

Abstract
Myf5, a member of the myogenic regulatory factor family, plays a major role in determining myogenic cell fate at the onset of skeletal muscle formation in the embryo. Spatiotemporal control of its expression during development requires multiple enhancer elements spread over >100 kb at the Myf5 locus. Transcription in embryonic limbs is regulated by a 145-bp element located at −57.5 kb from the Myf5 gene. In the present study we show that Myf5 expression is severely impaired in the limb buds of Six1−/− and Six1−/−Six4−/+ mouse mutants despite the presence of myogenic progenitor cells. The 145-bp regulatory element contains a sequence that binds Six1 and Six4 in electromobility shift assays in vitro and in chromatin immunoprecipitation assays with embryonic extracts. We further show that Six1 is able to transactivate a reporter gene under the control of this sequence. In vivo functionality of the Six binding site is demonstrated by transgenic analysis. Mutation of this site impairs reporter gene expression in the limbs and in mature somites where the 145-bp regulatory element is also active. Six1/4 therefore regulate Myf5 transcription, together with Pax3, which was previously shown to be required for the activity of the 145-bp element. Six homeoproteins, which also directly regulate the myogenic differentiation gene Myogenin and lie genetically upstream of Pax3, thus control hypaxial myogenesis at multiple levels.
Keywords: embryonic mouse limb muscle, Pax3, Six1
Skeletal muscles arise from myogenic progenitor cells present in the somites of the embryo. At the interlimb level, cells delaminate from the edges or lips of the epithelial dermomyotome, the dorsal part of the somite (1), to form the subjacent postmitotic myotome, which gives rise to trunk muscles. At the limb level, cells from the hypaxial (ventrolateral) lips of the dermomyotome delaminate and migrate into the limb bud to form the musculature of the limbs. This process depends on Pax3, a transcription factor present in the dermomyotome, which marks myogenic progenitor cells. Entry of progenitor cells into the myogenic program depends on the myogenic determination factors Myf5, MyoD, and MRF4, of which Myf5 and MyoD are expressed in the limbs, whereas differentiation of these cells into postmitotic muscle fibers in the limb bud is under the control of Myogenin, the fourth member of this family (see ref. 2). Myf5 is the first of the myogenic regulatory factors to be expressed in the mouse embryo, starting at embryonic day 8 (E8) in the epaxial (dorsomedial) lip of the dermomyotome, followed by its expression in the hypaxial dermomyotome and all subsequent skeletal muscles (3). At the limb bud level, it has been shown that myogenic progenitor cells do not activate Myf5 until they reach the limb (4). During limb bud development Myf5 is expressed before MyoD (5, 6), and in Myf5−/− embryos MyoD expression is delayed (7).
Mice and humans have six Six genes (Sine oculis homeobox genes, Six1 to Six6) in their genome. These homeogenes are expressed in several cell types during embryonic and adult life (see ref. 8) and are involved in different types of organogenesis (9–13). In the mouse, Six1, Six4, and Six5 genes are expressed from E8 in overlapping expression patterns in somites, limb buds, dorsal root ganglia, and branchial arches (14). Four Eya genes have been cloned in mice, humans, and chicks (15). Physical interactions between Six and Eya proteins, first described in Drosophila, are conserved in vertebrates and allow transcriptional synergy of the Six–Eya complex (16–19). Six1 is expressed throughout muscle development, from E8 in the mouse embryo to adult skeletal muscle (19). Six1−/− fetuses die at birth and display severe but selective muscle hypoplasia in the diaphragm, forelimb, distal ventral hindlimb, and abdomen (20). MyoD and Myogenin expression is delayed in forelimbs and hindlimbs of Six1−/− embryos, whereas their expression pattern in the trunk is reduced ventrally (20). Myogenin expression is directly controlled by Six proteins through a MEF3 site present in its promoter (21). Six1−/−Six4−/− embryos show an aggravation of the Six1−/− muscular phenotype. Notably, these double mutant embryos no longer have myogenic progenitor cells in their limb buds, resulting in muscle-less legs (22). We have shown that in both Six1−/−Six4−/− and Eya1−/−Eya2−/− double mutants, Pax3 expression in the hypaxial dermomyotome is lost, leading to cell misrouting and cell death, preventing muscle progenitor cell migration into the limbs (18, 22). We have thus established that these Six and Eya genes lie upstream of Pax3 in the genetic hierarchy of hypaxial myogenesis. In the trunk, Six1 and Six4 genes have been shown to control the expression of Mrf4, and Six1−/−Six4−/− embryos also have a reduced and delayed expression of MyoD, Myogenin, and myotomal markers, whereas the early activation of Myf5 in the epaxial somite still takes place (22).
The spatiotemporal expression pattern of the Myf5 gene is driven by multiple DNA elements dispersed throughout its locus. The onset of Myf5 expression in the limb bud depends on a DNA sequence located between −58 and −48 kb from the gene (23, 24). Within this sequence, a shorter region is both necessary and sufficient to drive the expression of a lacZ transgene in the limbs (23, 25). A 145-bp element within this region has been shown to drive Myf5 expression in the limb buds, and this sequence is directly under the control of the Pax3 homeodomain and paired domain protein (26). We show here that Six homeoproteins are also involved in the expression of Myf5 in limb buds through direct binding to a conserved MEF3 binding site present in the 145-bp regulatory sequence of Myf5, adjacent to the Pax3 binding site. Mutation of this MEF3 site abrogates the activity of this regulatory sequence without interfering with Pax3 binding. These results show that Six1/4 and Pax3 are involved in Myf5 transcription in the limb and further highlight the multiple steps controlled by Six proteins during muscle development.
Results
Myf5 Expression Is Altered in the Limbs of Six1 Mutant Embryos.
To address the requirement for Six1 in the control of Myf5 expression in the limb buds, we analyzed its expression in relation to Pax3, which marks myogenic progenitor cells. Although substantially reduced, Pax3-positive cells are still detectable within the forelimb bud and entering the hindlimb bud from the hypaxial dermomyotome of adjacent somite at E10.5 (Fig. 1A a–f). At this stage, Myf5 transcripts are not detectable in Six1−/− embryos (data not shown), whereas in controls they are already seen in the forelimb (4). Analysis of Myf5 expression by in situ hybridization at E11.5 [supporting information (SI) Fig. 6I], when Pax3-positive progenitor cells present in the forelimb and hindlimb buds have engaged the myogenic program in WT embryos (Fig. 1A g–j), shows that transcripts are undetectable in the few Pax3-positive cells of the forelimb and are reduced in the Pax3-positive cells of the hindlimb in Six1−/− embryos (Fig. 1A k–n). In Six1−/− Six4−/+ embryos, no Pax3-positive cells are detectable in the forelimb; however, at this stage a few Pax3-positive cells can be seen in the hindlimb, with barely detectable Myf5 hybridization (Fig. 1A o–r). To quantify the proportion of Myf5+/Pax3+ cells at E11.5, we performed serial vibratome sections through the limb buds of embryos in which in toto in situ hybridization had been performed with Pax3 and Myf5 probes (SI Fig. 6II). We evaluated the extent of the Myf5+ region compared with the Pax3+ region. From 115% in the hindlimbs of WT embryos, this ratio drops to 35% in Six1−/− hindlimbs and to 25% in Six1−/−Six4−/+ hindlimbs, suggesting that Six4 can partially compensate for the absence of Six1 in controlling Myf5 transcription in the limb. We also performed immunocytochemistry for Pax3 and Myf5 on sections of forelimb and hindlimb buds at E11.5. In Six1+/− embryos, most Pax3-positive cells are also Myf5-positive in the hindlimb (Fig. 1B a–c), whereas in the absence of Six1 most Pax3-positive cells do not express Myf5 (Fig. 1B d–f), and in the Six1−/− Six4+/− embryos Myf5-positive cells are absent (Fig. 1B h–j). These results are quantified in Fig. 1Bk for the hindlimb; by this approach, Myf5 was also detectable in a few Pax3-positive cells of Six1−/− forelimb (data not shown). At E12.5, Myf5 and Myod transcripts are present in the posterior region of Six1−/− forelimb but not in the forelimbs of Six1−/− Six4−/+ embryos, where no Pax3-positive cells can be detected (data not shown). Expression of Myf5 is severely reduced in the hindlimbs of Six1−/− embryos at E12.5, as compared with MyoD, which is still expressed in the dorsal part of the limb (Fig. 1C g and h and SI Fig. 6III).
Fig. 1.

Whole-mount in situ hybridization with Myf5, MyoD, and Pax3 probes and immunocytochemistry data performed with Myf5 and Pax3 antibodies. (A) Whole-mount in situ hybridization with Myf5 and Pax3 probes at E10.5 (a–f) and E11.5 (g–r) on WT (a, b, e, and g–j), Six1−/− (c, d, f, and k–n), and Six1−/−Six4−/+ (o–r) embryos. FL, forelimb; HL, hindlimb. Arrowheads in c and d show the reduced number of myogenic progenitors expressing Pax3 migrating into the limbs of Six1−/− embryos at E10.5. The arrowhead in l shows the few posterior cells with Pax3 transcripts observed in the Six1−/− forelimb, and the arrowhead in q shows the few cells with Myf5 transcripts present in the Six1−/−Six4−/+ hindlimb. (B) Coimmunocytochemistry on transverse sections at the level of the hindlimbs of Six1+/− (a–c), Six1−/− (d–f), and Six1−/−Six4−/+ (h–j) embryos at E11.5 using antibodies against Pax3 (green in a, c, d, f, h, and j) and Myf5 (red in b, c, e, f, i, and j). Nuclei are shown by DAPI staining (c, f, and j). Histograms indicate the number of Myf5+ cells compared with the number of Pax3+ cells in hindlimbs from Six1−/+, Six1−/−, or Six1−/−Six4−/+ embryos at E11.5 (k). (C) WT (a–d) and Six1−/− (e–h) embryos at E12.5 were hybridized with probes for Myf5 (a, c, e, and g) or MyoD (b, d, f, and h). Enlargements at the forelimb and hindlimb levels are shown. The arrowhead in c shows Myf5 expression in the dorsal region of WT hindlimb. The arrowheads in e and f show restriction of MyoD and Myf5 expression to the posterior domain of the forelimb. The arrowheads in g point to the reduced expression of Myf5 in the dorsal part of the hindlimb, compared with the expression of MyoD in this structure indicated by the arrowhead in h.
A Conserved MEF3 Binding Site Lies Within the 145-bp Sequence at −57.5 kb from the Myf5 Gene.
Myf5 expression in the embryonic limb has been shown to depend on a 145-bp element located at −57.5 kb from the Myf5 transcription start site (26). Examination of the 145-bp sequence revealed the presence of a GTAACTGGAGA motif, matching the MEF3 site consensus sequence, GWAANYNGANA, recognized by Six proteins (21, 27). Multiple sequence alignments of this site from several species show that the MEF3 motif at this position is 100% conserved (Fig. 2A), suggesting that it is functionally important. To test the binding of the Six1 protein to this enhancer, we performed gel electromobility shift assays using a 30-bp oligonucleotide containing this putative MEF3 site as a probe and in vitro translated Six1 or Six4 protein. Both Six1 and Six4 bind to the sequence (Fig. 2B and SI Fig. 7A). Antibodies raised against Six1 disrupt the formation of the Six1 DNA–protein complex (Fig. 2B). DNA competition experiments with the MEF3 site from the Myf5 regulatory region or from the previously characterized MEF3 site from the promoter of the Myogenin gene (21) showed the specificity of the binding; the Myf5 sequence with the MEF3 site mutated or a non-MEF3-containing sequence showed no competition (Fig. 2B). We next verified that the MEF3 mutation did not modify Pax3 binding to the adjacent Pax3 binding site that is essential for the activity of this Myf5 regulatory sequence (26). Individually each protein binds to an oligonucleotide containing both sites, and, using competition experiments, Pax3 was shown to bind equally well when the Six site is mutated (SI Fig. 7B). Mutation in this site therefore reflects a role for Six1, independent of Pax3, in activating the Myf5 enhancer. When Pax3 and Six1 are present together, a larger complex is formed and may correspond to both proteins binding to the same fragment (SI Fig. 7B).
Fig. 2.

The MEF3/Six binding site present in the 145-bp sequence is conserved between species and binds Six1 and Six4 proteins. (A) DNA sequence alignments, from different species, of the 145-bp Myf5 element (26), showing 100% sequence conservation of the putative MEF3 sites. MEF3 and Pax3 sequences are underlined. The MEF3 consensus is shown below the MEF3 box site. (B) Electromobility shift assays with a 30-bp oligonucleotide containing the Myf5 MEF3 site (lanes 1–11) or a mutant MEF3 site (lanes 12 and 13), incubated with recombinant Six1 protein (lanes 2–11 and 13) or with crude reticulocyte lysate (lanes 1 and 12). Six1 antibodies (lane 11) or a 66- or 200-fold excess of MEF3-Myf5 (lanes 3 and 4), MEF3-Myogenin (lanes 5 and 6), mutant MEF3-Myf5 (lanes 7 and 8), or NF1-Myogenin (lanes 9 and 10) unlabeled competitors was added in the mix. Note that Six1 is unable to recognize the mutant MEF3 oligonucleotide.
Forced Six1 Expression Triggers the Transcriptional Activity of the 145-bp Enhancer.
We next tested whether Six1 transactivates the 145-bp Myf5 element through its MEF3 site. We placed the WT 145-bp sequence or the sequence with its MEF3 site mutated upstream of a minimal TATA box and a luciferase reporter and cotransfected these plasmids into primary chick embryonic myoblasts, with increasing concentrations of Six1 expression vector. Luciferase activity was significantly increased by cotransfection with the Six1 vector (Fig. 3). In contrast, cotransfection of the vector with the MEF3 site mutated did not result in an increase in luciferase activity. Thus, the Six1 protein is able to transactivate the 145-bp element through binding to the MEF3 site in myogenic cells, suggesting that Six proteins may regulate its activity in vivo.
Fig. 3.

The 145-bp Myf5 element can be activated by forced Six1 expression. Primary chick myoblasts were cotransfected with a 145-tata-luci or a 145mut-tata-luci in the presence of increasing amounts of expression vectors coding for Six1. Relative luciferase activity was measured by normalizing the luciferase (Luc) 145-bp-dependent activity to the Renilla activity. ∗, P < 0.05; ∗∗, P < 0.01.
The Six1 Protein Is Loaded on the MEF3 Site of the 145-bp Myf5 Element in Vivo.
Six1 and Six4 are present in the nuclei of Pax3+ muscle progenitor cells of the limb buds as shown by coimmunochemistry (Fig. 4A and data not shown), allowing Six and Pax homeoproteins to activate Myf5 gene expression. To test whether Six binds to the Myf5 limb enhancer in cells, we first prepared chromatin from myogenic cells of the C2 skeletal muscle cell line and looked for selective enrichment, after immunoprecipitation with Six1 antibody, of the 145-bp element versus a control DNA sequence by semiquantitative PCR experiments. As shown in Fig. 4B, Six1 antibodies specifically immunoprecipitate the 145-bp sequence but not the histone H4 promoter sequence, showing that, in these cells, the MEF3 site in the Myf5 element is loaded with Six1 proteins. Chromatin was then prepared from E11.5 WT embryos, and selective enrichment of the 145-bp enhancer versus a control DNA sequence, located at 200 kb upstream of the 145-bp enhancer element, was assessed by quantitative PCR (Fig. 4C). We detected a 5-fold enrichment of the 145-bp sequence versus the sequence at −200 kb from the Myf5 gene, showing that Six1 proteins are bound to the 145-bp element in vivo. This enrichment is specific for Six1, because it was not seen when chromatin was immunoprecipitated with preimmune serum.
Fig. 4.

Six1 is bound to the 145-bp Myf5 element in vivo. (A) a double immunohistochemistry experiment performed on a section of a forelimb from a WT embryo at E10.5 revealing coexpression of Six1 (green) and Pax3 (red) in nuclei of myogenic progenitor cells. Cells expressing both proteins show a yellow nucleus (arrows). (B) ChIP assays with nuclear extracts from C2C12 myotubes. ChIP assays were performed with Six1 antibodies or IgG from preimmune goat serum. Immunoprecipitated chromatin was amplified by semiquantitative PCR to test for the selective enrichment of the 145-bp element compared with the Histone H4 promoter, used as negative control. An aliquot of input chromatin (before ChIP) was also amplified by the same primer pairs to ensure PCR efficiency. Input DNA underwent 22 cycles of PCR amplification, whereas ChIP samples underwent 35 cycles of PCR amplification. (C) ChIP assays using nuclear extracts from WT embryos at E11.5. ChIP was performed with anti-Six1 antibodies or IgGs from preimmune goat serum (T−). ChIP samples were challenged for the selective enrichment of the 145-bp enhancer vs. an irrelevant sequence located 200 kb upstream of the 145-bp sequence of the Myf5 gene by quantitative PCR. 145-bp quantification is normalized to that of the −200 kb amplicon.
Mutation of the MEF3 Site in the 145-bp Element Impairs Its Activity in Vivo.
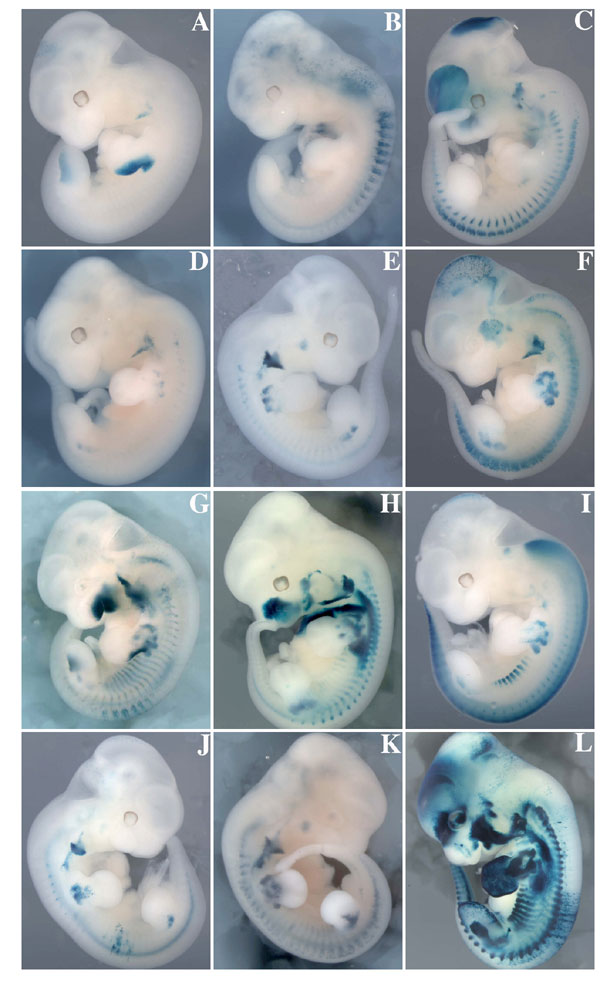
We next determined the importance of the MEF3 site in the 145-bp Myf5 element by transgenic analysis in vivo. We had previously shown that the 145-bp element directs transgene expression in limb buds and mature somites (26). To have maximally robust expression, we had mutated the Pax3 binding site in this element in the context of a larger −58/−57 fragment placed in front of a proximal 3-kb Myf5 promoter sequence, including the branchial arch regulatory element (28), as a positive control. This proximal region also directs some expression in the neural tube. We adopted the same strategy with a WT or mutated MEF3 site and monitored expression of the nlacZ reporter in transient transgenic experiments. Seven WT transgenic embryos all showed strong expression of nlacZ in both forelimbs and hindlimbs, as well as in the somites and branchial arches at E11.5 (Fig. 5A). Strong somitic expression is seen in the hypaxial and epaxial lips of the residual dermomyotome epithelium at this stage (23). Mutation of the MEF3 site impaired the transcriptional activity of the transgene in all Myf5-expressing territories except the branchial arch (Fig. 5 B and C and Table 1). Three out of 12 embryos showed no transgene expression in the limb buds (SI Fig. 8 A–C), and eight had incomplete expression mainly in the proximal part (SI Fig. 8 D–K). One embryo with a stronger expression in the limb buds also had extensive ectopic expression, indicating an integration site effect (SI Fig. 8L). Thus, in 11 of 12 embryos limb expression of the transgene was compromised, and we therefore conclude that the MEF3 site in the 145-bp element plays a role in its activity in vivo.
Fig. 5.

Mutation of the Six binding site in the 145-bp element within the larger Myf5 −58/−57 sequence compromises transgene expression. X-gal staining of transgenic embryos at E11.5 carrying a nlacZ reporter, regulated by −3 kb adjacent to the Myf5 gene, containing the promoter and the branchial arch sequence as a positive control, proceeded by a −58/−57 fragment containing the WT 145-bp element, −58/−57baMyf5nlacZ (A) or with the 145-bp MEF3 site mutated, −58/−57SixMutbaMyf5nlacZ (B and C). ba, branchial arch; fl, forelimb; hl, hindlimb.
Table 1.
Summary of the expression of the −58/−57baMyf5nlacZ and the −58/−57SixMutbaMyf5nlacZ transgenes in limb buds and mature somites at E11.5
| Territory | −58/−57 | −58/−57SixMut |
|
|---|---|---|---|
| Negative | Partial | ||
| Limb buds | 7/7 | 3/12 | 9/12 |
| Mature somites | 6 (1*)/7 | 6/12 | 6/12 |
Discussion
We had shown previously that Six homeoproteins are implicated in the regulation of the myogenic regulatory genes MyoD, Mrf4, and Myogenin. Their somitic expression is affected in Six1−/−Six4−/− double mutants (22). In the case of Myogenin, direct regulation of a MEF3 site by Six proteins has been demonstrated (21). In the context of hypaxial myogenesis, loss of myogenic progenitor cells in the hypaxial dermomyotome of the Six1−/−Six4−/− double mutant, probably partly due to negative effects on Pax3 expression, precludes analysis of the effects of Six on skeletal muscles, such as those in the limbs, that derive from this part of the somite. However, analysis of hindlimb myogenesis in the Six1−/− mutant now reveals that Myf5 expression, both transcript and protein, is severely reduced in Pax3+ progenitor cells in which MyoD activation still occurs. In the forelimb, which is more severely affected, Myf5 transcripts are not detectable in the residual Pax3+ cells at E11.5, whereas a low level of Myf5 expression can be detected at E12.5. This may reflect the sensitivity of detection or a delay in Myf5 activation. We had previously reported higher Myf5 expression in Six1−/− forelimbs (20), and we can only suppose that after more extensive breeding it reflects a change in genetic background or that Myf5 antibodies used in the previous study detected a protein unrelated to Myf5. Further specific reduction of Myf5 expression in the hindlimbs of Six1−/−Six4−/+ suggests that Six4, also expressed in myogenic progenitor cells, is responsible for the remaining Myf5 transcription in the Six1−/− mutant. Although MyoD expression is observed, it is delayed by 1 or 2 days in hindlimb or forelimb buds, respectively (ref. 20 and unpublished data). This may reflect a direct effect of reduced Six1/4 levels on the initiation of MyoD transcription or an indirect effect due to the absence or reduction of Myf5 expression, because a delay in MyoD activation was observed in the limb buds of Myf5−/− (Mrf4−/−) mutant embryos (7). In the trunk, in the absence of Six1, Six4, and Myf5, MyoD is not expressed (J.D., M.B., and P.M., unpublished data), whereas its activation is delayed in the trunk in the absence of Myf5 (30). This would suggest that Six1/4 may also have a role in regulating MyoD during limb myogenesis.
We demonstrate that Six1 and Six4 act directly on Myf5 activation through a MEF3 site present in the 145-bp regulatory element that directs Myf5 expression in the limb buds. Another regulatory element contributes to partial limb expression of Myf5 mainly in the hindlimbs, and this element may ensure some of the remaining expression of Myf5 observed in the hindlimb buds of Six1−/−. Mutation of the MEF3 site in the 145-bp element, which we show binds Six1/4 in vitro and in vivo, does not totally abolish its activity in all transgenic embryos. There is no apparent difference in the partial expression seen in E11.5 transgenic embryos between forelimbs and hindlimbs. The reduced expression of Myf5 seen in the Six1−/− mutant may reflect differences in Six4 levels, which fail to reach a threshold level for Myf5 expression in the forelimb. We had previously shown that the activity of the 145-bp element is totally dependent on a site to which Pax3 binds in vivo. Here we show that Pax3 binds efficiently to its site when the MEF3 site is mutated; activation by Pax3 may partially override the requirement for Six in some cells of the limbs of Six mutant embryos. Meox2 is another factor that affects Myf5 transcription, as shown by the phenotype of Meox2 mutant embryos in which Myf5 activation in the limb buds is delayed (31). It is not yet clear whether Meox2 also intervenes directly in the regulation of the 145-bp sequence. Myf5 is not expressed in WT myogenic progenitor cells before they reach the limb buds (3) despite the fact that they express both Pax3 and Six1/4. Wnt signaling may be important in regulating the activation of Myf5 in the limb bud, and Wnt6 is a candidate in this context (32). It remains to be seen whether Wnt signaling also directly impacts the 145-bp element.
The regulation of the 145-bp Myf5 element by Pax3 and Six1/4 provides an example of the participation of both types of transcription factor in the regulation of myogenic progenitor cells. This is seen at the level of their entry into the myogenic program, as shown here. Unlike Pax3, which is down-regulated after initial expression of the myogenic determination genes (see ref. 33), Six1/4 continue to be expressed in Myogenin-positive cells in muscle fibers, and indeed Six factors play an important role in the activation of fiber type-specific genes such as MCK and aldolase A (19, 34). Pax3 and Six1/4 play an important upstream role in myogenic progenitor cells in the somite, regulating their behavior in the dermomyotome and their migration from it (see refs. 16, 18, 22, and 29). Furthermore, there are complex interactions between Pax3 and Six1/4, such that Six1/4 regulate Pax3 expression in the hypaxial somite (22). The key roles of Six1/4 in many myogenic processes are summarized in SI Fig. 9.
Materials and Methods
Transfection Experiments.
Primary myogenic cells were obtained from E10 hindlimbs of chick embryos and were grown in MEM/E199 medium (Invitrogen, Cergy Pontoise, France) complemented with 10% FCS. Four hours after plating, cells were transfected with Lipofectamine 2000 (Invitrogen). The WT or MEF3 mutant 145-bp Myf5 limb element (26) was cloned upstream of the −35 to +45 minimal promoter of the aldolase A gene, linked to the firefly luciferase transgene (Promega, Charbonnières, France). Transfection efficiency was normalized by measuring the activity of the TK-Renilla transgene. Thirty-six hours after transfection, both luciferase and Renilla activities were determined on a Lumat LB 907 luminometer (Berthold, Thoiry, France).
ChIP.
ChIP with embryonic extracts was performed as described previously (18). ChIP from C2 cells was performed with cells after 2 days in differentiation medium. For PCR, 10 ng of genomic DNA was amplified as a positive control. The histone H4 promoter, devoid of MEF3 binding sites, was amplified as a ChIP negative control. Primers used for amplification of the 145-bp element, histone H4 promoter, and for the sequence at −200 kb 5′ to the 145-bp sequence are described in SI Text. For quantitative PCR experiments, immunoprecipitated DNA and input DNA were analyzed by real-time PCR using a Light Cycler Faststart DNA Master SYBR Green I Mix and a Light Cycler (Roche Applied Science, Meylan, France).
Supplementary Material
Acknowledgments
We thank Milan Esner and Catherine Baudin for their assistance. J.G. and L.B. were supported by a fellowship from the Ministère de la Recherche et de l'Education Nationale and from the Association Française Contre les Myopathies. Financial support for P.M.'s laboratory has been provided by the Institut National pour la Santé et la Recherche Médicale, the Association Française Contre les Myopathies, the Centre National de la Recherche Scientifique, Action Concertée Incitative 0220514, the Agence Nationale pour la Recherche, the Association pour la Recherche sur le Cancer, and the FP6 MYORES European Muscle Development Network of Excellence. The contribution of the Région Ile de France to the Institut Cochin animal care facility is also acknowledged. Work on skeletal myogenesis in M.B.'s laboratory is supported by the Pasteur Institute, the Centre National de la Recherche Scientifique, the Association Française Contre les Myopathies, and two European Union Networks of Excellence (Cells into Organs and MYORES).
Abbreviation
- En
embryonic day n.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0611299104/DC1.
References
- 1.Gros J, Scaal M, Marcelle C. Dev Cell. 2004;6:875–882. doi: 10.1016/j.devcel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 2.Buckingham M. Curr Opin Genet Dev. 2006;16:525–532. doi: 10.1016/j.gde.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Tajbakhsh S, Bober E, Babinet C, Pournin S, Arnold H, Buckingham M. Dev Dyn. 1996;206:291–300. doi: 10.1002/(SICI)1097-0177(199607)206:3<291::AID-AJA6>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 4.Tajbakhsh S, Buckingham ME. Proc Natl Acad Sci USA. 1994;91:747–751. doi: 10.1073/pnas.91.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bober E, Franz T, Arnold HH, Gruss P, Tremblay P. Development (Cambridge, UK) 1994;120:603–612. doi: 10.1242/dev.120.3.603. [DOI] [PubMed] [Google Scholar]
- 6.Delfini M, Hirsinger E, Pourquie O, Duprez D. Development (Cambridge, UK) 2000;127:5213–5224. doi: 10.1242/dev.127.23.5213. [DOI] [PubMed] [Google Scholar]
- 7.Kablar B, Krastel K, Ying C, Asakura A, Tapscott SJ, Rudnicki MA. Development (Cambridge, UK) 1997;124:4729–4738. doi: 10.1242/dev.124.23.4729. [DOI] [PubMed] [Google Scholar]
- 8.Kawakami K, Sato S, Ozaki H, Ikeda K. BioEssays. 2000;22:616–626. doi: 10.1002/1521-1878(200007)22:7<616::AID-BIES4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 9.Xu PX, Zheng W, Huang L, Maire P, Laclef C, Silvius D. Development (Cambridge, UK) 2003;130:3085–3094. doi: 10.1242/dev.00536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Laclef C, Souil E, Demignon J, Maire P. Mech Dev. 2003;120:669–679. doi: 10.1016/s0925-4773(03)00065-0. [DOI] [PubMed] [Google Scholar]
- 11.Zheng W, Huang L, Wei ZB, Silvius D, Tang B, Xu PX. Development (Cambridge, UK) 2003;130:3989–4000. doi: 10.1242/dev.00628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozaki H, Nakamura K, Funahashi J, Ikeda K, Yamada G, Tokano H, Okamura HO, Kitamura K, Muto S, Kotaki H, et al. Development (Cambridge, UK) 2004;131:551–562. doi: 10.1242/dev.00943. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Oghi KA, Zhang J, Krones A, Bush KT, Glass CK, Nigam SK, Aggarwal AK, Maas R, Rose DW, et al. Nature. 2003;426:247–254. doi: 10.1038/nature02083. [DOI] [PubMed] [Google Scholar]
- 14.Oliver G, Wehr R, Jenkins NA, Copeland NG, Cheyette BN, Hartenstein V, Zipursky SL, Gruss P. Development (Cambridge, UK) 1995;121:693–705. doi: 10.1242/dev.121.3.693. [DOI] [PubMed] [Google Scholar]
- 15.Xu PX, Woo I, Her H, Beier DR, Maas RL. Development (Cambridge, UK) 1997;124:219–231. doi: 10.1242/dev.124.1.219. [DOI] [PubMed] [Google Scholar]
- 16.Heanue TA, Reshef R, Davis RJ, Mardon G, Oliver G, Tomarev S, Lassar AB, Tabin CJ. Genes Dev. 1999;13:3231–3243. doi: 10.1101/gad.13.24.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ohto H, Kamada S, Tago K, Tominaga SI, Ozaki H, Sato S, Kawakami K. Mol Cell Biol. 1999;19:6815–6824. doi: 10.1128/mcb.19.10.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grifone R, Demignon J, Giordani J, Niro C, Souil E, Bertin F, Laclef C, Xu PX, Maire P. Dev Biol. 2007;302:602–616. doi: 10.1016/j.ydbio.2006.08.059. [DOI] [PubMed] [Google Scholar]
- 19.Grifone R, Laclef C, Spitz F, Lopez S, Demignon J, Guidotti JE, Kawakami K, Xu PX, Kelly R, Petrof BJ, et al. Mol Cell Biol. 2004;24:6253–6267. doi: 10.1128/MCB.24.14.6253-6267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laclef C, Hamard G, Demignon J, Souil E, Houbron C, Maire P. Development (Cambridge, UK) 2003;130:2239–2252. doi: 10.1242/dev.00440. [DOI] [PubMed] [Google Scholar]
- 21.Spitz F, Demignon J, Porteu A, Kahn A, Concordet JP, Daegelen D, Maire P. Proc Natl Acad Sci USA. 1998;95:14220–14225. doi: 10.1073/pnas.95.24.14220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grifone R, Demignon J, Houbron C, Souil E, Niro C, Seller M, Hamard G, Maire P. Development (Cambridge, UK) 2005;132:2235–2249. doi: 10.1242/dev.01773. [DOI] [PubMed] [Google Scholar]
- 23.Hadchouel J, Carvajal JJ, Daubas P, Bajard L, Chang T, Rocancourt D, Cox D, Summerbell D, Tajbakhsh S, Rigby PW, et al. Development (Cambridge, UK) 2003;130:3415–3426. doi: 10.1242/dev.00552. [DOI] [PubMed] [Google Scholar]
- 24.Hadchouel J, Tajbakhsh S, Primig M, Chang TH, Daubas P, Rocancourt D, Buckingham M. Development (Cambridge, UK) 2000;127:4455–4467. doi: 10.1242/dev.127.20.4455. [DOI] [PubMed] [Google Scholar]
- 25.Buchberger A, Nomokonova N, Arnold HH. Development (Cambridge, UK) 2003;130:3297–3307. doi: 10.1242/dev.00557. [DOI] [PubMed] [Google Scholar]
- 26.Bajard L, Relaix F, Lagha M, Rocancourt D, Daubas P, Buckingham M. Genes Dev. 2006;20:2450–2464. doi: 10.1101/gad.382806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pauli T, Seimiya M, Blanco J, Gehring W. Development (Cambridge, UK) 2005;132:2771–2782. doi: 10.1242/dev.01841. [DOI] [PubMed] [Google Scholar]
- 28.Summerbell D, Ashby PR, Coutelle O, Cox D, Yee S, Rigby PW. Development (Cambridge, UK) 2000;127:3745–3757. doi: 10.1242/dev.127.17.3745. [DOI] [PubMed] [Google Scholar]
- 29.Relaix F, Polimeni M, Rocancourt D, Ponzetto C, Schafer BW, Buckingham M. Genes Dev. 2003;17:2950–2965. doi: 10.1101/gad.281203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Cell. 1997;89:127–138. doi: 10.1016/s0092-8674(00)80189-0. [DOI] [PubMed] [Google Scholar]
- 31.Mankoo BS, Collins NS, Ashby P, Grigorieva E, Pevny LH, Candia A, Wright CV, Rigby PW, Pachnis V. Nature. 1999;400:69–73. doi: 10.1038/21892. [DOI] [PubMed] [Google Scholar]
- 32.Geetha-Loganathan P, Nimmagadda S, Huang R, Scaal M, Christ B. Anat Embryol (Berlin) 2006;211:183–188. doi: 10.1007/s00429-005-0069-6. [DOI] [PubMed] [Google Scholar]
- 33.Relaix F, Rocancourt D, Mansouri A, Buckingham M. Nature. 2005;435:948–953. doi: 10.1038/nature03594. [DOI] [PubMed] [Google Scholar]
- 34.Himeda CL, Ranish JA, Angello JC, Maire P, Aebersold R, Hauschka SD. Mol Cell Biol. 2004;24:2132–2143. doi: 10.1128/MCB.24.5.2132-2143.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}