

Abstract
1. S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide (also known as S-4) is a non-steroidal selective androgen receptor modulator demonstrating tissue-selective androgenic and anabolic effects. The purpose of the present study was to examine the systemic pharmacokinetics, elimination and oral bioavailability of S-4 in rats.
2. Thirty-five male Sprague–Dawley rats weighing approximately 250 g were randomly assigned to one of seven treatment groups. Intravenous doses of 0.5, 1, 10, and 30 mg kg−1 were given via a jugular catheter. Oral doses of 1, 10 and 30 mg kg−1 were administered via gavage. Plasma concentrations were determined using a validated high-performance liquid chromatography or by a high-performance liquid chromatography/mass spectrometry method.
3. Clearances ranged between 1.0 and 2.1 ml min−1 kg−1 and varied with dose. The volume of distribution was approximately 0.448 l kg−1 in all treatment groups. Oral bioavailability was also dose dependent, with the lower doses showing complete oral bioavailability. The half-life of S-4 over the dose range tested was between 2.6 and 5.3 h.
4. It was demonstrated that S-4 is rapidly absorbed, slowly cleared, and has a moderate volume of distribution in rats. The pharmacokinetics and oral bioavailability of S-4 indicate that it is an excellent candidate for clinical development.
Introduction
Synthesized steroidal androgens are used to treat a wide variety of conditions resulting from endogenous androgen deficiencies in both male and female patients. Unfortunately, virtually all current formulations have severe limitations (Wu 1992, Bhasin and Bremner 1997). Testosterone, in its pure form, suffers from low systemic bioavailability when administered orally (Handelsman et al. 1990). Testosterone esters (e.g. testosterone propionate, testosterone enanthate, testosterone cypionate and testosterone buciclate) were developed in order to achieve a more desirable pharmacokinetic profile. They are usually administered intramuscularly in oil. Since the esters are less polar than free testosterone, they are absorbed more slowly and therefore offer a prolonged duration of action (Wilson 1996). However, the esters produce erratic plasma concentrations of testosterone. 17-Alkylated testosterone derivatives (e.g. methyltestosterone and oxandrolone) are orally bioavailable, but they cause unacceptable hepatic toxicity, are less efficacious and are, therefore, not recommended for long-term administration (Heywood et al. 1977, Ishak and Zimmerman 1987, Velazquez and Bellabarba Arata 1998). Another drawback of testosterone administration is the cross-reactivity of both testosterone and its metabolites with other steroid hormone receptors (Wilson et al. 1980, Bhasin and Bremner 1997).
The present authors' laboratory was the first to report the discovery of a series of non-steroidal agonists for the androgen receptor (AR). These ligands bind the AR with an affinity similar to that of testosterone, and they mimic the effects of known steroidal agonists on receptor-mediated transcriptional activation (Dalton et al. 1998). However, extensive hepatic metabolism and poor oral bioavailability severely limit the in vivo androgenic activity of these compounds (Yin et al. 2003).
It was recently demonstrated that a series of structurally optimized non-steroidal AR agonists elicit in vivo androgenic and anabolic effects upon parenteral administration to rats (Yin et al. 2003). S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide (denoted S-4 herein and by Yin et al. 2003) emerged as the lead compound in this novel class of drugs. The structure of S-4 is shown in figure 1.
Figure 1.

Structure of S-4.
S-4 is a ligand for the AR with potent binding affinity that exhibits tissue-selective androgenic and anabolic effects in rats, identifying it as one of the first members of a new class of drugs known as selective AR modulators (SARMs). In castrated male rats, S-4 shows dose-dependent effects in the levator ani muscle. These effects are similar in potency and efficacy to those of testosterone propionate. However, S-4 is only a partial agonist in the prostate and seminal vesicles, restoring them to 33.8 and 28.2% of intact animals, respectively (Yin et al. 2003). These results clearly show that compound S-4 exhibits tissue-specific pharmacological activity and is a strong candidate for clinical development as the first non-steroidal SARM. Negro-Vilar (1999, p. 3460) defined the ideal anabolic SARM as ‘orally active, ideally with a pharmacokinetic profile consistent with once a day administration, with anabolic effects on muscle and bone, but lesser activity in the prostate and seminal vesicles’. An orally active anabolic SARM would be a valuable therapeutic agent for a variety of indications including muscle wasting diseases, male hypogonadism, male fertility, and osteoporosis. Given its non-steroidal structure, it was hypothesized that S-4 would be rapidly and completely absorbed after oral administration and avoid some of the historical drawbacks of testosterone administration. Yin et al. (2003) administered S-4 via subcutaneous osmotic pumps. Therefore, the purpose of the present study was to examine the pharmacokinetics and oral bioavailability of S-4 in rats, and further to demonstrate the promising preclinical properties of a representative SARM.
Materials and methods
Animals
The Institutional Laboratory Animal Care and Use Committee (ILACUC) of The Ohio State University approved the study. Thirty-five male Sprague–Dawley® rats, weighing approximately 250 g, were purchased from Harlan (Indianapolis, IN, USA). Eighteen hours before dosing, the animals were catheterized in the right jugular vein, and food (Harlan Teklad 22/5 rodent diet) was removed. The animals were provided water ad libitum and weighed immediately before dose administration. Food was returned 12 h after dosing.
Chemicals and formulations
S-4 was synthesized using the methods of Marhefka et al. (2003). Chemical purity was confirmed using elemental analysis, mass spectrometry and proton nuclear magnetic resonance. Dosing solutions were prepared immediately before administration and consisted of a co-solvent mixture of polyethylene glycol-300 (Sigma Chemical Co., St Louis, MO, USA; Lot 30K0174) and ethanol (Pharmco Products, Inc., Brookfield, CT, USA; Lot T022001).
Study design
Animals were randomized into seven groups, with five animals per group. Intravenous (i.v.) doses (0.5, 1, 10, 30 mg kg−1) were administered via the jugular catheter. Dosing solutions were prepared at an appropriate concentration to deliver the dose in a final volume of 0.2−0.3 ml. A 1 ml syringe graduated to 0.1 ml was used to deliver the dose volumetrically. After dose administration, the catheters were flushed with an aliquot (three times the volume of the administered dose) of sterile heparinized saline. Oral (p.o.) doses (1, 10, 30 mg kg−1) were introduced directly into the stomach via oral gavage in 0.2−0.3 ml. These doses were chosen to represent the range of S-4 doses used during preclinical pharmacology, safety and toxicology studies.
Sample collection and preparation
Blood samples (about 250 μl each) were withdrawn from the jugular vein catheter before each dose and at 5, 10, 20, 30, 60, 120, 240, 480, 720 and 1440 min after i.v. doses of 0.5, 1 and 10 mg kg−1, and before and at 30, 60, 90, 180, 240, 360, 480, 720, 1440 and 2160 min after dose administration for the 1 and 10 mg kg−1 p.o. doses. For the 30 mg kg−1 i.v. and p.o. dose groups, blood samples were drawn immediately before and at 20, 40, 60, 90, 120, 240, 360, 480, 720 and 1440 min after dose administration. A volume of heparinized saline (100 units ml−1) equal to the volume of blood removed was infused following each blood draw. Blood samples were collected into 1.5-ml heparinized microcentrifuge tubes and placed on ice until the plasma was separated by centrifugation at 800 g for 10 min at 4°C. Plasma samples were stored at −20°C until analysis. Metabolic cages were used to collect urine samples from the high-dose group over the 0−24 and 24−48 h following administration of the dose.
Plasma extraction for high-performance liquid chromatography (HPLC)
S-4 was extracted from plasma using a liquid/liquid extraction method. Aliquots of plasma (100 μl) were spiked with internal standard (2-ethyl-2-hydroxy-N-(4-nitro-3-trifluoromethyl-phenyl)-butyramide, MW 320, a structural analogue of S-4) and vortexed briefly. Plasma proteins were precipitated by the addition of acetonitrile (500 μl) and separated from the solution by centrifuging at 9300 g for 2 min. The supernatant was aspirated into 12-ml glass extraction tubes and 1.0 ml phosphate buffered saline, pH 7.4, and 7.0 ml ethyl acetate were added. Extraction tubes were shaken horizontally at 180 oscillations min−1 for 40 min on an Eberbach (Ann Arbor, MI, USA) reciprocating shaker. The mixture was then centrifuged at 1540 g for 10 min to facilitate phase separation. The organic phase was removed, placed in a glass centrifuge tube and evaporated to dryness under nitrogen. The residue was reconstituted in 150 μl mobile phase, centrifuged at 9300 g for 2 min to separate any residual particulate matter and 100 μl were injected into the HPLC.
HPLC plasma analysis
Plasma concentrations for the 10 mg kg−1 i.v. dose group and the 30 mg kg−1 i.v. and p.o. dose groups were determined using a validated HPLC method. HPLC analysis was performed on a Waters Chromatographic System (Waters Corporation, Milford, MA, USA) consisting of a model 510 pump, Nova-Pak® C18 3.9 × 150 mm × 4 μm column model 717 autosampler, and a model 486 ultraviolet light detector (Waters Corporation, Milford, MA, USA). The mobile phase consisted of acetonitrile:water (40:60) with 30 mM ammonium acetate at a flow rate of 1 ml min−1. The eluent was monitored for ultraviolet light absorbance at 270 nm. Standard curves were linear (R2≥0.989) over the concentration range 1−150 μg ml−1, and all quality control standards predicted values were within 85−115% of the actual concentration. Intra- and interday variabilities were less than 14.9 and 8.2%, respectively, at the lower limit of quantitation.
HPLC/mass spectrometry (MS) plasma and urine analysis
Plasma concentrations for the 0.5 and 1 mg kg−1 i.v. doses and the 1 and 10 mg kg −1 p.o. dose were determined using a validated HPLC/MS method. S-4 was extracted from plasma aliquots using a liquid/liquid extraction method. Aliquots of plasma (100 μl) were spiked with internal standard (deuterated compound S-4, MW 444) and vortexed briefly. Plasma proteins were precipitated by the addition of acetonitrile (500 μl) and separated from the solution by centrifuging at 16 100 g for 30 min. The supernatant was aspirated into 1.5-ml Eppendorf® tubes and evaporated to dryness under nitrogen. The residue was reconstituted in 150 μl mobile phase, centrifuged at 16 100 g for 5 min to separate any residual particulate matter, and 20 μl were injected into the HPLC/MS system column. HPLC analysis was performed on an Agilent 1100 Series System (binary pump, G1312A; degasser, G1379A; autosampler, G1367A; autosampler thermostat, G1330B; thermostatted column compartment, G1316A; Palo Alto, CA, USA) with a 2.1 × 50 mm, 5-μm particle size, Zorbax® SB-phenyl column (Agilent). The mass spectrometer was a single quadrupole mass selective detector (Agilent SL G1946D). Liquid chromatography (LC)/MS analyses were performed using an electrospray ionization source and the following conditions: negative single-ion mode; dry gas flow 10 l min−1; capillary voltage 2500 V; nebulizer pressure 25 psig; drying gas temperature 350°C; fragmentor voltage 174 V; peak width 0.15 min; time filter on; run time 12.3 min; post-run time 6 min; column temperature 28°C; flow rate 0.15 ml min−1. A gradient mobile phase consisting of two solutions was used. Solution A was an aqueous solution containing 5% acetonitrile, 10 mM ammonium formate and 0.1% formic acid. Solution B contained 95% acetonitrile, 5% water, 10 mM ammonium formate and 0.1% formic acid. The percentage of solution B in the mobile phase increased linearly from 40 to 100% during the first 6 min of each chromatographic run, was maintained at 100% from 6 to 12.0 min, and then decreased linearly to 40% at 12.2 min for the remainder of the run. S-4 was identified as the ion with 440.3 m/z while the deuterated compound S-4 was identified as the ion with 443.3 m/z. Data acquisition was performed with Agilent ChemStation Software (Version A.08.04). Standard curves for plasma were linear (R2 > 0.984) over the concentration range 0.005−0.050 μg ml−1, and all quality control standards predicted values were within 85−115% of the actual concentration. Rat urine samples were centrifuged at 800 g for 10 min to separate particulates, and then filtered through a 0.22-μm syringe filter. Filtered rat urine samples were diluted 200 times with HPLC-grade water and a 15-μl aliquot was injected into the LC/MS. The running conditions for the urine samples were identical to those used for plasma analysis. The standard curve for urine specimens was linear over the range 0.005−0.5 μg ml−1. Intra- and interday variabilities were less than 12 and 11%, respectively, at the lower limit of quantitation for these assays.
Pharmacokinetic data analysis
The plasma concentration–time data were analysed by non-compartmental methods using WinNonlin® (Version 3.1, Pharsight Corporation, Mountain View, CA, USA). The area under the plasma concentration–time curve from time zero to infinity (AUC) was calculated by the trapezoidal rule with extrapolation to time infinity. The terminal half-life (T½) was calculated as 0.693 λz−1, where λz was the terminal phase rate constant. The plasma clearance (CL) was calculated as CL=dosei.v. AUCi.v.−1, where dosei.v. and AUCi.v. are the i.v. dose and corresponding area under the curve from time 0 to infinity, respectively. The maximal plasma concentration (Cmax) and time at which it occurred (Tmax) for the p.o. doses were determined by visual inspection of the concentration–time profiles. The apparent volume of distribution at equilibrium (Vss) was calculated using:
where AUMC0–∞ is the area under the first moment of the plasma concentration–time curve extrapolated to infinity. The mean residence time (MRT) was calculated using:
Oral bioavailability (F) for each dose was calculated using:
where dosep.o., dosei.v., AUCi.v. and AUCp.o. are the mean oral dose, mean i.v. dose, and the corresponding mean areas under the curve from time 0 to infinity, respectively. Oral bioavailability for the 300 mg kg−1 p.o. dose was calculated using the CL of the 100 mg kg−1 i.v. dose.
Statistical analysis
Statistical analyses were performed by single-factor analysis of variance (ANOVA). p<0.05 was considered as statistically significant differences.
Results
Pharmacokinetics of compound S-4 after i.v. doses
S-4 achieved average maximal plasma concentrations of 1.6, 2.3, 28 and 168 μg ml−1 following i.v. doses of 0.5, 1, 10 and 30 mg kg−1, respectively (figure 2). The average steady-state volume of distribution for S-4 (0.45 l kg−1) was slightly less than total body water (0.67 l kg−1) (Davies and Morris 1993). CL remained relatively constant for the 0.5, 1 and 10 mg kg−1 doses at 1.92, 2.12 and 1.52 ml min−1 kg−1, respectively. However, the CL of S-4 was lower (1.00 ml min−1 kg−1, p<0.05) at the 30 mg kg−1 dose. Accordingly, the area under the plasma concentration–time curve increased proportionally with dose up to the 10 mg kg−1 dose, 0.24 mg min ml−1 at the 0.5 mg kg−1 dose, 0.46 mg min ml−1 at the 1mg kg−1 dose, and 6 mg min ml−1 at the 10 mg kg−1 dose. However, at an i.v. dose of 30 mg kg−1, the AUC increased disproportionately to 29 mg min ml−1. Urinary excretion data showed that less than 0.15% of the drug was excreted unchanged, indicating that renal clearance of S-4 was negligible. The T½ of S-4 was 154, 182, 223 and 316 min after doses of 0.5, 1, 10 and 30 mg kg−1, respectively. MRT increased from 222 and 240 min at the 0.5 and 1 mg kg−1 doses to 305 and 423 min following the 10 and 30 mg kg−1 doses, respectively. Pharmacokinetic parameters for the i.v. doses are summarized in table 1.
Figure 2.
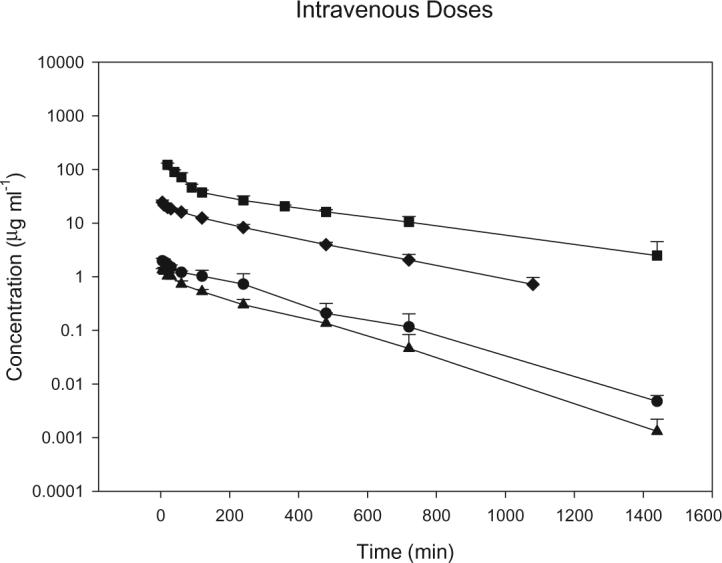
Mean plasma concentration–time profiles following intravenous doses of S-4 in male rats. Square, 30 mg kg−1; diamond, 10 mg kg−1; circle, 1 mg kg−1; triangle, 0.5 mg kg−1. Data are the mean ± SD for n = 5 in each group.
Table 1.
Pharmacokinetics of S-4 in rats after intraverous administration.
| 0.5 mg kg−1 | 1 mg kg−1 | 10 mg kg−1 | 30 mg kg−1 | |
|---|---|---|---|---|
| CL (ml min−1 kg−1) | 1.92 ± 0.20 | 2.12 ± 0.07 | 1.52 ± 0.12 | 1.00 ± 0.08 |
| Vss (1 kg−1) | 0.42 ± 0.06 | 0.48 ± 0.08 | 0.44 ± 0.04 | 0.42 ± 0.14 |
| λz (min−1) | 0.0045 ± 0.00046 | 0.0038 ± 0.00077 | 0.0031 ± 0.00044 | 0.0022 ± 0.0008 |
| MRT (min) | 222 ± 43 | 240 ± 45 | 305 ± 32 | 423 ± 158 |
| AUC (mg min ml−1) | 0.24 ± 0.03 | 0.46 ± 0.16 | 6.0 ± 0.5 | 28 ± 3 |
Data are mean ± SEM, with five animals per dose group.
Pharmacokinetics of S-4 after oral doses
S-4 achieved average maximal plasma concentrations of 1.4, 11 and 20 μg ml−1 following p.o. doses of 1, 10 and 30 mg kg−1, respectively (figure 3). The time to reach the maximal plasma concentration (Tmax) was 48, 84, and 336 min for the 1, 10 and 30 mg kg−1 doses, respectively. S-4 was completely bioavailable for the 1 and 10 mg kg−1 doses. However, following the 30 mg kg−1 dose, the bioavailability of S-4 decreased to 57%. The T1/2 of S-4 was 203, 173 and 266 min after doses of 1, 10 and 30 mg kg−1, respectively. Pharmacokinetic parameters for the p.o. doses are summarized in table 2.
Figure 3.
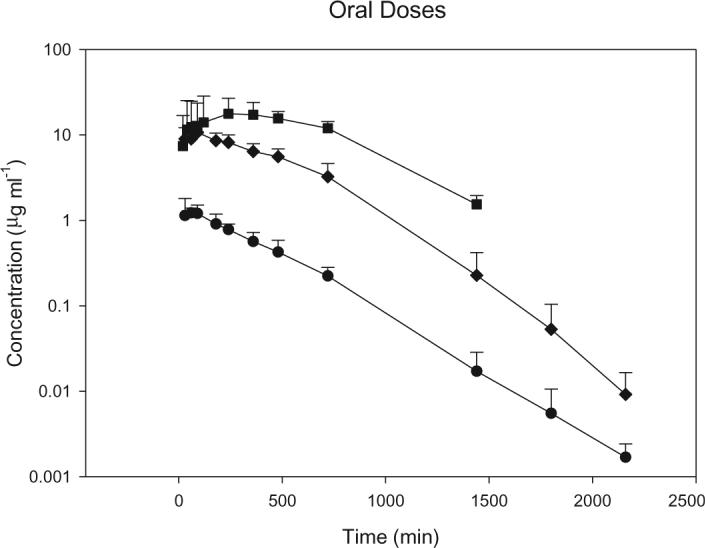
Mean plasma concentration–time profiles following oral doses of S-4 in male rats. Square, 30 mg kg−1; diamond, 10 mg kg−1; circle, 1 mg kg−1. Data are the mean ± SD for n = 5 in each group.
Table 2.
Pharmacokinetics of S-4 in rats after oral administration.
| 1 mg kg−1 | 10 mg kg−1 | 30 mg kg−1 | |
|---|---|---|---|
| λz (min−1) | 0.0034 ± 0.00032 | 0.004 ± 0.0004 | 0.0026 ± 0.00021 |
| Cmax (μg ml−1) | 1.39 ± 0.49 | 11.32 ± 3.7 | 20 ± 11 |
| Tmax (μg ml−1) | 48 ± 16 | 84 ± 61.5 | 336 ± 156 |
| AUC (mg min ml−1) | 0.55 ± 0.08 | 6.0 ± 1.6 | 16 ± 6 |
| F (%) | 120 | 100 | 57 |
Data are mean ± SEM, with five animals per dose group.
Discussion
Analysis of variance showed no significant difference in the CL of S-4 at doses of 0.5, 1 and 10 mg kg−1 ( p>0.05). Previous in vivo studies in the present authors' laboratory showed that the dose required to restore the levator ani muscle weight in castrated animals, an indicator of anabolic activity, compared with that of intact animals was less than 4 mg kg−1 day−1 (Yin et al. 2003). Thus, S-4 demonstrates linear pharmacokinetics within the dose range needed to exert maximal pharmacological effects.
The lack of parent drug in the urine suggests that S-4 is extensively metabolized. Assuming a hepatic blood flow of 13.8 ml min −1 in the rat (Davies and Morris 1993), the hepatic extraction ratio of S-4 would be less than 0.05. Based on this hepatic extraction ratio, a greater than 95% bioavailability (i.e. less than 5% of the drug would be removed by first-pass metabolism) is predicted. The present results confirmed this prediction, as S-4 was completely bioavailable following pharmacologically relevant doses (i.e. doses ≤ 10 mg kg−1).
Although S-4 is structurally similar to bicalutamide, a clinically used anti-androgen, its total body clearance was quite different. In rats, bicalutamide is eliminated with a half-life of 17−21 h (Boyle et al. 1993). S-4 is cleared much more rapidly with an average half-life of about 4 h. The p.o. bioavailability of bicalutamide (61%) following a 25 mg kg−1 p.o. dose (Boyle et al. 1993) is nearly identical to the p.o. bioavailability of S-4 (57%) following the 30 mg kg −1 dose. Like bicalutamide, a very small percentage (0.15%) of S-4 is excreted unchanged in the urine. As a whole, these data suggest that the primary difference in pharmacokinetic parameters for S-4 and bicalutamide is a result of differences in metabolism and not other factors such as drug absorption, distribution or renal excretion.
The observed decrease in p.o. bioavailability and the prolonged time to peak plasma concentration observed after 30 mg kg−1 p.o. doses of S-4 were most likely due to the poor aqueous solubility of the compound (about 80 μg ml−1). Similar behaviour was noted for bicalutamide during preclinical pharmacokinetic studies (Boyle et al. 1993). Decreased p.o. bioavailability at higher doses may serve as a protective mechanism given the potential for abuse of an anabolic drug such as S-4. However, it is important to note that S-4 demonstrated high p.o. bioavailabilty and a short time to peak plasma concentration at doses capable of eliciting a maximal pharmacological effect (Yin et al. 2003).
A similar trend was observed in half-life following i.v. administration. As the dose was increased, λz decreased ( p<0.001) and the half-life of S-4 increased ( p<0.01). This observation also supports the conclusion that the CL of the drug was diminished at the highest doses. The CL of S-4 at a dose of 0.5 mg kg−1 (1.92 ml min−1 kg−1) was significantly ( p<0.001) greater than that observed for the 30 mg kg−1 dose (1.00 ml min−1 kg−1). These data suggest that saturation of the drug-metabolizing enzymes might be occurring at this higher dose. Therefore, one would expect to see further suppression of CL following doses greater than 30 mg kg−1. However, due to the potency of S-4, the authors do not anticipate the need for such high doses during clinical use. Forthcoming data from the present authors' laboratory will provide needed information about the hepatic metabolism and pharmacokinetics of S-4 in this and other species.
The pharmacological activity and pharmacokinetics of S-4 in rats suggest that this compound has the properties of an ideal SARM as defined by Negro-Vilar (1999). It is rapidly absorbed following p.o. doses (tmax, 48−84 min), and it exerts tissue-specific anabolic effects in vivo, with anabolic effects in muscle and bone but lesser effects in the prostate and seminal vesicles (Kearbey et al. 2003, Yin et al. 2003). These properties coupled with forthcoming reports from the present authors' laboratory about the pharmacological effects of S-4 in other pertinent animal models and its pharmacokinetics and metabolism in dogs and humans, favour the continued development of S-4 as an orally bioavailable non-steroidal SARM.
Acknowledgements
Research was supported in part by grants from the NIH (R01 DK59800) and GTx, Inc. (Memphis, TN, USA).
References
- Bhasin S, Bremner WJ. Clinical review 85: Emerging issues in androgen replacement therapy. Journal of Clinical Endocrinology and Metabolism. 1997;82:3–8. doi: 10.1210/jcem.82.1.3640. [DOI] [PubMed] [Google Scholar]
- Boyle GW, McKillop D, Phillips PJ, Harding JR, Pickford R, McCormick AD. Metabolism of casodex in laboratory animals. Xenobiotica. 1993;23:781–798. doi: 10.3109/00498259309166784. [DOI] [PubMed] [Google Scholar]
- Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD. Discovery of nonsteroidal androgens. Biochemical and Biophysical Research Communications. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharmaceutical Research. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- Handelsman DJ, Conway AJ, Boylan LM. Pharmacokinetics and pharmacodynamics of testosterone pellets in man. Journal of Clinical Endocrinology and Metabolism. 1990;71:216–222. doi: 10.1210/jcem-71-1-216. [DOI] [PubMed] [Google Scholar]
- Heywood R, Chesterman H, Ball SA, Wadsworth PF. Toxicity of methyl testosterone in the beagle dog. Toxicology. 1977;7:357–365. doi: 10.1016/0300-483x(77)90053-1. [DOI] [PubMed] [Google Scholar]
- Ishak KG, Zimmerman HJ. Hepatotoxic effects of the anabolic/androgenic steroids. Seminars in Liver Disease. 1987;7:230–236. doi: 10.1055/s-2008-1040579. [DOI] [PubMed] [Google Scholar]
- Kearbey JD, Gao W, Miller DD, Dalton JT. Selective androgen receptor modulators inhibit bone resorption in rats. AAPS Pharm Sci. 2003;5 Abstract R6167. [Google Scholar]
- Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. Design, synthesis, and biological characterization of metabolically stable selective androgen receptor modulators. Journal of Medicinal Chemistry. 2003 doi: 10.1021/jm030336u. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negro-Vilar A. Selective androgen receptor modulators (SARMs): A novel approach to androgen therapy for the new millennium. Journal of Clinical Endocrinology and Metabolism. 1999;84:3459–3462. doi: 10.1210/jcem.84.10.6122. [DOI] [PubMed] [Google Scholar]
- Velazquez E, Bellabarba Arata G. Testosterone replacement therapy. Archives of Andrology. 1998;41(2):79–90. doi: 10.3109/01485019808987949. [DOI] [PubMed] [Google Scholar]
- Wilson JD. Androgens. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Gilman AG, editors. The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 1996. pp. 1441–1458. [Google Scholar]
- Wilson JD, Aiman J, MacDonald PC. The pathogenesis of gynecomastia. Advances in Internal Medicine. 1980;25:1–32. [PubMed] [Google Scholar]
- Wu FC. Testicular steroidogenesis and androgen use and abuse. Baillieres Clinical Endocrinology and Metabolism. 1992;6:373–403. doi: 10.1016/s0950-351x(05)80155-7. [DOI] [PubMed] [Google Scholar]
- Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT. Pharmacodynamics of selective androgen receptor modulators. Journal of Pharmacology and Experimental Therapeutics. 2003;304:1334–1340. doi: 10.1124/jpet.102.040840. [DOI] [PubMed] [Google Scholar]