1 maintained the weights of the prostate, seminal vesicles, and levator ani muscle at 14.9, 13.4, and 74.3%, respectively, of those observed in intact animals via subcutaneous osmotic pumps (Yin et al., 2003a). Compound S-1 and testosterone propionate showed a similar degree of anabolic activity, but S-1 demonstrated lesser androgenic activity in a rat model (Yin et al., 2003a). S-1 behaves as a partial agonist in androgenic tissues (i.e., prostate and seminal vesicle) and as a full agonist in anabolic tissues (i.e., levator ani muscle) in both castrated and intact male rats (Yin et al., 2003a; Gao et al., 2004). In intact male rats, S-1 selectively decreased prostate weight without affecting the levator ani muscle or increasing plasma levels of testosterone, luteinizing hormone, or follicle-stimulating hormone at doses of 5, 10, and 25 mg/kg (Gao et al., 2004). As such, S-1 may have clinical potential as a single agent or combination therapy for benign prostate hyper-plasia (Gao et al., 2004). Based on its tissue-specific pharmacologic activities, S-1 was selected as one of several lead compounds for potential development as an orally active SARM.
We previously showed that hepatic metabolism plays a major role in the in vivo pharmacologic activity of a structurally related class of nonsteroidal androgens (Yin et al., 2003b). Oxidation, hydrolysis, and sulfate conjugation contributed to high plasma clearance and lack of in vivo androgenic activity of a thio-ether-linked analog (i.e., acetothiolutamide). The ideal pharmacokinetic properties of an anabolic SARM would be high oral bioavailability, reasonably long half-life, linear kinetics, and low potential for drug-drug interactions. In the current study, we examined the pharmacokinetics, oral bioavailability, and in vivo metabolism of S-1 in rats and compare its pharmacokinetic properties with those of the structural analogs, androgen receptor antagonists (bicalutamide, flutamide, and nilutamide) and agonists [S-4 (i.e., 3-[4-(acetylamino)phenoxy]-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide) and C-6 (i.e., 3-(4-chloro-3-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide)] (Fig. 2), to better understand how its physicochemical properties and metabolic profile relate to its pharmacologic activity and the behavior of other SARMs. These are the first studies of the metabolism of this class of ether-linked SARMs and provide new knowledge regarding pharmacokinetic optimization of this new generation of propanamide derivatives, using S-1 as a model compound.
Fig. 2.

Structural analogs of S-1.
Materials and Methods
Chemicals and Reagents
S-1 (i.e., 3-(4-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide) was synthesized using previously described methods (Marhefka et al., 2004). Two internal standards, the 4-chloro and 4-bromo analogs of S-1, were also obtained using these procedures. 3-(4-Fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid was synthesized using similar procedures. Chemical purity was confirmed using elemental analysis, mass spectrometry, and proton nuclear magnetic resonance. HPLC-grade acetonitrile, water, and acetic acid were purchased from Fisher Scientific (Fair Lawn, NJ). Polyethylene glycol-300 (PEG-300) and dimethyl sulfoxide (DMSO) were obtained from Sigma Chemical Co. (St. Louis, MO). Ethanol was purchased from Pharmaco Products Inc. (Brookfield, CT).
Animals and Procedures
Male Sprague-Dawley rats from Harlan Bioscience (Indianapolis, IN), weighing approximately 250 g, were maintained in accordance with the animal protocol approved by the Institutional Laboratory Animal Care and Use Committee of The Ohio State University. Animals were kept on a 12-h light/dark cycle with food and water available ad libitum. Twenty hours before dosing, a catheter was implanted in the right external jugular vein of each rat, and food (Harlan Teklad 22/5 rodent diet) was removed. The rats were supplied with water ad libitum and weighed immediately before dose administration. Food was returned 12 h after dosing.
Forty male Sprague-Dawley rats were randomly assigned to treatment groups and received either an i.v. or a p.o. dose of S-1 at a dose level of 0.1, 1, 10, or 30 mg/kg. Dosing solutions were prepared in 5% DMSO in PEG-300 (v/v) 12 h before dosing and stored at −20°C. The jugular vein catheter was flushed with an aqueous solution of heparinized saline (100 U/ml, equal volume with the dosing solution) immediately after administration of the i.v. dose. Serial blood samples were collected at 5, 10, 20, 30, 60, 120, 240, 480, 720, and 1440 min after administration via the i.v. route, whereas blood samples were obtained at 30, 60, 90, 180, 240, 360, 480, 720, 1440, 1800, and 2160 min after dosing by oral gavage. Plasma was separated immediately by centrifugation (800g for 10 min at 4°C), and samples were stored at −20°C until analysis. Dosing solution, comprising 5% DMSO in PEG-300 (v/v) and 5 or 10% ethanol in PEG-300, was used at the dose level of 10 mg/kg via i.v. and p.o. routes to examine the effect of solubility or vehicle on oral absorption or clearance of S-1.
For metabolic profiling, an i.v. dose of S-1 (50 mg/kg) was administered to male Sprague-Dawley rats (n = 2). Urine and fecal specimens were collected in 6- to 12-h intervals for up to 72 h, using metabolic cages, and combined before analyses to provide sufficient volumes of urine and metabolite concentrations for analysis and to protect against degradation at room temperature. All the urinary and fecal specimens were stored at −80°C until analysis.
Extraction Procedure of S-1 for HPLC Method
Aliquots of rat plasma (100 μl) were spiked with internal standard (30 μl of a solution containing the 4-bromo analog of S-1 at a concentration of 15 μg/ml) and mixed with 1 ml of acetonitrile. Samples were centrifuged at 16,100g for 10 min. The supernatant was removed and evaporated to dryness under nitrogen in a clean centrifuge tube. The residue was reconstituted with 150 μl of the HPLC mobile phase, centrifuged at 16,100g for 5 min, and an aliquot of 100 μl was injected to the HPLC.
HPLC-UV Measurement of S-1 in Plasma
Plasma concentrations for the 10 and 30 mg/kg i.v. and p.o. dose groups were determined using a validated HPLC method. HPLC analysis was performed using a model 515 HPLC pump (Waters, Milford, MA), a model 717 plus autosampler (Waters), and a model 486 absorbance detector (Waters). HPLC separation was conducted using a mobile phase of acetonitrile/H2O (54:46 v/v) on a Waters Nova-pak C18 column (150 × 3.9 mm, 4 μ) at a flow rate of 1 ml/min, with detection wavelength set at 297 nm. Analytical data were acquired by Millennium software (Waters). The limit of quantitation of the HPLC assay was 0.05 μg/ml. Calibration standard curves were constructed over 0.05 to 100 μg/ml. Within- and between-day precision was within 1.8 to 18.2% coefficient of variation, and the accuracy was 90.0 to 92.4% of the nominal concentrations. The relative recoveries of S-1 in rat plasma ranged from 90.5 to 97.5%.
Extraction Procedure of S-1 for LC-MS Assay
Aliquots of rat plasma (100 μl) were spiked with internal standard (30 μl of a solution containing the 4-chloro analog of S-1 at a concentration of 0.01 μg/ml) and mixed with 1 ml of acetonitrile. Samples were centrifuged at 16,100g for 10 min. The supernatant was removed and mixed with 1 ml of water before extraction with ethyl acetate (7.5 ml) in a 13-ml extraction tube. Samples were shaken at room temperature for 10 min and then centrifuged at 1540g for 10 min. The organic supernatant was removed and evaporated to dryness under nitrogen in a clean test tube. The residue was reconstituted with 150 μl of the initial mobile phase, centrifuged at 16,100g for 5 min, and an aliquot of 100 μl was injected into the LC-mass spectrometer.
LC-MS Measurement of S-1 in Plasma
Plasma concentrations for the 0.1 and 1.0 mg/kg i.v. and p.o. dose groups were determined using a validated LC-MS method. LC-MS (Agilent 1100 series; Agilent, Palo Alto, CA) analyses were performed using an electrospray ionization source and the following conditions: dry gas flow 12 l/min, nebulizer pressure 45 psi, dry gas temperature 350°C, capillary voltage 1500 V, and fragmentor voltage 180 V. All other LC-MS parameters were set at default. Single ion monitoring at m/z 401.10 and 417.10 in the negative ion mode was used for detection of S-1 and the internal standard, respectively. Individual samples were injected onto a monolithic column (SpeedRod RP 18e, 50 × 4.6 mm; Merck KGaA, Darmstadt, Germany) maintained at 30°C during analysis. Compounds of interest were separated from interference using a gradient mobile phase composed of acetonitrile (A) and 0.1% acetic acid/water (B) at a flow rate of 1 ml/min. The mobile phase was composed of a 50:50 mixture of components A and B for the first 5 min of each chromatographic run, increased to 100% B in a linear gradient from 5.1 to 7.5 min, and then returned to 50% B at 7.6 min. The equilibration time for the column with the initial mobile phase was 1.5 min. Analytical data were acquired by ChemStation software (Agilent). The limit of quantitation of the LC-MS assay was 0.3 ng/ml. Calibration standard curves were constructed over a concentration range of 0.3 to 300 ng/ml. Within- and between-day precision was within 0.4 to 12.4% coefficient of variation and the accuracy was 87.1 to 104.8% of the nominal concentrations. The relative recoveries of S-1 in rat plasma ranged from 99.4 to 105.7%.
Pharmacokinetic Data Analysis
Plasma concentration-time data were analyzed by noncompartmental analysis using WinNonlin 4.0 (Pharsight Corporation, Mountain View, CA). The area under the plasma concentration-time curve from time 0 to infinity (AUC0-∞) was calculated by the trapezoidal rule with extrapolation to time infinity. The terminal half-life (t1/2) was calculated as 0.693/λz, where λz is the terminal phase elimination constant. The plasma clearance (CL) was calculated as CL = dosei.v./AUCi.v., 0-∞, where dosei.v. and AUCi.v., 0-∞ are the i.v. dose and corresponding area under the curve from time 0 to infinity, respectively. The maximum plasma concentration (Cmax) and the time when it occurred (Tmax) in p.o. dose groups were obtained by visual inspection of the plasma concentration-time curves. The apparent volume of distribution at equilibrium (Vdss) was calculated as Vdss = CL · MRT, where MRT is the mean residence time after the i.v. bolus dose. MRT was calculated as MRT = (AUMCi.v., 0-∞)/(AUCi.v., 0-∞), where AUMCi.v., 0-∞ is the area under the first moment of the plasma concentration-time curve extrapolated to infinity. Oral bioavailability (F) for each dose was calculated using F = (AUCp.o.× dosei.v.)/(AUCi.v.× dosep.o.), where dosep.o., dosei.v., AUCi.v., and AUCp.o. are the mean oral dose, mean i.v. dose, and the corresponding mean areas under the curve from time 0 to infinity, respectively, at each dose.
Statistical Analysis
Statistical analyses were performed using single-factor analysis of variance. p < 0.05 was considered as a statistically significant difference.
Identification of Metabolites
Urinary specimens were thawed and extracted with ethyl acetate at a volume 5 times that of the urinary samples. The extraction procedure was repeated twice, and the combined organic and combined aqueous phases were evaporated separately on a rotary evaporator at 35°C. Fecal specimens of 0 to 24 h (5.996 g) and 24 to 72 h (14.673 g) were thawed and extracted with methanol at a ratio of 6 ml/g. Methanolic fractions were centrifuged at 1540g for 10 min, and supernatant was evaporated to dryness with nitrogen. The residues were dissolved with 3 ml of methanol/H2O (50:50) and extracted with 7.5 ml of ethyl acetate. The extraction procedure was repeated twice. The combined organic and combined aqueous phases were evaporated to dryness using nitrogen. The residues from the organic phase and the aqueous phase were dissolved using acetonitrile/H2O (50:50) and acetonitrile/H2O (10:90), respectively. Each solution was filtered through an Acrodisc syringe filter (0.2 μm, 13 mm; Pall Corporation, East Hills, NY). Twenty microliters of each fraction was injected directly on a Thermo Finnigan LCQDECA quadrupole ion trap mass spectrometer (Thermo Electron, Franklin, MA) coupled with a Surveyor HPLC (Thermo Electron) using the negative-ion electrospray ionization mode. HPLC separation was performed on a Waters XTerra C18 column (150 × 2.1 mm, 3.5 μ) with an XTerra guard column (10 × 2.1 mm, 3.5 μ) at a flow rate of 0.2 ml/min using a gradient mobile phase comprising acetonitrile (A) and water (B). The mobile phase comprised a 90:10 mixture of components A and B for the first 10 min of each chromatographic run, increased to 60% B in a linear gradient from 10 to 60 min, then further increased to 95% B from 60 to 65 min and maintained for 10 min, and finally returned to 10% B at 76 min. The column was equilibrated with the initial mobile phase for 10 min. A second mobile phase system that included 0.1% acetic acid in both A and B was used in some instances. The same gradient program was used in both mobile phase systems. The capillary heater was set to 180 or 225°C, and spray voltage was 3.6 kV. Full-scan analysis was programmed to scan from m/z 100 to 900 every second.
LC-MS Measurement of M-1 in Urine and Feces
Urinary and fecal concentrations of M-1 were determined using a validated LC-MS method. LC-MS (Agilent 1100 series) analyses were performed using an electrospray ionization source and the following conditions: dry gas flow 8 l/min, nebulizer pressure 30 psi, dry gas temperature 350°C, capillary voltage 2000 V, and fragmentor voltage of 130 V. All other LC-MS parameters were set at default. Single ion monitoring at m/z 213.10 and 417.10 in the negative ion mode was used for detection of M-1 and the internal standard, respectively. HPLC separation was performed using the same column and gradient program as listed above. Analytical data were acquired using ChemStation software (Agilent).
Fecal samples from each rat were collected in 50-ml plastic tubes. Five milliliters of distilled water (pH 3.0, by acetic acid) and 30 μl of internal standard (0.1 μg/ml) were added into each tube and samples were homogenized with a glass rod. Thirty milliliters of methanol were then added into each tube at a ratio of 6 ml/g. These tubes were shaken at room temperature for 60 min. After shaking, tubes were centrifuged for 5 min at 16,100g, the supernatant was transferred into a clean tube, and solvent was removed under a stream of nitrogen. The residue was dissolved in 3 ml of methanol/water (pH 3.0, by acetic acid) (1:1, v/v). An aliquot of 20 μl was injected into the LC-mass spectrometer. Urine samples were not extracted before analysis, but were simply diluted with internal standard immediately before analysis. One hundred microliters of each urinary sample was spiked with the internal standard, and an aliquot of 20 μl was injected directly into the LC-mass spectrometer.
The limits of quantitation of M-1 in feces and urine were 0.05 and 0.01 μg/ml, respectively. Calibration standard curves were constructed over 0.01 to 10 μg/ml. Within- and between-day precision was within 5.3 to 14.8% coefficient of variation, and the accuracy was 89.9 to 115.4% of the nominal concentrations. The relative recovery of M-1 in rat urine and feces ranged from 88.6 to 107.2%.
Results
Pharmacokinetics of S-1 in Male Sprague-Dawley Rats
Plasma concentration-time curves after i.v. and p.o. administration of S-1 are shown in Fig. 3. The corresponding pharmacokinetic parameters of S-1 at different dose levels are listed in Table 1. Plasma concentrations declined in a multiexponential manner after i.v. administration of S-1. The systemic clearance (CL) of S-1 was 5.2, 4.4, 4.0, and 3.6 ml/min/kg at the dose levels of 0.1, 1, 10, and 30 mg/kg by i.v. administration, respectively. Despite a trend of lower CL values at higher doses, statistical analysis revealed no significant difference among CL values from the four dose levels, demonstrating that the CL of S-1 was not dose-dependent over the dose range (i.e., 0.1–30 mg/kg) studied. Previous in vivo studies conducted in our laboratory showed that doses required to exhibit anabolic effects in castrated rats or selectively decrease the prostate weight in intact rats were less than 25 mg/kg/day via subcutaneous osmotic pumps or daily subcutaneous injection, respectively. Therefore, S-1 exhibited linear pharmacokinetics within the pharmacological dose range. The steady-state volume distribution (Vss) of S-1 was approximately 1.5 l/kg at the four i.v. dose levels examined, suggesting that S-1 was moderately distributed. Urinary and fecal excretion data showed that less than 0.4% of the dose was excreted unchanged, indicating that renal and biliary elimination of S-1 is negligible. Based on a CL of S-1 as 5.2 ml/ min/kg and rat hepatic blood flow as 55.2 ml/min/kg (Davies and Morris, 1993), the hepatic extraction ratio of S-1 is less than 0.1. This suggested that first-pass hepatic metabolism would not significantly limit exposure to S-1 after oral administration.
Fig. 3.
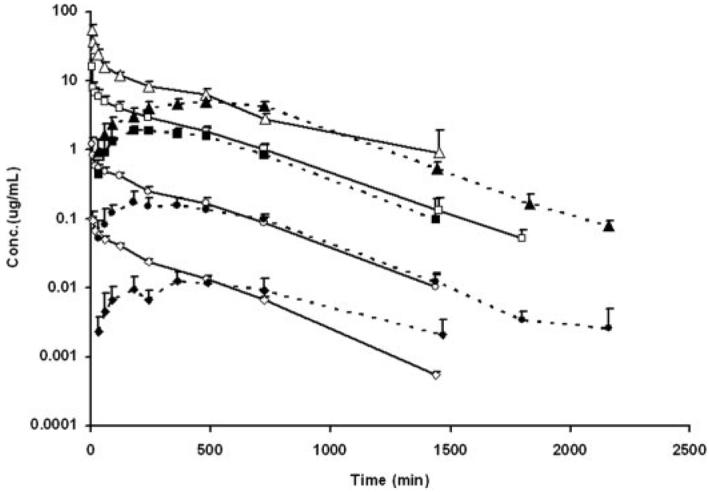
Plasma concentration-time profiles of S-1 after i.v. and p.o. administration in male Sprague-Dawley rats (n = 5/dose group). Solid symbols indicate doses via the p.o. route, whereas open symbols indicate doses via the i.v. route. Triangle, 30 mg/kg; square, 10 mg/kg; circle, 1 mg/kg; diamond, 0.1 mg/kg.
TABLE 1. Pharmacokinetics of S-1 in male Sprague-Dawley rats after i.v. and p.o. administration.
Values are mean ± S.E. (n = 5/dose group).
| Pharmacokinetics | Unit | 0.1 mg/kg | 1 mg/kg | 10 mg/kg | 30 mg/kg |
|---|---|---|---|---|---|
| i.v. | |||||
| t1/2 | min | 217 | 241 | 248 | 315 |
| MRT | min | 301 ± 20 | 348 ± 49 | 363 ± 51 | 442 ± 170 |
| AUC | min · μg/ml | 20.4 ± 0.7 | 228 ± 32 | 2549 ± 267 | 8738 ± 2053 |
| CL | ml/min/kg | 5.2 ± 0.2 | 4.4 ± 0.7 | 4.0 ± 0.5 | 3.6 ± 0.7 |
| Vss | ml/kg | 1564 ± 149 | 1532 ± 227 | 1458 ± 352 | 1512 ± 216 |
| p.o. | 0.1 mg/kg | 1 mg/kg | 10 mg/kg | 30 mg/kg | |
| t1/2 | min | 322 | 255 | 219 | 254 |
| MRT | min | 685 ± 131 | 542 ± 28 | 481 ± 43 | 625 ± 39 |
| Tmax | min | 347 ± 126 | 276 ± 138 | 288 ± 133 | 510 ± 151 |
| Cmax | μg/ml | 0.014 ± 0.004 | 0.18 ± 0.06 | 2.12 ± 0.19 | 5.01 ± 0.72 |
| AUC | min · μg/ml | 12 ± 4 | 136 ± 19 | 1412 ± 218 | 4771 ± 350 |
| CL/F | ml/min/kg | 9.5 ± 3.3 | 7.6 ± 1.0 | 7.2 ± 1.0 | 6.3 ± 0.5 |
| Vz/F | ml/kg | 4699 ± 2546 | 2840 ± 468 | 2294 ± 383 | 2361 ± 468 |
| Bioavailability | % | 59.6 | 58.0 | 54.9 | 55.7 |
The Tmax of S-1 ranged from 4.6 to 8.5 h after oral administration, indicating that S-1 was slowly absorbed. The terminal half-lives of S-1 after oral administration were comparable to that observed after i.v. administration of S-1 at the corresponding dose level. Oral bio-availability of S-1 was approximately 60% and did not vary with dose. Comparison of the effect of dosing vehicle, using ethanol instead of DMSO with PEG-300, on pharmacokinetic parameters of S-1 was conducted at the dose level of 10 mg/kg. Plasma concentration versus time profiles are shown in Fig. 4. The pharmacokinetic data are listed in Table 2. Use of ethanol in the dosing solution did not affect the CL or Vss of S-1, but decreased Tmax from 4.8 to 1.77 h and increased the bioavailability from 54.9 to 95.9%, suggesting that solubility is an important factor governing the oral absorption of S-1.
Fig. 4.
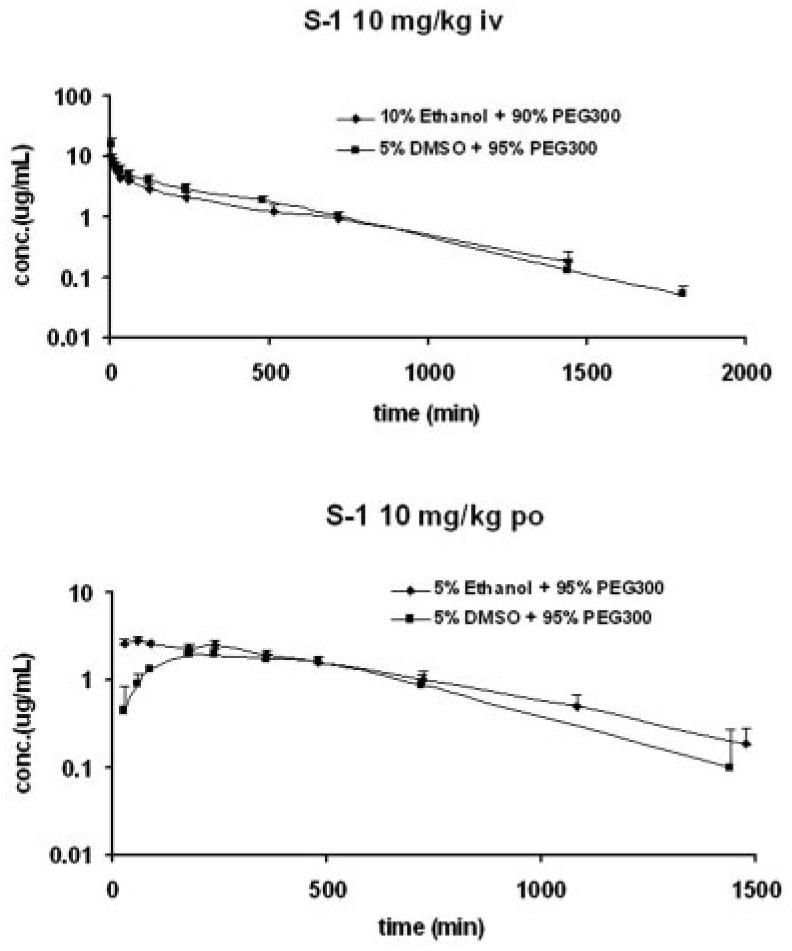
Comparison of effects of dosing formulation on pharmacokinetic profiles of S-1 at the dose level of 10 mg/kg in male Sprague-Dawley rats (n = 5/dose group).
TABLE 2. Effects of dosing vehicle on pharmacokinetic parameters of S-1 at the dose level of 10 mg/kg in male Sprague-Dawley rats (n = 5/dose group).
| Dosing Vehicle | Unit | 10 mg/kg |
|
|---|---|---|---|
| 10% Ethanol in PEG-300 | 5% DMSO in PEG-300 | ||
| i.v. | |||
| t1/2 | min | 330 | 248 |
| MRT | min | 434 ± 63 | 363 ± 51 |
| AUC | min · μg/ml | 2009 ± 413 | 2549 ± 267 |
| CL | ml/min/kg | 5.1 ± 1.1 | 4.4 ± 0.5 |
| Vss | ml/kg | 2214 ± 486 | 1458 ± 352 |
| 10 mg/kg |
|||
| 5% Ethanol in PEG-300 | 5% DMSO in PEG-300 | ||
| p.o. | |||
| t1/2 | min | 283 | 219 |
| MRT | min | 502 ± 88 | 481 ± 43 |
| Tmax | min | 106 ± 89 | 288 ± 133 |
| Cmax | μg/ml | 2.97 ± 0.15 | 2.12 ± 0.19 |
| AUC | min · μg/ml | 1860 ± 249 | 1412 ± 218 |
| CL/F | ml/min/kg | 5.3 ± 0.7 | 7.2 ± 1.0 |
| Vz/F | ml/kg | 2155 ± 179 | 2294 ± 383 |
| Bioavailability | % | 95.9 | 54.9 |
Identification and Quantitation of Metabolites of S-1 in Rat Urine and Feces
Metabolism studies were performed to identify the major metabolic pathways and metabolites of S-1, especially those of chemically reactive metabolites, and to further evaluate the clinical potential of S-1. In addition, a complete metabolic profile of S-1 will facilitate structure modification on the propanamide template to obtain more potent and metabolically stable propanamide compounds functioning as SARMs.
The mass spectra of S-1 fragmentation is shown in Fig. 5. The S-1 fragmentation pathway under conditions of collision-induced dissociation is proposed in Fig. 6. Identification of the metabolites of S-1 was based on the understanding of the fragmentation pathway of the parent compound. Metabolites of S-1 identified from rat urine and feces are listed in Tables 3 and 4, respectively. S-1 eluted at 58.39 min under both of the mobile phase systems used. S-1 was stable under acidic conditions used in extraction. A total of 40 phase I and phase II metabolites of S-1 were found in the urine and feces of male Sprague-Dawley rats that received 50 mg/kg S-1 via the i.v. route.
Fig. 5.

S-1 fragmentation mass spectra. A, MS2 (m/z 401); B, MS3 (m/z 261).
Fig. 6.
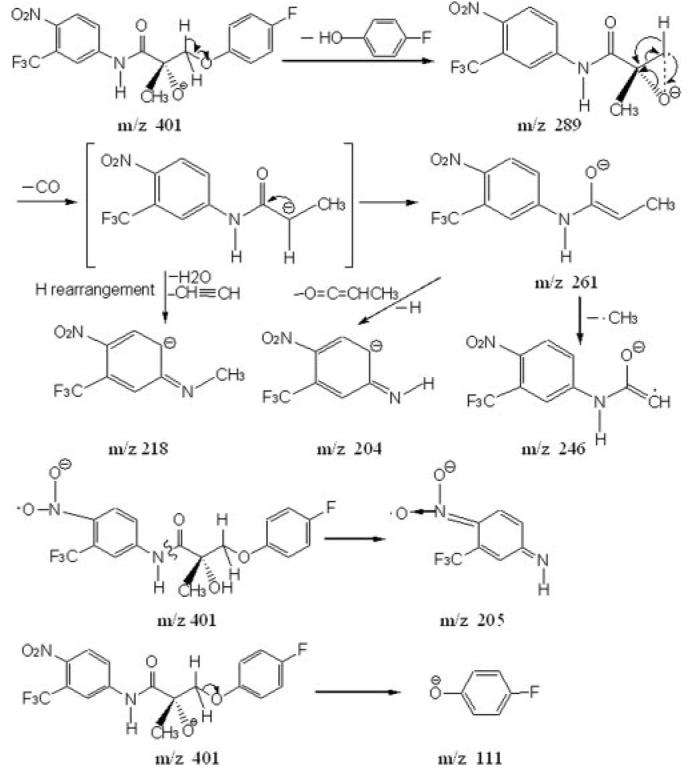
Proposed fragmentation pathway of S-1 under conditions of collision-induced dissociation.
TABLE 3. Metabolites of S-1 identified in rat urine of 0 to 24 h (using mobile phase system 1).
Numbers in bold are values of m/z for basic mass peaks in the corresponding MS spectrum.
| Metabolite Identification |
Retention Time |
[M - H]− | MS/MS | Sites of Biotransformation or Confirmed Structure |
|---|---|---|---|---|
| min | M/z | m/z | ||
| 1* | 15.56 | 213 | 111 | 3-(4-Fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid |
| 2 | 20.87 | 591 | 415; MS3 of 415→287, 127; MS4 of 287→229, 215, 203, 197 | Nitro reduction, di-methylation on N (phenylamine or amide) or hydroxyl group connected to the chiral center, mono-hydroxylation on the B-ring and glucuronidation |
| 3 | 21.66 | 579 | 403; MS3 of 403→143 | Nitro reduction, di-hydroxylation on the B-ring and glucuronidation |
| 4 | 21.69 | 563 | 387; MS3 of 387→259, 245, 127 | Nitro reduction, mono-hydroxylation on the B-ring and glucuronidation |
| 5 | 28.17 | 575 | 399; MS3 of 399→287, 259 203; MS4 of 287→229 | Nitro reduction, di-methylation on N or O (containing H) and glucuronidation |
| 6 | 29.00 | 301 | 221; MS3 of 221→3191, 171 | Mono-hydroxylation on 4-nitro-3-trifluoromethyl-phenylamine and sulfation |
| 7 | 29.18 | 415 | 287, 203, 127; MS3 of 287→229, 215, 197; MS4 of 229→172 | Nitro reduction, di-methylation on N (phenylamine or amide) and/or hydroxyl group connected to the chiral center or mono-methylation with oxidation to ketone on alkyl C adjacent to the ether linkage and mono-hydroxylation on the B-ring |
| 8 | 29.44 | 547 | 435, 371, 259, 231, 201, 175; MS3 of 435→277, 231, 175; MS4 of 277→231, 171 | Nitro reduction and glucuronidation |
| 9 | 30.59 | 467 | 387, 127; MS3 of 387→259, 127 | Nitro reduction, mono-hydroxylation on the B-ring and sulfation |
| 10 | 31.86 | 415 | 287, 259, 229, 203; MS3 of 287→244, 229, 215, 203, 189; MS4 of 229→201, 189 | Nitro reduction, di-methylation on N (phenylamine or amide) and/or hydroxyl group connected to the chiral center or mono-methylation with oxidation to ketone on alkyl C adjacent to the ether linkage and mono-hydroxylation on the B-ring |
| 11 | 32.30 | 483 | 403; MS3 of 403→143 | Nitro reduction, di-hydroxylation on the B-ring and sulfation |
| 12 | 32.43 | 467 | 387; MS3 of 387→259, 127 | Nitro reduction, mono-hydroxylation on the B-ring and sulfation |
| 13 | 34.65 | 483 | 403; MS3 of 403→275, 259, 221; MS4 of 275→255, 201 | Nitro reduction, mono-hydroxylation on the A-ring or phenylamine on the A-ring, mono-hydroxylation on the B-ring and sulfation |
| 14 | 34.90 | 263 | 221; MS3 of 221→191, 171 | Mono-hydroxylation on N in the acetamide group or on the benzene ring of N-(4-nitro-3-trifluoromethyl-phenyl)-acetamide |
| 15 | 35.03 | 343 | 263; MS3 of 263→221; MS4 of 221→191, 171 | Mono-hydroxylation on N in the acetamide group or on the benzene ring of N-(4-nitro-3-trifluoromethyl-phenyl)-acetamide and sulfation |
| 16 | 36.5 | 467 | 387; MS3 of 387→275, 255, 191, 171, 111; MS4 of 275→255, 191 | Nitro reduction, mono-hydroxylation on the A-ring or phenylamine on the A-ring and sulfation |
| 17 | 36.66 | 387 | 307, 275, 259, 127 | Nitro reduction and mono-hydroxylation on the B-ring |
| 18 | 37.20 | 263 | 221; MS3 of 221→191, 171 | Mono-hydroxylation on N in the acetamide group or on the benzene ring of N-(4-nitro-3-trifluoromethyl-phenyl)-acetamide |
| 19 | 38.07 | 513 | 433; MS3 of 433→307, 289, 261, 227, 205, 143 | Di-hydroxylation on alkyl C adjacent to the ether linkage and the B-ring and sulfation |
| 20 | 38.55 | 415 | 287, 127; MS3 of 287→259, 245, 229, 217, 203, 201; MS4 of 229→189 | Nitro reduction, di-methylation on N (phenylamine or amide) and/or hydroxyl group connected to the chiral center or mono-methylation with oxidation to ketone on alkyl C adjacent to the ether linkage and mono-hydroxylation on the B-ring |
| 21 | 38.69 | 387 | 259, 127; MS3 of 259→229, 201, 175 | Nitro reduction and mono-hydroxylation on the B-ring |
| 22 | 39 | 513 | 433; MS3 of 433→275, 204 | Nitro reduction, mono-hydroxylation on the A-ring or phenylamine on the A-ring, di-hydroxylation on the B-ring with methylation on one hydroxyl group and sulfation |
| 23 | 40.43 | 513 | 433; MS3 of 433→289, 261, 227, 205, 143 | Di-hydroxylation on the B-ring and sulfation |
| 24 | 40.91 | 577 | 401; MS3 of 401→289, 261, 205; MS4 of 261→246, 218, 204, 190 | Glucuronidation of S-1 |
| 25 | 41.74 | 247 | 205 | |
| 26 | 41.89 | 513 | 433; MS3 of 433→289, 275, 227, 205, 183, 143 | Nitro reduction, mono-hydroxylation on the A-ring or phenylamine on the A-ring, methylation on N in amide bond, di-hydroxylation on the B-ring and sulfation |
| 27 | 42.78 | 403 | 143 | Nitro reduction and di-hydroxylation on the B-ring |
| 28 | 43.64 | 385 | 273, 245, 225, 217, 189; MS3 of 245→225, 205, 203; MS4 of 225→205 | Nitro reduction, methylation on N or O (containing H) |
| 29 | 44.5 | 387 | 259, 201, 127; | Nitro reduction and mono-hydroxylation on the B-ring |
| 30 | 45.49 | 497 | 417; MS3 of 417→289, 205, 127; MS4 of 205→175 | Mono-hydroxylation on the B-ring and sulfation |
| 31 | 45.52 | 387 | 275, 255, 191, 111; MS3 of 275→255 | Nitro reduction and mono-hydroxylation on the A-ring or phenylamine on the A-ring |
| 32 | 46.62 | 371 | 259, 231, 111; MS3 of 259→229, 201, 175 | Nitro reduction |
| 33 | 47.31 | 385 | 273, 245, 225, 217, 189; MS3 of 245→225, 203 | Nitro reduction, methylation on N or O (containing H) |
| 34 | 47.9 | 563 | 307; MS3 of 307→289, 261, 205 | Sulfation and glucuronidation of 2,3-dihydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide |
| 35 | 51.50 | 417 | 289, 261, 205; MS3 of 289→261; MS4 of 261→246, 218, 204, 190 | Mono-hydroxylation on alkyl C adjacent to the ether linkage |
| 36 | 55.55 | 417 | 305, 289, 261, 205, 127; MS3 of 261→246, 218, 204, 190 | Mono-hydroxylation on the B-ring |
| 37 | 66.26 | 452 | 407, 289, 261; MS3 of 289→261, 244, 233, 231, 205 | O-Dephenylation (the B-ring) and mercapturic acid conjugation [2-Acetylamino-3-[1-hydroxy-2-(4-nitro-3-(trifluoromethyl)phenylcarbamoyl)-propylsulfanyl]-propanoic acid] |
indicates that the structure of the metabolite has been confirmed using a synthetic standard in LC-MSn.
TABLE 4. Metabolites of S-1 identified in rat feces of 0 to 24 h (using mobile phase system 2).
Numbers in bold are values of m/z for basic mass peaks in the corresponding MS spectrum.
| Metabolite Identification |
Retention Time |
[M - H]− | MS/MS | Sites of Biotransformation or Confirmed Structure |
|---|---|---|---|---|
| min | m/z | m/z | ||
| 1* | 26.62 | 213 | 111 | 3-(4-Fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid |
| 9 | 29.40 | 467 | 387, 127; MS3 of 387→259, 201, 175 127; MS4 of 259→229, 209, 201, 175 | Nitro reduction, mono-hydroxylation on the B-ring and sulfation |
| 17 | 32.34 | 387 | 259, 127; MS3 of 259→201, 175 | Nitro reduction and mono-hydroxylation on the B-ring |
| 12 | 32.55 | 467 | 387, 127; MS3 of 387→259, 201, 175 127; MS4 of 259→230, 201, 175 | Nitro reduction, mono-hydroxylation on the B-ring and sulfation |
| 6 | 38 | 301 | 221; MS3 of 221→191, 171 | Mono-hydroxylation on 4-nitro-3-trifluoromethyl-phenylamine and sulfation |
| 38 | 38.14 | 467 | 387, 127; MS3 of 387→259, 201, 175 127; MS4 of 259→229, 209, 201, 175 | Nitro reduction, mono-hydroxylation on the B-ring and sulfation |
| 21 | 39.01 | 387 | 259, 201,127; MS3 of 259→229, 201, 175 | Nitro reduction and mono-hydroxylation on the B-ring |
| 39 | 40.18 | 451 | 371; MS3 of 371→259, 231, 201, 175, 111; MS4 of 259→201, 175 | Nitro reduction and sulfation |
| 29 | 44.84 | 387 | 259, 127 | Nitro reduction and mono-hydroxylation on the B-ring |
| 31 | 45.52 | 387 | 275, 255, 111; MS3 of 275→255 | Nitro reduction and mono-hydroxylation on the A-ring or phenylamine on the A-ring |
| 32 | 47.19 | 371 | 259, 231, 201, 175, 111; MS3 of 259→229, 201, 175 | Nitro reduction |
| 24 | 50.01 | 577 | 401, 307, 261, 205, 175 | Glucuronidation of S-1 |
| 40 | 52.56 | 307 | 289, 245, 205; MS3 of 289→245, 204 | O-Dephenylation (the B-ring) {2,3-dihydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)-phenyl]-propanamide} |
indicates that the structure of the metabolite has been confirmed using a synthetic standard in LC-MSn.
The two major urinary metabolites of S-1 were a carboxylic acid (M1, m/z 213) and a sulfate-conjugate of 4-nitro-3-trifluoromethylphenylamine (M6, m/z 301) that arose from amide hydrolysis of S-1 or its metabolites. M1 was confirmed as 3-(4-fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid by showing the same chromatographic (i.e., retention time) and mass (i.e., molecular mass and fragmentation pattern) behavior as those of the synthetic standard (Fig. 7). M-1 excreted in rat urine was estimated as 25.3, 15.7, and 6.2% of the dose administrated i.v. in rats at 0- to 12-, 12- to 24-, and 24- to 72-h time intervals, respectively. M-1 excreted in rat feces was also calculated as 1.5, 0.1, and 0.03% of the dose given i.v. in rats at 0- to 12-, 12- to 24-, and 24- to 72-h time intervals, respectively. M-1 accounted for 48.83% of the total dose given i.v. in rats during the first 72 h. There were more metabolites identified qualitatively in urine than in feces, indicating that biliary excretion of metabolites occurred.
Fig. 7.

Comparison of chromatographic and mass behavior of M1 and synthetic standard, 3-(4-fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid (using mobile phase 2). A, rat urinary samples of 0–12 h; B, synthetic standard.
It is common that fragmentation limitations apply to ions around m/z 200 using the quadrupole ion trap mass spectrometer (i.e., LCQDECA). Therefore, the structure of one-ring metabolites, elucidated through MS1 or MS2 mode using LCQDECA and proposed in Figs. 3 and 4, need to be further confirmed either by using synthetic standards for comparison or by obtaining NMR data for confirmation after isolation and purification. However, these one-ring metabolites clearly appeared in rat urinary and fecal samples after dosing with S-1, comparing with the blank urine and feces collected from the same rat. Based on our recent findings regarding negative-ion electrospray ionization efficiency in the presence of weak acid (Wu et al., 2004), metabolites of S-1 containing only a single phenyl ring demonstrate less ionization efficiency than metabolites of S-1 maintaining both the A- and B-phenyl rings, especially if the metabolites containing a single phenyl ring are more lipophilic. We compared the ionization efficiency of the available synthetic standard (M1) with that of the parent compounds under the same chromatographic and mass conditions (data not shown) and confirmed the above conclusion. Although it is possible to quantitate all the metabolites with standards using LC-MS, it is also reasonable to estimate the relative amount based on the understanding of chromatographic separation and ionization efficiency. Metabolites consisting of a single phenyl ring (e.g., M1, M6, M14, etc.) showed signal intensities similar to or even higher than metabolites of S-1 maintaining the two phenyl rings, suggesting that they were present at significantly higher concentrations. Based on the fact that 48.83% of the total dose administered was biotransformed to M-1 in rats during 0 to 72 h, M6 was deduced as another major metabolite showing the highest signal intensities among those metabolites with a single phenyl ring (A-ring).
Phase I metabolites arising from A-ring nitro reduction to an aromatic amine and B-ring hydroxylation were also identified in the urinary and fecal samples of rats. Furthermore, a variety of phase II metabolites arising from sulfation, glucuronidation, or methylation were also found. In addition to the hydrolysis metabolites mentioned above, nitro reduction on the A-ring as well as hydroxylation on the B-ring play an important role in the biotransformation of S-1, since the majority of the metabolites maintaining both phenyl rings incorporated nitro reduction, including hydroxylamine intermediates, and/or hydroxylation on the B-ring. In summary, S-1 was susceptible to three phase I metabolic routes: hydrolysis of the amide bond, nitro reduction on the A-ring, and hydroxylation on the B-ring, with hydrolysis as a major metabolic pathway in rats. Phase II metabolic routes of S-1 in rats included sulfation, glucuronidation, and methylation.
Discussion
These studies demonstrate that S-1 is rapidly absorbed, slowly cleared, moderately distributed, and extensively metabolized in rats.
Effects of Dosing Vehicle on the Pharmacokinetic Profile
For the oral dose, the ethanol vehicle (for 10 mg/kg p.o.) apparently promoted the absorption of S-1, leading to faster absorption and higher bioavailability, as observed for the 10 mg/kg p.o. group. These observations suggest that the increased bioavailability of S-1 after administration in ethanol is due to enhanced gastrointestinal absorption, although other effects of ethanol on S-1 pharmacokinetics (e.g., increasing transcellular permeability or reducing intestinal first-pass metabolism) cannot be excluded.
Ethanol was reported to alter the pharmacokinetic profile of other drugs by increasing the gastrointestinal absorption of several drugs in the rat (Magnussen, 1968), increasing transcellular permeability, changing plasma protein binding by different mechanisms (Seller and Holloway, 1978), and inhibiting the mixed function oxidase enzyme in vitro and in vivo in laboratory animals (Lane et al., 1985). DMSO acts as an excellent solvent for some organic compounds with properties being highly polar, hygroscopic, and completely miscible with water (Elzinga et al., 1989; Ali, 2001). Ethanol and DMSO might also have an effect on intestinal first-pass metabolism. Further study is needed to sort out possible mechanisms of the vehicle effect of ethanol and DMSO on S-1 pharmacokinetics.
Major Metabolic Pathways and Chemically Reactive Metabolites
Major metabolic pathways of S-1 are outlined in Fig. 8. There are three major metabolic pathways in the metabolism of S-1: nitro reduction, hydroxylation on the B-ring, and hydrolysis of the amide bond. Although slow, amide hydrolysis can occur by the action of nonspecific plasma esterases. More likely, the amide bond of S-1 can be hydrolyzed by liver amidase (Gordon Gibson and Skett, 1994). However, amidase was found to be ubiquitously expressed in every tissue and physiological fluid (Atkinson et al., 2001). Therefore, amidase in intestinal wall might have contributed to the formation of M-1 during intestinal first-pass metabolism when S-1 was administered orally. P450 could also be responsible for the reduction of the nitro group, but other enzymes (e.g., xanthine oxidase, microsomal NADPH-cytochrome c) might also be involved (Purohit and Basu, 2000). In addition, reduction can be carried out by reductase enzymes in intestinal anaerobic bacteria for orally administered drugs, or accumulated drug or its metabolites, in intestine through biliary elimination when drugs are given intravenously (Atkinson et al., 2001; Ueda, 2001).
Fig. 8.

Proposed major metabolism pathways of S-1 in male Sprague-Dawley rats.
Drugs containing a primary amine or aromatic nitro are usually associated with a high incidence of idiosyncratic drug reactions (Hornsten et al., 1990; Jorga et al., 1999). From Figs. 3 and 4, metabolites produced by hydrolysis and nitro reduction (e.g., M6, 13, 14, 15, etc.) might be considered as chemically reactive metabolites. In addition, observed M34 and M40 led to the production of another chemically reactive metabolite in urinary samples. A Michael acceptor (m/z 289) can also be produced through losing a molecule of water in M40. A stable product resulting from glutathione trapping was observed at m/z 452 (M37), which is a mercapturic acid conjugate with this Michael acceptor, indicating that Michael acceptors react with sulfhydryl groups easily as soft electrophiles. These findings confirmed that a Michael acceptor (m/z 289) existed in vivo, although only the mercapturic acid conjugate form was observed. This Michael acceptor (m/z 289) might be formed through O-dealkylation by the microsomal mixed-function oxidase system in liver, kidney, lung, and intestine (Gordon Gibson and Skett, 1994). The low signal intensity of M37 might indicate a very low concentration of this Michael acceptor (m/z 289) and its glutathione conjugates in vivo, compared with those of M-1. However, it is important to note that the metabolism studies were conducted in rats at superpharmacologic doses.
Clinical studies with flutamide and nilutamide (Fig. 2), two structurally related androgen receptor ligands that include an A-ring nitro group, showed that most of the adverse effects (e.g., gynecomastia, breast pain, etc.) were associated with expected antiandrogenic effects of drugs (Mahler et al., 1998). It is known that intermediate metabolites of aryl amines are susceptible to extensive redox cycling leading to methemoglobinemia and hemolytic anemia (Mahmud et al., 1997). However, this is rarely a clinical problem for flutamide (Mahler et al., 1998). Although hydroxylamines and aryl amines have been the primary metabolites of nilutamide observed in human, increases in hepatic transaminases and hepatitis were only reported in 8% and less than 1% of the treated patients, respectively (Mahler et al., 1998).
Knowing the interspecies differences in metabolism of bicalutamide, flutamide, and nilutamide (Neri, 1989; Creaven et al., 1991; Boyle et al., 1993; McKillop et al., 1993), additional in vitro and in vivo studies of S-1 should be considered to evaluate the potential toxicity of S-1 and its metabolites in different species. Although in vitro and preclinical measurements cannot be interpreted directly into prediction of drug-induced toxicity, either in animals or in humans (Baillie et al., 2002), choices can be made in selection of lead compounds in preclinical or clinical development.
Structure-Pharmacokinetics and Metabolism Relationship
The physicochemical properties of small-molecule drugs play an important role in determining their absorption, distribution, metabolism, excretion, and toxicity. Comparison of physicochemical properties, metabolism profiles, pharmacokinetics of S-1, and structurally related SARMs shed light on structural modification of this new class of propanamides for better pharmacokinetic characteristics.
S-1 displayed linear pharmacokinetics over the 0.1 to 30 mg/kg i.v. dose range with 55 to 60% oral bioavailability over the oral dose range from 0.1 to 30 mg/kg in rats. S-4, a lead compound investigated as a SARM, is a structural analog of S-1 having the same chemical moieties and backbone as S-1, with the only exception being the incorporation of an acetamido group instead of a fluoro at the para position of the B-ring (Fig. 2). S-4 showed decreased p.o. bioavail-ability at higher doses in rats, with 120, 100, and 57% at a dose level of 1, 10, and 30 mg/kg, respectively (Kearbey et al., 2004). Bicalutamide, a nonsteroidal antiandrogen, has a structure similar to that of S-1, with a cyano group instead of the nitro group on the A-ring and a sulfonyl linkage instead of an ether linkage to the B-ring (Fig. 2.). Bicalutamide demonstrated linear pharmacokinetics over the i.v. range of 0.5 to 2 mg/kg in rats (Cockshott et al., 1991). Oral bioavail-ability of bicalutamide decreased with dose in rats, with 72, 71.5, and 35.7% at a dose level of 1, 10, and 30 mg/kg, respectively (Cockshott et al., 1991). The nonlinear pharmacokinetics of bicalutamide could be to due to a solubility issue at the high dose level. Linear pharmacokinetics of S-1 may help determine the clinical dose regimen.
Bicalutamide exhibited two major metabolic pathways: hydrolysis of the amide bond and hydroxylation of the B-ring in rats (Boyle et al., 1993). During pharmacokinetics studies of bicalutamide in rats, the half-life, CL, and V of racemic bicalutamide were 17.7 h, 0.80 ml/min/kg, and 1.23 l/kg, respectively, at a dose level of 0.5 mg/kg (Cockshott et al., 1991). S-1 had a V similar to that of bicalutamide, but an approximately 6 times higher CL with a 5 times shorter half-life. This phenomenon could be explained by the different metabolism of the two compounds. A large number of nitro-reduced metabolites of S-1 were observed in rats, suggesting that this pathway may contribute greatly to the rapid CL of the compound. Furthermore, the presence of the nitro substituent in the A-ring may affect the rate of amide hydrolysis and further contribute to the more rapid metabolism of S-1. In comparison, the sulfonyl linkage present in bicalutamide is a strong electron-withdrawing group which probably deactivates the B-ring and makes it less susceptible to oxidation, whereas the cyano-substituent in the A-ring of bicalutamide is less susceptible to reduction. Thus, bicalutamide exhibits a much longer half-life than S-1, with a volume of distribution value similar to that of S-1. As such, nitro reduction and hydroxylation of the B-ring probably play more important roles, in addition to hydrolysis, during in vivo metabolism of S-1.
Nilutamide showed less clearance (2.5 ml/min/kg) and a longer half-life (7 h) than S-1 when it was given to rats by oral administration of 10 mg/kg, where bioavailability of nilutamide was complete (Creaven et al., 1991). Flutamide was rapidly converted to 2-hydroxyflutamide, an active and major metabolite of flutamide in rats (Neri, 1989). Hydroxyflutamide showed a clearance of 3 ml/min/kg in rats receiving 25 mg/kg via the i.v. route (Xu and Li, 1999). Compared with S-1, both nilutamide and 2-hydroxyflutamide have lower log P because of the fact that they only contain one phenyl group in both of their structures. In vitro microsomal studies showed that lipophilicity of compounds correlates well with their Km values, indicating that lipophilicity directly influences the affinity of compounds to the active sites of enzymes (Martin and Hansch, 1971). Log P can be used as an indicator of lipophilicity since Log P is well correlated with lipophilicity. Reduction of lipophilicity normally accompanies a reduced rate of metabolic clearance. As such, reduced metabolic clearance of nilutamide and 2-hydroxyflutamide may explain the lower clearance and longer half-life of both drugs when compared with S-1.
Pharmacokinetic studies of S-4 showed that it has a shorter half-life, smaller volume of distribution, and lower clearance than S-1 (Kearbey et al., 2004). The higher log P value of S-1 and lesser plasma protein binding might explain its higher volume distribution compared with that of S-4. Lesser electronegativity of the acetamido substitute as compared with the fluoro group might lead to more rapid oxidation of the B-ring of S-4. However, deacetylation and acetylation of the acetamido substituent during biotransformation give rise to complexity in predicting the pharmacokinetics of S-4 using structural information.
Halogen groups are often used to block metabolism or potentially deactivate ring systems (Birnbaum, 1985). C-6, another structural analog of S-1, has chloro and fluoro groups at para and meta sites, respectively, on the B-ring, and shares the same chemical backbone and other moieties as S-1 (Fig. 2). Pharmacokinetic studies of C-6 showed that it has a longer half-life, smaller volume of distribution, and lower clearance than S-1 (Chen et al., 2005). The lower clearance of C-6 might be explained by the ability of its two halogen substituents to sterically and electronically prevent metabolism. Surprisingly, C-6, having a higher log P than S-1, showed a lower volume of distribution. This observation might be explained by higher plasma protein binding of C-6. The higher log P value of C-6 probably contributed to its better absorption and higher AUC after i.v. and p.o. administration. Different substitutions (i.e., number and position) of halogen atoms on the B-ring would be expected to further block oxidative metabolism to a larger degree if substituted propanamides are pharmacologically active.
S-1, having a high degree of efficacy and potency in animal models, acceptable pharmacokinetics in preclinical species, and appropriate physicochemical properties, is a promising drug candidate SARM for clinical development. However, more potent and metabolically stable SARMs can be further explored based on the molecular information obtained in the pharmacological, pharmacokinetics, and metabolism studies on several SARMs intensively investigated in our laboratory. Additional structural modification of SARMs to reduce or avoid amide hydrolysis and metabolism to an aromatic amine are warranted and the subject of ongoing studies in our laboratories.
Acknowledgments
WinNonlin software was generously provided through an Academic License by Pharsight Corporation.
These studies were supported by a grant from the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK59800-06).
ABBREVIATIONS
- SARM
selective androgen receptor modulator
- S-1
3-(4-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)-phenyl]-propanamide
- S-4
3-[4-(acetylamino)phenoxy]-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide)
- C-6
3-(4-chloro-3-fluorophenoxy)-2-hydroxy-2-methyl-N-[4-nitro-3-(trifluoromethyl)phenyl]-propanamide
- M-1
3-(4-fluorophenoxy)-2-hydroxy-2-methyl-propanoic acid
- PEG-300
polyethylene glycol-300
- DMSO
dimethyl sulfoxide
- AUC
area under the plasma concentration-time curve
- AUMC
area under the first moment of the plasma concentration-time curve
- Cmax
maximal drug plasma concentration
- Tmax
time to reach the maximal drug concentration (at the Cmax)
- Vss
steady-state volume of distribution
- CL
clearance
- MRT
mean residence time
- V
volume of distribution
- HPLC
high-performance liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
References
- Ali BH. Dimethyl sulfoxide: recent pharmacological and toxicological research. Vet Hum Toxicol. 2001;43:228–231. [PubMed] [Google Scholar]
- Atkinson AJ Jr, Daniels CE, Dedrick RL, Grudzinskas CV, Markey SP, editors. Principles of Clinical Pharmacology. Academic Press; San Diego: 2001. Pathways of drug metabolism; pp. 123–142. [Google Scholar]
- Baillie TA, Cayen MN, Fouda H, Gerson RJ, Green JD, Grossman SJ, Klunk LJ, LeBlanc B, Perkins DG, Shipley LA. Drug metabolites in safety testing. Toxicol Appl Pharmacol. 2002;182:188–196. doi: 10.1006/taap.2002.9440. [DOI] [PubMed] [Google Scholar]
- Bhasin S, Bremner WJ. Clinical review 85: emerging issues in androgen replacement therapy. J Clin Endocrinol Metab. 1997;82:3–8. doi: 10.1210/jcem.82.1.3640. [DOI] [PubMed] [Google Scholar]
- Birnbaum LS. The role of structure in the disposition of halogenated aromatic xenobiotics. Environ Health Perspect. 1985;61:11–20. doi: 10.1289/ehp.856111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle GW, McKillop D, Phillips PJ, Harding JR, Pickford R, McCormick AD. Metabolism of casodex in laboratory animals. Xenobiotica. 1993;23:781–798. doi: 10.3109/00498259309166784. [DOI] [PubMed] [Google Scholar]
- Brown TR. Nonsteroidal selective androgen receptors modulators (SARMs): designer androgens with flexible structures provide clinical promise. Endocrinology. 2004;145:5417–5419. doi: 10.1210/en.2004-1207. [DOI] [PubMed] [Google Scholar]
- Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. A selective androgen receptor modulator for hormonal male contraception. J Pharmacol Exp Ther. 2005;312:546–553. doi: 10.1124/jpet.104.075424. [DOI] [PubMed] [Google Scholar]
- Cockshott ID, Plummer GF, Cooper KJ, Warwick MJ. The pharmacokinetics of casodex in laboratory animals. Xenobiotica. 1991;21:1347–1355. doi: 10.3109/00498259109043209. [DOI] [PubMed] [Google Scholar]
- Creaven PJ, Pendyala L, Tremblay D. Pharmacokinetics and metabolism of nilut-amide. Urology. 1991;37(Suppl 2):13–19. doi: 10.1016/0090-4295(91)80096-p. [DOI] [PubMed] [Google Scholar]
- Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD. Discovery of nonsteroidal androgens. Biochem Biophys Res Commun. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res (NY) 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Elzinga LW, Bennett WM, Barry JM. The effect of dimethyl sulfoxide on the absorption of cyclosporine in rats. Transplantation. 1989;47:394–395. [PubMed] [Google Scholar]
- Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Comparison of the pharmacological effects of a novel selective androgen receptor modulator, the 5α-reductase inhibitor finasteride and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia. Endocrinology. 2004;145:5420–5428. doi: 10.1210/en.2004-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon Gibson G, Skett P, editors. Introduction to Drug Metabolism. Blackie Academic & Professional; New York: 1994. Pathways of drug metabolism; pp. 1–33. [Google Scholar]
- Handelsman DJ, Conway AJ, Boylan LM. Pharmacokinetics and pharmacodynamics of testosterone pellets in man. J Clin Endocrinol Metab. 1990;71:216–222. doi: 10.1210/jcem-71-1-216. [DOI] [PubMed] [Google Scholar]
- Hornsten P, Keisu M, Wiholm B. The incidence of agranulocytosis during treatment of dermatitis herpetiform with dapsone as reported in Sweden 1972 through 1988. Arch Dermatol. 1990;126:919–922. doi: 10.1001/archderm.1990.01670310081011. [DOI] [PubMed] [Google Scholar]
- Ishak KG, Zimmerman HJ. Hepatotoxic effects of the anabolic/androgenic steroids. Semin Liver Dis. 1987;7:230–236. doi: 10.1055/s-2008-1040579. [DOI] [PubMed] [Google Scholar]
- Jorga K, Fotteler B, Heizmann P, Gasser R. Metabolism and excretion of tolcapone, a novel inhibitor of catechol-o-methyltransferase. Br J Clin Pharmacol. 1999;48:513–520. doi: 10.1046/j.1365-2125.1999.00036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearbey JD, Wu D, Gao W, Miller DD, Dalton JT. Pharmacokinetics of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide in rats, a non-steroidal selective androgen receptor modulator. Xenobiotica. 2004;34:273–280. doi: 10.1080/0049825041008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane EA, Guthrie S, Linnoila M. Effects of ethanol on drug and metabolite pharmacokinetics. Clin Pharmacokinet. 1985;10:228–247. doi: 10.2165/00003088-198510030-00003. [DOI] [PubMed] [Google Scholar]
- Magnussen MP. The effect of ethanol on the gastrointestinal absorption of drugs in the rat. Acta Pharmacol Toxicol. 1968;26:130–144. doi: 10.1111/j.1600-0773.1968.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Mahler C, Verhelst J, Denis L. Clinical pharmacokinetics of the antiandrogens and their efficacy in prostate cancer. Clin Pharmacokinet. 1998;34:405–417. doi: 10.2165/00003088-199834050-00005. [DOI] [PubMed] [Google Scholar]
- Mahmud R, Tingle MD, Maggs JL, Cronin MTD, Dearden JC, Park BK. Structural basis for the haemotoxicity of dapsone: the importance of the sulphonyl group. Toxicology. 1997;117:1–11. doi: 10.1016/s0300-483x(96)03548-2. [DOI] [PubMed] [Google Scholar]
- Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. Design, synthesis and biological characterization of metabolically stable selective androgen receptor modulators. J Med Chem. 2004;47:993–998. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin YC, Hansch C. Influences of hydrophobic character on the relative rate of oxidation of drugs by rat liver microsomes. J Med Chem. 1971;14:777–779. doi: 10.1021/jm00291a600. [DOI] [PubMed] [Google Scholar]
- McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA. Metabolism and enantioselective pharmacokinetics of Casodex in man. Xenobiotica. 1993;23:1241–1253. doi: 10.3109/00498259309059435. [DOI] [PubMed] [Google Scholar]
- Mooradian AD, Morley JE, Korenman SG. Biological actions of androgens. Endocr Rev. 1987;8:1–28. doi: 10.1210/edrv-8-1-1. [DOI] [PubMed] [Google Scholar]
- Neri R. Pharmacology and pharmacokinetics of flutamide. Urology. 1989;34(Suppl 4):19–21. doi: 10.1016/0090-4295(89)90230-6. [DOI] [PubMed] [Google Scholar]
- Purohit V, Basu AK. Mutagenicity of nitroaromatic compounds. Chem Res Toxicol. 2000;13:673–692. doi: 10.1021/tx000002x. [DOI] [PubMed] [Google Scholar]
- Seller EM, Holloway MR. Drug kinetics and alcohol ingestion. Clin Pharmacokinet. 1978;3:440–452. doi: 10.2165/00003088-197803060-00002. [DOI] [PubMed] [Google Scholar]
- Ueda O. Metabolism of 2-nitrofluorene, 2-aminofluorene and 2-acylaminofluorenes in rat and dog and the role of intestinal bacteria. Xenobiotica. 2001;31:33–49. doi: 10.1080/00498250010031647. [DOI] [PubMed] [Google Scholar]
- Wilson JD. Androgens. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, editors. The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 1996. pp. 1441–1458. [Google Scholar]
- Wilson JD, Aiman J, MacDonald PC. The pathogenesis of gynecomastia. Adv Intern Med. 1980;25:1–32. [PubMed] [Google Scholar]
- Wu Z, Gao W, Phelps MA, Wu D, Miller DD, Dalton JT. Favorable effects of weak acids on negative-ion electrospray ionization mass spectrometry. Anal Chem. 2004;76:839–847. doi: 10.1021/ac0351670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Li D. Pharmacokinetics of 2-hydroxyflutamide, a major metabolite of flutamide, in normal and CCl4-poisoned rats. Acta Pharmacol Sinica. 1999;20:655–658. [PubMed] [Google Scholar]
- Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT. Pharmacodynamics of selective androgen receptor modulators. J Pharmacol Exp Ther. 2003a;304:1334–1340. doi: 10.1124/jpet.102.040840. [DOI] [PubMed] [Google Scholar]
- Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol Pharmacol. 2003b;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- Yin D, Xu H, He Y, Kirkovsky LI, Miller DD, Dalton JT. Pharmacology, pharmacokinetics and metabolism of acetothiolutamide, a novel nonsteroidal agonist for the androgen receptor. J Pharmacol Exp Ther. 2003c;304:1323–1333. doi: 10.1124/jpet.102.040832. [DOI] [PubMed] [Google Scholar]
- Zhi L, Martinborough E. Selective androgen receptor modulators (SARMs) Annu Rep Med Chem. 2001;36:169–180. [Google Scholar]