

Abstract
Genetic studies show that TGFβ signaling is essential for vascular development, although the mechanism through which this pathway operates is incompletely understood. Here we demonstrate that the TGFβ auxiliary coreceptor endoglin (eng, CD105) is expressed in a subset of neural crest stem cells (NCSCs) in vivo and is required for their myogenic differentiation. Overexpression of endoglin in the neural crest caused pericardial hemorrhaging, correlating with altered vascular smooth muscle cell investment in the walls of major vessels and upregulation of smooth muscle α-actin protein levels. Clonogenic differentiation assay of NCSCs derived from neural tube explants demonstrated that only NCSC expressing high levels of endoglin (NCSCCD105+) had myogenic differentiation potential. Furthermore, myogenic potential was deficient in NCSCs obtained from endoglin null embryos. Expression of endoglin in NCSCs declined with age, coinciding with a reduction in both smooth muscle differentiation potential and TGFβ1 responsiveness. These findings demonstrate a cell autonomous role for endoglin in smooth muscle cell specification contributing to vascular integrity.
Keywords: Endoglin, Neural Crest, Transforming growth factor beta, Myogenesis, cardiovascular
Introduction
Endoglin is an auxiliary receptor that interacts with the TGFβ signaling receptors (Barbara et al., 1999; Cheifetz et al., 1992). Endoglin null embryos die by E11.5 due to defects in vessel formation (Arthur et al., 2000; Bourdeau et al., 1999; Li et al., 1999) characterized by a failure of vascular smooth muscle cell (SMC) recruitment and loss of expression of the SMC differentiation marker smooth muscle alpha actin (α-SMA) (Li et al., 1999), whereas endothelial cells appear normal. Endoglin is expressed in stem cell populations including hematopoietic, mesenchymal, and neural stem cells (Chen et al., 2002; Vogel et al., 2003; Zvaifler et al., 2000). Furthermore, endoglin is upregulated in SMCs in response to vascular injury (Ma et al., 2000) and in human atherosclerotic vascular SMCs (Conley et al., 2000). Mutations in both endoglin and the type I TGFβ receptor ALK1 cause hereditary hemorrhagic telangiectasia (HHT) types 1 and 2 respectively, disorders characterized by vessel fragility and arteriovenous malformation (McAllister et al., 1994; Pece et al., 1997) suggesting that these proteins play a critical role in vascular homeostasis.
TGFβ ligands and receptors play important roles in the specification and development of neural crest-derived cell types (Choudhary et al., 2006; Kaartinen et al., 2004; Shah et al., 1996; Wurdak et al., 2005). The neural crest is a heterogeneous cell population derived from the neural tube and contributes to multiple vital developmental processes. Within the neural crest, a population of cells expressing low affinity nerve growth factor receptor (p75) and lacking expression of myelin P0 protein are referred to as neural crest stem cells (NCSCs). NCSCs differentiate into a variety cell types including neurons, glia, melanocytes, SMCs, and head cartilage and bone (Anderson, 1997; Bronner-Fraser, 1995). A number of growth factors instruct differentiation of NCSCs from explants of embryonic neural tube, sciatic nerve, or intestine. These factors include BMP2 and BMP4, which instruct neurogenic differentiation (Shah et al., 1996), the soluble Notch ligand, Delta-Fc (Morrison et al., 2000) and neuregulin (Shah et al., 1994), which instruct gliogenic differentiation, and TGFβ1, which instructs SMC differentiation (Chen et al., 2006; Shah and Anderson, 1997).
Previously, we reported that NCSCs become progressively more gliogenic and less neurogenic with age (Kubu et al., 2002). This transition reflects a decrease in sensitivity to BMP2, and an increase in sensitivity to Delta-Fc. These changes involve altered expression of the BMP type I receptor ALK3 and Notch1 (Kubu et al., 2002; White et al., 2001). Interestingly, NCSCs retain SMC differentiation potential in the presence of both BMP and Delta-Fc, demonstrating that the altered balance between gliogenic and neurogenic fates does not affect SMC differentiation potential. While the age-related loss of neurogenic potential is characterized, the effects of age on SMC myogenic potential of NCSCs remain unknown.
Based on the report of SMC defects in endoglin null embryos combined with the previous observation of upregulation of endoglin expression in diseased vascular SMCs and in response to injury, we investigated the hypothesis that endoglin plays a cell autonomous role in vascular SMC development and maintenance of vascular integrity.
Materials and Methods
Endoglin knockout and transgenic mouse studies
Endoglin null C57Bl6 mice were kindly provided by Dr. D. Marchuk (Arthur et al., 2000). Transgenic mouse embryos used in this study were maintained on an FVB background. Mice were maintained according to the NIH standards established in the “Guidelines for the Care and Use of Experimental Animals”. Protocols and procedures were approved by the Institutional Animal Care and Use Committee at the Maine Medical Center Research Institute. Full length human endoglin cDNA was blunt end ligated into the CAGCAT vector (Yamauchi et al., 1999). Microinjection into fertilized FVB oocytes and other surgical procedures were performed as described (Hofmann et al., 1988). The genotypes of all offspring were analyzed by PCR as well as by chloramphenicol acetyl transferase (CAT) enzyme-linked immunosorbent assay, which was used to normalize the transgene locus CAT activity levels prior to establishment of mouse lines.
The Wnt1cre driver mouse was used to achieve conditional expression of endoglin in neural crest stem cells (Jiang et al., 2000). Mating of Wnt1cre with TgEngloxP generated Wnt1cre;TgEngloxP embryos that were genotyped using PCR primers as previously described (Chytil et al., 2002). Briefly, yolk sac genomic DNA was amplified for 35 cycles (94°C for 1 min, 60°C for 1.5 min, 72°C for 2 min) in a thermal cycler. The primers were GCTGGTTAGCACCGCAGGTGTAGAG and CGCCATCTTCCAGCAGGCGCACC in the cre coding region and CAGAGTGTCCCCATCCGT and TGGGTATGGGTACTGTGTAGAAGT in the endoglin coding region.
Conditional inactivation of Smad4 with simultaneous activation of human endoglin was achieved by breeding Wnt1cre;TgEngloxP on to the Smad4 heterozygous background (Wnt1cre;TgEngloxP;Sm4n/+), and subsequently intercrossing this strain with the Smad4 conditional strain (SM4c/c) (Chu et al., 2004). Embryos of two different genotypes resulting from this mating were analyzed: Wnt1cre;TgEngloxP;Sm4c/+ and Wnt1cre;TgEngloxP;Sm4n/c. The latter configuration is sensitized to conditional inactivation as Wnt1cre-mediated recombination of only a single allele is required to render the Smad4 locus null. This strategy was previously used for complete inactivation of the Smad4 locus in embryonic tissues (Chu et al., 2004).
Histological analysis
X-gal staining was performed according to standard procedures (Hogan et al., 1994). For hematoxylin and eosin (H&E) staining and immunohistochemistry, embryos were fixed in 4% paraformaldehyde overnight and subsequently dehydrated through a graded ethanol series and paraffin embedded. Five μm sections were stained with H&E for morphological analysis or immunohistochemistry was performed using the following antibodies: PECAM (BD Pharmingen) 1:3000, AP-conjugated α-SMA (Sigma) 1:50, SOX-10 (1:1), and HNK-1 (1:1) (Anderson and Axel, 1986). PECAM staining was performed using the TSA Biotin kit (PerkinElmer) with trypsin antigen retrieval per manufacturer’s instructions. IHC for endoglin was performed using α-human endoglin (SN6h) 1:500 (Dako). This anti-human endoglin antibody was raised against human endoglin, and was selected for its preferential recognition of human endoglin. This reagent preferentially detects human endoglin (Rodriguez-Pena et al., 2002), which was utilized to generate the transgene. Histological staining for α-SMA, SOX-10, and HNK-1 was performed using the Avidin/Biotin kit (Vector Labs) with citrate buffer antigen retrieval (Dako) following the manufacturer’s instructions. SOX-10 immunofluorescence was conducted using Alexa-645 labeled anti-mouse antibody (Molecular Probes) at 1:100 dilution for 1 hour. Fluorescence was imaged using an Axioskop 2 Plus microscope fitted with an AxioCam MRm camera (Zeiss). Immunohistochemistry using α-phospho Smad 1/5/8 (anti-pSmad 1/5/8) and α-phospho Smad 2/3 antibody (anti-pSmad 2/3, Cell Signaling Technology) 1:1000 was performed with citrate retrieval and the TSA signal amplification system (Perkin Elmer).
Immunoprecipitation and Western Blot
Embryos were harvested at E11.5. Whole embryos were dissociated by passing through a 1 mL syringe into RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 300 mM sucrose, 1.0% Triton-X100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 10 mM β-glycerol phosphate) supplemented with a protease inhibitor cocktail (Roche) and processed as described (Koleva et al., 2006). Bound antibodies were visualized by chemiluminescence (ECL, Amersham). Anti-human endoglin antibody Clone 35 at 1:2000 (BD Transduction) does not efficiently detect mouse endoglin (Rodriguez-Pena et al., 2002), and was used to demonstrate cre-recombination and over expression of the human endoglin transgene in the TgEngloxP embryos. Endogenous levels of α-SMA and Flk-1 were detected using α-SMA antibody at 1:1000 (Sigma) and α-Flk-1 antibody at 1:200 (Santa Cruz) respectively.
Neural Crest Cell culture
NCSCs were isolated by the method of Stemple (Stemple and Anderson, 1992). In short, neural tubes from E10.5 rat or E9.5 mouse embryos were isolated between presumptive limb buds and plated on 35 mm fibronectin-coated dishes. Crest cells were allowed to migrate away from the tube overnight in a defined medium containing 10% chick embryo extract (CEE). Neural tubes were then removed using a tungsten needle (Morrison et al., 2000). Explant cells were trypsinized and plated at clonal density (50 cells per 35 mm dish) in a growth factor rich medium containing 15% CEE (Morrison et al., 2000) that enhances autonomic phenotype production (referred to as Morrison medium). To identify NCSCs, the cells were live labeled with the 192IgG p75 antibody (Chandler et al., 1982), and the P0 monoclonal antibody against the peripheral myelin protein, P0, was conjugated to phycoerythrin (gift of J. Achelos, Munich). cells (migrating NCSCs) were identified and used for analysis. Once all non- cells were removed from the dish with a tungsten needle, the remaining cells were restained with anti-CD105 antibody conjugated to PE (anti-CD105-PE, BioCytex) to delineate the NCSCCD105+ and NCSCCD105- stem cells. NCSCCD105+ and NCSCCD105- cell designations were based on strong or undetectable anti-CD105-PE antibody immunoreactivity, respectively, as determined by immunofluorescence microscopy.
In polymerase chain reaction (PCR) experiments, trypsinized crest explants were plated at 500 cells per dish and labeled with the p75 and P0 antibodies.
E14 sciatic nerve-derived NCSCs were isolated and cultured according to the method of Morrison (Morrison et al., 1999). Sciatic nerves isolated from E14 embryos, P1 rat pups, and adult (greater than 6 weeks) rats were enzymatically dissociated and labeled with the p75 antibody conjugated to fluorescein and the P0 monoclonal antibody conjugated to phycoerythrin. Cells were sorted using a FACSVantage flow cytometer. The fraction (NCSCs) was plated at clonal density for clonogenic assays in Morrison medium, or at higher density for reverse transcriptase (RT)-PCR studies after staining for CD105-PE to identify NCSCCD105+ and NCSCCD105- subpopulations.
Clonogenic assays
Clonogenic assays were performed according to the original method of Stemple (Stemple and Anderson, 1992). Briefly, NCSCs were plated at clonal density and cultured for up to 14 days in Morrison medium. The resulting clones were fixed in acid ethanol and stained with antibodies against peripherin (Chemicon AB1530, Temecula, CA) to detect neurons (N), α-SMA (Sigma A-2547, St. Louis, MO) to detect myofibroblasts (M), and anti-glial fibrillary acidic protein (GFAP) antibody (Chemicon AB 5040) to detect Schwann cells (S). Clones were scored based on the appearance of just one phenotypic representative (Stemple and Anderson, 1992). The number and percentage of clones containing neurons only (N), glial cells only (G), myofibroblasts only (M), combinations of (N+G), (N+M), (N+G+M), and (G+M) were scored at day 14 unless stated otherwise. Also scored was the production of each representative phenotype in each clone to complement our clonal population assay.
Retroviral mediated gene transfer and reconstitution studies
Over expression of the genes of interest in NCSCs was achieved by retroviral-mediated gene transfer as described (Verdi and Anderson, 1994). Endoglin was cloned into the pWzl retrovirus (Pear et al., 1993), as previously described (Conley et al., 2004). Virus was produced using the EcoPac-293 packaging cell line (ClonTech). On the day before transfection, the EcoPac-293 cells were split and plated at 2×106 cells per 60 mm dish in DMEM plus 10% fetal bovine serum. Effectine (Promega) mediated transfection was used to introduce constructs harboring the genes of interest into the packaging cell line. Virus was collected for the next 72 hours, at which time the virus-containing medium was divided in half and concentrated according to the method of Cepko (Cepko et al., 1998). The resulting viral pellet was resuspended in Morrison medium minus CEE. The concentrated viral stock was added to the NCSC cultures in the presence of 4 μg/ml polybrene. Two 2-hour rounds of infection were conducted. Thereafter, the cells were washed, split to clonal density, Morrison medium was applied and clonogenic differentiation assays were conducted. Infection efficiencies as measured by GFP or RFP expression ranged from a low of 3% to a high of over 12%.
RT-PCR
Total RNA was isolated using the RNA micro-isolation kit (Stratagene). The RNA was treated with DNAse to remove potential genomic contamination and reprecipitated. The resulting RNA was oligo-dT primed and reverse transcribed using superscript II as previously described (Verdi and Anderson, 1994). PCR was performed in the linear range of amplification for each primer set, as empirically determined using total RNA from 50 and 500 cell equivalents. Primers were chosen to have an annealing temperature of approximately 55°C with a 50-60% GC content. The primers were chosen to span an intron/exon border to avoid amplification of any residual genomic DNA
Results
Endoglin is expressed in the neural crest, in areas along the A/P axis that give rise to vascular smooth muscle cells
Endoglin targeted (eng-/-) embryos die by E11.5 due to defects in angiogenesis and cardiac morphogenesis resulting in septation defects suggesting a loss of endocardial to mesenchymal transitions and a possible absence of vascular SMCs (Li et al., 1999). This interpretation was suggested by a loss of α-SMA staining in eng-/- embryos, however α-SMA is first detected in wild type embryos at E11.5 and therefore it is unclear whether loss of endoglin results in a delay or loss of SMC specification and/or differentiation. Since many of the SMCs that invest the large vessels and form the cardiac cushions are derived from the neural crest (Jiang et al., 2000), we sought to characterize a role for endoglin in the neural crest by determining whether endoglin is expressed in neural crest cells in vivo. Using the heterozygous eng mouse with the β-galactosidase cDNA expressed from the eng locus (Arthur et al., 2000), we observed endoglin-expressing cells surrounding the neural tube, a location typical for neural crest cells (Fig. 1A). To determine if these endoglin-expressing cells were neural crest cells, we performed immunohistochemistry for two neural crest markers, HNK-1 (Minarcik and Golden, 2003) and SOX-10 (Young et al., 2004) on β-galactosidase-stained sections through the neural tube at E9.5 (Figs. 1B, 1D). Endoglin in E9.5 eng+/- embryos was co-expressed with both HNK-1 and SOX-10 in some but not all such cells, indicating that endoglin is expressed in a subset of neural crest cells. To confirm these results, we isolated NCSCs (Morrison et al., 1999) and demonstrated by RT-PCR analysis that isolated NCSCs expressed endoglin, as well as the TGFβ type I receptors ALK1 and ALK5 (Fig. 1E). Because NCSCs display a graded loss in developmental potential along the rostrocaudal axis (Lwigale et al., 2004), we examined endoglin expression along the rostrocaudal axis. We found that endoglin expression correlated with areas of the neural crest that give rise to vascular SMCs; β-galactosidase-stained sections representing an anterior to posterior series through the cardiac crest, suggested that neural crest cells in this region expressed endoglin. However, endoglin was not expressed in more posterior regions of the neural crest that do not give rise to smooth muscle, but was restricted to the vasculature derived from the mesoderm in this area (Figs. 1F-J). Taken together, these results demonstrate that endoglin is spatially expressed in the neural crest, in regions that give rise to vascular SMCs.
Figure 1. Endoglin is expressed in the neural crest in vivo.
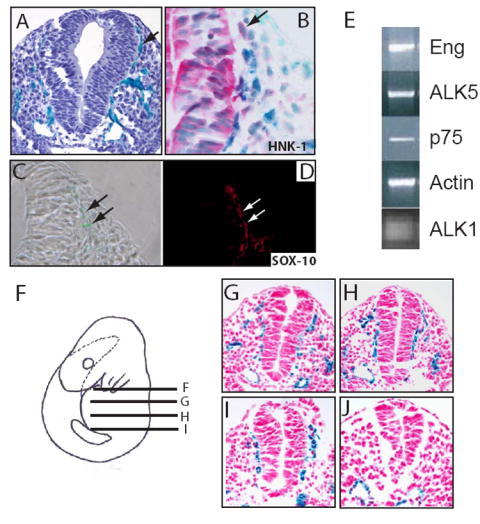
(A) Section of embryo in which LacZ is inserted into the endoglin locus are stained for β-galactosidase activity. Cells expressing β-galactosidase under control of the endogenous endoglin promoter and indicating endoglin gene transcriptional activity around the neural tube. (B) β-galactosidase staining of eng+/- embryos in combination with HNK-1 immunostaining. Arrow indicates an example of a cell expressing β-Galactosidase (Blue) under the control of the endoglin promoter and HNK-1 immunostaining (Red). (C,D) β-galactosidase staining of eng+/- embryo section (C) in combination with SOX-10 immunofluorescence (D). Arrows indicate examples of cells expressing both β-Galactosidase (green cells, panel C) and which exhibit immunofluorescent staining for SOX-10 (red cells, panel D). (E) RT-PCR analysis of NCSC isolated from E10.5 rat neural tube for the TGFβ receptors ALK1, ALK5, and endoglin, as well as the low-affinity nerve growth factor, p75. β-actin serves as a control. (F-J) Schematic of an eng+/- E9.5 embryo (F) and corresponding sections stained for β-galactosidase (G-J) demonstrating endoglin expression in the neural crest along the anterior-posterior axis. Note that endoglin expression is restricted to the vasculature in more posterior regions of the neural crest.
Vascular defects are associated with endoglin over expression in the neural crest
To determine if eng-/- embryos exhibit defective SMC specification or differentiation, we performed converse in vivo gain of function analysis to determine if increased endoglin expression affected the specification or differentiation of SMCs derived from the neural crest. For these studies, we generated a cre-inducible transgenic mouse line conditionally expressing endoglin (TgEngloxP) (Fig. 2A). TgEngloxP mice were bred to Wnt1cre mice to induce expression of the TgEngloxP transgene in cells of the neural crest (Jiang et al., 2002). In vivo cre-recombination and subsequent expression of the eng transgene was confirmed by immunoprecipitation and immunoblotting for endoglin protein from whole Wnt1cre;TgEngloxP embryos (Fig. 2B). Immunohistochemistry for transgenic endoglin expression in the Wnt1cre;TgEngloxP embryos, conducted using an anti-endoglin antibody that preferentially recognizes human endoglin (Rodriguez-Pena et al., 2002), confirmed that endoglin was upregulated in neural crest-derived tissues relative to wild type (Figs. 2C,D). Wnt1cre;TgEngloxP embryos consistently displayed dilated cranial vessels and bleeding in the thoracic cavity (Figs. 2E,G). Lethality was observed in the majority of Wnt1cre;TgEngloxP embryos by E18.5.
Figure 2. Over expression of endoglin in the neural crest in vivo resulted in vascular hemorrhage.
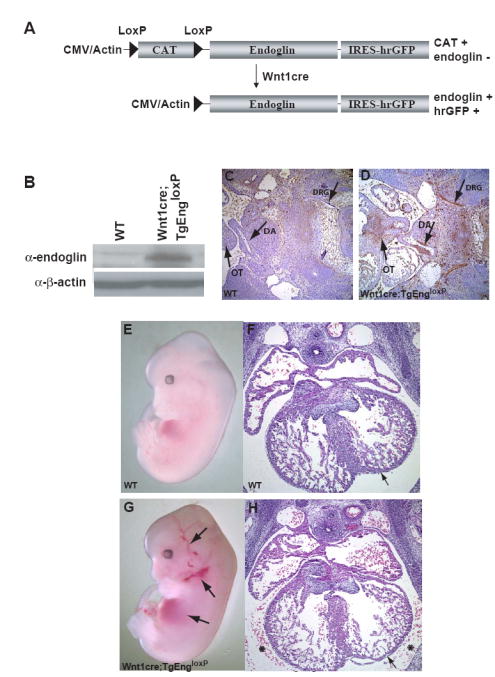
(A) Schematic of transgenic strategy and resulting construct upon Wnt1cre-mediated recombination. (B) Immunoprecipitation of human endoglin followed by immunoblot analysis of E11.5 wild type and Wnt1cre;TgEngloxP embryos. (C,D) α-human endoglin antibody immunohistochemistry was conducted using the human endoglin-specific SN6h anti-endoglin antibody (Bodey et al., 1998; Rodriguez-Pena et al., 2002) on sections from E12.5 wild type and Wnt1cre;TgEngloxP transgenic embryos. Arrows indicate that increased endoglin expression was restricted to neural crest-derived structures. Neural crest-derived structures are: OT=outflow tract; DRG=dorsal root ganglia; DA=ductus arteriosus. (E,G) E12.5 wild type and Wnt1cre;TgEngloxP transgenic whole embryos. Arrows indicate areas of hemorrhaging in the cranial vasculature and heart in Wnt1cre;TgEngloxP embryos. (F,H) H&E staining of transverse heart sections from E12.5 wild type (F) and Wnt1cre;TgEngloxP (H) embryos. Asterisks indicate areas of hemorrhaging in the pericardial cavity; Arrows indicate areas of atrial wall thinning.
Histological analysis of serial transverse sections through the outflow tract and major vessels revealed hemorrhaging into the pericardial cavity (Figs. 2F,H). While no gross malformations were observed in the heart that resulted in hemorrhaging, the overall morphology of the Wnt1cre;TgEngloxP heart exhibited thinning of the atrial walls (Figs. 2H versus 2F). TUNEL staining did not indicate increased cell death in the Wnt1cre;TgEngloxP compared to wild type mice (data not shown). Additionally, phospho Histone H3 staining did not reveal differences in proliferation in the heart and surrounding vasculature (data not shown). These data suggested that vascular leakage in the Wnt1cre;TgEngloxP mouse was due to a loss of vascular structural integrity, potentially involving neural crest-derived SMCs.
Endoglin over expression does not effect the migration of NCSCs
Previous work implicates endoglin in the regulation of cell migration (Conley et al., 2004; Guerrero-Esteo et al., 1999), and therefore we determined if the vascular defects were due to inhibition of migration of neural crest-derived SMC precursors to major vessels and the outflow tract of the developing heart. TgEngloxP mice were bred with R26R mice bearing a cre-inducible LacZ reporter (TgEngloxP;R26R) (Jiang et al., 2000; Soriano, 1999). These mice were subsequently bred to the Wnt1cre driver, to label endoglin-over expressing neural crest cells and their derivatives. Analysis of transverse sections through the heart region of E12.5 embryos revealed little effect of endoglin over expression on the number of neural crest cells in this region using this technique (Figs. 3A-E), indicating that a defect in cell migration was not likely the cause of this phenotype.
Figure 3. Vascular hemorrhaging in vivo is correlated with an increase in disorganized vascular SMCs.
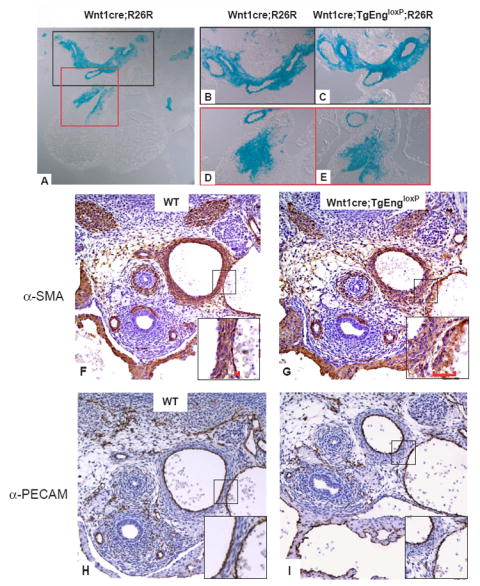
(A) β-galactosidase staining of a transverse section through the anterior region the heart of an E12.5 Wnt1cre;R26R embryo reveals the structures arising from NCSCs, including the major vessels (black box) and outflow tract (red box). (B, C) Higher magnification of Wnt1cre;R26R and Wnt1cre;TgEngloxP;R26R highlights major vessels and (D, E) cardiac outflow tract. (F, G) α-SMA immunohistochemical analysis of major vessels of E12.5 Wnt1cre;TgEndloxP embryos compared to wild type. The higher magnification view shows the layer of SMCs was thicker and less compact in Wnt1cre;TgEndloxP vessels and the individual SMCs demonstrated a rounded morphology (note areas delimited by red bars within black box). (H, I) Immunohistochemical staining for PECAM in sections adjacent to (F, G).
Endoglin over expression in NCSCs results in aberrant vascular SMC development
The loss of blood vessel integrity in Wnt1cre;TgEngloxP embryos could be due to a disturbance in development of the vascular SMC layer. Indeed, examination of the major vessels surrounding the hearts of E12.5 Wnt1cre;TgEngloxP;R26R embryos using the SMC marker, α-SMA and the endothelial specific marker, PECAM revealed disorganization of SMCs (Figs. 3F,G) with normal endothelial morphology (Figs. 3H, I). Furthermore, the SMC layer in the vessel was broader and less compact in the Wnt1cre;TgEngloxP embryos compared to wild type (insets, Figs. 3F,G). The SMCs had a rounded morphology, which is indicative of defects in cell spreading and adhesion (Foo et al., 2006). These data suggest that the embryonic defects result from aberrant SMC differentiation and organization. However, while the number of neural crest cells was similar between the wild type and Wnt1cre;TgEngloxP embryos (Figure 3B-E), α-these data suggested that there was enhancement of smooth muscle cell differentiation in Wnt1cre;TgEngloxP embryos.
The effects of endoglin over expression on neural crest-derived SMCs are potentially Smad-independent
Neural crest smooth muscle specification is thought to be regulated by TGFβ- and Smad-dependent signaling (Chen and Lechleider, 2004). Because endoglin acts as an auxiliary cell-surface receptor that interacts with TGFβ and BMP ligands (Barbara et al., 1999), we wanted to ascertain whether the cardiovascular defects in Wnt1cre;TgEngloxP embryos could be directly due to hyperactivation of TGFβ or BMP signaling. Therefore, we looked to see if endoglin was effecting Smad activation. The degree of activation of BMP and TGFβ signaling in neural crest-derived structures was assayed by immunohistochemical staining for phosphorylated forms of the Smad transcription factors, which are the most thoroughly characterized effectors of TGFβ signaling. Immunohistochemistry using an antibody that recognizes the phosphorylated receptor-restricted Smads, pSmad 1/5/8, which mediate BMP signaling (Figs. 4A,B) or pSmad 2/3, which mediate TGFβ signaling (Figs. 4C,D), revealed no differences in Smad phosphorylation in neural crest-derived structures between Wnt1cre;TgEngloxP and wild type embryos. Importantly, there was no alteration in Smad phosphorylation in the aberrant SMCs in the Wnt1cre;TgEngloxP embryos. Thus, over expression of endoglin in NCSCs does not lead to increased levels of phosphorylated Smads characteristic of amplified pathway activation.
Figure 4. Over expression of endoglin in vivo results in aberrant neural crest-derived SMCs independently of Smads.
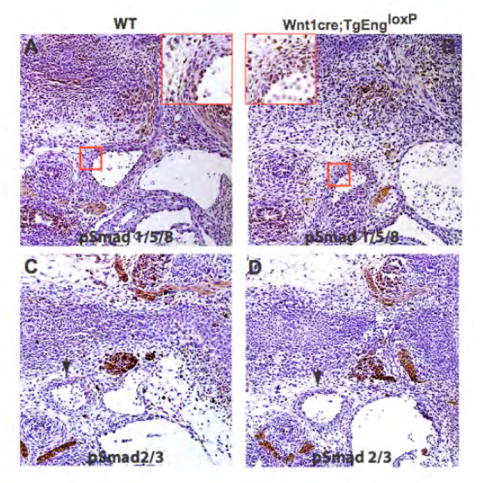
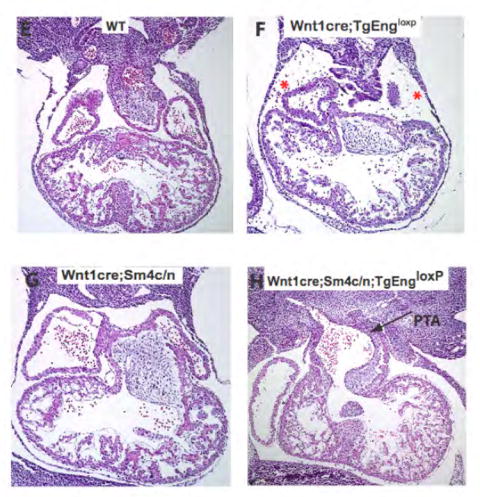

(A,B) α-phospho Smad 1/5/8 and (C,D) α-phospho Smad 2/3 antibody immunohistochemistry in E12.5 Wnt1cre;TgEngloxP (B,D) compared to wild type (A,C) embryos. Note that there is a similar percentage of pSmad 1/5/8-expressing cells in the wall of the vessel (red boxes in A,B) and no ectopic expression of pSmad 2/3 in the neural crest-derived vessels (arrows, C,D), as revealed by . (E-H) H&E analysis of sections through the heart region of E11 embryos which are either (E) wild type, (F) Wnt1cre;TgEngloxP, (G) Smad4 conditional allele on the heterozygous null background (Wnt1cre;Sm4c/n) alone, or (H) in compound mutant mouse embryos in which endoglin is over expressed and Smad4 is simultaneously inactivated in neural crest cells (Wnt1cre;Sm4c/n;TgEngloxP). Asterisks indicate hemorrhaging (F). Arrow indicates region of persistent truncus arteriosus (PTA) in panel H which is absent in panels E-G. (I) Immunoblot analysis of total α-SMA (vascular smooth muscle) and Flk-1 (endothelium) in E11.5 Wnt1cre;TgEngloxP embryos compared to wild type. (J) Endoglin immunoprecipitation followed by immunoblot analysis of endoglin expression upon cre-recombination in wild type versus SM22αcre;TgEngloxP transgenic embryos. (K) Immunoblot analysis of total α-SMA in E11.5 SM22αcre;TgEngloxP whole embryos compared to wild type embryos.
To verify that the effect of endoglin over expression in the neural crest is independent of hyperactivation of the canonical TGFβ and BMP signaling pathways, we generated compound mutant mice in which endoglin was over expressed and Smad4 was simultaneously inactivated in neural crest cells (Wnt1cre;Sm4c/n;TgEngloxP) (Chu et al., 2004). Smad4 is required for the canonical signaling responses to the TGFβ-, activin- and BMP-related ligands (Shi and Massague, 2003), and represents a tractable target for conditional inactivation of Smad-dependent TGFβ receptor signaling. If endoglin over expression hyperactivated canonical TGFβ signaling, we predicted that inactivation of Smad4 should ameliorate the cardiovascular defects seen when endoglin is over expressed in NCSCs. However, if endoglin were operating in a distinct pathway, inactivation of Smad4 would either have no effect on, or could exacerbate the Wnt1cre;TgEngloxP phenotype. Indeed, we found exacerbation of the Wnt1cre;TgEngloxP phenotype upon simultaneous inactivation of Smad4. Wnt1cre;Sm4c/n;TgEngloxP embryos demonstrated characteristic defects in the outflow tract, persistent truncus arteriosus, in which there was a loss of septation of the aorta and pulmonary trunk which form the outflow tract (Fig. 4H). This defect was not observed in wild type (Fig. 4E) or Wnt1cre;Sm4c/n embryos (Fig. 4G). Consistent with the data shown in Figure 2F, hemorrhaging was observed in the Wnt1cre;TgEngloxP sections (Fig. 4F). As shown in Figure 4E-H, hearts of compound Wnt1cre;Sm4c/n;TgEngloxP embryos displayed a malformation more severe than in Wnt1cre;TgEngloxP embryos. Collectively, these results strongly suggest that endoglin is operating in a pathway or pathways separate from or overlapping with canonical TGFβ receptor signaling.
Endoglin expression affects SMC specification
To further understand the role of endoglin in SMC differentiation from neural crest, we performed whole embryo immunoprecipitation of α-SMA at E11.5 followed by immunoblot analysis. Immunoblotting demonstrated that total protein levels of α-SMA were consistently higher in the Wnt1cre;TgEngloxP embryos compared to the wild type control embryos (Fig. 4I). Consistent with immunohistochemical data, no differences were observed in total protein levels of the endothelial cell marker Flk-1 (Fig. 4I). This result contrasted with use of the SM22αcre driver to activate the expression of the TgEngloxP transgene in specified SMC, where total protein levels of α-SMA were similar in SM22αcre;TgEngloxP embryos compared to wild type (Fig. 4K). We confirmed that in vivo cre-recombination and subsequent expression of the endoglin transgene also occurred using the SM22αcre driver (Fig. 4J). Because SM22α expression occurs in committed SMC (Solway et al., 1995), whereas Wnt1cre activates endoglin expression in a progenitor population (Choudhary et al., 2006), these data provide confirmation that neural crest-expressed endoglin affected SMC specification.
Endoglin is required for SMC differentiation potential of NCSCs
To further understand the functional role of endoglin in SMC differentiation from neural crest stem cells, we performed clonogenic differentiation assays (Stemple and Anderson, 1992). The SMC differentiation potential of NCSCs isolated from explants of rat E10.5 neural tubes were evaluated by two parameters: i) the generation of SMC-only containing clones (M-only), defined by expression of α-SMA, and ii) the production of clones containing SMCs (i.e. M-Containing) as well as glia or neuronal cells (N-Containing), or both as defined by expression of the markers GFAP and peripherin, respectively. Isolated NCSCs could be divided into two populations based on endoglin expression determined by the presence or absence of CD105 staining (data not shown), hereafter referred to as NCSCCD105+ and NCSCCD105-, which was consistent with our expression data in embryos showing that only a subpopulation of migrating neural crest cells express endoglin (Figure 1). The NCSCCD105- population of cells constituted approximately 25% of the total NCSCs isolated. We therefore explored the possibility that differences in SMC differentiation potential existed between NCSCCD105+ and NCSCCD105-. Lack of endoglin expression in the NCSCCD105- population correlated with impaired SMC differentiation potential as demonstrated by a decrease in the generation of M-Containing clones (Fig. 5A), which are defined as clones containing at least one SMC actin-positive cell (Kubu et al., 2002; Shah and Anderson, 1997). NCSCCD105- and NCSCCD105+ were equally capable of producing clones containing neuronal cells, indicating that this was not due to an overall decrease in differentiation capacity. SMC differentiation potential of NCSCCD105+ was enhanced by treatment with 5 pM TGFβ1. However, NCSCCD105- failed to show enhanced M-Containing clone production in response to TGFβ1 (Fig. 5A), even when the concentration of TGFβ1 was increased by one order of magnitude (data not shown).
Figure 5. Endoglin is required for myogenic differentiation of NCSCs.
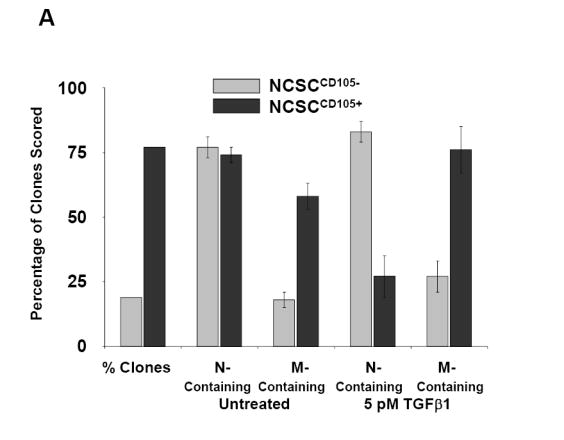
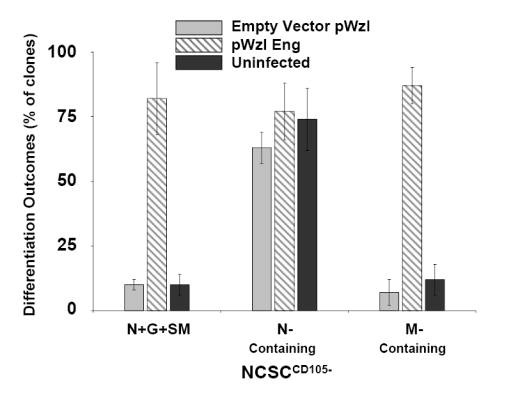
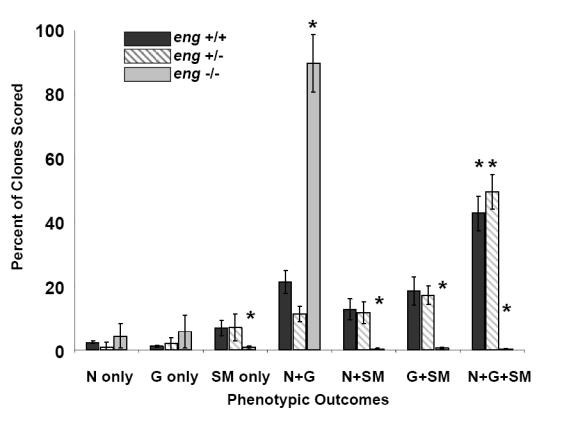
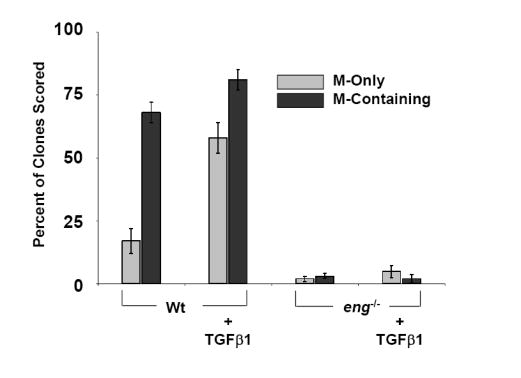
(A) Clonogenic differentiation assays from E10.5 rat neural tube explants can be divided into two populations based on endoglin expression. Presented are the percentages of clones scored that were neural-containing (N-Containing) or smooth muscle-containing (M-Containing), which were defined as clones containing one or more cells of that type, as determined by marker analysis. NCSCCD105+ relative to NCSCCD105- exhibit a higher myogenic differentiation potential. Myogenic differentiation in NCSCCD105- can not be restored to that of NCSCCD105+ by treatment with TGFβ1. Presented are the means, ± standard deviation, of three independent experiments. (B) Reconstitution of endoglin expression in NCSCCD105- via retroviral transduction. Presented are the means ± standard deviation of four experiments originating from distinct cell isolations, identifications, and retroviral transduction. (C) Clonogenic differentiation assay of NCSCs isolated from E9.5 wild type eng+/+, heterozygous eng+/-, and eng null eng-/- embryos. (D) Clonogenic differentiation assay of NCSCs isolated from E9.5 eng+/+ and eng-/- embryos treated with TGFβ1. Values presented are the means ± the standard deviation of three separate experiments.
Reconstitution of endoglin expression restores SMC differentiation potential in NCSCCD105-
The observation that NCSCCD105- were impaired in their capacity for SMC differentiation established a correlation between the levels of endoglin expression and SMC differentiation potential. Interestingly, reconstitution of endoglin expression in NCSCCD105- by retroviral transduction restored SMC differentiation potential, compared to either control-transduced or uninfected NCSCCD105- (Fig. 5B). In addition to demonstrating a causal relationship between endoglin expression and SMC differentiation potential, this experiment indicated that NCSCCD105+ population contained the necessary signaling components for SMC differentiation.
Endoglin null NCSCs exhibit loss of SMC potential
We next sought to determine whether endoglin was required for the myogenic differentiation potential of NCSCs. To address this question, NCSCs were isolated from E9.5 eng-/-, eng+/-, and wild type (eng+/+) neural tube explants. Clonogenic differentiation assays demonstrated that eng-/- NCSCs were deficient in their SMC differentiation potential as compared to eng+/+ and eng+/- clones (Fig. 5C). Clonogenic differentiation assays for SMC potential of eng-/- NCSCs in response to TGFβ1 also demonstrated minimal SMC progeny (Fig. 5D). These results provide strong support for the hypothesis that endoglin is required for SMC differentiation from NCSCs.
NCSCs undergo age-related loss of SMC differentiation potential and endoglin expression
As previously described, the neurogenic potential of NCSCs decreases as gliogenic potential increases during development (Kubu et al., 2002; White et al., 2001). Therefore, we determined whether myogenic potential of NCSCs was also affected during development and in the adult. Comparison of the myogenic potential of NCSCs derived from E10.5 neural tube explants and E12.5 and adult (>6 weeks) sciatic nerve explants showed that in adult NCSCs, there was a significant (p≤.025) decline in ability to form M-Containing clones into adulthood compared with those isolated at E10.5. This result suggested that myogenic potential decreased with age (Fig. 6A). To assess the sensitivity of NCSCs to TGFβ1 as a function of age, we examined the ability of TGFβ1 to instruct SMC differentiation over a developmental time course (Figs. 6B,C). Interestingly, the frequency of NCSCs generating M-Only clones steadily declined with age, even in the presence of increasing doses of TGFβ1 (Fig. 6B). The concentration that produced a half-maximal response [EC50] increased from 500 pM at E10.5 to 650 pM at E14, and to 1600 pM in NCSCs in the adult (Fig. 6B). Whereas TGFβ1 enhanced the overall production of α-SMA-positive clones, the myogenic activity of TGFβ1 significantly decreased with age; Overall, 88 ± 6% of E10.5 NCSCs were competent to respond to TGFβ1 and produce myogenic clones, compared to 48 ± 12% of adult cells (Fig. 6C).
Figure 6. Myogenic differentiation potential of NCSCs decrease with age with a concomitant decrease in endoglin expression and responsiveness to TGFβ1.
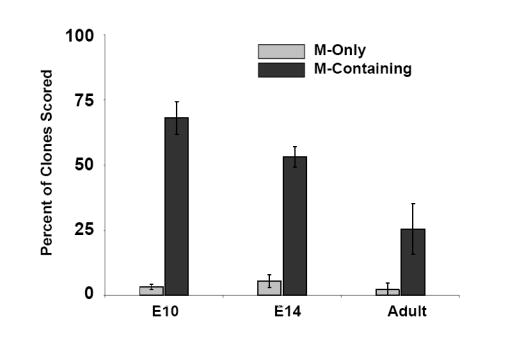
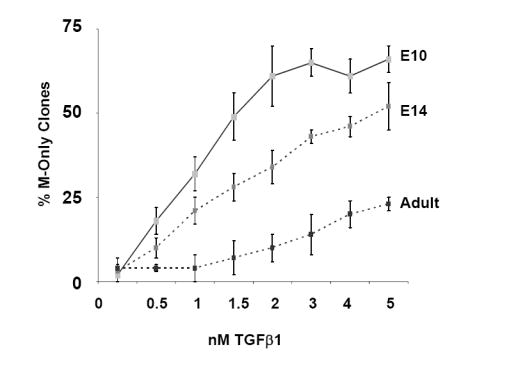
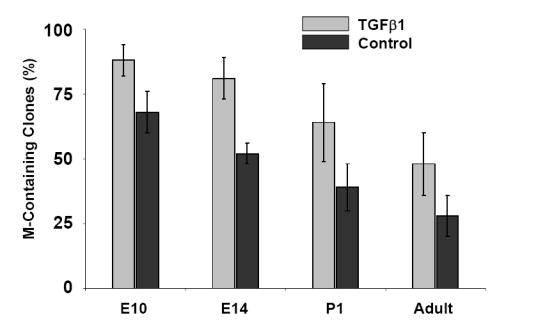
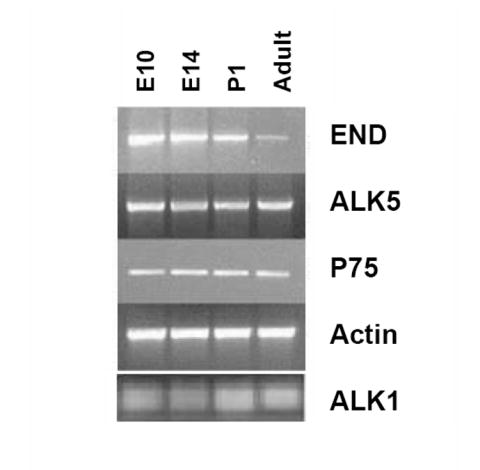
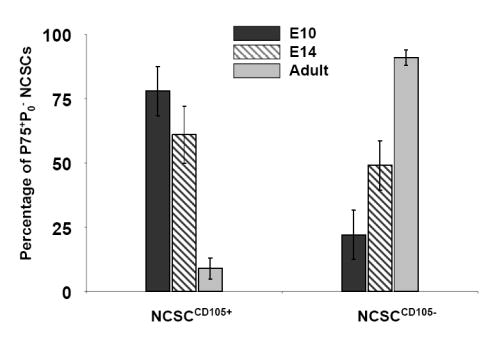
(A) Clonogenic differentiation assays conducted using NCSC isolated from E10.5 neural tube, and E14 and adult sciatic nerve. Production of muscle-only (M-Only) nd M-Containing clones is compared. Presented are the means, ± standard deviation of three experiments. (B,C) NCSCs isolated from E10.5, E14, and adult animals are evaluated for the production of clones containing (B) only myogenic progeny (M-Only) or (C) containing at least one differentiated myogenic cell (M-Containing) in response to TGFβ1. Presented are the means ± standard deviation of three experiments representing a minimum of 200 clones scored. (D) RT-PCR of NCSCs isolated at different time points for the TGFβ type I receptors ALK1 and ALK5, and the low affinity nerve growth factor receptor (p75). Levels of β-actin serve as a control. (E) NCSCs were identified from E10 neural tube explants 12 hours after isolation and culturing in Morrison media. Non-NCSCs were removed from the dish and the remaining cells stained for Endoglin expression. A similar strategy was used on FACS isolated NCSC from the sciatic nerve of E14 and adult rats. Presented are the means ± St.Dev on two experiments originating from distinct isolations.
Studies addressing NCSC maturation demonstrated quantitative changes in the expression of genes in the pathways involved in instructive signaling events (Kubu et al., 2002; White et al., 2001). The loss of neurogenic potential is associated with a loss of the expression of the BMP type I receptor ALK3, and enhanced gliogenic differentiation is correlated with an increase in Notch expression (Kubu et al., 2002; Morrison et al., 1999; White et al., 2001). Therefore, we asked whether there is a similar age-related decline in TGFβ signaling components and subsequent SMC differentiation potential of NCSCs. Interestingly, the age-related loss of SMC differentiation potential in NCSCs did not correlate with changes in mRNA levels of the type I receptors for TGFβ1, ALK1, or ALK5 which remained unchanged (Fig. 6D). In contrast, endoglin mRNA expression in NCSCs decreased with age (Fig. 6D) and was accompanied by corresponding changes in endoglin protein expression. Approximately 75-85% of NCSCs that were isolated from E10.5 neural tube expressed endoglin compared with 10-20% from the adult sciatic nerve (Fig. 6E). As scored by the number of clones present at end of experiment (day 3), this number never dropped below 95% and there was no significant difference between any of the conditions. Thus, clone survival was not a significant contributor to these results.
Taken together, these findings demonstrate that endoglin expression decreases with age in NCSCs, which correlates with a decrease in responsiveness to TGFβ1 and SMC differentiation potential.
Discussion
Appropriate specification of stem cell fates is essential for normal vertebrate development. NCSCs give rise to a variety of cell types, including neurons, glia, and the SMCs of the major vessels and outflow tract of the heart (Fig. 7). NCSCs undergo age-related changes in their ability to respond to instructive signals (Kubu et al., 2002). For example, a decline in neuronal versus glial differentiation of NCSCs with age is well established. Neurogenic specification is instructed by BMP2 and BMP4, and age-related decreases in neurogenic differentiation potential can be attributed to decreases in the levels of the BMP type I receptor, ALK3. TGFβ1 was identified as an instructive myogenic signal for NCSCs (Chen and Lechleider, 2004; Gadson et al., 1997); however, the mechanisms governing NCSC differentiation potential remain poorly understood. Clonogenic assay of neural crest stem cells demonstrated a requirement for endoglin in SMC differentiation from NCSCs, because SMC differentiation was blocked in the absence of endoglin, even in the presence of TGFβ1. We note that subtle differences in cell proliferation between populations would not contribute to the observed changes in clone composition because situations in which there was either 1 neuron and 1000 glia or 500 neurons and 5000 glia were still recorded as a neuron-containing or glia-containing clone. Similarly, significant differences in cell survival in the clonogenic assay could be excluded because of the absence of α-SMA-positive and annexin V-positive cells. Additionally, we also used Hoescht staining to look for nuclear fragmentation of α-SMA-positive cells and this result was also negative (data not shown).
Figure 7. Endoglin is required for the myogenic differentiation potential of neural crest stem cells.
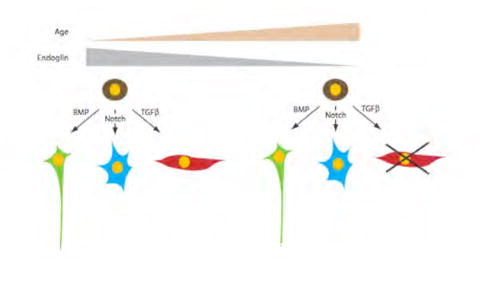
The schematic summarizes the findings suggesting that NCSCs undergo age-related loss of endoglin expression. Loss of endoglin expression is accompanied by a decrease in their responsiveness to the myogenic instructive ligand TGFβ, resulting in impaired myogenic potential, whereas other cell fates are not affected.
Furthermore, we showed that similar to neurogenic differentiation potential of NCSCs, SMC differentiation potential decreased with age. However, the loss of potential correlated with a loss of endoglin expression, not with loss of the TGFβ type I receptors ALK1 or ALK5. These data agree with recent findings that human mesenchymal stem cells expressing endoglin demonstrated the capacity for SMC differentiation potential (Roura et al., 2006), suggesting that endoglin may have a general role in SMC differentiation of stem cell populations.
Endoglin has been extensively studied in endothelial cells; however, an important role in SMC differentiation is suggested by the phenotype of the null mouse (Arthur et al., 2000; Bourdeau et al., 1999; Li et al., 1999). Eng-/- embryos die by E11.5 due to defects in angiogenesis characterized by failure of SMC differentiation and investment accompanied by normal endothelial development (Arthur et al., 2000; Bourdeau et al., 1999; Li et al., 1999). From analysis of the endoglin loss of function phenotype, it is unclear whether endoglin plays an essential cell autonomous role in SMC, or if impaired SMC development is secondary to a lack of endoglin expression in the endothelium. For the first time, data presented in this study demonstrate an intrinsic, cell autonomous role for endoglin in SMC precursors.
Over expression of endoglin in neural crest cells resulted in pronounced hemorrhaging in cranial vessels and the pericardial cavity. Hemorrhaging and aberrant SMC’s in the wall of the vessels did not appear to be associated with cell death or proliferation as measured by TUNEL and phospho histone H3 staining, respectively. Hemorrhaging into the pericardial sac could have resulted from defects in the junction between the smooth muscle and the myocardium, arising from perturbation of the secondary heart field with altered patterning of the atrial pole (Waldo et al., 2005). Lineage tracing studies demonstrated that migration of endoglin over expressing NCSCs to vessel walls is comparable with wild type, arguing against a migration defect as the origin of the vascular phenotype. Furthermore, skewing of NCSC fates secondary to endoglin over expression is unlikely because we show that NCSCCD105+ and NCSCCD105- were both capable of producing neuronal and glial cells. This finding was corroborated in vivo because endoglin specifically affected neural crest-derived SMC; whereas other neural crest derivatives including craniofacial structures, branchial arches, and dorsal root ganglion (data not shown) were grossly normal.
Importantly, hemorrhaging was not observed in the trunk vessels, where the contribution of NCSCs to SMC is absent. Hemorrhaging was ostensibly due to decreased vascular integrity secondary to disorganization of SMCs in the walls of major vessels, an area populated by Wnt1-expressing cells. In addition, SMC disorganization was accompanied by an apparent increase in the number of SMCs. Upregulation of α-SMA expression in endoglin-expressing NCSCs is consistent with data suggesting enhanced SMC differentiation. We cannot exclude the possibility of an increase in total α-SMA protein expression in the same number of SMC. However, over expression of endoglin using the SM22αcre driver had no effect on total protein levels of α-SMA. This observation suggests that the effect of endoglin expression on SMC differentiation is specific to stem cell populations and does not impact committed progenitors.
An explanation for the disorganization and rounded morphology of SMCs secondary to endoglin over expression is alteration in the cell adhesive properties. Previously, we demonstrated: i) a role for endoglin in regulating cell adhesion through interactions with the focal adhesion protein, zyxin, and ii) subsequent modulation of the levels of p130Cas and Crk in sites of focal adhesion (Conley et al., 2004). Like endoglin, Ephrin B2 is highly expressed in vascular endothelial cells with lower levels of expression seen in vascular SMCs. Conditional inactivation of Ephrin B2 in SMCs results in impaired mural cell attachment surrounding the vessels due to an apparent lack of focal adhesions and altered Crk-p130Cas signaling (Foo et al., 2006). More recently, mice in which Notch signaling was specifically inactivated in derivatives of the neural crest exhibited aortic arch patterning defects, pulmonary artery stenosis, and ventricular septal defects (High et al., 2007). Immunohistochemical examination of mutant arteries revealed disruption of the smooth muscle architecture that is similar to that seen in Figure 3. Similarly, over expression of endoglin may modulate interactions with proteins such as zyxin and zyxin-related protein-1 (Conley et al., 2004; Sanz-Rodriguez et al., 2004), thus modifying adhesive properties of SMCs. Therefore, we propose that the observed cardiovascular phenotype of the Wnt1cre;TgEngloxP might be due to a combination of enhanced SMC differentiation potential accompanied by defects in cellular adhesion. These results are consistent with a cell autonomous role for endoglin in the maintenance of vascular integrity through SMC differentiation and adhesion.
Although TGFβ1 is implicated in SMC differentiation in vitro (Chen and Lechleider, 2004; Shah and Anderson, 1997), recent studies in vivo suggest TGFβ1 signaling in neural crest does not specify SMCs. Surprisingly, conditional inactivation of ALK5 and TβRII in the neural crest results in normal SMC differentiation (Choudhary et al., 2006; Wang et al., 2006). Thus in vivo, it appears that factors other than TGFβ1 specify SMC from neural crest. Interestingly, Wnt1cre conditional inactivation of the putative BMP receptor ALK2 results in impaired SMC differentiation (Kaartinen et al., 2004). We showed by anti-phospho Smad 2/3 and anti-phospho Smad 1/5/8 antibody immunostaining that endoglin over expression had no apparent effect on canonical TGFβ or BMP receptor signaling. Furthermore, a compound mutant in which the Smad4 transcription factor is inactivated simultaneously with endoglin over expression suggests that there is no epistatic relationship between these two genes, indicating that they operate in distinct pathways. In this regard, our data suggesting the lack of involvement of canonical signaling is consistent with recent studies indicating that α-SMA expression in response to TGFβ involves Smad-independent signaling (Chen et al., 2006). These results support our hypothesis that endoglin plays a Smad-independent role in SMC differentiation from NCSCs in vivo.
Involvement of endoglin in non-canonical signaling was previously suggested based on the phenotypic comparison between the eng-/- and TAK1-/- mouse strains (Jadrich et al., 2006). TAK1 is a non-canonical Smad-independent effector of TGFβ signaling. Similar to the eng-/- mouse, SMC development was impaired with normal endothelial cell development (Jadrich et al., 2006), thereby raising the possibility that TAK1 may act downstream of endoglin.
Accumulating evidence suggests that the precursors of HHT lesions may occur early in human development (Mei-Zahav et al., 2006; Morgan et al., 2002). Our data suggest that vascular smooth muscle precursor cells that are deficient in endoglin may contribute to the formation of lesions that can appear later in life. By elucidating the molecular mechanisms through which endoglin functions, we will better understand the process of SMC differentiation and homeostasis. These findings may provide further insight into the vascular disorder HHT, which is caused by haploinsufficiency of endoglin with resulting vascular fragility. Moreover, we propose that these insights may be applicable to other stem cell populations with SMC differentiation potential such as mesenchymal stem cells, which also express endoglin (Vogel et al., 2003; Zvaifler et al., 2000) and are involved in vasculogenesis (Gojo et al., 2003).
Acknowledgments
The authors thank Anne Harrington and the Mouse Transgenic and Magnetic Resonance Imaging Core Facility for production of the transgenic mice, and Kathleen Carrier and the Histology Core Facility for expert preparation of embryo sections. We thank Dr. D. Anderson for anti-SOX-10 and anti-HNK-1 antibodies. This work was supported in part by NIH/NCRR grants P20 RR 18789 (JMV and LHO), and P20 RR 15555 (CPHV) from the COBRE program of the National Center for Research Resources, and R01-HL083151 from the NIH National Heart , Lung, and Blood Institute (CPHV).
Abbreviations
- BMP
bone morphogenetic protein
- eng
endoglin gene
- SMC
smooth muscle cell
- Eng
endoglin protein
- (α-SMA)
smooth muscle alpha actin
- HHT
hereditary hemorrhagic telangiectasia
- p75
low affinity nerve growth factor receptor
- Po
myelin P0 protein
- GFAP
glial fibrillary acidic protein
- NCSC
neural crest stem cell
- TGFβ1
transforming growth factor β1
- ALK
activin receptor-like kinase
- TβRII
transforming growth factor beta type 2 receptor
- RT-PCR
reverse transcriptase polymerase chain reaction
- TUNEL
terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson DJ. Cellular and molecular biology of neural crest cell lineage determination. Trends Genet. 1997;13:276–80. doi: 10.1016/s0168-9525(97)01187-6. [DOI] [PubMed] [Google Scholar]
- Anderson DJ, Axel R. A bipotential neuroendocrine precursor whose choice of cell fate is determined by NGF and glucocorticoids. Cell. 1986;47:1079–90. doi: 10.1016/0092-8674(86)90823-8. [DOI] [PubMed] [Google Scholar]
- Arthur HM, Ure J, Smith AJ, Renforth G, Wilson DI, Torsney E, Charlton R, Parums DV, Jowett T, Marchuk DA, Burn J, Diamond AG. Endoglin, an ancillary TGFbeta receptor, is required for extraembryonic angiogenesis and plays a key role in heart development. Dev Biol. 2000;217:42–53. doi: 10.1006/dbio.1999.9534. [DOI] [PubMed] [Google Scholar]
- Barbara NP, Wrana JL, Letarte M. Endoglin Is an Accessory Protein That Interacts with the Signaling Receptor Complex of Multiple Members of the Transforming Growth Factor- beta Superfamily. J Biol Chem. 1999;274:584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Upregulation of endoglin (CD105) expression during childhood brain tumor-related angiogenesis. Anti-angiogenic therapy. Anticancer Res. 1998;18:1485–500. [PubMed] [Google Scholar]
- Bourdeau A, Dumont DJ, Letarte M. A murine model of hereditary hemorrhagic telangiectasia. J Clin Invest. 1999;104:1343–1351. doi: 10.1172/JCI8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner-Fraser M. Origins and developmental potential of the neural crest. Exp Cell Res. 1995;218:405–17. doi: 10.1006/excr.1995.1173. [DOI] [PubMed] [Google Scholar]
- Cepko CL, Ryder E, Austin C, Golden J, Fields-Berry S, Lin J. Lineage analysis using retroviral vectors. Methods. 1998;14:393–406. doi: 10.1006/meth.1998.0594. [DOI] [PubMed] [Google Scholar]
- Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, Letarte M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- Chen CZ, Li M, de Graaf D, Monti S, Gottgens B, Sanchez MJ, Lander ES, Golub TR, Green AR, Lodish HF. Identification of endoglin as a functional marker that defines long-term repopulating hematopoietic stem cells. Proc Natl Acad Sci U S A. 2002;99:15468–73. doi: 10.1073/pnas.202614899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Crawford M, Day RM, Briones VR, Leader JE, Jose PA, Lechleider RJ. RhoA Modulates Smad Signaling during Transforming Growth Factor-beta-induced Smooth Muscle Differentiation. J Biol Chem. 2006;281:1765–70. doi: 10.1074/jbc.M507771200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–9. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Chu GC, Dunn NR, Anderson DC, Oxburgh L, Robertson EJ. Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development. 2004;131:3501–12. doi: 10.1242/dev.01248. [DOI] [PubMed] [Google Scholar]
- Chytil A, Magnuson MA, Wright CV, Moses HL. Conditional inactivation of the TGF-beta type II receptor using Cre:Lox. Genesis. 2002;32:73–5. doi: 10.1002/gene.10046. [DOI] [PubMed] [Google Scholar]
- Conley BA, Koleva RI, Smith JD, Kacer D, Zhang D, Bernabeu C, Vary CPH. Endoglin Controls Cell Migration and Composition of Focal Adhesions. J Biol Chem. 2004;279:27440–27449. doi: 10.1074/jbc.M312561200. [DOI] [PubMed] [Google Scholar]
- Conley BS, Smith JD, Guerrero-Esteo M, Bernabeu C, Vary CPH. Endoglin, a TGF-beta receptor-associated protein, is expressed by smooth muscle cells in human atherosclerotic plaques. Atherosclerosis. 2000;153:323–335. doi: 10.1016/s0021-9150(00)00422-6. [DOI] [PubMed] [Google Scholar]
- Foo SS, Turner CJ, Adams S, Compagni A, Aubyn D, Kogata N, Lindblom P, Shani M, Zicha D, Adams RH. Ephrin-B2 controls cell motility and adhesion during blood-vessel-wall assembly. Cell. 2006;124:161–73. doi: 10.1016/j.cell.2005.10.034. [DOI] [PubMed] [Google Scholar]
- Gadson PF, Jr, Dalton ML, Patterson E, Svoboda DD, Hutchinson L, Schram D, Rosenquist TH. Differential response of mesoderm- and neural crest-derived smooth muscle to TGF-beta1: regulation of c-myb and alpha1 (I) procollagen genes. Exp Cell Res. 1997;230:169–80. doi: 10.1006/excr.1996.3398. [DOI] [PubMed] [Google Scholar]
- Gojo S, Gojo N, Takeda Y, Mori T, Abe H, Kyo S, Hata J, Umezawa A. In vivo cardiovasculogenesis by direct injection of isolated adult mesenchymal stem cells. Exp Cell Res. 2003;288:51–9. doi: 10.1016/s0014-4827(03)00132-0. [DOI] [PubMed] [Google Scholar]
- Guerrero-Esteo M, Lastres P, Letamendia A, Perez-Alvarez MJ, Langa C, Lopez LA, Fabra A, Garcia-Pardo A, Vera S, Letarte M, Bernabeu C. Endoglin overexpression modulates cellular morphology, migration, and adhesion of mouse fibroblasts. European Journal of Cell Biology. 1999;78:614–623. doi: 10.1016/S0171-9335(99)80046-6. [DOI] [PubMed] [Google Scholar]
- High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–63. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann SL, Russell DW, Brown MS, Goldstein JL, Hammer RE. Overexpression of low density lipoprotein (LDL) receptor eliminates LDL from plasma in transgenic mice. Science. 1988;239:1277–81. doi: 10.1126/science.3344433. [DOI] [PubMed] [Google Scholar]
- Hogan BL, Beddington RS, Costantini F, Lacy E. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1994. [Google Scholar]
- Jadrich JL, O’Connor M B, Coucouvanis E. The TGF{beta} activated kinase TAK1 regulates vascular development in vivo. Development. 2006;133:1529–41. doi: 10.1242/dev.02333. [DOI] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–16. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–16. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–90. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- Koleva RI, Conley BA, Romero D, Riley KS, Marto JA, Lux A, Vary CP. Endoglin Structure and Function: Determinants of Endoglin Phosphorylation by Transforming Growth Factor-beta Receptors. J Biol Chem. 2006;281:25110–23. doi: 10.1074/jbc.M601288200. [DOI] [PubMed] [Google Scholar]
- Kubu CJ, Orimoto K, Morrison SJ, Weinmaster G, Anderson DJ, Verdi JM. Developmental changes in Notch1 and numb expression mediated by local cell-cell interactions underlie progressively increasing delta sensitivity in neural crest stem cells. Dev Biol. 2002;244:199–214. doi: 10.1006/dbio.2002.0568. [DOI] [PubMed] [Google Scholar]
- Li DY, Sorensen LK, Brooke BS, Urness LD, Davis EC, Taylor DG, Boak BB, Wendel DP. Defective angiogenesis in mice lacking endoglin. Science. 1999;284:1534–7. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- Lwigale PY, Conrad GW, Bronner-Fraser M. Graded potential of neural crest to form cornea, sensory neurons and cartilage along the rostrocaudal axis. Development. 2004;131:1979–91. doi: 10.1242/dev.01106. [DOI] [PubMed] [Google Scholar]
- Ma X, Labinaz M, Goldstein J, Miller H, Keon WJ, Letarte M, O’Brien E. Endoglin Is overexpressed after arterial injury and is required for transforming growth factor-ss-induced inhibition of smooth muscle cell migration. Arterioscler Thromb Vasc Biol. 2000;20:2546–2552. doi: 10.1161/01.atv.20.12.2546. [DOI] [PubMed] [Google Scholar]
- McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- Mei-Zahav M, Letarte M, Faughnan ME, Abdalla SA, Cymerman U, MacLusky IB. Symptomatic children with hereditary hemorrhagic telangiectasia: a pediatric center experience. Arch Pediatr Adolesc Med. 2006;160:596–601. doi: 10.1001/archpedi.160.6.596. [DOI] [PubMed] [Google Scholar]
- Minarcik JC, Golden JA. AP-2 and HNK-1 define distinct populations of cranial neural crest cells. Orthod Craniofac Res. 2003;6:210–9. doi: 10.1046/j.1601-6335.2003.00268.x. [DOI] [PubMed] [Google Scholar]
- Morgan T, McDonald J, Anderson C, Ismail M, Miller F, Mao R, Madan A, Barnes P, Hudgins L, Manning M. Intracranial hemorrhage in infants and children with hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome) Pediatrics. 2002;109:E12. doi: 10.1542/peds.109.1.e12. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Perez SE, Qiao Z, Verdi JM, Hicks C, Weinmaster G, Anderson DJ. Transient Notch activation initiates an irreversible switch from neurogenesis to gliogenesis by neural crest stem cells. Cell. 2000;101:499–510. doi: 10.1016/s0092-8674(00)80860-0. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–49. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci U S A. 1993;90:8392–6. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece N, Vera S, Cymerman U, White RI, Jr, Wrana JL, Letarte M. Mutant endoglin in hereditary hemorrhagic telangiectasia type 1 is transiently expressed intracellularly and is not a dominant negative. J Clin Invest. 1997;100:2568–2579. doi: 10.1172/JCI119800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Pena A, Eleno N, Duwell A, Arevalo M, Perez-Barriocanal F, Flores O, Docherty N, Bernabeu C, Letarte M, Lopez-Novoa JM. Endoglin upregulation during experimental renal interstitial fibrosis in mice. Hypertension. 2002;40:713–20. doi: 10.1161/01.hyp.0000037429.73954.27. [DOI] [PubMed] [Google Scholar]
- Roura S, Farre J, Soler-Botija C, Llach A, Hove-Madsen L, Cairo JJ, Godia F, Cinca J, Bayes-Genis A. Effect of aging on the pluripotential capacity of human CD105(+) mesenchymal stem cells. Eur J Heart Fail. 2006 doi: 10.1016/j.ejheart.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Sanz-Rodriguez F, Guerrero-Esteo M, Botella LM, Banville D, Vary CPH, Bernabeu C. Endoglin Regulates Cytoskeletal Organization through Binding to ZRP-1, a Member of the Lim Family of Proteins. J Biol Chem. 2004;279:32858–32868. doi: 10.1074/jbc.M400843200. [DOI] [PubMed] [Google Scholar]
- Shah NM, Anderson DJ. Integration of multiple instructive cues by neural crest stem cells reveals cell-intrinsic biases in relative growth factor responsiveness. Proc Natl Acad Sci U S A. 1997;94:11369–74. doi: 10.1073/pnas.94.21.11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah NM, Groves AK, Anderson DJ. Alternative neural crest cell fates are instructively promoted by TGFbeta superfamily members. Cell. 1996;85:331–43. doi: 10.1016/s0092-8674(00)81112-5. [DOI] [PubMed] [Google Scholar]
- Shah NM, Marchionni MA, Isaacs I, Stroobant P, Anderson DJ. Glial growth factor restricts mammalian neural crest stem cells to a glial fate. Cell. 1994;77:349–60. doi: 10.1016/0092-8674(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Solway J, Seltzer J, Samaha FF, Kim S, Alger LE, Niu Q, Morrisey EE, Ip HS, Parmacek MS. Structure and expression of a smooth muscle cell-specific gene, SM22 alpha. J Biol Chem. 1995;270:13460–9. doi: 10.1074/jbc.270.22.13460. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stemple DL, Anderson DJ. Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell. 1992;71:973–85. doi: 10.1016/0092-8674(92)90393-q. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Anderson DJ. Neurotrophins regulate sequential changes in neurotrophin receptor expression by sympathetic neuroblasts. Neuron. 1994;13:1359–72. doi: 10.1016/0896-6273(94)90421-9. [DOI] [PubMed] [Google Scholar]
- Vogel W, Grunebach F, Messam CA, Kanz L, Brugger W, Buhring HJ. Heterogeneity among human bone marrow-derived mesenchymal stem cells and neural progenitor cells. Haematologica. 2003;88:126–33. [PubMed] [Google Scholar]
- Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Wang J, Nagy A, Larsson J, Dudas M, Sucov HM, Kaartinen V. Defective ALK5 signaling in the neural crest leads to increased postmigratory neural crest cell apoptosis and severe outflow tract defects. BMC Dev Biol. 2006;6:51. doi: 10.1186/1471-213X-6-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White PM, Morrison SJ, Orimoto K, Kubu CJ, Verdi JM, Anderson DJ. Neural crest stem cells undergo cell-intrinsic developmental changes in sensitivity to instructive differentiation signals. Neuron. 2001;29:57–71. doi: 10.1016/s0896-6273(01)00180-5. [DOI] [PubMed] [Google Scholar]
- Wurdak H, Ittner LM, Lang KS, Leveen P, Suter U, Fischer JA, Karlsson S, Born W, Sommer L. Inactivation of TGFbeta signaling in neural crest stem cells leads to multiple defects reminiscent of DiGeorge syndrome. Genes Dev. 2005;19:530–5. doi: 10.1101/gad.317405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212:191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- Young HM, Bergner AJ, Anderson RB, Enomoto H, Milbrandt J, Newgreen DF, Whitington PM. Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Dev Biol. 2004;270:455–73. doi: 10.1016/j.ydbio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Zvaifler NJ, Marinova-Mutafchieva L, Adams G, Edwards CJ, Moss J, Burger JA, Maini RN. Mesenchymal precursor cells in the blood of normal individuals. Arthritis Res. 2000;2:477–88. doi: 10.1186/ar130. [DOI] [PMC free article] [PubMed] [Google Scholar]