

Abstract
Microbial pathogens use a variety of mechanisms to disrupt the actin cytoskeleton during infection. Vibrio parahaemolyticus (V. para) is a Gram-negative bacterium that causes gastroenteritis, and new pandemic strains are emerging throughout the world. Analysis of the V. para genome revealed a type III secretion system effector, VopL, encoding three Wiskott–Aldrich homology 2 domains that are interspersed with three proline-rich motifs. Infection of HeLa cells with V. para induces the formation of long actin fibers in a VopL-dependent manner. Transfection of VopL promotes the assembly of actin stress fibers. In vitro, recombinant VopL potently induces assembly of actin filaments that grow at their barbed ends, independent of eukaryotic factors. Vibrio VopL is predicted to be a bacterial virulence factor that disrupts actin homeostasis during an enteric infection of the host.
Keywords: actin assembly, microbial pathogenesis, virulence, stress fibers, WH2 domains
Vibrio parahaemolyticus (V. para) is a Gram-negative bacterium that causes gastroenteritis after consumption of undercooked or raw shellfish. New pandemic strains of this pathogen are emerging throughout the world (1). Sequencing of the V. para genome identified two pathogenicity islands encoding type III secretion systems, TTSS1 and TTSS2, the latter associated with virulent, clinical isolates of V. para (2). Iida and colleagues (3, 4) have demonstrated that the TTSS on chromosome one (TTSS1) is linked to cytotoxicity in infected tissue culture cells, whereas the TTSS on chromosome two (TTSS2) is associated with enterotoxicity in the rabbit ileal loop model.
TTSSs transfer virulence factors (also called effectors) from the bacterial cell to the cytoplasm of the infected cell, resulting in disruption of a variety of eukaryotic signaling pathways (5). Each effector mimics or captures the activity of a eukaryotic protein and alters the signaling machinery in the target cell to give an advantage to the pathogen during infection. The effectors remain quiescent inside the pathogen because of the presence of a chaperone or because the bacterium lacks a specific substrate or activator (6, 7). Ultimately, utilization of TTSS effector proteins to manipulate eukaryotic signaling systems ensures the bacterium's survival.
Two major pathways targeted by these pathogens are the innate immune system and the actin cytoskeleton (6, 8). The innate immune system is a key target for bacterial pathogens because attenuating this system provides an obvious advantage for the pathogen in the initial stages of infection. Bacterial pathogens also use and manipulate the actin cytoskeleton, but for diverse reasons. They can manipulate the actin cytoskeleton to prevent (5, 6) or induce (9) their own phagocytosis. Other pathogens manipulate actin assembly to facilitate their movement into, out of, and within infected host cells (10).
Actin plays a key role in cellular motility and is a major determinant of the shape of a eukaryotic cell (11). Actin is bound to ATP or ADP and is found as a monomer (G-actin) or as a filamentous polymer (F-actin) in a cell. The actin cytoskeleton in a cell is highly dynamic with continuous assembly and disassembly of actin filaments, thus allowing the cell to rapidly change morphology in response to external cues (12). The assembly of actin fibers is a complex process that involves an initial nucleation step requiring three or more actin monomers that then serve as a priming site for further polymerization of an actin filament (12). The actin filament is polarized, with a slow-growing “pointed” end and a fast-growing “barbed” end. A variety of proteins manipulate the actin assembly process, resulting in an acceleration or inhibition of each of the several steps involved in actin polymerization. One of these proteins, profilin, is extremely abundant and associated with the majority of G-actin in a eukaryotic cell (13). Profilin-bound actin cannot spontaneously nucleate, nor can it be added to the pointed end of a filament (13).
A key step in actin dynamics is nucleation of new filaments (12). Eukaryotic cells have specialized machineries to accelerate this process. The Arp2/3 complex nucleates filaments that grow from the side of existing filaments, creating branched networks, whereas formins and SPIRE nucleate unbranched filaments (14–18). A common mechanistic feature of all three systems is the ability to assemble actin or actin-like proteins into an arrangement that can serve as a template for growth of a new filament. The Arp2/3 complex contains two actin-related subunits, which form a pseudoactin trimer with an actin monomer provided by activators in the Wiskott–Aldrich syndrome protein (WASP) family. Formins bind two actin monomers and are thought to position them appropriately for filament growth (14). SPIRE proteins have multiple repeats of Wiskott–Aldrich homology 2 (WH2) domains that bind actin and appear to create a three-actin template for filament extension (17). All three of these systems manipulate the structure and dynamics of the actin cytoskeleton.
Because of the complexity of this system, pathogens are able to target and manipulate the formation and dissolution of the actin cytoskeleton using diverse mechanisms. By influencing these systems, bacteria are able to survive an infection and ensure their transmission to another host. Herein we describe VopL, a type III effector protein used by V. para to manipulate actin assembly. VopL is a bacterial effector that mimics a eukaryotic actin-nucleating protein by containing all of the necessary domains to enable it to potently and directly facilitate the assembly of actin. VopL-mediated actin assembly occurs in a manner that is independent of any other eukaryotic factor.
Results
Vibrio Effector VopL Contains WH2 Domains and Proline-Rich Repeats.
We used a bioinformatic approach to identify effectors on TTSS2 that encode domains similar to those found in other effectors or eukaryotic proteins. The first effector that we characterized, VopA, was identified as a YopJ-like protein and, subsequently, was shown to inhibit MAPK signaling pathways (19). Another gene, VopL (Vibrio outer protein L, VPA1370, 483 aa), was also identified as a candidate TTSS2 effector protein. It encodes three closely spaced WH2 domains that are known to bind actin and, in some cases, have been shown to promote actin nucleation (17, 20) (Fig. 1A). Interspersed with the WH2 domains are three proline-rich motifs (PRMs). PRMs have many potential interacting partners, including WW domains, SH3 domains, and profilin (21) (Fig. 1A). The PRMs in VopL closely resemble those found in the FH1 domains of formins, which are known to bind profilin and profilin–actin complexes (21).
Fig. 1.

VopL, a WH2- and PRM-containing protein, is secreted in a TTSS2-dependent manner from V. para. (A) Schematic diagram and amino acid sequence of VopL indicating the three PRMs (blue) and the three WH2 domains (red). (B) Secretion of VopL by the V. para strains POR-1, POR-2, and POR-3 detected by analysis of secreted proteins that were TCA-precipitated from culture supernatants and analyzed by immunoblotting with a rabbit anti-VopL antibody. HeLa cells were either mock-infected (C and D) or infected with POR-1 (E and F), POR-2 (G and H), or POR-3 (I and J) at a multiplicity of infection of 25 and analyzed by confocal microscopy using rhodamine–phalloidin to stain actin and Hoechst to stain nuclei and bacteria. (Magnification: C–J, ×100.).
VopL Is Secreted in a TTSS2-Dependent Manner from V. para.
To determine whether VopL is a candidate TTSS effector protein, we analyzed TTSS-dependent secretion from V. para. We used a series of strains (POR-1, POR-2, and POR-3) that have been characterized previously by Iida and colleagues (22). POR-1 is a derivative of the pathogenic KP-positive RIMD2210633 strain with deletions in the two thermostable direct hemolysin genes (tdhA and tdhS) (3). The second strain (POR-2) is a derivative of the POR-1 strain that encodes an additional deletion in vcrD1, an essential inner-membrane component of TTSS1 (22). The third strain (POR-3) is also a derivative of POR-1 but encodes a deletion in the vcrD2, an essential inner-membrane component of TTSS2 (22). We predicted that VopL is secreted only by the POR-1 and POR-2 strains because these two strains encode a functional TTSS2. Analysis of proteins secreted from the three strains reveals that POR-1 and POR-2, but not POR-3, secrete VopL (Fig. 1B). These data support the hypothesis that VopL is secreted from V. para in a TTSS2-dependent manner (Fig. 1B).
Induction of Actin Stress Fibers During Infection with V. para Is VopL-Dependent.
Using the POR strains to infect HeLa cells, we observed that both POR-1 (Fig. 1 E and F) and POR-3 (Fig. 1 I and J) cause a dramatic cytotoxic affect by 2 h. The POR-2 strain causes dramatic changes in the actin cytoskeleton, inducing long actin filaments that traverse the cell (Fig. 1 G and H). These structures have similar morphology to actin stress fibers. We hypothesized, based on its domain architecture, that VopL might be the effector that induced the observed changes in the actin cytoskeleton. Therefore, we deleted VopL from the POR-2 strain and infected HeLa cells with the mutant POR-2 strain [supporting information (SI) Fig. 7]. After infection of HeLa cells with the POR-2-ΔVopL strain, we observed a small reduction of stress fibers in infected cells when compared with POR-2-infected cells (SI Fig. 7 E and F and SI Fig. 7 C and D, respectively). We predict that this is due to a combination of confounding effects caused by the presence of additional effectors/virulence factors and generalized cellular toxicity caused by exposure of the cells to endotoxin (23, 24). Interestingly, the POR-2-ΔVopL strain caused the cells to shrink (SI Fig. 7) and more cells to die, relative to the POR-2-infected cells. When we reconstituted POR-2-ΔVopL with pLM1877-VopL and used this strain to infect HeLa cells, the phenotype of the infected cells reverted to that of the POR-2-infected cells, with both increased stress fibers and cell viability (SI Fig. 7 G and H). No change in infection phenotype was observed with the POR-2-ΔVopL strain containing an empty vector (SI Fig. 7 I and J). Based on the molecular Koch's postulate, it appears that VopL is a Vibrio TTSS effector that contributes to changes in cellular morphology, viability, and the cytoskeleton (25).
Transfection of VopL Induces Actin Stress Fibers in HeLa Cells.
Many bacterial pathogens use TTSS virulence factors to manipulate the structure and dynamics of actin filament networks (10). We speculated that VopL may be like the Drosophila protein SPIRE, which similarly contains multiple WH2 domains and induces the formation of actin clusters when transfected into NIH 3T3 cells (17). VopL, like SPIRE, causes an increase in cellular F actin upon transfection of eukaryotic cells (17). However, the actin morphology induced in NIH 3T3 cells by the two factors is quite different. SPIRE creates actin clusters, whereas VopL creates long parallel fibers that span transfected NIH 3T3 cells (SI Fig. 8). This is not cell-type-specific because we also see the induction of long actin fibers in HeLa cells (Fig. 2D). Staining of VopL-transfected cells for actin and vinculin reveals that VopL induces actin fibers, the ends of which colocalize with vinculin (Fig. 2 E and G). Vinculin staining is also evident in vector-transfected cells, but the vinculin puncta are distributed evenly around the periphery of the cells rather than concentrated at opposite ends of the cell as is seen for VopL-transfected cells. In addition, staining with anti-VopL antibody reveals localization of VopL along the length of these actin structures (Fig. 2 F and H). Staining of VopL-transfected HeLa cells with myosin IIA antibody shows that myosin localizes to the actin fibers induced by VopL (Fig. 2 M, N, and P) and that VopL localizes to these same actin structures (Fig. 2 L, N, and O). Myosin colocalizes to actin structures in vector-transfected cells as well (Fig. 2 I–K).
Fig. 2.

Transfection of VopL into HeLa cells induces formation of actin stress fibers. (A–H) HeLa cells were transfected with either empty vector (A–C) or pSFFV-VopL-FLAG (D–H). The cells were then analyzed by confocal microscopy using rhodamine–phalloidin to stain for actin (A, D, and G), mouse anti-vincullin with a FITC-conjugated secondary antibody (B, E, G, and H), and anti-VopL with an Alexa Fluor 680-conjugated secondary antibody (F and H). (I–P) HeLa cells were transfected with either empty vector and GFP as a marker of transfection (I–K) or pSFFV-VopL-FLAG (no GFP). The cells were then analyzed by confocal microscopy using rhodamine–phalloidin to stain for actin (I, L, and O), rabbit polyclonal anti-non-muscle myosin IIA (J, M, and P), and mouse anti-FLAG antibody to stain for VopL-FLAG (N and P). (Magnification: ×100.)
VopL Induces Stress Fibers in the Presence of Dominant Negative Small Rho-Like GTPases.
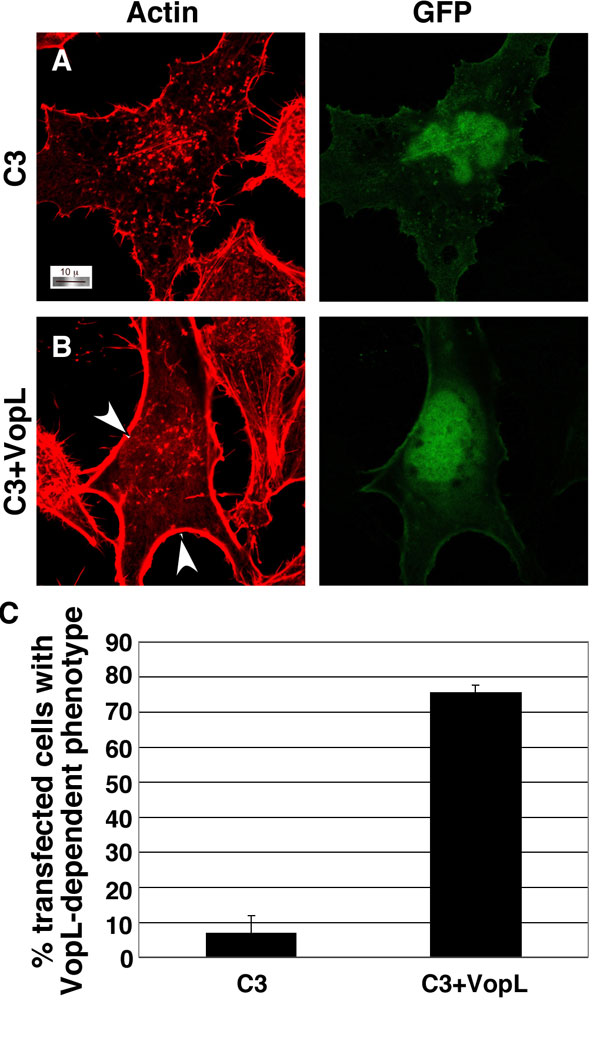
Many bacterial virulence factors target the small Rho family of GTPases including RhoA, Rac, and Cdc42, which are involved in the formation of stress fibers, lamellipodia, and filapodia, respectively (11). Bacterial pathogens manipulate these proteins so that infected cells exhibit either a dominant active phenotype (constitutive induction of stress fiber or membrane ruffling) or a dominant negative phenotype (unable to induce stress fibers or membrane ruffling) (26). In HeLa cells, RhoA activation is associated with the induction of actin stress fibers; therefore, we hypothesized that VopL might use RhoA to induce changes in the cytoskeleton of a transfected cell. We cotransfected VopL with dominant negative RhoA-T19N (RhoDN), which is known to prevent stress fiber formation induced by upstream stimuli (26). In the presence of RhoDN, stress fibers are not observed in HeLa cells (Fig. 3C). Upon coexpression of VopL with RhoDN, transfected cells contain long actin fibers, similar to those observed in cells transfected with VopL alone (Fig. 3 B and D). VopL activity is also observed in cells cotransfected with dominant negative Rac1 or Cdc42 and VopL (SI Fig. 9). We also analyzed the ability of VopL to induce the formation of stress fibers in the presence of Clostridium C3 exoenzyme (C3), which potently inactivates RhoA, RhoB, and RhoC by ADP-ribosylating an asparagine residue located adjacent to the switch I region (27, 28). Cells expressing C3 alone lack stress fibers and have only weak actin staining at the cell periphery. In contrast, coexpression of VopL and C3 produces a profound phenotype wherein actin is assembled around the inner rim of the cell membrane (SI Fig. 10). Interestingly, VopL-induced actin fibers that stretch across the cell are not observed, indicating that localization of RhoA may play a role in this activity. These observations support the hypothesis that VopL acts independent of the Rho family GTPases to induce actin assembly, but the presence of endogenous wild-type RhoA may play a role in VopL-induced stress fibers that extend cross the cell.
Fig. 3.

VopL activity in the presence of dominant negative RhoA. Shown are images from confocal microscopy using rhodamine–phalloidin to stain for actin in HeLa cells cotransfected with peGFP-N1 (GFP) and empty vector (A), pSFFV-VopL-Flag (B), pcDNA3-(HA)3-RhoA T19N (dominant negative) (C), and RhoA T19N with VopL-Flag (D). (Magnification: ×100.) (E) Quantitation of transfected cells with increased actin fibers in a double blind study.
VopL-Induced Actin Assembly Depends on the WH2 Domains.
Because WH2 domains are linked with actin assembly, we assessed whether the WH2 domains of VopL are necessary for the formation of the actin fibers in VopL-transfected cells. We mutated the WH2 domains by substituting four highly conserved amino acids with alanine residues, as indicated by asterisks below mutated residues in Fig. 4B. These amino acids have been identified as being important for actin binding to WH2 domains (17, 20). When all three WH2 domains are mutated, the resulting mutant VopL-WH2*×3 has no effect on the actin cytoskeleton (Fig. 4A and SI Fig. 11G). However, mutation of any single WH2 domain has only a small effect on the actin phenotype as observed in HeLa cells, with the most C-terminal WH2 mutant (WH2–3*) showing the greatest decrease in activity (Fig. 4A and SI Fig. 11 C–E). Mutation of two WH2 domains (VopL-WH2–1*+2*) further decreases actin fiber formation in transfected HeLa cells (Fig. 4A and SI Fig. 11F), demonstrating that one WH2 domain is sufficient, although not as potent as molecules with two or more functional WH2 domains. Interestingly, VopL does not contain a linker-3 sequence like that found in SPIRE, nor does it have similarity with Arp2/3 or its related subunits. Fig. 4B shows conservation in the WH2 domains from VopL compared with WH2 domains from the human WASP and WAVE proteins, Drosophila SPIRE, Chlamydia TARP, and Vibrio cholerae VopF. The WH2 domains in VopL maintain most of the important hydrophobic residues in the N-terminal α-helical region of WH2, and all have the downstream LKKV motif, both of which are important for actin binding (Fig. 4B) (29).
Fig. 4.

Mutation of the WH2 domains affects VopL activity in HeLa cells. (A) Quantitation of transfected cells with increased actin fibers in a double blind study. (B) An alignment of the WH2 domains (with National Center for Biotechnology Information accession numbers) from human WASP (AF115549), N-WASP (D88460), WAVE1 (D87459), WAVE2 (AB026542), WAVE3 (AB020707), Drosophila melanogaster Spire (AF184975), Chlamydia TARP (YP_328278), V. cholerae VopF (AAZ32252), and V. para VopL (NP_800881). A consensus sequence is shown below the alignment, and the residues in VopL that were mutated to alanines in the WH2 domain mutants are indicated by asterisks below the consensus.
VopL Nucleates New Actin Filaments That Grow from Their Barbed Ends.
To test whether VopL is able to directly induce the assembly of actin monomers into filaments, we expressed and purified recombinant wild-type VopL and the VopL-WH2×3* mutant and analyzed their activity in an in vitro actin assembly assay (Fig. 5A) (30). When increasing amounts of purified rVopL (0.1–5 nM) are added to 4 μM 5% pyrene-labeled actin, the rate of actin assembly, indicated by an increase in pyrene fluorescence, is correspondingly and greatly accelerated over the spontaneous rate (Fig. 5A) (30). The potency of VopL in this assay is appreciably higher than Arp2/3 complex maximally activated by the VCA domain of WASP (Fig. 5 A and B). Identical rates of actin assembly are observed with a truncated, more soluble form of VopL, VopL-WH2C (90–483 aa), which is missing its putative secretion/translocation signal sequence (data not shown).
Fig. 5.

rVopL enhances actin filament assembly in vitro. (A) A rVopL (0.1–5 nM) or rVopL-WH2×3* mutant was incubated with 4 μM actin (5% pyrene-labeled), and changes in fluorescence were measured over time. (B) Comparison of rVopL (5 nM) and Arp2/3 (5 nM) maximally activated with WASP VCA (500 nM). The proteins were incubated with 4 μM actin (5% pyrene-labeled), and changes in fluorescence were measured over time. “Control” refers to the spontaneous assembly of 4 μM actin (5% pyrene-labeled). (C) VopL-nucleated filaments grow at their barbed ends. Shown are pyrene actin polymerization assays in the presence of actin (0.5 μM 20% pyrene), 5 nM VopL-WH2C (WH2C), and mouse capping protein α1β2 (CP) from 0.2 nM to 10 nM. At 10 nM, CP completely inhibits barbed-end filament growth. (D) VopL has no effect on filament elongation. Shown is elongation of 1 μM phalloidin-stabilized actin filaments with 0.5 μM actin monomer (40% pyrene) in the presence and absence of 3 nM VopL-WH2C (WH2C). (E) VopL binds filament sides. Shown are filament binding assays with GST-VopL-WH2C (GST-WH2C) and 5 μM phalloidin-stabilized actin filaments. In this assay, GST-VopL-WH2C is used so that it can be distinguished from actin by SDS/PAGE. Lane 1, 1 μM GST-VopL-WH2C alone; lanes 2–6, 5 μM actin filaments and 1, 2.5, 5, 7.5, and 10 μM GST-VopL-WH2C, respectively; lane 7, 5 μM actin filaments and 10 μM GST; lane 8, 5 μM actin filaments and 10 μM CP. (F) Coomassie-stained SDS/PAGE gel of rVopL (wt, 1 μg) and rVopL-WH2×3* (mt, 1 μg).
The effect of VopL on filament assembly could arise from increased filament nucleation or an increased elongation rate of existing filaments. However, at concentrations up to 3 nM, VopL-WH2C has no effect on the initial rate of barbed-end elongation of actin filament seeds (Fig. 5D). Thus, VopL most likely accelerates assembly through increasing the efficiency of filament nucleation de novo. The VopL-WH2C-nucleated filaments grow at their barbed ends, because assembly of 0.5 μM actin is substantially blocked by 5 nM capping protein (Fig. 5C). Inhibition is identical whether capping protein is added to the assay before or after assembly is induced by VopL-WH2C (data not shown). At higher actin concentrations, where filaments can also grow at their pointed ends, capping protein also substantially inhibits VopL-WH2C-induced assembly (SI Fig. 12). However, inhibition is not complete under these conditions because of the filament-nucleating activity of capping protein at concentrations >5 nM (31). Because of this complication, we do not yet know whether VopL-nucleated filaments can grow from their pointed ends, or whether the pointed ends are blocked as in Arp2/3-nucleated filaments (16). As a positive control in these assays, we used purified Arp2/3 complex maximally activated by the VCA domain of WASP (Fig. 5B) (32, 33). For similar maximal rates, VopL assembly shows a shorter lag phase relative to Arp2/3 complex maximally activated by the VCA domain of WASP, suggesting a different dependence on actin filaments of the two nucleation systems (Fig. 5 A and B) (32, 33). The VopL-WH2×3* mutant does not promote assembly of actin in vitro (Fig. 5A) consistent with the idea that the WH2 domains are required for effects of VopL on the cytoskeleton in vivo (Fig. 4 and SI Fig. 11). Finally, we found that VopL-WH2C binds to actin filaments sedimented by centrifugation (Fig. 5E). However, compared with capping protein, which binds only the filament barbed end, the stoichiometry of VopL-WH2C binding is much higher, suggesting that VopL can bind filament sides (Fig. 5E). We do not yet know the relationship, if any, of this filament side binding to VopL-mediated nucleation.
Discussion
We have identified and characterized VopL, a V. para TTSS effector protein. The VopL gene is encoded within a pathogenicity island that correlates with both the virulent strains of V. para and the emerging pandemic strains of V. para. We observe VopL-dependent formation of actin stress fibers in infected and transfected HeLa cells. In vitro, the assembly of actin by VopL is direct and extremely potent. Interestingly, strains of another pathogenic bacterium, V. cholerae, possess a TTSS that encodes a protein with significant similarity to VopL (34). Tam et al. (35) recently demonstrated that this effector from V. cholerae is able to induce actin polymerization in vivo and in vitro but that the phenotype in cells is distinct from that observed with VopL.
Vibrio appears to have usurped eukaryotic WH2 and PRM domains to create VopL, a TTSS effector that can nucleate actin filaments in vitro and generate actin stress fibers in vivo. The phenotype of cells transfected with VopL supports the hypothesis that VopL induces the formation of unbranched actin structures rather than the branched networks induced by Arp2/3 complex. We observe a similar rate of actin assembly when using 0.5 nM VopL and 5 nM maximally activated Arp2/3 complex, indicating that VopL is a more potent activator than the Arp2/3 complex (33). VopL is also more potent than SPIRE, which requires a >500 nM concentration for efficient actin assembly (17). VopL, Arp2/3 complex, and SPIRE all require WH2 domains for activity. In SPIRE, a minimum of one WH2 domain plus a small peptide sequence called “linker 3” is needed in vitro, but a nucleation mechanism has been proposed in which three of SPIRE's WH2 domains organize actin monomers along a long pitch actin strand (17). In Arp2/3 complex, only a single WH2 domain, provided by activating VCA peptides, is needed for nucleation (36). It is believed that during nucleation this WH2 serves to recruit the first actin monomer to the pseudoactin dimer created by the actin-related subunits of Arp2/3 complex (18). Like SPIRE, VopL contains multiple WH2 domains, and in principle these domains could act in SPIRE-like fashion to assemble an actin trimer representing one strand of an actin filament (17). However, our in vivo data indicate that only one WH2 domain of VopL is necessary to induce actin stress fiber formation, albeit with reduced efficiency relative to the wild-type protein. Additional studies are needed to elucidate the molecular mechanism of VopL-mediated actin assembly.
VopL contains all of the predicted properties of a TTSS effector, including mimicking or capturing a eukaryotic activity, higher potency than its functional eukaryotic counterparts, and interacting partners that are found only in eukaryotes (37). First, VopL utilizes multiple eukaryotic WH2 domains to manipulate the actin cytoskeleton. Second, VopL encodes an activity that is orders of magnitude more efficient than its predicted eukaryotic counterparts, thereby enabling the pathogen to tip the balance in a host/microbe interaction in favor of the invading pathogen (6, 38). Third, VopL uses a substrate (actin) that is found only in the eukaryotic host, which supports the hypothesis that this extremely active protein is quiescent in the pathogen because of lack of substrate (5, 6).
Bacterial pathogens use a number of mechanisms to manipulate the actin cytoskeleton, including GTPase exchange factors and GTPase-activating proteins, proteases, and kinases (Fig. 6). Still other bacterial pathogens use virulence factors, such as ActA from Listeria, which encodes multiple PRMs to manipulate the eukaryotic nucleating factor Arp2/3 (39, 40). The TARP protein from Chlamydia contains only one WH2 domain and is less potent than VopL in in vitro actin assembly assays (41). In addition, a family of bacterial G protein mimics have been identified that replicate signaling properties of Rho-like GTPase in manipulating the actin cytoskeleton (42).
Fig. 6.

Bacterial virulence factors that manipulate eukaryotic machinery involved in assembly of actin filaments. The formation of actin filaments can be stimulated by activation of Rho-like GTPases that can directly or indirectly activate nucleation factors, which control the rate-limiting step. Bacterial effectors (red) manipulate the state of GTPases by mimicking GTPase-activating proteins (Salmonella SptP and Yersinia YopE), GTPase exchange factors (Salmonella SopE), or even GTPases themselves (WxxxE). They can sever the activity of G proteins using proteolytic (Yersinia YopT) or kinase activity (Yersinia YpkA). Effectors (Listeria ActA, Rickettsia RickA, and Shigella IscA) can also hijack the Arp2/3 nucleation factor complex. Other effectors (Salmonella SipC and Chlamydia TARP) appear to induce actin assembly, albeit inefficiently, and others induce the formation of actin bundles (Salmonella SipA and SipC). VopL is a bacterial effector that appears to have usurped all of the domains necessary to be an extremely efficient nucleation factor for actin and profilin-bound actin.
Herein we present studies on VopL, a nucleating factor functioning independent of other eukaryotic proteins that promotes actin assembly in a manner even more efficient than its eukaryotic counterparts. We propose that V. para uses VopL to disrupt actin homeostasis in the epithelial cells of the gut during infection, thereby initiating an enterotoxic effect in the intestine. Support for this hypothesis comes from observations by Nelson and colleagues (43) that demonstrate that even slight changes in the actin cytoskeleton by Rho-like GTPases in a polarized epithelium can disrupt the integrity of the monolayers.
Identifying and characterizing bacterial effector proteins is important, not only for our understanding of new and emerging pandemic bacterial strains, but also for identifying and characterizing key components and mechanisms of eukaryotic signaling systems. Future microbial, biochemical, and biophysical studies are aimed at elucidating the mechanism of actin assembly that VopL has so efficiently usurped.
Materials and Methods
Details.
Methods for the following are in SI Text: bioinformatics, Vibrio secretion, infections, gene disruption, confocal microscopy, protein purification, quantitation of transfections, pyrene actin assembly assays, and barbed-end elongation assays.
Vectors and Strains.
pSFFV-VopL-Flag, pSFFV-VopL-WH2C (90–483 aa), pGEX-rTEV-VopL, pGEX-rTEV VopL-WH2C, and WH2 mutants (SI Figs. 11 C–G and 12) were made by using standard molecular biology techniques (44, 45). pCMV5-C3 was a kind gift from P. C. Sternweis (University of Texas Southwestern Medical Center). V. para strains POR-1, POR-2, and POR-3 were obtained from T. Iida and T. Honda (Osaka University, Osaka, Japan) (3, 22). Expression plasmids containing dominant negative [pcDNA3-(HA)3-RhoA T19N] RhoA, Rac1 T17N, and CDC42 T17N were obtained from the Guthrie cDNA Resource Center. pEGFP-N1 was obtained from BD Biosciences Clontech. pLM1877 was obtained from L. McCarter (University of Iowa, Ames, IA) (46). See SI Text for specifics.
Cell Culture and Transfections.
HeLa and NIH 3T3 cells were obtained from American Type Culture Collection (Manassas, VA), passaged under standard conditions, and transfected with FuGENE (Roche Diagnostics). See SI Text for specifics.
Pyrene Actin Assembly Assays.
Actin polymerization assays were performed as described previously (36). See SI Text for specifics.
Actin Filament Binding Assays.
Filament binding assays were conducted as described previously (47, 48). Briefly, phalloidin-stabilized actin filaments were incubated with various concentrations of GST-VopL-WH2C, GST, or capping protein in KMEI buffer for 30 min before centrifugation at 200,000 × g for 20 min. Supernatants and pellets were analyzed by SDS/PAGE and Coomassie staining.
Supplementary Material
Acknowledgments
We thank Drs. T. Iida and T. Honda for their generosity in supplying Vibrio strains. For their kind support and assistance, we thank Dr. Michelle Laskowski-Arce and other members of the K.O. laboratory, Drs. Linda McCarter and Igor Rybkin. K.O., D.L.B., and J.E.T. are supported by National Institute of Allergy and Infectious Diseases Grants R01-AI056404 and R21-DK072134 and Welch Research Foundation Grant I-1561. A.D.B.L. and M.L.Y. are supported by National Institutes of Health Training Grant T32-GM08203-18. H.-C.C. and D.W.L. are supported by National Institutes of Health Grants R01-GM056322 and P01-GM066311 and Welch Research Foundation Grant I-1544. M.K.R. is a Howard Hughes Medical Institute Investigator. K.O. is a Burroughs Wellcome Investigator in Pathogenesis of Infectious Disease and a C. C. Caruth Biomedical Scholar.
Abbreviations
- PRM
proline-rich motif
- WH2
Wiskott–Aldrich homology 2
- V. para
Vibrio parahaemolyticus
- TTSS
type III secretion system
- WASP
Wiskott–Aldrich syndrome protein.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0703196104/DC1.
References
- 1.Morris JG., Jr Clin Infect Dis. 2003;37:272–280. doi: 10.1086/375600. [DOI] [PubMed] [Google Scholar]
- 2.Makino S, Tobe T, Asakura H, Watarai M, Ikeda T, Takeshi K, Sasakawa C. J Clin Microbiol. 2003;41:2341–2347. doi: 10.1128/JCM.41.6.2341-2347.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Park KS, Ono T, Rokuda M, Jang MH, Iida T, Honda T. Microbiol Immunol. 2004;48:313–318. doi: 10.1111/j.1348-0421.2004.tb03512.x. [DOI] [PubMed] [Google Scholar]
- 4.Makino K, Oshima K, Kurokawa K, Yokoyama K, Uda T, Tagomori K, Iijima Y, Najima M, Nakano M, Yamashita A, et al. Lancet. 2003;361:743–749. doi: 10.1016/S0140-6736(03)12659-1. [DOI] [PubMed] [Google Scholar]
- 5.Ghosh P. Microbiol Mol Biol Rev. 2004;68:771–795. doi: 10.1128/MMBR.68.4.771-795.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orth K. Curr Opin Microbiol. 2002;5:38–43. doi: 10.1016/s1369-5274(02)00283-7. [DOI] [PubMed] [Google Scholar]
- 7.Feldman MF, Cornelis GR. FEMS Microbiol Lett. 2003;219:151–158. doi: 10.1016/S0378-1097(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 8.Cornelis GR. Nat Rev Mol Cell Biol. 2002;3:742–752. doi: 10.1038/nrm932. [DOI] [PubMed] [Google Scholar]
- 9.Patel JC, Galan JE. Curr Opin Microbiol. 2005;8:10–15. doi: 10.1016/j.mib.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Aktories K, Barbieri JT. Nat Rev. 2005;3:397–410. doi: 10.1038/nrmicro1150. [DOI] [PubMed] [Google Scholar]
- 11.Raftopoulou M, Hall A. Dev Biol. 2004;265:23–32. doi: 10.1016/j.ydbio.2003.06.003. [DOI] [PubMed] [Google Scholar]
- 12.Pollard TD, Borisy GG. Cell. 2003;112:453–465. doi: 10.1016/s0092-8674(03)00120-x. [DOI] [PubMed] [Google Scholar]
- 13.Higgs HN. Trends Biochem Sci. 2005;30:342–353. doi: 10.1016/j.tibs.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 14.Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK. Nature. 2005;433:488–494. doi: 10.1038/nature03251. [DOI] [PubMed] [Google Scholar]
- 15.Robinson RC, Turbedsky K, Kaiser DA, Marchand JB, Higgs HN, Choe S, Pollard TD. Science. 2001;294:1679–1684. doi: 10.1126/science.1066333. [DOI] [PubMed] [Google Scholar]
- 16.Mullins RD, Heuser JA, Pollard TD. Proc Natl Acad Sci USA. 1998;95:6181–6186. doi: 10.1073/pnas.95.11.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinlan ME, Heuser JE, Kerkhoff E, Mullins RD. Nature. 2005;433:382–388. doi: 10.1038/nature03241. [DOI] [PubMed] [Google Scholar]
- 18.Dayel MJ, Mullins RD. PLoS Biol. 2004;2:E91. doi: 10.1371/journal.pbio.0020091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trosky JE, Mukherjee S, Burdette DL, Roberts M, McCarter L, Siegel RM, Orth K. J Biol Chem. 2004;279:51953–51957. doi: 10.1074/jbc.M407001200. [DOI] [PubMed] [Google Scholar]
- 20.Hertzog M, van Heijenoort C, Didry D, Gaudier M, Coutant J, Gigant B, Didelot G, Preat T, Knossow M, Guittet E, et al. Cell. 2004;117:611–623. doi: 10.1016/s0092-8674(04)00403-9. [DOI] [PubMed] [Google Scholar]
- 21.Holt MR, Koffer A. Trends Cell Biol. 2001;11:38–46. doi: 10.1016/s0962-8924(00)01876-6. [DOI] [PubMed] [Google Scholar]
- 22.Park KS, Ono T, Rokuda M, Jang MH, Okada K, Iida T, Honda T. Infect Immun. 2004;72:6659–6665. doi: 10.1128/IAI.72.11.6659-6665.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips JR, Nadim HS, Layman DL. J Periodontal Res. 1990;25:339–346. doi: 10.1111/j.1600-0765.1990.tb00925.x. [DOI] [PubMed] [Google Scholar]
- 24.Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. J Trauma. 2003;54:104–113. doi: 10.1097/00005373-200301000-00013. [DOI] [PubMed] [Google Scholar]
- 25.Falkow S. Nat Rev. 2004;2:67–72. doi: 10.1038/nrmicro799. [DOI] [PubMed] [Google Scholar]
- 26.Hall A. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 27.Sekine A, Fujiwara M, Narumiya S. J Biol Chem. 1989;264:8602–8605. [PubMed] [Google Scholar]
- 28.Vogelsgesang M, Pautsch A, Aktories K. Naunyn-Schmiedeberg's Arch Pharmacol. 2007;374:347–360. doi: 10.1007/s00210-006-0113-y. [DOI] [PubMed] [Google Scholar]
- 29.Chereau D, Kerff F, Graceffa P, Grabarek Z, Langsetmo K, Dominguez R. Proc Natl Acad Sci USA. 2005;102:16644–16649. doi: 10.1073/pnas.0507021102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pollard TD, Cooper JA. Biochemistry. 1984;23:6631–6641. doi: 10.1021/bi00321a054. [DOI] [PubMed] [Google Scholar]
- 31.Caldwell JE, Heiss SG, Mermall V, Cooper JA. Biochemistry. 1989;28:8506–8514. doi: 10.1021/bi00447a036. [DOI] [PubMed] [Google Scholar]
- 32.Higgs HN, Pollard TD. J Biol Chem. 1999;274:32531–32534. doi: 10.1074/jbc.274.46.32531. [DOI] [PubMed] [Google Scholar]
- 33.Leung DW, Rosen MK. Proc Natl Acad Sci USA. 2005;102:5685–5690. doi: 10.1073/pnas.0406472102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dziejman M, Serruto D, Tam VC, Sturtevant D, Diraphat P, Faruque SM, Rahman MH, Heidelberg JF, Decker J, Li L, et al. Proc Natl Acad Sci USA. 2005;102:3465–3470. doi: 10.1073/pnas.0409918102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tam VC, Serruto D, Dziejman M, Brieher W, Mekalanos JJ. Cell Host Microbe. 2007;1:95–107. doi: 10.1016/j.chom.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Higgs HN, Blanchoin L, Pollard TD. Biochemistry. 1999;38:15212–15222. doi: 10.1021/bi991843+. [DOI] [PubMed] [Google Scholar]
- 37.Orth K. Microbe. 2007;2:183–186. [Google Scholar]
- 38.Zhang ZY, Clemens JC, Schubert HL, Stuckey JA, Fischer MW, Hume DM, Saper MA, Dixon JE. J Biol Chem. 1992;267:23759–23766. [PubMed] [Google Scholar]
- 39.Geese M, Schluter K, Rothkegel M, Jockusch BM, Wehland J, Sechi AS. J Cell Sci. 2000;113:1415–1426. doi: 10.1242/jcs.113.8.1415. [DOI] [PubMed] [Google Scholar]
- 40.Smith GA, Portnoy DA. Trends Microbiol. 1997;5:272–276. doi: 10.1016/S0966-842X(97)01048-2. [DOI] [PubMed] [Google Scholar]
- 41.Jewett TJ, Fischer ER, Mead DJ, Hackstadt T. Proc Natl Acad Sci USA. 2006;103:15599–15604. doi: 10.1073/pnas.0603044103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alto NM, Shao F, Lazar CS, Brost RL, Chua G, Mattoo S, McMahon SA, Ghosh P, Hughes TR, Boone C, et al. Cell. 2006;124:133–145. doi: 10.1016/j.cell.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 43.Jou TS, Schneeberger EE, Nelson WJ. J Cell Biol. 1998;142:101–115. doi: 10.1083/jcb.142.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Palmer LE, Pancetti AR, Greenberg S, Bliska JB. Infect Immun. 1999;67:708–716. doi: 10.1128/iai.67.2.708-716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chook YM, Blobel G. Nature. 1999;399:230–237. doi: 10.1038/20375. [DOI] [PubMed] [Google Scholar]
- 46.Boles BR, McCarter LL. J Bacteriol. 2000;182:1035–1045. doi: 10.1128/jb.182.4.1035-1045.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato M, Takenawa T. Biochem Biophys Res Commun. 2005;328:1058–1066. doi: 10.1016/j.bbrc.2005.01.058. [DOI] [PubMed] [Google Scholar]
- 48.Harris ES, Rouiller I, Hanein D, Higgs HN. J Biol Chem. 2006;281:14383–14392. doi: 10.1074/jbc.M510923200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}