

Abstract
Neural crest cells (NCCs) are essential components of the sympathetic nervous system, skin, craniofacial skeleton, and aortic arch. It has been known for many years that perturbation of migration, proliferation, and/or differentiation of these cells leads to birth defects such as cleft palate and persistent truncus arteriosus (PTA). Previously, we had shown that disruption of the platelet derived growth factor receptor (PDGFR) α in NCCs resulted in defects in craniofacial and aortic arch development, the latter with variable penetrance. Because we observed ventricular septal defects in embryos that are null for the PDGFRβ, we hypothesized that both PDGF receptors are involved in NCC formation. Here, we show that both receptors are expressed in cardiac NCCs and that the combined loss of the PDGFRα and PDGFRβ in NCCs resulted in NCC-related heart abnormalities, including PTA and a ventricular septal defect (VSD). Using NCC lineage tracing, we observed that loss of PDGF receptor signaling resulted in reduced NCCs in the conotruncus region, leading to defects in aortic arch septation. These results indicate that while PDGFRα plays a predominant role in NCC development, the PDGFRβ is expressed by and functions in cardiac NCCs. Combined PDGF receptor signaling is required for sufficient recruitment of cardiac NCCs into the conotruncal region and for formation of the aortico-pulmonary and ventricular septum.
Keywords: platelet derived growth factor, PDGF, migration, aortic arch, neural crest, mouse
INTRODUCTION
Cardiac NCCs delaminate from the dorsal neural tube between the midotic placode and the third somite and migrate ventrally to form a component of the vascular smooth muscle of the aortic arch as well as the aortico-pulmonary and ventricular septum. Classic experiments in the chick (Kirby et al., 1983) and multiple mutations in the mouse (Stoller and Epstein, 2005) have demonstrated that defects in this cell population cause heart malformations including persistent truncus arteriosus (PTA), ventricular septal defects (VSD), and aortic arch artery abnormalities. It is clear from these results that cardiac NCCs play an important part in the dynamic morphogenesis that occurs in the remodeling of the outflow tract into the asymmetric aortic arch, but their exact mode of action is not known. As the cardiac NCCs migrate along the arch arteries into the conotruncal region of the heart, some cells differentiate into vascular smooth muscle cells (VSMC) during this process. Other cells continue to migrate into the aortic sac where they proliferate and contribute to the aortico-pulmonary septum. Many cellular signals are involved in controlling NCC delamination, adhesion, migration, proliferation, remodeling, and survival, and defects in any of these signals can result in congenital heart defects (reviewed by (Hutson and Kirby, 2003b; Maschhoff and Baldwin, 2000; Stoller and Epstein, 2005)).
It has been established that disruption of PDGFRα signaling causes defects in cardiac and cranial NCC populations, including melanocytes and cells of the palate and aortic arch (Ding et al., 2004; Klinghoffer et al., 2002; Morrison-Graham et al., 1992; Soriano, 1997; Tallquist and Soriano, 2003). Previous analyses of the PDGFRα null and Patch (Ph) mutant allele mice (a naturally occurring PDGFRα deletion) suggested that cell survival or matrix deposition is the primary function of PDGFRα signaling (Morrison-Graham et al., 1992; Soriano, 1997), but no increase in apoptosis nor decrease in proliferation was observed in the NCC specific deletion of the PDGFRα (Tallquist and Soriano, 2003). These findings suggest that the survival and potentially the matrix defects observed in the PDGFRα null may have resulted from loss of PDGFRα in non-neural crest cells.
One difficulty in identifying the cellular cause of the aortic arch defects in PDGFRα mutants is the incomplete penetrance of the phenotype. This phenotypic variability may be caused by genetic modifiers or compensatory signaling in NCCs. A candidate protein that could compensate for loss of the PDGFRα is the PDGFRβ. PDGFRβ is expressed in many mesenchymal cells in the early embryo (Shinbrot et al., 1994). In addition, both receptors can bind the PDGFBB ligand and signal through many similar intracellular pathways. Mice lacking PDGFRβ or PDGFB die perinatally from vascular defects as a result of insufficient expansion of VSMC and pericyte formation (Leveen et al., 1994; Soriano, 1994). No overt craniofacial defects have been identified, although a dilation of the heart was observed in embryos deficient for the PDGFB ligand (Bjarnegard et al., 2004; Leveen et al., 1994). Interestingly, previous analysis of embryos possessing hypomorphic alleles of both PDGF receptors demonstrated that these two receptors can interact genetically (Klinghoffer et al., 2002).
To investigate this potential interaction we analyzed embryos with tissue specific deletion of both PDGF receptors in NCCs. Here, we observed that loss of the PDGFRβ exacerbated the previously described PDGFRα phenotype in the cardiac NCCs and resulted in PTA with 100% penetrance. While the initial delamination, migration, proliferation, and differentiation of NCCs was normal, the cause of PTA was the reduced ability of the PDGF receptor deficient cells to populate the outflow tract. These results suggest that PDGF receptor signaling instructs migration of the cardiac NCC population into the conotruncus region and demonstrate that these receptors are not required for the differentiation of the cardiac NCCs to form vascular smooth muscle.
MATERIALS AND METHODS
Mice
The PDGFRα NCC conditional/R26R-LacZ mouse line (PDGFRαfl/fl; R26R-LacZtg/tg) was described previously (Tallquist and Soriano, 2003). Mice possessing the PDGFRβ conditional (PDGFRβfl) and PDGFRα locus driven nuclear-localized green fluorescent protein (Hamilton et al., 2003) (PDGFRαGFP) alleles were kindly provided by P. Soriano (Fred Hutchinson Cancer Research Center). The PDGFRβ conditional animals will be described elsewhere in detail. Briefly, following Cre-mediated recombination, exons 1−6 are excised, and a deletion similar to the PDGFRβ null allele is generated (Soriano, 1994). Wnt1-CreTg mice were kindly provided by A. McMahon (Harvard University). To generate animals which lacked PDGF receptors in NCC populations, female mice were maintained as PDGFRαfl/fl; PDGFRβfl/fl; R26R-LacZtg/tg mice and crossed to PDGFRαfl/+; PDGFRβfl/+; Wnt1-Cretg/+ male mice. From these crosses, we cannot generate bona fide wild type littermate controls, therefore, we use embryos that are heterozygous for the floxed alleles as controls. The two genotypes used most often as controls were (PDGFRαfl/+; PDGFRβfl/+, and PDGFRαfl/+; PDGFRβfl/+; Wnt1-Cretg/+). Embryos bearing these genotypes were similar to wild type embryos.
Western Blot Analysis
Protein was isolated from the first and second branchial arches of control and NCC conditional E10.5 embryos. Actin and Erk1/2 antibodies were used as loading controls. The antibodies were obtained from the following sources: Erk 1/2, 06−182, (Upstate Biotechnology); PDGFRα, SSC-20, (Santa Cruz); cytoskeletal actin, (Novus Biologicals); and PDGFRβ, 90A, (a kind gift from A. Kazlauskas, Harvard).
Resin Corrosion Casting
Resin casting was done using Batson’s #17 plastic replica and corrosion kit (Polysciences; Warrington, PA). The protocol was carried out on E18.5 embryos as described previously (Tallquist and Soriano, 2003).
Histology and Immunohistochemistry
Hematoxylin and eosin staining were performed on paraffin sections according to standard procedures. PDGFRβ expression was detected on 10 μm frozen sections using 1:200 dilution of the CD140b antibody (ebioscience). αSMA expression was detected on paraffin sections with a 1:1000 dilution of the Clone 1A4 antibody (Sigma). Secondary antibodies were anti-rat and anti-mouse Alexafluor 594 (Molecular Probes), respectively. For the E10.5 PDGFRαGFP images an anti-GFP antibody (Clontech, JL-8) was used to detect the GFP protein and anti-mouse Alexafluor 498 (Molecular Probes) was used for the secondary antibody. For TUNEL staining, the embryos were paraffin embedded and serial sectioned. TUNEL labeling was carried out as previously described (Tallquist and Soriano, 2003).
BrdU (Sigma), 100 μg/g of body weight, was injected (i.p) into timed pregnant females. 2 hours later embryos were fixed with 4% paraformaldehyde overnight. Embryos were paraffin embedded, sectioned and prepared for immunohistochemsitry. After antigen retrieval, sections were incubated with anti-BrdU antibody at a 1:50 dilution (Pharmingen) and an anti-mouse Alexafluor 594 secondary antibody (Molecular Probes) was used as a secondary. In addition, sections were stained for DAPI. Nine sections from one embryo of each genotype were quantified at 20X field of view. Outflow tract regions from E10.5-E12.5 embryos were examined.
NCC lineage tracing
For the NCC lineage tracing we used the Wnt1-CreTg, R26R-LacZ reporter system has been previously described (Jiang et al., 2000). For whole mount staining, the ventral thoracic region of the embryo was removed to reveal the aortic arch. The samples were fixed in 2% formaldehyde/0.2% glutaraldehyde in PBS at 4° C for 20 minutes and incubated in X-gal buffer containing 4% X-gal overnight at room temperature. The embryos were post-fixed in 10% buffered formalin for 20 minutes at room temperature. Residual tissue surrounding the aortic arch and heart were removed for imaging. For transverse images of the aortic arch at E12.5 and E13.5, embryos were fixed for 1 hour in 4% paraformaldehyde, equilibrated in 10% sucrose, and embedded in OCT for frozen sectioning. For X-gal staining, 10 μm sections were washed in PBS + 0.05% Tween 20 for 20 minutes at room temperature and incubated in X-gal and buffer overnight. Frozen sections were counterstained with nuclear fast red (Fisher).
RESULTS
Ventricular septal defects in PDGFRβ null hearts
A direct role for the PDGFRα in cardiac NCCs has been established, but variability in the phenotype of embryos lacking PDGFRα signaling in the NCCs has prohibited the identification of the cellular function that is responsible for this phenotype. One explanation for the incomplete penetrance of the phenotype is the possibility that the PDGFRβ can partially compensate for loss of the PDGFRα. To investigate this possibility we examined PDGFRβ null embryos for NCC defects. One of the most common cardiac NCC defects is failure to form the membranous portion of the ventricular septum, and this defect was observed in embryos bearing the PDGFRα Ph/Ph allele (Schatteman et al., 1995). The frequency of this defect has not been documented for PDGFRα deficient embryos, and this may be because heart development is delayed in many of these embryos (Fig. 1B). Although it has been reported previously that PDGFRβ and PDGFB null embryos exhibit VSD (Betsholtz et al., 2001; Van Den Akker et al., 2005), the rate and cause of this defect has not been discussed. Therefore, we investigated the occurrence of VSD in PDGFRβ null embryos. We found membranous VSD in 8 out of 10 PDGFRβ null embryos (Fig. 1C). In the NCC tissue-specific deletion of the PDGFRα we also observed VSD as previously reported (Tallquist and Soriano, 2003), and the penetrance of this defect was less than 100% (Fig. 1D and Table I). Examination of other neural crest derivatives in PDGFRβ null embryos, such as aortic arch VSMC, cranial bones, and the thymic lobes revealed no obvious abnormalities (data not shown). Nonetheless, the presence of the VSD prompted us to investigate a potential role for the PDGFRβ in cardiac NCC development.
Fig. 1.
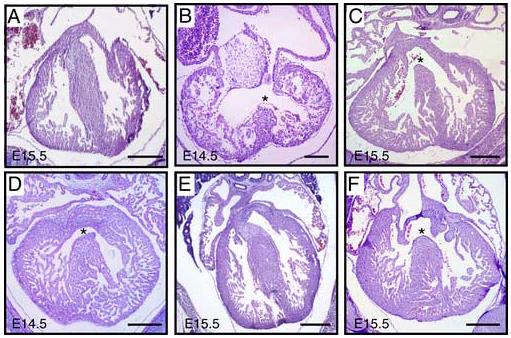
Ventricular septal defects are observed in PDGFRβ null and PDGF receptor NCC conditional animals Histological sections of control and PDGF receptor mutant embryos at E14.5 and E15.5 as indicated. (A) Control embryo with intact ventricular septum. (B) PDGFRα−/−, and (C) PDGFRβ−/− embryos exhibit VSD. Note that the PDGFRα null heart was developmentally delayed. (D) PDGFRα NCC conditional, (E) PDGFRβ NCC conditional, and (F) PDGFRα/β NCC conditional hearts. Asterisk indicates the VSD. Scale bar: 200μm.
Table 1.
Summary of cardiac NCC phenotypes
| Control (n=8) | PDGFRβ null (n=10) | PDGFRα NCC conditional (n=4) | PDGFRβ NCC conditional (n=13) | PDGFRα/β NCC conditional (n=25) | |
|---|---|---|---|---|---|
| PTA | n.d. | n.d. | 25% | n.d. | 100% |
| REO | n.d. | n.d. | 50% | n.d. | 100% |
| VSD | n.d. | 80% | 75% | n.d. | 100%* (22/22) |
| Thymus# | - | - (3/3) | Hypoplastic (7/7) | ||
| Thyroid# | - | - (3/3) | - (7/7) |
Data obtained from embryos E14.5-E18.5. Dash indicates organ was phenotypically normal, n.d.-none detected, PTA-persistent truncus arteriosus, REO-retroesophageal origin of the right subclavian artery, and VSD-ventricular septal defects. Controls were littermates that were heterozygous for the PDGFRα conditional and all combinations of PDGFRβ conditional and Wnt1CreTg. *n=22 for VSD because we did not evaluate VSD in the corrosion resin cast aortic arches. #PDGFRβ NCC conditional and PDGFRα/β NCC conditional embryos between E14.5 and E16.5.
PDGFRα and PDGFRβ expression in cardiac NCCs
To gain a better understanding of the extent of PDGF receptor expression in the cardiac NCC, we examined the receptors’ expression at several stages of cardiac development. Although expression analysis has been accomplished for each receptor previously (Morrison-Graham et al., 1992; Orr-Urtreger et al., 1992; Shinbrot et al., 1994), no reports exist for the co-expression of the two receptors in the conotruncal region. Fig. 2 demonstrates that while both receptors were expressed in cardiac NCCs, they did not have a completely overlapping profile. At E10.5 and E11.5 the two PDGF receptors were expressed in the NCCs migrating through the branchial arches. The PDGFRβ expression was highest in regions surrounding the arch arteries, but both receptors were detected in most NCC populations including the mesenchyme around the aortic sac and within the aortico-pulmonary spiral septum (Fig. 2 and data not shown). The PDGFRα had broader expression than the PDGFRβ as it was detected in the sclerotome and dermatome in these sections. By E13.5 the two receptors were no longer similarly expressed. While the PDGFRβ was robustly expressed in both NCC and non-NCC-derived VSMC surrounding the aorta, a more restricted pattern of expression was observed for the PDGFRα. By E15.5 the PDGFRα was down-regulated in the aortic VSMC but was still present in the adventitial fibroblasts. PDGFRβ expression remained in the VSMC region of the aorta and pulmonary trunk. This expression data supported a potential role for the PDGFRβ functioning redundantly with the PDGFRα in cardiac NCCs as they begin to invade the conotruncal region of the heart.
Fig. 2.
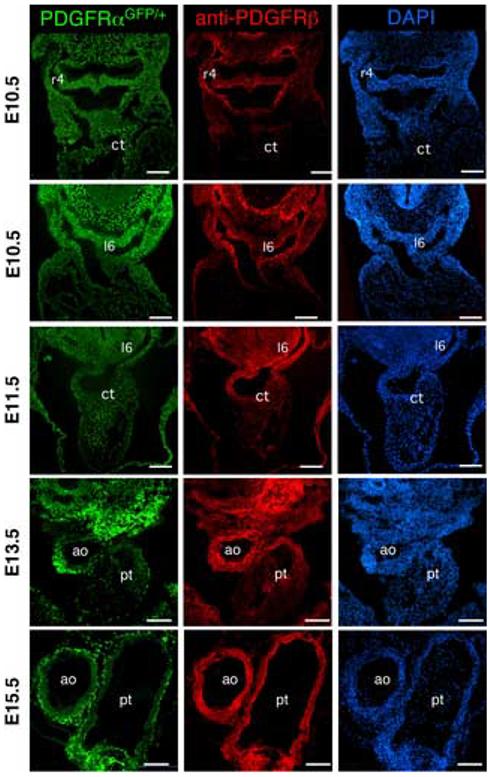
PDGFRα and PDGFRβ expression in the cardiac outflow tract PDGFRα expression was tracked using embryos that possessed a PDGFRαGFP allele and PDGFRβ was examined using immunohistochemistry. All three images at each stage are the same section. Embryonic stage is indicated at the left. Two E10.5 sections were imaged to illustrate PDGF receptor expression during migration of the cardiac NCC. r4, right fourth arch artery; l6 left sixth arch artery; ct, conotruncus; ao, aorta; and pt, pulmonary trunk. Note that the PDGFRα expression is followed using a nuclear localized GFP. Scale bar: 200μm.
Phenotypic analysis of PDGFRα/β NCC conditional embryos
Based upon the presence of a VSD in the PDGFRβ null animals we determined if the PDGFRβ was required for NCC development by generating mice that possessed a tissue specific deletion of the PDGFRβ in NCCs using the Cre/loxP recombination system (see materials and methods). Mice possessing the Wnt1-CreTg express the Cre recombinase in the premigratory NCCs and have been used extensively to generate tissue specific ablation in NCCs and for lineage tracing experiments (Danielian et al., 1998; Jiang et al., 2000). PDGFRβfl/fl; Wnt1-CreTg animals were viable and exhibited no VSD (Fig. 1E) or any other overt NCC defect (data not shown). These data indicated that either the VSD in the PDGFRβ null embryos was not cell autonomous to NCCs or that the deletion of the PDGFRβ in the NCC population was incomplete in our conditional animals. Western blot analysis and immunohistochemistry on tissue from conditional mutants confirmed that the PDGFRβ expression was greatly reduced in the branchial arches and the aortic arch in the presence of the Wnt1-CreTg (Fig. 3). The deletion in the branchial arches demonstrated not only that the tissue specific deletion of the PDGFRβ occurred, but also that the PDGFRβ was expressed abundantly in the NCC component of the first and second branchial arches (Fig. 3B). NCC deletion of the PDGFRβ resulted in loss of PDGFRβ from the NCC-derived VSMC but retention of the receptor in the splanchnic mesoderm-derived VSMC population surrounding the aorta and pulmonary trunk (Fig. 3C).
Fig. 3.
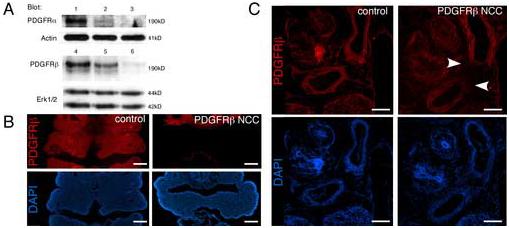
Efficient deletion of the loxP flanked alleles in the PDGFRα/β NCC conditional embryos (A) Western blot analysis of PDGF receptor expression in E10.5 branchial arches in (lane 1, PDGFRαfl/fl; PDGFRβfl/fl); (lane 2, PDGFRαfl/+; PDGFRβfl/+; Wnt1CreTg); (lane 3, PDGFRαfl/fl; PDGFRβfl/+; Wnt1CreTg); (lane 4, PDGFRαfl/fl;PDGFRβfl/fl); (lane 5, PDGFRαfl/+; PDGFRβfl/+; Wnt1CreTg); and (lane 6, PDGFRαfl/+; PDGFRβfl/fl; Wnt1CreTg). Detection of actin and erk1/2 was used as a loading control. (B-C) PDGFRβ immunohistochemistry on frozen sections of (B) E10.5 second branchial arches and (C) E14.5 aortic arch region demonstrating deletion of the PDGFRβ in NCCs. Arrowheads indicate loss of PDGFRβ from the VSMC of the aorta. Scale bar: 200μm.
Crosses of hypomorphic alleles of the two PDGF receptors suggested that there were genetic interactions of the two receptors in the formation of the cranial NCCs (MT and AR, unpublished observation, 2005), therefore, we hypothesized that the presence of the PDGFRα could compensate for the loss of PDGFRβ in some NCC populations. To test this hypothesis we generated mice doubly homozygous for PDGF receptor conditional alleles and also expressing the Wnt1-CreTg (PDGFR α/β NCC conditional). The efficient deletion of the PDGFRα in NCC was determined by western blotting (Fig. 3A) and has also been demonstrated previously (Tallquist and Soriano, 2003). PDGFRα/β NCC conditional embryos were recovered at the expected Mendelian ratios throughout embryogenesis, but 100% of these embryos died at birth. At birth these animals appeared cyanotic, exhibited a cleft palate similar to that observed in the PDGFRα NCC conditional embryos (Tallquist and Soriano, 2003), and likely died due to cardiovascular abnormalities and the inability to breathe. Further analysis revealed that loss of the PDGFRβ caused significantly more severe craniofacial (to be described elsewhere) and cardiac NCC phenotypes than what had been reported in the PDGFRα NCC conditional.
Loss of PDGFRα and PDGFRβ results in severe cardiac NCC defects
Defective thyroid and thymus formation have been associated with aberrant NCC development (Hutson and Kirby, 2003a), and recently it has been shown that PDGFRα-expressing, NCC-derived mesenchyme provides important signals for thymus expansion in the embryo (Jenkinson et al., 2007). In the mouse, NCCs begin to populate the stroma, lobe septae, and the capsule of the thymus at E11.5 (Jiang et al., 2000; Yamazaki et al., 2005). Therefore, we examined serial histological sections of the neck and upper thoracic region for thymus and thyroid development. Normal development of both thyroid and thymus were detected in PDGFRβ NCC conditional embryos (Table I), and when we examined PDGFRα/β NCC conditional embryos, we found that the thyroid gland was present and normal in size. In contrast, the development of the thymus was disrupted in the PDGFRα/β NCC conditional embryos. In 7/7 embryos the thymus was either absent or hypoplastic and in the neck region of the embryos. Four out of seven of these thymuses were located unilaterally (Table I). These data suggest that either PDGF receptor signaling was required for proper thymus development or was required for migration of NCCs to the thymic primordia.
To further investigate the development of the cardiac NCC we examined PDGF receptor NCC conditional animals by resin corrosion casting and histology, and we found defects in cardiac NCC derivatives including VSD (Fig. 1F), retroesophageal origin of the right subclavian artery (REO), and PTA (Fig. 4) in the mutant embryos. PTA is caused by a failure of NCCs to either migrate into or remodel the vascular arteries. The end result of this failure is that the aorta and pulmonary trunk fail to separate and remain as the primitive truncus arteriosus. The aberrant origin of the right subclavian is caused by abnormal regression of the right fourth arch artery. We observed PTA and REO in every PDGFRα/β NCC conditional embryo that we examined (25/25; Table 1) compared to the 25% and 50%, occurrence, respectively, that we observed in the PDGFRα NCC conditional. These phenotypes were consistent with a defect in NCCs that contributed to the cardiac outflow tract. The exacerbated phenotypes provided strong evidence that the PDGFRβ functions cooperatively with the PDGFRα during cardiac NCC development.
Fig. 4.
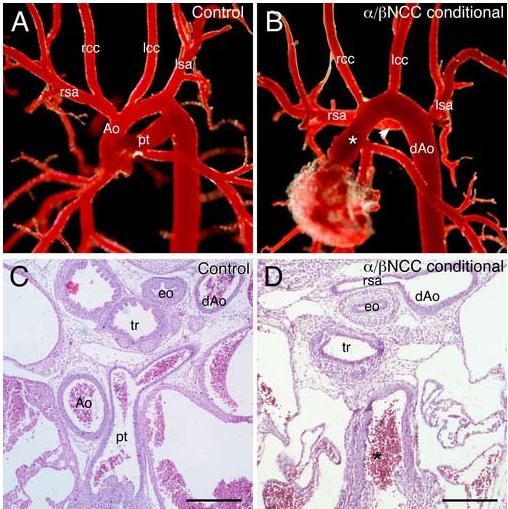
Aortic arch defects in PDGFRα/β NCC conditional embryos (A-B) Resin corrosion casts of E18.5 aortic arch vessels. (A) Control (PDGFRαfl/+; PDGFRβfl/+; Wnt1-CreTg) embryos exhibited an aortic arch architecture indistinguishable from that of wild type embryos while (B) PDGFRα/β NCC conditional (PDGFRαfl/fl; PDGFRβfl/fl;Wnt1-CreTg) embryos possessed persistent truncus arteriosus (asterisk) and retroesophageal origin of the right subclavian artery (arrowhead). Note: The rcc originated from the ascending truncus. (C-D) Transverse histological sections through E14.5 aortic arch regions demonstrated the same defects in the mutant embryo. Ao, aorta; dAo, descending aorta; eo, esophagus; lcc, left common carotid artery; lsa, left subclavian artery; pt, pulmonary trunk; rcc, right common carotid artery; rsa, right subclavian artery; and tr, trachea. Scale bar: 200μm.
The PDGFRβ has a well-established role in the proliferation of VSMC (Betsholtz et al., 2001), and recent evidence suggests that NCC differentiation into aortic arch smooth muscle is required for the morphogenesis of the symmetric arch arteries into the mature aortic arch (Kaartinen et al., 2004; Li et al., 2005; Oh et al., 2005). Therefore, we examined the VSMC component of the aortic arch in the PDGF receptor NCC conditional embryos at E11.5 to determine if failure in terminal differentiation of NCCs might be the cause of the aortic arch defects. The presence of α smooth muscle actin (αSMA) -positive cells around the aortic arch arteries demonstrated that NCCs were present and capable of differentiating into VSMC in the absence of PDGF receptor expression (Fig. 5). At E12.5 and 13.5 we also observed a normal pattern of smooth muscle cells surrounding the pharyngeal arch arteries (data not shown). These data demonstrated that PDGF receptor expression was not required for differentiation of NCCs into VSMC of the aortic arch and that the aortic arch defects observed could not be explained by absence of this cell lineage.
Fig. 5.
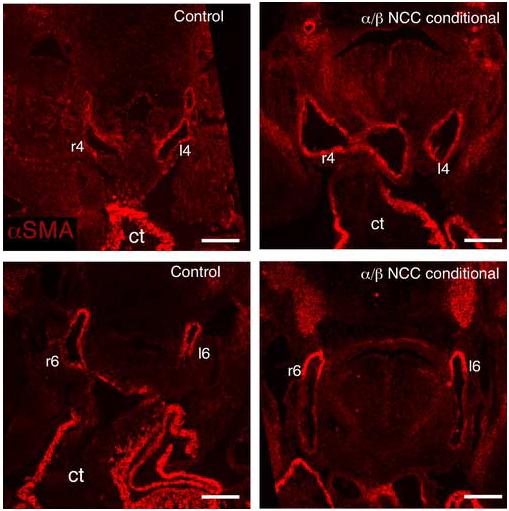
Normal smooth muscle cell distribution in PDGFRα/β NCC conditional embryo arch arteries Transverse sections through E11.5 control and PDGFRα/β NCC conditional embryos were stained for expression of α smooth muscle actin (αSMA) Scale bar: 200μm.
NCC migration defects occur in the conotruncus region of the aortic arch
We next analyzed the candidate cellular responses controlled by PDGF receptor signal transduction using common embryological techniques for proliferation, apoptosis, and migration. Defects in proliferation, migration, and apoptosis did not contribute to the observed phenotypes of the PDGFRα NCC conditional animals (Tallquist and Soriano, 2003). However, because PDGFRβ signaling in NCCs may have masked disruptions in these processes, we reexamined these potential cellular defects in our double NCC conditional embryos. Proliferation in the aortic sac was analyzed using BrdU incorporation of proliferating cells. Fate mapping of the cardiac neural crest in the mouse has shown that cells begin to migrate as two symmetrical prongs to the contotruncus region around E10.0, and septation of the aortico-pulmonary region begins around E12.0 (Jiang et al., 2000). Therefore, we analyzed proliferation of cells within the conotruncal region of embryos between the stages of E10.5 and E11.5 to assess the proliferation rate of NCCs in our PDGFRα/β NCC conditional embryos. The proliferation index of cells in outflow tract region of PDGFRα/β NCC conditional embryos demonstrated that there was no significant change in the number of proliferating cells compared to littermate controls (E10.5: control 33% ± 3%, mutant 36% ± 7%; E11.5: control 26% ± 4, mutant 23% ± 2%). Fig. 6 demonstrates that a similar rate of proliferation was observed in the outflow tract cushion of mutant and control embryos at E10.5. Finally, we also examined NCC populations for apoptosis in the conotruncal region at E10.5-E12.5, and PDGFRα/β NCC conditional mutants contained no increase in TUNEL positive cells in the conotruncal region compared to litter mate controls (data not shown). Taken together, these data demonstrated that proliferation and survival were normal in NCCs that lack both PDGFRα and PDGFRβ.
Fig. 6.
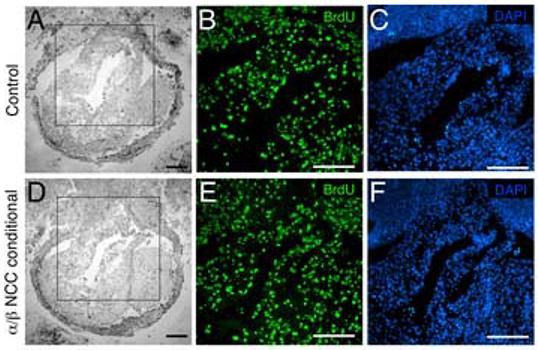
NCC proliferation remains unchanged in the outflow tract of PDGFRα/β NCC conditional mutants Transverse sections of E10.5 aortic sac stained for BrdU incorporation. (A-C) Control (PDGFRαfl/+; PDGFRβfl/+; Wnt1-CreTg) and (D-F) PDGFRα/β NCC conditional (PDGFRαfl/fl; PDGFRβfl/fl>;Wnt1-CreTg) embryos. (A, D) Brightfield images. Boxed region indicates close-up area for fluorescence detection. (B, E) BrdU positive nuclei and (C, F) DAPI stained nuclei. (ao) aorta, (es) esophagus (tr) trachea, (ta) truncus arteriosus. Scale bar: 200μm.
Because neither proliferation nor apoptosis were defective in the PDGFRα/β NCC conditional embryonic hearts, and the NCC defects that we observed were concentrated in the proximal outflow tract we examined NCC migration. NCCs were traced using mice that possess the R26R-LacZ and Wnt1-CreTg (Jiang et al., 2000). In these embryos, β-galactosidase is expressed in migrating NCCs and their descendents. Whole mount images of E16.5 hearts that possessed NCCs tagged by β-galactosidase expression revealed a decrease in the apparent number of NCCs in the aortic arch in PDGFRα/β NCC conditional compared to control and PDGFRα NCC conditional hearts (Fig. 7A-C). To determine when the cellular reduction in NCCs occurred, we examined embryos at earlier stages of development. At E10.5 and E11.5 we observed normal migration of cells from the neural tube (Fig. D-G). At E10.5 two streams of NCC were present in both the control and PDGFRα/β NCC conditional outflow tracts, but in PDGFRα/β NCC conditionals the NCC had not migrated as far into the outflow tract as the control NCCs. At E11.5 NCCs had entered the aortic sac but were less abundant in PDGFRα/β NCC conditional embryos (Fig. F-G and F’-G’). By E12.5 and E13.5 PDGFRα/β NCC conditional embryos possessed far fewer NCCs in the regions of the aortico-pulmonary septum when compared to littermate controls (Fig. 7 H-K). Fig. 7H’-K” demonstrate the location and numbers of the NCCs at the levels of the aortico-pulmonary septum and aortic valve leaflets. The controls had abundant numbers of NCCs surrounding both the aorta and the pulmonary trunk, while the PDGFRα/β NCC conditional embryos contained considerably fewer cells. To determine if the cause of thymic abnormalities was failure of NCCs from the third branchial pouch to invest in the thymic primordium, we examined E13.5 control and PDGFRα/β NCC conditional embryos for β-galactosidase expression. We found that in the control β-galactosidase expressing cells surrounded the thymic epithelium, but no β-galactosidase expressing cells were observed in the PDGFRα/β NCC conditional embryo (data not shown). This suggested that NCCs also failed to migrate to the thymic epithelium resulting in aberrant thymus development. Interestingly, cranial, thoracic, and enteric NCC migration was unperturbed in PDGFRα/β NCC conditional embryos (data not shown). We infer from this data that PDGF receptors are important for cardiac NCC migration into the outflow tract and that the reduction in NCCs reaching the outflow tract resulted in PTA and VSD.
Fig. 7.
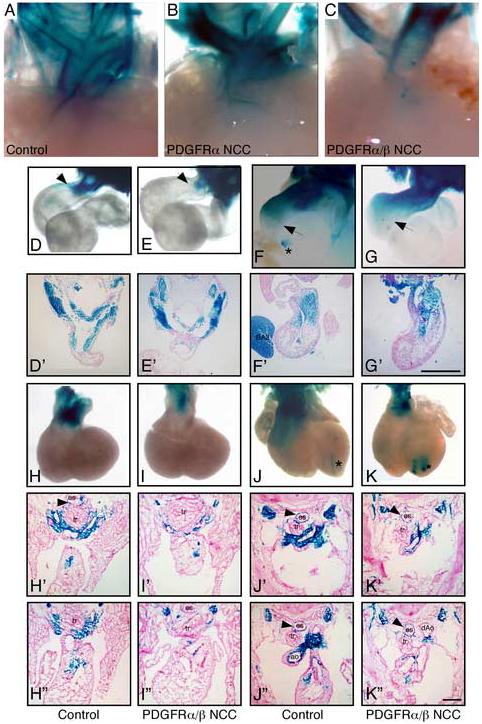
Reduced NCC migration into the conotruncus Detection of NCCs via the R26R-LacZ/Wnt1-CreTg lineage marker. Whole mount β-galactosidase activity of aortic arches at (A-C) E16.5, (D, E) E10.5, (F, G) E11.5, (H, I) E12.5, (J, K) E13.5. Asterisk indicates aberrant Wnt1-Cre activity that has been previously reported (Stottmann et al., 2004). Arrows point to furthest migration point of β-galacotosidase-tagged NCC. (D’-K’ and H”-K”) figures were transverse sections from control and mutant embryos. E10.5 and E11.5 sections were generated from the whole mount stained embryos. E12.5 and E13.5 were independent embryos that were frozen embedded, sectioned, and stained for β-galactosidase activity at the level of the outflow tract. Single prime (’) images were anterior to the double prime (”) images. A marked reduction in NCCs in the aortico-pulmonary septum of the aortic arch can be observed in the PDGFRα/β NCC conditional embryos at E12.5. Genotypes are as indicated. For whole mount images surrounding tissues were removed from the embryos to enhance visualization of the aortic arch region. BA2; second branchial arch; es, esophagus; Ao, aorta; dAo, descending aorta; and tr, trachea.
DISCUSSION
Our study has revealed that PDGF receptor signaling directs NCC function specifically in the conotruncal region of the aortic arch. Previous analyses of the PDGFRβ have demonstrated a role for this receptor in mesoderm derived VSMC and pericytes, but nothing was known about the requirement for PDGFRβ signal transduction in NCCs. These experiments demonstrate that the PDGFRβ provides signals to NCCs and that loss of both receptors causes a severe aortic arch phenotype, PTA. Our results suggest that NCCs respond to signals of either of the PDGF receptors. Therefore, presence of one receptor can partially compensate for loss of the other receptor family member.
Requirement for the PDGFRβ in cardiac NCCs
Tissue specific ablation in NCCs of the PDGFRβ alone did not cause any overt NCC phenotypes. This may be because the Wnt1Cre transgene did not delete the PDGFRβ gene efficiently or early enough, resulting in expression of the PDGFRβ that could rescue the VSD. In contrast, we have demonstrated that loss of the PDGFRβ in combination with the PDGFRα in NCCs lead to a phenotype more severe than the loss of the PDGFRα alone. In the absence of receptor activation, reduced numbers of NCCs migrated to the outflow tract. Loss of both receptors in NCCs resulted in PTA with 100% penetrance. These results provide genetic evidence that either of the PDGF receptors can direct NCC activities and that signaling downstream of these receptors is important for the formation of the NCC-derived components of the aortic arch. Despite the known requirement for PDGFRβ in many VSMC populations, we did not observe any defects in the PDGF receptor deficient NCCs’ ability to differentiate into αSMA-expressing cells. These data demonstrate that NCCs do not require PDGF receptor activity for formation nor proliferation of arch artery smooth muscle. Potentially, other signaling molecules are required in this particular cell population.
Our findings are somewhat surprising because the PDGFRα has such a dominant cranial NCC phenotype, and no definitive defects have been reported in the NCC derivatives in the PDGFRβ null embryos. It is likely that the apparent dominance of the PDGFRα is due to the profile of PDGF ligands that are expressed in the tissues surrounding the cranial and cardiac NCC populations. The PDGFRα can be activated by all PDGF ligands except for PDGFDD (Fredriksson et al., 2004). In contrast, the PDGFRβ can only bind PDGFBB and PDGFDD. The PDGFRα/β heterodimer can be activated by either PDGFBB or PDGFAB dimers. Based on the phenotypes of the PDGFRα and PDGFRβ NCC conditional embryos, our analysis suggests that both PDGFA and PDGFB may be the important ligands for signaling to NCC. The expression profile of the relevant ligands at the time of NCC migration into the outflow tract is dynamic. PDGFA expression in the mouse has been localized to epithelial tissues adjacent to PDGFRα-expressing mesenchyme (Orr-Urtreger and Lonai, 1992; Schattemann et al., 1996). No expression has been reported in the outflow tract, although PDGFA expression has been noted in the endocardial cushions in the avian system (Van Den Akker et al., 2005). PDGFB, which binds homo and heterodimers of the receptors, is expressed in all endothelial cells of the developing embryo (Hellstrom et al., 1999; Lindahl et al., 1997) and has been reported in the avian ventricular septum (Van Den Akker et al., 2005). During embryogenesis PDGFD is found in the myocardium at E12.5, and at E13.5 PDGFD is expressed by developing arterial VSMC (Ponten et al., 2005). While PDGFC expression has been shown to play a role in cranial NCC development (Ding et al., 2004), PDGFC mRNA is not expressed in the heart nor the vascular endothelium (Aase et al., 2002; Ding et al., 2000). Taken together, these data indicate that PDGFA and PDGFB may play a predominant role in the NCC migration and function in the developing outflow tract. In support of this idea, simultaneous deletion of PDGFA and PDGFC resulted in an atrial septal defect, but no aorticopulmonary nor ventricular septal defect was reported (Ding et al., 2000).
PDGF receptor-driven migration
Migration is one of the characteristic features of NCCs and failure of this cell population to reach its destination results in a variety of craniofacial and cardiac defects (Kirby et al., 1983; Phillips et al., 1987; Waldo et al., 1999). Although many receptors and matrix molecules have been implicated in NCC guidance and migration, little is known about how these signals direct aortic arch morphogenesis. Disruption of neuropilin1, plexin A2, or semaphorin 3C leads to a similar reduction in NCCs in the proximal outflow tract (Brown et al., 2001; Feiner et al., 2001; Kawasaki et al., 1999), but PTA occurs with a decreased frequency compared to the PDGFRα/β NCC conditional embryos. Some members of the TGFβ super family have also been implicated in NCC formation of the outflow tract. NCC specific ablation of BMPRIA (Stottmann et al., 2004), TGFβRII (Wurdak et al., 2005), ALK5 (Wang et al., 2006) or ALK2 (Kaartinen et al., 2004) results in defects of the aortic arch, but only the ALK2 mutation leads to defective NCC migration. Similar to what we observe in our PDGFRα/β NCC conditional embryos, ALK2 NCC conditional animals initiate migration but fail to migrate to the proximal zone of the outflow tract. It is of interest that both the ALK2 and PDGF receptor NCC conditional embryos exhibit PTA with 100% penetrance. Because these two receptors transduce signals through different intracellular components, it will be important to discover how the signaling pathways downstream of each receptor direct NCC migration.
Although one of the predominant actions of the PDGFRα and PDGFRβ in vitro is migration, and migration has been suggested as one of the mechanisms for guiding pericytes to endothelial cells, only two other examples of PDGF-directed migration have been demonstrated in vivo. In Drosophila, the PDGF receptor homolog, PVR, directs border cell migration to the oocyte (Duchek et al., 2001). In this system the target cell, the oocyte, expresses the PVF1 ligand, and the receptor-expressing cells are guided to the target. In Xenopus, PDGFAA expression directs the migration of PDGFRα-expressing mesodermal cells towards the animal pole (Nagel et al., 2004). A similar situation could be occurring in the region of the conotruncus.
Our NCC cell tracing indicated that PDGF signal transduction resulted in reduced NCCs in the proximal outflow tract, but our data does not rule out the possibility that PDGF stimulation could also affect outflow tract remodeling. The migration of NCCs is dependent on numerous extracellular matrix (ECM) proteins, and it may be that loss of receptor signaling causes a change in expression of various ECM proteins or matrix metalloproteases (Maschhoff and Baldwin, 2000). These disruptions could result in loss of cell adhesion or an increase of ECM, respectively. Therefore, PDGF receptors could additionally affect NCC migration by altering gene expression or metalloprotease activities. In support of this additional possibility, there is evidence that MMP-2 expression is lost in NCCs in Ph/Ph mutant mice. Recently, loss of PINCH1 or N-cadherin in NCC has been demonstrated to lead to a failure in aorticopulmonary septation that may result from inadequate adhesion or ECM in the outflow tract region. Further studies on the NCC conditional and signaling point mutants of the PDGF receptors (Heuchel et al., 1999; Klinghoffer et al., 2002) will allow us to determine the signaling pathways involved in directing cardiac NCC migration in vivo.
The combined function of PDGF receptor signal transduction in vivo
Understanding the combined functions of the PDGF receptors during embryogenesis and organogenesis is a complicated endeavor. Because homozygous nulls of each receptor results in embryonic lethality (Soriano, 1994; Soriano, 1997), the expected frequency of double homozygous null embryos is only 1 out of 16 by double heterozygous crosses. These analyses are further complicated by the fact that a high percentage of PDGFRα null embryos die before E10.5 likely due to defects in the spongiotrophoblasts cells (Hamilton et al., 2003). A hint that the receptors can cooperate during embryogenesis was revealed when mice carrying hypomorphic alleles of both receptors were generated (Klinghoffer et al., 2002). When either PDGF receptor is mutated such that it can no longer signal through the PI3K pathway, early embryonic lethality is circumvented, but when embryos are homozygous for both mutant receptors embryos die between E10.5 and E13.5 and exhibit a phenotype similar to the PDGFRα null embryos (Klinghoffer et al., 2002). These data suggested that the PDGFRβ is expressed in several of the same cell populations as the PDGFRα and can partially compensate for PDGFRα signal transduction when PDGFRα signaling is impaired. Our results are in agreement with these studies and specifically demonstrate that the two receptors are required in the cardiac NCC population. Our data illustrates the utility of conditional analysis and suggests that these two receptors have both overlapping and unique cellular roles. In NCCs, the PDGFRβ appears to play a minor role in the formation of the cranial bones but plays an essential role in migration of the cardiac neural crest.
In this study we have demonstrated that loss of both PDGF receptors consistently leads to PTA and VSD. Because the two PDGF receptors are on different chromosomes it is unlikely that simultaneous recessive mutations in these genes are responsible for NCC-related diseases, but disruption of molecules downstream of both receptors may lead to phenotypes similar to those that we observe. A thorough analysis of the signal transduction and gene induction downstream of the receptors will provide essential information regarding some of the candidate molecules that may lead to these common birth defects.
ACKNOWLEDGEMENTS
This work was supported by a MARC pre-doctoral fellowship F31 GM73417-01 to AMR and supported in part by research grants to MDT from the March of Dimes Birth Defects Foundation (5-FY2003-132), the American Heart Association (Grant 0330351N) and NIH (HL074257). We would like to thank Elizabeth Nichols and Haley Newton for their technical support. We would like to thank Phil Soriano for providing the PDGFRβ conditional animals prior to publication and for helpful comments on the manuscript. We would also like to thank Alisha Tiznor for her assistance on the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aase K, Abramsson A, Karlsson L, Betsholtz C, Eriksson U. Expression analysis of PDGF-C in adult and developing mouse tissues. Mech Dev. 2002;110:187–91. doi: 10.1016/s0925-4773(01)00560-3. [DOI] [PubMed] [Google Scholar]
- Betsholtz C, Karlsson L, Lindahl P. Developmental roles of platelet-derived growth factors. Bioessays. 2001;23:494–507. doi: 10.1002/bies.1069. [DOI] [PubMed] [Google Scholar]
- Bjarnegard M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fassler R, Betsholtz C. Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development. 2004;131:1847–57. doi: 10.1242/dev.01080. [DOI] [PubMed] [Google Scholar]
- Brown CB, Feiner L, Lu MM, Li J, Ma X, Webber AL, Jia L, Raper JA, Epstein JA. PlexinA2 and semaphorin signaling during cardiac neural crest development. Development. 2001;128:3071–80. doi: 10.1242/dev.128.16.3071. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–6. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O'Rourke M, Koh GY, Soriano P, Betsholtz C, Hart TC, Marazita ML, Field LL, Tam PP, Nagy A. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004;36:1111–6. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- Ding H, Wu X, Kim I, Tam PP, Koh GY, Nagy A. The mouse Pdgfc gene: dynamic expression in embryonic tissues during organogenesis. Mech Dev. 2000;96:209–13. doi: 10.1016/s0925-4773(00)00425-1. [DOI] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- Feiner L, Webber AL, Brown CB, Lu MM, Jia L, Feinstein P, Mombaerts P, Epstein JA, Raper JA. Targeted disruption of semaphorin 3C leads to persistent truncus arteriosus and aortic arch interruption. Development. 2001;128:3061–70. doi: 10.1242/dev.128.16.3061. [DOI] [PubMed] [Google Scholar]
- Fredriksson L, Li H, Eriksson U. The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004;15:197–204. doi: 10.1016/j.cytogfr.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003;23:4013–25. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Kalen M, Lindahl P, Abramsson A, Betsholtz C. Role of PDGF-B and PDGFR-beta in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development. 1999;126:3047–55. doi: 10.1242/dev.126.14.3047. [DOI] [PubMed] [Google Scholar]
- Heuchel R, Berg A, Tallquist M, Ahlen K, Reed RK, Rubin K, Claesson-Welsh L, Heldin CH, Soriano P. Platelet-derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3' kinase signaling. Proc Natl Acad Sci U S A. 1999;96:11410–5. doi: 10.1073/pnas.96.20.11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural Crest and Cardiovascular Development: A 20-Year Perspective. Birth Defects Research. 2003a;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Hutson MR, Kirby ML. Neural crest and cardiovascular development: a 20-year perspective. Birth Defects Res C Embryo Today. 2003b;69:2–13. doi: 10.1002/bdrc.10002. [DOI] [PubMed] [Google Scholar]
- Jenkinson WE, Rossi SW, Parnell SM, Jenkinson EJ, Anderson G. PDGFRalpha-expressing mesenchyme regulates thymus growth and the availability of intrathymic niches. Blood. 2007;109:954–60. doi: 10.1182/blood-2006-05-023143. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–90. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–61. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell. 2002;2:103–13. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev. 1994;8:1875–87. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- Li J, Zhu X, Chen M, Cheng L, Zhou D, Lu MM, Du K, Epstein JA, Parmacek MS. Myocardin-related transcription factor B is required in cardiac neural crest for smooth muscle differentiation and cardiovascular development. Proc Natl Acad Sci U S A. 2005;102:8916–21. doi: 10.1073/pnas.0503741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–5. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- Maschhoff KL, Baldwin HS. Molecular determinants of neural crest migration. Am J Med Genet. 2000;97:280–8. doi: 10.1002/1096-8628(200024)97:4<280::aid-ajmg1278>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Morrison-Graham K, Schatteman GC, Bork T, Bowen-Pope DF, Weston JA. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development. 1992;115:133–42. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- Nagel M, Tahinci E, Symes K, Winklbauer R. Guidance of mesoderm cell migration in the Xenopus gastrula requires PDGF signaling. Development. 2004;131:2727–36. doi: 10.1242/dev.01141. [DOI] [PubMed] [Google Scholar]
- Oh J, Richardson JA, Olson EN. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc Natl Acad Sci U S A. 2005;102:15122–7. doi: 10.1073/pnas.0507346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Do MS, Eisenbach L, Lonai P. Developmental expression of the alpha receptor for platelet-derived growth factor, which is deleted in the embryonic lethal Patch mutation. Development. 1992;115:289–303. doi: 10.1242/dev.115.1.289. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Lonai P. Platelet-derived growth factor-A and its receptor are expressed in separate, but adjacent cell layers of the mouse embryo. Development. 1992;115:1045–58. doi: 10.1242/dev.115.4.1045. [DOI] [PubMed] [Google Scholar]
- Phillips MT, Kirby ML, Forbes G. Analysis of cranial neural crest distribution in the developing heart using quail-chick chimeras. Circ Res. 1987;60:27–30. doi: 10.1161/01.res.60.1.27. [DOI] [PubMed] [Google Scholar]
- Ponten A, Folestad EB, Pietras K, Eriksson U. Platelet-derived growth factor D induces cardiac fibrosis and proliferation of vascular smooth muscle cells in heart-specific transgenic mice. Circ Res. 2005;97:1036–45. doi: 10.1161/01.RES.0000190590.31545.d4. [DOI] [PubMed] [Google Scholar]
- Schatteman GC, Motley ST, Effmann EL, Bowen-Pope DF. Platelet-derived growth factor receptor alpha subunit deleted Patch mouse exhibits severe cardiovascular dysmorphogenesis. Teratology. 1995;51:351–66. doi: 10.1002/tera.1420510602. [DOI] [PubMed] [Google Scholar]
- Schattemann GC, Loushin C, Li T, Hart CE. PDGF-A is required for normal murine cardiovascular development. Dev Biol. 1996;176:133–42. doi: 10.1006/dbio.1996.9988. [DOI] [PubMed] [Google Scholar]
- Shinbrot E, Peters KG, Williams LT. Expression of the platelet-derived growth factor beta receptor during organogenesis and tissue differentiation in the mouse embryo. Dev Dyn. 1994;199:169–75. doi: 10.1002/aja.1001990302. [DOI] [PubMed] [Google Scholar]
- Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev. 1994;8:1888–96. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Stoller JZ, Epstein JA. Cardiac neural crest. Semin Cell Dev Biol. 2005;16:704–15. doi: 10.1016/j.semcdb.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Stottmann RW, Choi M, Mishina Y, Meyers EN, Klingensmith J. BMP receptor IA is required in mammalian neural crest cells for development of the cardiac outflow tract and ventricular myocardium. Development. 2004;131:2205–18. doi: 10.1242/dev.01086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–18. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- Van Den Akker N, Lie-Venema H, Maas S, Eralp I, Deruiter M, Poelmann R, AC G-D. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Developmental Dynamics. 2005 doi: 10.1002/dvdy.20476. [DOI] [PubMed] [Google Scholar]
- Waldo KL, Lo CW, Kirby ML. Connexin 43 expression reflects neural crest patterns during cardiovascular development. Dev Biol. 1999;208:307–23. doi: 10.1006/dbio.1999.9219. [DOI] [PubMed] [Google Scholar]
- Wang J, Nagy A, Larsson J, Dudas M, Sucov HM, Kaartinen V. Defective ALK5 signaling in the neural crest leads to increased postmigratory neural crest cell apoptosis and severe outflow tract defects. BMC Dev Biol. 2006;6:51. doi: 10.1186/1471-213X-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19:530–5. doi: 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki H, Sakata E, Yamane T, Yanagisawa A, Abe K, Yamamura K, Hayashi S, Kunisada T. Presence and distribution of neural crest-derived cells in the murine developing thymus and their potential for differentiation. Int Immunol. 2005;17:549–58. doi: 10.1093/intimm/dxh237. [DOI] [PubMed] [Google Scholar]