

SUMMARY
Paramecium bursaria chlorella virus-1 encodes at least 5 putative glycosyltransferases that are probably involved in the synthesis of the glycan components of the viral major capsid protein. The 1.6 Å crystal structure of one of these glycosyltransferases (A64R) has a mixed α/β fold containing a central, six-stranded β-sheet flanked by α-helices. Crystal structures of A64R, complexed with UDP, CMP, or GDP, established that only UDP bound to A64R in the presence of Mn2+, consistent with its high structural similarity to glycosyltransferases which utilize UDP as the sugar carrier. The structure of the complex of A64R, UDP-glucose, and Mn2+ showed that the largest conformational change occurred when hydrogen bonds were formed with the ligands. Unlike UDP-glucose, UDP-galactose and UDP-GlcNAc did not bind to A64R, suggesting a selective binding of UDP-glucose. Thus, UDP-glucose is most likely the sugar donor for A64R, consistent with glucose occurring in the virus major capsid protein glycans.
INTRODUCTION
Paramecium bursaria chlorella virus-1 (PBCV-1), a member of the family Phycodnaviridae, is a large, icosahedral (diameter of ~1900 Å), plaque-forming, double-stranded DNA-containing virus that infects the green alga Chlorella NC64A (Van Etten et al., 1991; Yamada et al., 2006). The 331kb PBCV-1 genome has ~365 protein-encoding genes and 11 tRNA-encoding genes (Van Etten, 2003). The PBCV-1 virion is a multi-layered structure composed of the genome, an inner protein core, a lipid bilayer membrane and an outer icosahedral capsid shell (Yan et al., 2000). The virus major capsid protein, Vp54, has two “jelly-roll” domains with two O-linked and four N-linked glycans (Nandhagopal et al., 2002). Identification of the sequence of sugar moieties at each of the glycosylated sites is uncertain and is based merely on the crystallographic structure. Furthermore, the disorder of the six Vp54 glycans limits the number of observable sugar units.
None of the N-linked glycans occur at NX(S/T) sites that are commonly recognized by eukaryotic cellular enzymes involved in N-linked protein glycosylation. This finding, along with other observations such as the absence of amino sugars in the glycans, led to the prediction that PBCV-1 encodes most, if not all, of the machinery to glycosylate Vp54 (Van Etten, 2003). Consistent with this hypothesis, PBCV-1 encodes at least five putative glycosyltransferases. None of these five proteins have an identifiable signal peptide that would target them to the endoplasmic reticulum (ER). Furthermore, four of these five proteins are predicted to be cytoplasmic and the fifth is predicted to be in a membrane. A series of genetic experiments established that one of these five putative glycosyltransferases (A64R) was involved in Vp54 glycosylation (Graves et al., 2001; Wang et al., 1993).
Glycosyltransferase-encoding genes are rare in viruses but they have been reported in a few bacteriophages, poxviruses, herpesviruses, and baculoviruses (Markine-Goriaynoff et al., 2004). In some, if not all, of these viruses the enzymes are involved in biological processes other than proteinglycosylation. For instance, some phage-encoded glycosyltransferases modify virus DNA to protect it from host restriction endonucleases and a glycosyltransferase encoded by baculoviruses modifies a host insect ecdysteroid hormone leading to its inactivation (Markine-Goriaynoff et al., 2004).
Typically, viral structural proteins are glycosylated by host-encoded glycosyltransferases located in the ER and Golgi and then transported to a host membrane (Doms et al., 1993; Olofsson and Hansen, 1998). Nascent viruses acquire the glycoprotein(s) and only become infectious by budding through the membrane, usually as they are released from the cell. Consequently, the glycan portion of these virus glycoproteins is host specific. However, as noted above, glycosylation of the chlorella virus PBCV-1 major capsid protein differs from this paradigm because the virus appears to encode most, if not all, of its protein glycosylation machinery (Van Etten, 2003).
Glycosyltransferases transfer sugars from a donor substrate, usually a nucleotide-diphospho-sugar, to a polysaccharide, lipid, DNA, or protein acceptor. Most eukaryotic glycosyltransferases reside in either the endoplasmic reticulum (ER) or the Golgi as type II membrane proteins with a short N-terminal cytoplasmic tail, a membrane-spanning region, a stem, and a C-terminal catalytic domain (Paulson and Colley, 1989). Glycosyltransferases can be classified into either retaining or inverting enzymes (Figure 1), based on whether the anomeric configuration of the product is the same as or different from that of the donor substrate. By analogy with glycosidases, inverting glycosyltransferases probably follow a direct displacement mechanism (Davies and Henrissat, 1995; Davies, 2001), in which a general base assists in the deprotonation of the reactive hydroxyl of the sugar acceptor and then acts as the nucleophile to attack the sugar donor (Figure 1A). In retaining glycosyltransferases, the reaction involves a double displacement with the formation of a covalent intermediate (Figure 1B). Another mechanism proposed for retaining glycosyltransferases suggests that the enzyme utilizes an SNi transition state in which the approach of the attacking acceptor and the leaving donor are on the same side of the sugar ring (Figure 1C) (Persson et al., 2001; Sinnot, 1990).
Figure 1. Schematic diagram showing possible mechanisms for glycosyltransferases that utilize a nucleotide-diphospho-sugar as a donor substrate [Adapted from (Davies, 2001)].
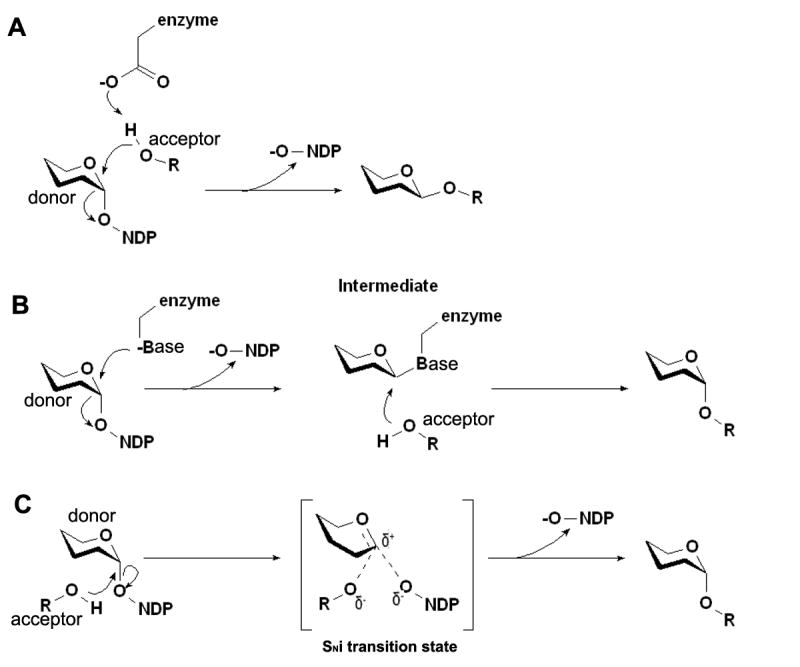
Inverting glycosyltransferases probably follow a direct displacement mechanism in which a general base assists in the deprotonation of the reactive hydroxyl of the sugar acceptor and then acts as the nucleophile to attack the sugar donor (Figure 1A). In retaining glycosyltransferases, the reaction involves a double displacement with the formation of a covalent intermediate (Figure 1B). Another mechanism proposed for retaining glycosyltransferases suggests that the enzyme utilizes an SNi transition state in which the approach of the attacking acceptor and the leaving donor are on the same side of the sugar ring (Figure 1C)
Two major structural folds, GT-A and GT-B, have been observed in glycosyltransferases (Breton et al., 2006) despite major differences in amino acid sequence. The GT-A fold is characterized by an N-terminal Rossmann-like fold*. A divalent cation, typically Mn2+, is coordinated by the aspartic acid side chains of a conserved DXD motif and the phosphate groups of the donor substrate. The GT-B fold is characterized by two similar Rossmann-like folds, with the N-terminal domain responsible for the acceptor binding and the C-terminal one for donor binding. The PBCV-1 A64R gene product could not be assigned to either the GT-A or GT-B class by sequence analysis. Glycosyltransferases have also been grouped into 87 different families based on their primary sequence similarities (http://afmb.cnrs-mrs.fr/CAZY/fam/acc_GT.html; (Coutinho et al., 2003)).
Sequence comparisons of the A64R protein (638 amino acid residues) indicate that it contains at least two domains with the N-terminal domain resembling a glycosyltransferase and the C-terminal domain an O-methyltransferase. The GlobPlot program (Linding et al., 2003) indicated that the A64R N-terminal fragment resembled glycosyltransferases in subfamily 34. Members in this subfamily are retaining glycosyltransferases, including some galactosyltransferases and xylosyltransferases. However, no three-dimensional structures exist for members of this family. None of the other putative glycosyltransferases coded by the PBCV1 genome belong to this subfamily.
Here, we report the crystal structures of the N-terminal glycosyltransferase fragment (residue 1 to 211) of the PBCV-1 A64R gene product. The apo-enzyme with Mn2+, with Mn2+ and uridine-5’-diphosphate (UDP), as well as with Mn2+ and UDP-glucose were determined to 1.6 Å, 2.3 Å, and 2.0 Å resolution, respectively. The crystal structures indicate that UDP, but not cytidine-5’-monophosphate (CMP) or guanidine-5’-diphosphate (GDP), bind to the A64R protein, suggesting that UDP is the carrier of the activated sugar donor. This conclusion is consistent with the structural comparison to other known glycosyltransferases. Furthermore, the crystal structure of A64R in complex with UDP-glucose indicates that UDP-glucose is the most likely donor sugar.
RESULTS AND DISCUSSION
Overall Protein Structure
A portion of the A64R gene (encoding amino acid residues 1-211) was cloned into expression plasmid PTYB1. Recombinant protein containing two extra residues at the N-terminus from the cloning vector was used for crystallization experiments. [Note: the amino acid numbers referred to in the text ignore these two extra residues.] The crystals, except those soaked with UDP-glucose, had a space group of P21 with one molecule in each asymmetric unit. The protein structure was determined to 1.6 Å resolution, using the multiple wavelength anomalous dispersion method (see Experimental Procedures). Of the 213 amino acids in the polypeptide chain, 206 (residues 4-209 in the ORF) could be visualized in the electron density map. One citrate ion and one Mn2+ ion, derived presumably from either the crystallization buffer or protein storage buffer, were also well-defined in the density map and consistent with the coordination geometry.
The structure had a mixed α/β fold containing a central, six-stranded β-sheet flanked by α-helices and a small, two-stranded β-sheet (Figure 2). The overall fold is very similar to catalytic domains of glycosyltransferases in the GT-A group, although the amino acid sequence similarity between them is very low (less than 14% for equivalent Cα atoms). The central sheet has a topology order of β2, β1, β3, β6, β5, and β7 with all strands being parallel to each other except β6. There are three cysteins in the sequence but, as expected for a cytoplasmic protein, no disulfide bonds are formed between them. Four cis-peptide bonds occur at Phe13-Pro14, Ala111-Pro112, Ala143-Pro144, and Phe148-Pro149. A Mn2+ ion interacts with the DXD motif in an extended, largely-opened cleft that lies between the central and small β-sheets, suggesting that this cleft is the enzyme’s active center. The Mn2+ ion is octahedrally coordinated by the ε nitrogen atom of His193, three oxygen atoms from an anionic citrate, and two oxygen atoms of the Asp in the DXD motif.
Figure 2. Crystal structure of the glycosyltransferase domain of A64R.

(A) Ribbon diagram with bound Mn2+ and citrate ions. Mn2+ and citrate ions are represented in ball-and-stick representation. N, C, O, and Mn atoms are colored blue, grey, red, and purple, respectively.
(B) Topology diagram of the A64R structure. The DXD motif is indicated.
Nucleotide Binding
UDP, GDP, and CMP are the most common nucleotides used as monosaccharide donors (although a viral-encoded glycosylation system might utilize other nucleotides). Therefore, crystals of the nucleotide-free enzyme were soaked with UDP, GDP, or CMP. The difference map (see Experimental Procedures) between a UDP-soaked crystal and the crystal of the native enzyme showed a strong, positive density peak with magnitudes roughly 1.8 times as large as any other positive or negative peak. This peak, presumably corresponding to UDP, was located in the cleft where the Mn2+ and citrate ions were bound to the native enzyme. However, no significant density was observed in the difference map between the GDP- or CMP-soaked crystals and the native enzyme, suggesting that UDP is most likely the carrier of the donor substrate for the A64R glycosyltransferase.
The overall fold of the protein is not perturbed by the binding of UDP (Figure 3A), indicated by a root-mean-square-deviation (RMSD) of only 0.61 Å between equivalent Cα atoms. The major conformational change after binding UDP occurs in loop regions surrounding the uracil and ribose rings of UDP (Figure 3). The largest of these changes is for Phe13 which moves toward the uracil ring. In addition, the UDP binding displaces the side chain of His54 towards UDP and the surface of the protein, forming a hydrogen bond with the UDP ribose (Figure 3B), thereby creating a larger space, presumably to accommodate the donor sugar.
Figure 3. The structure of A64R complexed with UDP.
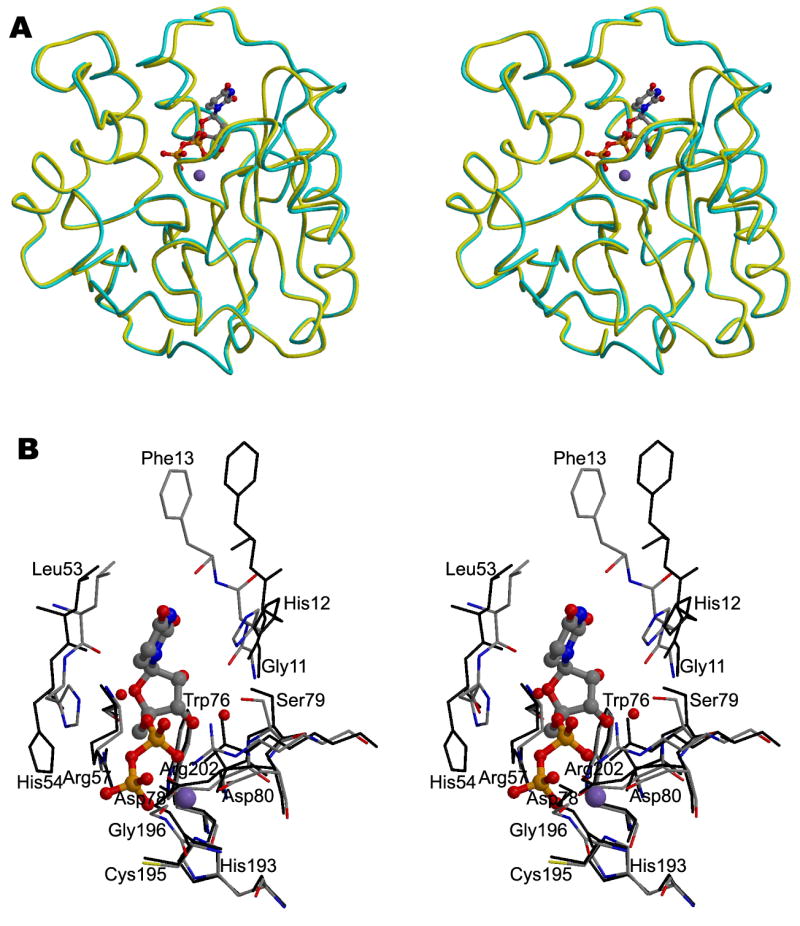
(A) Stereodiagram showing the superposition of the Cα traces of A64R with (yellow) or without UDP (cyan) bound. UDP is shown in a ball-and-stick representation and colored the same as in Figure 2. P atoms are colored in orange.
(B) Stereodiagram showing the interaction between UDP/Mn2+ and protein residues within 4 Å of UDP. Water molecules which form hydrogen bonds with UDP are also shown. UDP is shown in a ball-and-stick representation. The structure of A64R when UDP bound is color-coded according to the nature of the atoms. The A64R structure without bound UDP is colored black.
The citrate ion in the native enzyme was displaced by UDP in the UDP-soaked crystal. The UDP binding is stabilized by several hydrogen bonds and Mn2+-oxygen interactions (Figure 3B). The uracil base interacts primarily with the main chain atoms of Phe13 within the loop connecting β1 and α1. The ribose is in the 3’-endo conformation and forms hydrogen bonds with residues Gly11, Asp78, and Ser79. The oxygen atoms of the diphosphate group are hydrogen-bonded to residues Asp80, His193, Gly196, and Arg202, in addition to coordinating the bound Mn2+ ion. Model building shows that the inability of GDP to bind to A64R may be due to the steric hindrance between the guanidine base and the surrounding protein. In addition, the inability of CMP to bind to A64R is probably caused by the reduced number of phosphate groups and the inappropriate hydrogen-bonding properties of cytosine.
Comparison to Other Known Glycosyltransferases
The glycosyltransferases with greatest structural similarity to A64R, as found by the DALI program (Holm and Sander, 1995), are galactosyltransferase LgtC (Persson et al., 2001), bovine alpha1,3-galactosyltransferase (Gastinel et al., 2001) and glycogenin (Gibbons et al., 2002). The average RMSD between about 180 equivalenced Cα atoms was about 3.2 Å for these comparisons. The above structures share a similar fold and belong to the GT-A family with a 6- to 8-stranded central β-sheet, although there is no apparent amino acid sequence similarity. These three proteins all utilize UDP as the carrier for the activated galactose or glucose donor, consistent with the finding that UDP, but not GDP or CMP, could bind to A64R. The three proteins with the highest similarity to A64R, although belonging to different subfamilies, are all retaining glycosyltransferases. Furthermore, members in the same subfamily 34 as A64R are also retaining enzymes (Edwards et al., 1999; Faik et al., 2002), providing further support that the A64R protein is a retaining glycosyltransferase.
Substrate Binding
A crystal of A64R, when soaked with UDP-glucose, diffracted to 2.0 Å resolution and had a space group of C2. The self-rotation function showed that the two-fold non-crystallographic symmetry axis was parallel to the crystallographic ac plane and oriented at an angle of ~20° with respect to the crystallographic a axis. The two molecules in the crystallographic asymmetric unit formed a tail-to-tail dimer. A (Fo-Fc) difference density map showed a positive peak with the density height that was 1.9 times as large as the next largest peak in the map. This difference density was located in the active center of one of the two molecules within the asymmetric unit and was consistent with the structure of UDP-glucose. However, the difference map did not show any significant density peak at the corresponding position in the other molecule. In the UDP-glucose bound molecule, residue Phe13 moves toward UDP-glucose and forms a pair of hydrogen bonds with the uracil ring (Figures 4A and 4B). In the other molecule this residue forms a crystal contact with its crystallographic 2-fold symmetry mate. This stabilization of the apo-conformation presumably prevents this residue from undergoing the conformational changes required to complex UDP-glucose, highlighting its importance in binding the donor substrate. In contrast to UDP-glucose, neither UDP-galactose nor UDP-GlcNAc bound to A64R, even when they were co-crystallized with A64R (Table 1). Selective binding of UDP-glucose to A64R suggests that UDP-glucose is probably the donor substrate or that the sugar donor is in a gluco conformation. The preference for binding UDP-glucose as opposed to UDP-galactose is the result of the formation of a hydrogen bond between Gln152 and 4-OH of glucose. The inability to bind UDP-GlcNAc is probably the result of the steric hinderance with the NAc group.
Figure 4. The structure of A64R complexed with UDP-glucose.

(A) Stereodiagram showing the interaction between UDP-glucose/Mn2+ and residues within 4 Å of UDP-glucose. Water molecules which form hydrogen bonds with UDP-glucose are also shown. UDP-glucose is shown in a ball-and-stick representation. The electron density shown is a (Fo-Fc) map contoured at 2.5σ.
(B) Schematic diagram showing the interactions between A64R and UDP-glucose/Mn2+. Distances are in Å.
(C) Stereodiagram showing the molecular surface of A64R. UDP-glucose is shown in a ball-and-stick representation and color-coded as above. The black arrow points to the putative binding site of an acceptor substrate. The surface C, O, N, and S atoms are colored brown, red, blue and yellow, respectively.
Table 1.
Data Collection and Structure Refinement Statistics
| Se-Met | Native | UDP | CMP | GDP | UDP-glucose | UDP-GlcNAc | UDP-gal | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Peak | Inflection | Remote | ||||||||
| Data collection | ||||||||||
| Wavelength(Å) | 0.97915 | 0.97945 | 0.94916 | 1.542 | 1.006 | 0.979 | 1.006 | 1.542 | 1.542 | 1.543 |
| Resolution(Å) | 40.0-2.0 | 40.0-2.0 | 40.0-2.0 | 30.0-1.6 | 50.0-2.3 | 50.0-1.5 | 50-1.9 | 20.0-2.0 | 30.0-1.6 | 30.0-2.2 |
| Space group | P21 | P21 | P21 | P21 | P21 | P21 | P21 | C2 | P21 | P21 |
| Unit cell | ||||||||||
| a(Å) | 43.3 | 43.3 | 43.4 | 43.2 | 43.4 | 43.2 | 43.3 | 126.4 | 43.2 | 42.9 |
| b(Å) | 63.2 | 63.3 | 63.3 | 63.0 | 63.5 | 63.2 | 63.2 | 61.8 | 63.0 | 62.9 |
| c(Å) | 44.8 | 44.8 | 44.9 | 44.8 | 44.8 | 44.7 | 44.8 | 76.1 | 44.8 | 44.6 |
| β | 115.5 | 115.5 | 115.5 | 115.4 | 114.0 | 116.1 | 115.2 | 126.6 | 115.4 | 115.3 |
| Completeness(%)a | 99.6(96.4) | 99.6(96.1) | 99.7(97.9) | 90.6(73.7) | 97.7(92.7) | 99.6(98.3) | 97.6(83.2) | 94.5(93.2) | 96.0(86.5) | 99.1(96.4) |
| Redundancy | 7.0 | 7.0 | 7.2 | 3.4 | 3.1 | 3.5 | 3.6 | 3.3 | 3.7 | 2.3 |
| Rsym a,b(%) | 7.5(13.5) | 7.2(15.9) | 8.5(22.5) | 6.7(35.8) | 9.1(35.8) | 9.1(41.9) | 7.5(30.3) | 7.2(42.8) | 6.1(38.0) | 9.8(45.7) |
| Soaking/Cocrystalliz. | Soaking | Soaking | Soaking | Soaking | Co-crystalliz. | Co-crystalliz. | ||||
| Ligand binding | Yes | No | No | Yes | No | No | ||||
| Refinement Statistics | ||||||||||
| Rwork/Rfree c | 17.3/19.5 | 19.0/24.3 | 20.7/23.3 | |||||||
| R.m.s. deviations | ||||||||||
| Bonds(Å) | 0.010 | 0.011 | 0.013 | |||||||
| Angles(°) | 1.397 | 1.282 | 1.274 | |||||||
| Ramachandran plot (% residues) | ||||||||||
| Most favored | 90.2 | 92.4 | 92.7 | |||||||
| Additional allowed | 9.2 | 7.1 | 6.7 | |||||||
| Disallowed | 0.5 | 0.5 | 0.6 |
Values in parentheses are for the highest-resolution shells.
Rsym=ΣhΣi ∣Ii(h)-<I(h)>∣/ ΣhΣi Ii(h), where Ii(h) is the ith observation and <I(h)> is the mean of all measurements of Ii(h).
R=Σ∣Fo-Fc∣/Σ∣Fo∣, where Fo and Fc are the observed and calculated amplitudes of the structure factor.
The structure of A64R in complex with UDP-glucose is very similar to that when complexed with UDP, with a RMSD of 0.3 Å between equivalent Cα atoms. The side chains of Phe13 and His54 in the native enzyme were displaced in the same way by UDP and UDP-glucose (Figure 3B). The conformation of the uracil and ribose rings was the same in the UDP-complexed and in the UDP-glucose-complexed structures. However, the β-phosphate moved away from the Mn2+ ion when glucose was bound to UDP, thus accommodating the interaction between the glucose moiety and its surrounding residues His54, Arg57, and Gln152 (Figures 4A and 4B). The conformational change of His54 upon binding UDP-glucose might suggest that His54 could be the required catalytic base. The ε atom of this histidine is 4.5 Å from the C1 atom of the glucose which might be close enough to help stabilize a partial positive charge on the C1 atom. Binding of UDP-glucose alters the coordination of the Mn2+ as two water molecules are brought into the active center to coordinate with the metal ion. The weaker interaction with the metal ion puts a stronger emphasis on the interaction with the sugar molecule, which might provide a means of discriminating between cognate and non-cognate UDP-donor sugars. The glucose moiety adopts a different conformation to that found in a structure of the glycogenin enzyme, which belongs to the GT-A family and utilizes UDP-glucose as the donor substrate (Gibbons et al., 2002) (PDB accession code, 1LL2) (Figure 5). The different conformation is probably due to the difference in residues around His54 in the glycogenin active site as compared to A64R. Even after the UDP-glucose is bound into the cleft that forms the A64R active center, a substantial cavity remains on the surface of the enzyme in the vicinity of the glucose moiety (Figure 4C). This could be the binding site for the acceptor molecule, consistent with the location of the acceptor substrate found in galactosyltransferase LgtC (Persson et al., 2001).
Figure 5. Stereoview of the active center showing the superposition of A64R (black) and glycogenin (red).

The superposition was based on the main chain atoms of the DXD motif and the Mn2+ ion. UDP-glucose molecules are displayed in ball-and-stick formats and color-coded the same as the corresponding proteins. The Mn atoms are shown in purple. Residues in the glycogenin structure are labeled.
Implications for Viral Glycosylation
Mutations in the conserved DXD motif (S79L, D80N) and also mutation G121E result in smaller glycan(s) on the major capsid glycoprotein (Graves et al., 2001). All three of these residues are located in the active center of A64R. Ser79 and Asp80 form hydrogen bonds with UDP and interact with the Mn2+ ion, respectively (Figure 4B). Gly121 is about 5 Å away from both the Mn2+ ion and the closest glucose atom. Therefore, replacing Gly121 with a residue which has a longer side chain could produce steric hindrance with the UDP-glucose ligand. All three point mutations appear to interfere with the binding of UDP-glucose, thus abolishing A64R glycosyltransferase activity.
The crystal structure showed that A64R can bind UDP-glucose and possibly utilize it as the donor substrate; however, definitive proof will require a more complete biochemical analysis. This structure is reasonable because, unlike mature eukaryotic N- or O-glycoproteins that typically lack glucose, glucose is one of the carbohydrate components of the PBCV-1 major capsid protein (Wang et al., 1993). In addition accumulating evidence indicates that glycosylation of the PBCV-1 major capsid protein probably occurs in the cytoplasm, not in the ER or Golgi (Markine-Goriaynoff et al., 2004; Nandhagopal et al., 2002; Van Etten, 2003). Consistent with this hypothesis, A64R lacks an N-terminal signal sequence that would target it to the ER. Thus, PBCV-1 requires its own glycosyltransferases, such as A64R, to glycosylate its capsid protein.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The virus PBCV-1 genomic DNA fragment, corresponding to amino acid residues 1-211 in the a64r gene, was amplified via PCR and was cloned into the PTYB1 expression plasmid (New England Biolabs Inc.) by using NheI/SapI cloning sites. This construct encodes a fusion protein composed of the target gene, an intein tag and a chitin-binding domain. There are three additional residues (MAS) at the N terminus of the target protein derived from the cloning vector. The constructed plasmid was transformed into E. coli BL21-CodonPlus®(DE3)-RIL cells (Stratagene), which were grown at 37°C until the OD600 reached ~0.8. Expression was then induced by adding isopropyl β-D-1-thiogalactopyranoside to a final concentration of 0.25 mM, followed by growing the culture at 20°C for 16 hrs. The cells were harvested and resuspended in a lysis buffer containing 20 mM Tris, pH 8.0, 0.5 M NaCl, 10% glycerol, 1 mM Tris(hydroxypropyl)phosphine (THP), and the complete EDTA-free protease inhibitor mixture (Roche Molecular Biochemicals). Cells were lysed by sonication and the lysate was cleared by centrifugation at 25,000 × g for 20 min. The soluble fraction of the lysate was passed through a chitin bead column (New England Biolabs), allowing the fusion protein to bind to the resin via its chitin-binding domain. The resin was then washed with 15-column volumes of lysis buffer with a higher concentration of NaCl (2.0 M). To induce the intein-mediated cleavage, the beads were incubated in lysis buffer containing 30 mM dithiothreitol for 16 hrs at room temperature. Appropriate fractions were further purified through a Superdex 75 column (GE Healthcare) and concentrated in a buffer containing 20 mM Tris, pH 8.0, 0.15 M NaCl ,10 mM THP, and 2 mM MnCl2. The protein was more than 95% pure as judged by a coomassie-stained SDS-PAGE. Mass spectrometry analysis of the purified protein showed that the first Met was removed, possibly by an N terminal Met-specific peptidase during protein expression.
Selenomethionine (SeMet) was incorporated into the protein by metabolic inhibition using the standard procedure (Van Duyne et al., 1993). SeMet-labeled protein was prepared using a similar procedure as described above.
Crystallization and Data Collections
The A64R protein, at a concentration of ~13 mg/ml, was crystallized using the hanging drop method. The protein was diluted 1:1 with mother liquor consisting of 20-26% polyethylene glycol 3350, 0.2 M NaCl, and 50 mM sodium citrate, pH 6.0. Crystals were grown at 20°C and then soaked in a solution containing the mother liquor, plus 10% glycerol, followed by flash-freezing with a stream of nitrogen gas at 100 K. X-ray diffraction data of the native protein were collected using a Rigaku X-ray generator and an Raxis IV detector. Data of the SeMet-labeled protein were collected at beamline ID23B at the Advanced Photon Source (Argonne National Laboratory, USA). Crystals of the native protein and SeMet-labeled protein diffracted to 1.6 Å and 2.0 Å resolution, respectively. Data were processed and scaled using the HKL2000 package (Otwinowski and Minor, 1997) (Table 1).
Structure Determination
Data at three different wavelengths (0.97915, 0.97945, and 0.94916 Å, determined from an X-ray fluorescence scan) were collected for the SeMet-labeled protein and scaled using the program SCALEIT in the CCP4 program suite (Collaborative Computational Project Number 4, 1994). The positions of two Se atoms were determined unambiguously by inspection of the Bijvoet anomalous difference Patterson map (Rossmann, 1961) and the isomorphous Patterson map (Rossmann, 1960). The program SHARP (Vonrhein et al., 2006) was used for the refinement of heavy-atom parameters and the phase calculation, yielding an averaged figure of merit of 0.6 to 2.0 Å resolution. The initial electron-density map was solvent-flattened, histogram-matched, and phase-extended to 1.6 Å resolution using the program DM (Cowtan, 1994). ARP/wARP was then used to automatically build the atomic structure into the electron density map. Manual correction of the model building was performed using the graphics program COOT (Emsley and Cowtan, 2004). The structure was refined using native data in the resolution range of 20-1.6 Å with the program REFMAC (Murshudov et al., 1997) (Table 1). Cycles of manual rebuilding and structure refinement were continued until no further improvement, indicated by the convergence of the Rfree factor (Brünger, 1992).
Complexes with Donor Substrate
UDP (Sigma), GDP (Sigma), CMP (Sigma) and UDP-glucose (Sigma), at a concentration of 10 mM, were independently incubated with ‘native’ crystals in the mother liquor for ~16 hrs prior to data collection (Table 1). Ten mM UDP-galactose (Sigma) and UDP-GlcNAc (Sigma) were co-crystallized with the A64R protein under similar crystallization conditions. Crystals, except for the crystal of A64R complexed with UDP-glucose, were isomorphous with the native structure, permitting the calculation of Fourier difference maps using (Fcomp-Fnat)*e-iα coefficients, where α is the phase of the native enzyme structure factors. Fcomp and Fnat represent the amplitudes of the structure factors of the ligand-complexed crystal and the crystal of the native enzyme, respectively. However, because the anionic citrate was also present in the active site of the native enzyme, the above difference map is not as sensitive as the difference map using (Fcomp-Fp,nat)*e-iα as the Fourier coefficient, where α and Fp,nat represent the calculated phase and amplitude of the native structure without the citrate ion, respectively. Structure amplitudes used in these difference maps were scaled together by a least squares procedure using a single scale factor and a Gaussian “temperature” correction.
The native protein structure was used as the starting model for manual rebuilding and refinement of the UDP-soaked crystal. Following refinement of the protein component, a model of UDP and water molecules were built into the (2Fo-Fc) electron density map. The refinement was continued until convergence of the Rfree factor (Brünger, 1992) was reached.
The crystal of A64R when soaked with UDP-glucose has a space group of C2, different from the native crystal (Table 1). Initial phases were obtained by molecular replacement with the program MOLREP (Vagin and Teplyakov, 1997), using the previous refined native structure as a starting model. Refinement of this structure followed the same procedure as that for the UDP-soaked crystal.
Structure coordinates of the native protein, the complex with UDP, and the complex with UDP-glucose were deposited with the protein data bank (accession number 2P6W, 2P73 and 2P72, respectively).
Acknowledgments
Figures 2A and 3 were prepared with the program Molscript (Kraulis, 1991) and Raster3D (Merritt and Bacon, 1997). Figures 4A, 4C, and 5 were made with the PyMOL program (http://www.pymol.org). We thank Gideon Davies and Bärbel Kaufmann for many helpful discussions. We are grateful to Sheryl Kelly, Cheryl Towell, and Sharon Wilder for the preparation of the manuscript. The work was supported by NIH grants AI 11219 (MGR) and GM 32441 (JVE) as well as grant P20-RR15635 from the Centers of Biomedical Research Excellence Program of the National Center for Research Resources (JVE).
Footnotes
* The term “Rossmann fold” is used throughout the literature describing glycosyltransferases and other related structures. For clarity and consistency we adhere to this convention here.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Breton C, Snajdrová L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology. 2006;16:29R–37R. doi: 10.1093/glycob/cwj016. [DOI] [PubMed] [Google Scholar]
- Brünger AT. Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature (London) 1992;355:472–475. doi: 10.1038/355472a0. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Coutinho PM, Deleury E, Davies GJ, Henrissat B. An evolving hierarchical family classification for glycosyltransferases. J Mol Biol. 2003;328:307–317. doi: 10.1016/s0022-2836(03)00307-3. [DOI] [PubMed] [Google Scholar]
- Cowtan KD. ‘dm’: an automated procedure for phase improvement by density modification. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:34–38. [Google Scholar]
- Davies G, Henrissat B. Structures and mechanisms of glycosyl hydrolases. Structure. 1995;3:853–859. doi: 10.1016/S0969-2126(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Davies GJ. Sweet secrets of synthesis. Nat Struct Biol. 2001;8:98–100. doi: 10.1038/84198. [DOI] [PubMed] [Google Scholar]
- Doms RW, Lamb RA, Rose JK, Helenius A. Folding and assembly of viral membrane proteins. Virology. 1993;193:545–562. doi: 10.1006/viro.1993.1164. [DOI] [PubMed] [Google Scholar]
- Edwards ME, Dickson CA, Chengappa S, Sidebottom C, Gidley MJ, Reid JS. Molecular characterization of a membrane-bound galactosyltransferase of plant cell wall matrix polysaccharide biosynthesis. Plant J. 1999;19:691–697. doi: 10.1046/j.1365-313x.1999.00566.x. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan KD. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004;D60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Faik A, Price NJ, Raikhel NV, Keegstra K. An Arabidopsis gene encoding an α-xylosyltransferase involved in xyloglucan biosynthesis. Proc Natl Acad Sci U S A. 2002;99:7797–7802. doi: 10.1073/pnas.102644799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastinel LN, Bignon C, Misra AK, Hindsgaul O, Shaper JH, Joziasse DH. Bovine α 1,3-galactosyltransferase catalytic domain structure and its relationship with ABO histo-blood group and glycosphingolipid glycosyltransferases. EMBO J. 2001;20:638–649. doi: 10.1093/emboj/20.4.638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons BJ, Roach PJ, Hurley TD. Crystal structure of the autocatalytic initiator of glycogen biosynthesis, glycogenin. J Mol Biol. 2002;319:463–477. doi: 10.1016/S0022-2836(02)00305-4. [DOI] [PubMed] [Google Scholar]
- Graves MV, Bernadt CT, Cerny R, Van Etten JL. Molecular and genetic evidence for a virus-encoded glycosyltransferase involved in protein glycosylation. Virology. 2001;285:332–345. doi: 10.1006/viro.2001.0937. [DOI] [PubMed] [Google Scholar]
- Holm L, Sander C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Linding R, Russell RB, Neduva V, Gibson TJ. GlobPlot: exploring protein sequences for globularity and disorder. Nucleic Acids Res. 2003;31:3701–3708. doi: 10.1093/nar/gkg519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markine-Goriaynoff N, Gillet L, Van Etten JL, Korres H, Verma N, Vanderplasschen A. Glycosyltransferases encoded by viruses. J Gen Virol. 2004;85:2741–2754. doi: 10.1099/vir.0.80320-0. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Meth Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;D53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nandhagopal N, Simpson AA, Gurnon JR, Yan X, Baker TS, Graves MV, Van Etten JL, Rossmann MG. The structure and evolution of the major capsid protein of a large, lipid-containing DNA virus. Proc Natl Acad Sci U S A. 2002;99:14758–14763. doi: 10.1073/pnas.232580699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson S, Hansen JE. Host cell glycosylation of viral glycoproteins - a battlefield for host defence and viral resistance. Scand J Infect Dis. 1998;30:435–440. doi: 10.1080/00365549850161386. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Meth Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Paulson JC, Colley KJ. Glycosyltransferases. Structure, localization, and control of cell type-specific glycosylation. J Biol Chem. 1989;264:17615–17618. [PubMed] [Google Scholar]
- Persson K, Ly HD, Dieckelmann M, Wakarchuk WW, Withers SG, Strynadka NCJ. Crystal structure of the retaining galactosyltransferase LgtC from Neisseria meningitidis in complex with donor and acceptor sugar analogs. Nat Struct Biol. 2001;8:166–175. doi: 10.1038/84168. [DOI] [PubMed] [Google Scholar]
- Rossmann MG. The accurate determination of the position and shape of heavy-atom replacement groups in proteins. Acta Crystallogr. 1960;13:221–226. [Google Scholar]
- Rossmann MG. The position of anomalous scatterers in protein crystals. Acta Crystallogr. 1961;14:383–388. [Google Scholar]
- Sinnot ML. Catalytic mechanism of enzymic glycosyl transfer. Chem Rev. 1990;90:1171–1202. [Google Scholar]
- Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J Mol Biol. 1993;229:105–124. doi: 10.1006/jmbi.1993.1012. [DOI] [PubMed] [Google Scholar]
- Van Etten JL. Unusual life style of giant chlorella viruses. Annu Rev Genet. 2003;37:153–195. doi: 10.1146/annurev.genet.37.110801.143915. [DOI] [PubMed] [Google Scholar]
- Van Etten JL, Lane LC, Meints RH. Viruses and viruslike particles of eukaryotic algae. Microbiol Rev. 1991;55:586–620. doi: 10.1128/mr.55.4.586-620.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated sructure solution with autoSHARP. Methods Mol Biol. 2006;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Wang I, Li Y, Que Q, Bhattacharya M, Lane LC, Chaney WG, Van Etten JL. Evidence for virus-encoded glycosylation specificity. Proc Natl Acad Sci U S A. 1993;90:3840–3844. doi: 10.1073/pnas.90.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Onimatsu H, Van Etten JL. Chlorella viruses. In: Maramorosch K, Shatkin AJ, editors. Advances in Virus Research. Academic Press; 2006. pp. 293–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Olson NH, Van Etten JL, Bergoin M, Rossmann MG, Baker TS. Structure and assembly of large lipid-containing dsDNA viruses. Nat Struct Biol. 2000;7:101–103. doi: 10.1038/72360. [DOI] [PMC free article] [PubMed] [Google Scholar]