

Abstract
The homochirality of biomolecules is a prerequisite for the origin and evolution of terrestrial life. The unique selection of D-monosaccharides, in particular, D-ribose in RNA and D-deoxyribose in DNA, leads to the construction of proteins by L-amino acids. This points to the exclusive role of stereoselectivity in the most important physiological processes. So far, there is no experimental confirmation for the theoretical calculations of the energy differences between enantiomers used for the explanation of the stereoselection of biomolecules. Therefore, the question of why nature prefers one configuration over the other still lacks a definitive answer. Here, we present the first experimental evidence that the D-enantiomer of RNA has a different electronic structure compared to the corresponding L-enantiomer. When varying the incident photon energy of the ultraviolet Raman probe across 5 eV, D- and L-isomers of the RNA duplex with the sequence [r(CUGGGCGG).r(CCGCCUGG)] show differences in the intensity of the vibrational modes with energies of 124.0 meV to 210.8 meV. The intensity difference of these vibrational modes can be traced back to energy differences in the electronic levels of D- and L-RNA leading to the preferential stabilization of the naturally occurring D-configuration of RNA over the L-configuration.
Keywords: homochirality, RNA stabilization, Raman spectroscopy, enantiomer, stereoselection
INTRODUCTION
The homochirality of biomolecules is directly connected to the origin and evolution of terrestrial life. However, the reasons for the symmetry breaking and preference of one stereoisomer over another in nature still continues to be an unanswered question.
Chirality is not only an inherent property of matter (Davankov 2006), but also a common property of all informational and functional biomolecules: DNA, RNA, proteins, and carbohydrates. L-α-amino acids are chemically related to D-sugars. This was demonstrated by the transformation of D-glucosamine to L-alanine (Wolfrom et al. 1949). Moreover, amino acids catalyze asymmetric de novo synthesis of hexoses through cross-aldol reactions that proceed with high stereoselectivity (Cordova et al. 2005a,b). In this way, an asymmetric synthesis of deoxysugars with >99% enantiomeric excess has been achieved (Cordova et al. 2005a). It is evident that the building blocks of the naturally occurring proteins, carbohydrates, and nucleic acids are structurally related.
Giant biomolecules are involved in processes that are essential for living organisms and outline the extreme importance of homochirality for life. In particular, RNA plays an intermediate function between DNA and proteins. RNA possesses functions typical for both types of biopolymers, as an informational carrier and biocatalyst (Zaug and Cech 1986; Cech 1989). As an example, the hydrolytic stability of heterochiral RNA is reduced in comparison to that of the pure homochiral form (Urata et al. 2005). It can be supposed that the homochirality of RNA affords possibilities for the chiral selection of amino acids during the aminoacylation of nucleic acids, which is the first step of protein synthesis. Experiments with RNA constructs have shown that the α-amino group of amino acids does not play any role in the chiral selection (Tamura and Schimmel 2006). These investigators supposed that the chiral preference of RNA constructs for L-amino acids depends on avoiding a sugar-pucker-sensitive steric clash between a pendant group of a base and the amino acid side chain.
The experimental work connected with the origin of homochirality has been addressed mainly to the chiral building blocks of biopolymers, for example, amino acids and sugars (Tranter 1985, and citations therein, 1987; Urata 1999; Hazen et al. 2001; Meierhenrich and Thiemann 2004; Urata et al. 2005; Nanita and Cooks 2006). Theoretical calculations of the parity violating energy differences (PVEDs) were used to explain the homochiral selection of monomeric biomolecules with small differences in the energy (ΔE PV) of enantiomers (Yamagata 1966; Hazen et al. 2001; Faglioni et al. 2005, and citations therein). Here, we present the first experimental evidence that RNA in the D-configuration has a different energy level scheme than the corresponding L-form, leading to an improved stability of the naturally occurring D-configuration.
RESULTS AND DISCUSSION
We have analyzed the electronic energy levels associated with excitations of vibrations employing ultraviolet resonance Raman spectroscopy (Schulz et al. 2005) on the D- and L-enantiomers of an RNA duplex with the sequence [r(CUGGGCGG).r(CCGCCUGG)]. The sequence of this RNA fragment corresponds to domain E of Thermus flavus 5S rRNA. The spectra obtained at excitation energies close to 5 eV are near the absorption maxima of the free bases adenine, guanine, cytosine, and uracil. A difference between the Raman spectra of the two enantiomers was observed when changing the incident photon energy through the resonance level. Figure 1a shows spectra taken with incident photon energies (wavelength) 5.2094 eV (238.1 nm), 5.0834 eV (243.9 nm), and 4.8205 eV (257.2 nm). The spectra were obtained from the D- and L-isomers for comparison. All measurements were done with three accumulations. The received spectra were identical for all accumulations, therefore no time dependence, and accordingly no effect of denaturation, was observed. Whereas the spectra of D- and L-RNA show hardly any difference for incident photon energy of 4.8205 eV, we observed significant differences in the spectra taken with 5.0834 eV and 5.2094 eV. This difference is plotted as Raman difference spectra in Figure 1b. Differences in the intensity of the vibrational spectra were observed in the region between 124.0 meV and 210.8 meV with maxima at 186.0 meV, 192.2 meV, and 200.9 meV. Clearly, the spectra depend on the energy of the incident photons, and the intensity of the band changes with the wavelength of the excitation light.
FIGURE 1.

(a) Raman spectra of RNA duplexes in D- and L-configuration with the sequence [r(CUGGGCGG).r(CCGCCUGG)] at excitation wavelength of the incident photons of 5.2094 eV, 5.0834 eV, and 4.8205 eV. Each spectrum corresponds to the median of three accumulations. (b) Difference spectra at incident photon energies of 5.2094 eV, 5.0834 eV, and 4.8205 eV. The gray dotted background line, which is shown for the difference spectra at 4.8205 eV, demonstrates the size of the experimental error, valid and representative for all three measurements.
Figure 2a shows that resonance Raman scattering is a two-step process. An electronic transition creates a dipole moment, which is modulated by an elementary excitation such as a vibration. This implies that incoming light is absorbed by the electronic system. The coupling of the vibration and the electronic system of the molecule yields a scattered and frequency-shifted Raman line that is strongly dependent on the energy of the incident photons. This is due to the fact that the absorption varies strongly as a function of the incident energy. Therefore, differences in the intensities could be due to a different strength in the absorption or due to an energy difference of the electronic levels seen in the absorption process. However, both mechanisms can be discriminated, as shown in Figure 2, b and c, by the fact that a change in the oscillator strength yields only a negative or positive difference in the difference spectra but never both. Due to the fact that we observe positive and negative difference spectra when subtracting L-D-RNA spectra, we can conclude that the electronic levels coupling the vibrational excitations are shifted in energy (Fig. 2d). The magnitude of the shift is in the order of a few millielectron volts, which is still several orders of magnitude larger than expected by theory (Faglioni et al. 2005). Assuming that the energy difference ΔE is at least in the order of a few millielectron volts, and taking into account that the room temperature corresponds to an energy of ∼25 meV, it can be concluded that the energy difference clearly indicates an evolutionary preference of D- over L-RNA:
FIGURE 2.

(a) Feynman-Graph of the resonance Raman process. Light is absorbed by the electronic system, which, in turn, couples to the vibrational system of a molecule. (b) Raman difference spectra when subtracting D-RNA from L-RNA, when the strength of the absorption process of the electric levels is different. (c) Intensity of the Raman difference spectra when the electronic levels are shifted in energy. The diagram shows the Resonance profiles of D-RNA (red) and L-RNA (blue) as a function of the incident photon energy. The points and their corresponding error bars indicate positions of the three measurements at 4.8205 eV, 5.0834 eV, and 5.2094 eV. (d) Term scheme as a possible explanation of homochirality due to a shift in the energy levels between L-RNA and D-RNA.
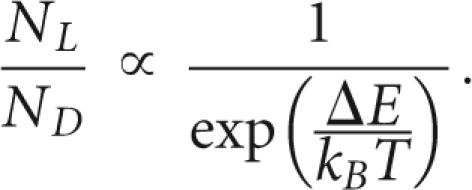 |
This points toward the possibility that the different orientations of the base pairs in the RNA enantiomers have an effect on the electronic energy levels and correspondingly on the intensity of the vibrational Raman spectra.
The observed relatively large energy shift points toward a novel mechanism involving a strong coupling of the electronic energy levels with the vibrational system. This effect, even though being somewhat anticipated by theory, requires a significant modification of many calculations (Bonner 1999; Faglioni et al. 2005).
As was mentioned above, only theoretical calculations of PVEDs between enantiomers of amino acids and ribose were used for the explanation of the homochirality of biomolecules. Although the calculated values are exceedingly low, some investigators seem to accept that PVEDs are a valid reason for the observed homochirality (Kondepudi and Nelson 1985). For others (Bonner 1999), these differences are too small to promote a preferred chirality. In any case, so far there is no experimental confirmation of the theoretical calculations. This is the first study that provides experimental evidence at the macromolecular level that the D-enantiomer of RNA has a different energy level scheme from its counterpart in the L-configuration.
MATERIALS AND METHODS
Synthesis of D- and L-enantiomers of an RNA duplex
The oligoribonucleotides 5′-CUGGGCGG-3′ and 5′-CCGCCUGG-3′ have been synthesized in D- and L-configuration and purified as described previously (Perbandt et al. 2001; Vallazza et al. 2004). The D-RNA and the L-RNA helices were separately annealed (Perbandt et al. 2001), and HPLC analysis showed homogeneity of both samples.
Raman spectroscopy
Raman spectra were recorded with three accumulations using a custom-made UV resonance Raman spectrometer as described previously (Schulz et al. 2005). The instrument has an all-reflective design and is set up in a clean room with constant temperature (22.0°C ± 0.5°C) and humidity (40% ± 3%). The samples were measured in water (concentration: 2 mg/mL), in a cuvette with a Suprasil window under continuous mixing. The sample volume was 100 μL in each case. The radiant flux of the incident laser beam was ∼1 mW for the applied excitation wavelengths, and the diameter of the beam and the focal depth were 15 μm. The illuminated volume of the sample was 2.7 × 10−6 μL, which is only 2.7 × 10−8 times the overall sample volume. In this way, a possible denaturation effect caused by the illumination was eliminated. The obtained spectra were background corrected and normalized to 1 mW power as well as to an integration time of 1 sec. Measurements with D- and L-RNA were done repeatedly at each of the three wavelengths, with equal acquisition times and identical photon power. The spectra show no time dependence, and moreover the applied mixing procedure provided a sufficient material exchange in the measured volume so that RNA material is continuously transported and exchanged in the sampling volume. The instrument was calibrated for its spectral response.
ACKNOWLEDGMENTS
We thank RiNA GmbH, Berlin, for the financial support; Stefan Vonhoff of NOXXON Pharma for providing the oligonucleotides; and M.V. Klein for discussions.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.564507.
REFERENCES
- Bonner, W.A. Enantioselective autocatalysis. V. The spontaneous resolution of tri-O-thymotide. Orig. Life Evol. Biosph. 1999;29:317–328. doi: 10.1023/a:1006593011038. [DOI] [PubMed] [Google Scholar]
- Cech, T. Ribozyme self-replication? Nature. 1989;339:507–508. doi: 10.1038/339507a0. [DOI] [PubMed] [Google Scholar]
- Cordova, A., Ibrahem, I., Casas, J., Sunden, H., Engqvist, M., Reyes, E. Amino acid catalyzed neogenesis of carbohydrates: A plausible ancient transformation. Chemistry. 2005a;11:4772–4784. doi: 10.1002/chem.200500139. [DOI] [PubMed] [Google Scholar]
- Cordova, A., Enqvist, M., Ibrahem, I., Casas, J., Sunden, H. Plausible origins of homochirality in the amino acid catalyzed neogenesis of carbohydrates. Chem. Commun. 2005b;20:2047–2049. doi: 10.1039/b500589b. [DOI] [PubMed] [Google Scholar]
- Davankov, V. Chirality as an inherent general property of matter. Chirality. 2006;18:459–461. doi: 10.1002/chir.20271. [DOI] [PubMed] [Google Scholar]
- Faglioni, F., Passalacqua, A., Lazzeretti, P. Parity violation energy of biomolecules. I: Polypeptides. Orig. Life Evol. Biosph. 2005;35:461–475. doi: 10.1007/s11084-005-3511-0. [DOI] [PubMed] [Google Scholar]
- Hazen, R., Filley, T., Goodfriend, G. Selective adsorption of L- and D-amino acids on calcite: Implications for biochemical homochirality. Proc. Natl. Acad. Sci. 2001;98:5487–5490. doi: 10.1073/pnas.101085998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondepudi, D.K., Nelson, G.W. Weak neutral currents and the origin of biomolecular chirality. Nature. 1985;314:438–441. [Google Scholar]
- Meierhenrich, U., Thiemann, W. Photochemical concepts on the origin of biomolecular asymmetry. Orig. Life Evol. Biosph. 2004;34:111–121. doi: 10.1023/b:orig.0000009832.71546.1d. [DOI] [PubMed] [Google Scholar]
- Nanita, S., Cooks, R. Serine octamers: Cluster formation, reactions, and implications for biomolecule homochirality. Angew. Chem. Int. Ed. Engl. 2006;45:554–569. doi: 10.1002/anie.200501328. [DOI] [PubMed] [Google Scholar]
- Perbandt, M., Vallazza, M., Lippmann, C., Betzel, Ch., Erdmann, V.A. Structure of an RNA duplex with an unusual G·C pair in wobble-like conformation at 1.6 Å resolution. Acta Crystallogr. D Biol. Crystallogr. 2001;57:219–224. doi: 10.1107/s0907444900017042. [DOI] [PubMed] [Google Scholar]
- Schulz, B., Bäckström, J., Budelmann, D., Maeser, R., Rübhausen, M., Klein, M.V., Schoeffel, E., Mihill, A., Yoon, S. Fully reflective deep ultraviolet to near infrared spectrometer and entrance optics for resonance Raman spectroscopy. Rev. Sci. Instrum. 2005;76:1–12. [Google Scholar]
- Tamura, K., Schimmel, P.R. Chiral-selective aminoacylation of an RNA minihelix: Mechanistic features and chiral suppression. Proc. Natl. Acad. Sci. 2006;103:13750–13752. doi: 10.1073/pnas.0606070103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tranter, G.E. The parity violating energy differences between the enantiomers of α-amino acids. Mol. Phys. 1985;56:825–838. [Google Scholar]
- Tranter, G.E. The enantio-preferential stabilization of D-ribose from parity violation. Chem. Phys. Lett. 1987;135:279–282. [Google Scholar]
- Urata, H. Effect of chirality of ribose on nucleic acid structure and function. Yakugaku Zasshi. 1999;119:689–709. doi: 10.1248/yakushi1947.119.10_689. [DOI] [PubMed] [Google Scholar]
- Urata, H., Sasaki, R., Morita, H., Kusumoto, M., Ogawa, Y., Mitsuda, K., Akagi, M. Kinetic analysis of hydrolytic reaction of homo- and heterochiral adenylyl (3′–5′) adenosine isomers: Breaking homochirality reduces hydrolytic stability of RNA. Chem. Commun. 2005;20:2578–2580. doi: 10.1039/b500673b. [DOI] [PubMed] [Google Scholar]
- Vallazza, M., Perbandt, M., Klussmann, S., Rypniewski, W., Einspahr, H.M., Erdmann, V.A., Betzel, Ch. First look at RNA in-L-configuration. Acta Crystallogr. D Biol. Crystallogr. 2004;60:1–7. doi: 10.1107/s0907444903027690. [DOI] [PubMed] [Google Scholar]
- Wolfrom, M., Lemieux, R., Olin, S. Configurational correlation of L-(levo)-glyceraldehyde with natural (dextro)-alanine by a direct chemical method. J. Am. Chem. Soc. 1949;71:2870–2873. [Google Scholar]
- Yamagata, Y. A hypothesis for the asymmetric appearance of biomolecules on Earth. J. Theor. Biol. 1966;11:495–498. doi: 10.1016/0022-5193(66)90110-x. [DOI] [PubMed] [Google Scholar]
- Zaug, A., Cech, T. The intervening sequence RNA of tetrahymena is an enzyme. Science. 1986;231:470–475. doi: 10.1126/science.3941911. [DOI] [PubMed] [Google Scholar]