

Abstract
Src homology 2 (SH2) domains mediate protein-protein interactions by recognizing short phosphotyrosyl (pY) peptide motifs in their partner proteins. Protein tyrosine phosphatases (PTPs) catalyze the dephosphorylation of pY proteins, counteracting the protein tyrosine kinases. Both types of proteins exhibit primary sequence specificity, which plays at least a partial role in dictating their physiological interacting partners or substrates. A combinatorial peptide library method has been developed to systematically assess the sequence specificity of SH2 domains and PTPs. A “one-bead-one-compound” pY peptide library is synthesized on 90-μm TenteGel beads and screened against an SH2 domain or PTP of interest for binding or catalysis. The beads that carry the tightest binding sequences against the SH2 domain or the most efficient substrates of the PTP are selected by an enzyme-linked assay and individually sequenced by a partial Edman degradation/mass spectrometry technique. The combinatorial method has been applied to determine the sequence specificity of 8 SH2 domains from Src and Csk kinases, adaptor protein Grb2, and phosphatases SHP-1, SHP-2, and SHIP1 and a prototypical PTP, PTP1B.
Keywords: SH2 domain, PTP, combinatorial peptide library, sequence specificity, partial Edman degradation, phosphotyrosine, protein-protein interaction, signal transduction
1. Introduction
Reversible phosphorylation of proteins on tyrosyl residues is one of the key events that mediate the execution and regulation of many cellular processes. For example, phosphorylation can change the conformation and/or catalytic activity of a protein. It can also promote protein-protein interactions by recruiting Src homology 2 (SH2) or phosphotyrosine-binding (PTB) domain-containing proteins. A proper level of phosphorylation is critical for these processes and is controlled by the opposing action of protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs).
SH2 domains are ~100-aa modules present in many signaling proteins. The human genome encode 120 SH2 domains in 110 distinct proteins [1]. Numerous biochemical and structural studies of different SH2 domains have shown that they all bind to their partner proteins by recognition of short linear phosphotyrosyl (pY)-containing sequence motifs [2–5]. The pY peptide typically binds to the SH2 domain surface in an extended conformation [2]. A key interaction, which provides the majority of the binding energy (ΔG) and is common to all SH2 domains, is the insertion of the pY side chain into a deep pocket in the SH2 domain, where an invariant arginine residue (Arg βB5) forms a pair of hydrogen bonds with the pY phosphate group. This ensures that the SH2 domain acts as a phosphorylation-dependent molecular switch. The SH2-pY peptide interaction is sequence specific; each SH2 domain recognizes a specific subset of pY peptide sequences [4]. The specificity is dictated by the interactions between amino acids adjacent to pY and the less conserved regions of the SH2 domain surface. Many SH2 domains contain a second, relatively deep pocket, which recognizes the side chain of pY+3 residue (relative to pY, which is defined as position 0). The rest of the peptide residues (pY−2, pY−1, pY+2, pY+4, and pY+5) presumably make specific contacts with surface residues on the SH2 domain. Thus, determination of the sequence specificity of these SH2 domains is an important first step towards identifying their in vivo binding partners and understanding their physiological functions. Such information will also facilitate the development of specific SH2 domain inhibitors as research tools and therapeutic agents [6].
PTPs are also abundant in eukaryotic cells; the human genome encode 107 PTPs [7]. All known PTPs belong to the same family with a conserved catalytic domain of ~250 amino acid residues. They all share a common catalytic mechanism, in which catalysis involves nucleophilic attack on the tyrosyl phosphate by the thiolate of a conserved active-site cysteine, leading to the formation of a covalent phosphocysteinyl intermediate [8]. Subsequent hydrolysis of the phosphoenzyme intermediate by an activated water molecule regenerates the thiolate group, thereby concluding the catalytic cycle. PTPs were initially thought as promiscuous “housekeeping” enzymes that simply oppose the action of PTKs. Recent work indicates that PTPs play active roles in a wide variety of cellular processes and that PTPs exhibit exquisite substrate specificity in vivo [9]. However, in contrast to our advanced knowledge of PTP catalytic mechanism, many questions remain regarding their precise mechanism of action in vivo. How does a PTP recognize its specific substrate(s), given that many PTPs co-exist with numerous pY proteins in a single cell? What is the physiological substrate(s) and function of a PTP? How is the activity of PTPs regulated inside the cell? It is believed that the in vivo substrate specificity of PTPs is controlled by two factors. The first is the presence of targeting domains, which direct the phosphatase activity to their physiological substrates or proper cellular locations [10, 11]. Many PTPs (but not all) indeed contain other structural elements (e.g., SH2 domains and ER localization signals) in addition to the catalytic domain. The second is the intrinsic sequence specificity of the PTP domain. In vitro studies with a limited set of synthetic pY peptides indicated that the kinetic constants (kcat/KM) toward the same PTP differ by several orders of magnitude [12–18]. There is also in vivo evidence for the existence of substrate specificity. For example, by using catalytically inactive mutants, Tonks and coworkers were able to fish out the specific protein substrates of a few PTPs by “substrate trapping” [19, 20]. A chimeric protein containing the SH2 domains of SHP-1 but the catalytic domain of SHP-2 failed to dephosphorylate EGF receptor in vivo, as did wild-type SHP-1 [21]. There is growing evidence that PTP activity and specificity are controlled by a combination of both factors [9]. Thus, sequence specificity data of a PTP will help identify its physiological substrates. It will facilitate the design of specific PTP inhibitors. Efficient substrates would also facilitate the kinetic assays of these enzymes.
In recent years, considerable efforts have been made by many laboratories to define the sequence specificity of SH2 domains and PTPs. Cantley and co-workers employed a GST-SH2 affinity column to enrich the SH2-binding sequences from a pY peptide library [4]. The enriched peptides were then sequenced as a mixture by conventional Edman degradation. A variation of this method involved screening resin-bound libraries against a fluorescently labeled SH2 domain [22]. The positive beads with the bound SH2 were removed from the library using a fluorescence-activated cell sorter and again sequenced as a pool by Edman degradation. Other methods included phage display [23, 24] and blotting position-scanning sublibraries on filter paper with SH2 domains of interest (OPAL) [25]. For PTPs, earlier studies employed synthetic pY peptides derived from known phosphorylation sites in proteins to define the substrate specificity of PTPs [12–18]. This method is inherently limited because the PTP active site interacts with 3–5 residues on either the N- or C-terminal side of pY [26, 27]. A complete characterization of each PTP would require the synthesis and assay of a prohibitive number of single peptides (206–2010). Therefore, more recent efforts have been focused on various combinatorial approaches. However, due to the lack of stable association between an active PTP and a pY peptide, some investigators have screened pY peptide libraries against a catalytically impaired PTP mutant or employed a non-hydrolyzable pY analogue [28–31]. Other investigators have employed inverse alanine scanning [32], ECLPISE [33], or cleavage of PTP product with α-chymotrypsin [34]. Each of these methods has its own limitations and consequently, none of the previous studies have led to comprehensive specificity data on any PTP.
We have recently developed a combinatorial library method that can be used to determine the sequence specificity of modular domains (e.g., SH2 domain) and PTPs. In this method, pY peptides are chemically synthesized on individual beads to give a “one-bead-one-compound” library. The library is directly screened against an SH2 domain or PTP to identify the positive beads that have undergone binding and/or catalysis. The peptides on the positive beads were individually sequenced by partial Edman degradation/mass spectrometry (PED/MS), a high-throughput peptide sequencing technique developed in our laboratory [35, 36]. In the following section, we will provide the detailed experimental procedures for the method and its application to several SH2 domains and PTP1B.
2. Experimental procedures
2.1. Library synthesis on solid phase
2.1.1 Materials
TentaGel S NH2 resin (90 μm, 0.3 mmol/g) were from Advanced ChemTech (Louisville, KY). 9-Fluorenylmethoxycarbonyl (Fmoc)-amino acids with standard side-chain protecting groups, O-benzotriazole-N, N, N′, N′-tetramethyluronium hexafluorophosphate (HBTU), and N-hydroxybenzotriazole (HOBt) were obtained from Advanced ChemTech, Peptides International (Louisville, KY) or NovaBiochem (San Diego, CA). All other chemical reagents were from Sigma-Aldrich (St Louis, MO).
2.1.2 Protocols
The “split-and-pool” synthesis method was employed to generate “one-bead one-compound (OBOC)” peptide libraries [37, 38]. In such libraries, each bead displays multiple copies of a unique peptide sequence (~100 pmol of peptides on a 90-μm bead). Since SH2 domains typically make contact with pY and 2 to 3 residues flanking the pY residue [2–6], they are usually first screened against a pY peptide library containing 5 randomized positions, TAXXpYXXXLNBBRM-resin [where X represents any of the 18 proteinogenic amino acids (except for Cys and Met) plus norleucine (Nle) and L-α-aminobutyric acid (Abu) as Met and Cys surrogates, respectively; B is β-alanine]. The N-terminal dipeptide (TA) helps reduce any potential bias caused by electrostatic interactions between an SH2 protein and the positively charged N-terminus (which is required for peptide sequencing). The C-terminal methionine permits the release of peptides from the resin by CNBr treatment prior to sequencing, while the arginine increases peptide solubility and ionization efficiency during PED/MS sequencing. The two β-alanines add flexibility to the peptides, making them more accessible to a protein receptor. The dipeptide LN is added to increase the masses of the peptides to >600 Da, so that the peptide peaks do not overlap with those from the MALDI matrix. Methionine is excluded from the random positions to avoid internal peptide cleavage during CNBr treatment and is replaced by the isosteric Nle residue. The theoretical diversity of the library is 205 or 3.2 × 106.
The library was typically synthesized on 5 g of TentaGel S NH2 resin using standard Fmoc chemistry and HBTU/HOBt/4-methylmorpholine as the coupling reagents. The linker and other invariant residues (LNBBRM, pY and TA) were synthesized in a 90-mL glass vessel, with 4 equivalents of Fmoc-amino acids, HOBt and HBTU, and 8 equivalents of 4-methylmorpholine. The vessel was clipped onto a rotary shaker (Figure 1) and shaken for ~1 h at room temperature. After the coupling reaction was complete (as judged by ninhydrin tests), the vessel was removed from the shaker and attached to a vacuum manifold (Figure 1). The reaction solution was drained by vacuum suction and the resin was washed with DMF. Deprotection of the Fmoc group was performed using 20% piperidine in DMF (5 + 15 min). The resin was washed with DMF and dichloromethane (DCM) and dried overnight in air. The resin was split into 20 equal portions (by weight) and transferred into 20 5-mL glass vessels. A different Fmoc-amino acid (5 equivalents) was added into each reaction vessel, together with 5 equivalents of HOBt and HBTU, 10 equivalents of 4-methylmorpholine, and 3 mL of DMF. The reaction vessels were capped and clipped onto the rotary shaker and incubated at room temperature for 1 h. The coupling reaction was repeated once using fresh reagents to ensure complete reaction. To distinguish amino acids of degenerate masses during peptide sequencing by PED/MS, 5% CD3CO2D (mol/mol) was added to the coupling reactions of leucine and lysine, whereas 5% CH3CD2CO2D was added to the coupling reaction of norleucine. After the coupling reactions were complete, the resin from 20 reaction vessels was combined, mixed, and treated with 20% piperidine to remove the N-terminal Fmoc group. The resin was again split into 20 portions for the addition of the next random residue. After the library synthesis was complete and removal of the N-terminal Fmoc group, the resin was exhaustively washed with DCM and the side-chain protecting groups were removed by treatment with a modified reagent K (7.5% phenol, 5% ddH2O, 5% thioanisole, 2.5% ethanedithiol, and 1% anisole in TFA) for 1 h at room temperature. The resin was washed with TFA and DCM, incubated for 10 min in 5% diisopropylethylamine in DCM, and washed again with DCM. The resin was then dried in air and stored at −20 °C.
Fig. 1.

Apparatus for peptide library synthesis. It consists of a dry-ice cold trap connected to house vacuum (part A), a vacuum manifold with a solvent reservoir (part B), and a rotary shaker (part C).
2.2 Purification and Labeling of SH2 domain and PTP
The SH2 domain used in library screening must be labeled by a fluorescent group or biotin. We have employed two different labeling methods. In the first method, the SH2 domain (or other modular domains) is expressed as a maltose-binding protein (MBP) fusion, which is directly treated with 1.5–2 molar equivalents of N-hydroxysuccinimidobiotin or the N-hydroxysuccinimidyl ester of a fluorescent probe. Because MBP contains 38 lysine residues (compared to ~5 lysines in a typical SH2 domain), the label is predominantly added to the MBP portion. While this method is experimentally straightforward, the labeled product is heterogeneous in nature and any labeling in the SH2 domain region may change its binding properties. In the second method, an SH2 domain is expressed with an 11-residue peptide called the “ybbR tag” (DSLEFIASKLA) fused to its N- or C-terminus (Figure 2) [39]. A six-histidine tag is usually also added to either terminus to facilitate its purification. The ybbR tag is specifically recognized by Sfp phosphopantetheinyl transferase, which catalyzes the transfer of a phosphopantetheinyl group from CoA to a serine in the ybbR tag (the underlined residue) [39]. The Sfp enzyme readily accepts CoA analogues that are modified at the thiol group as substrates, thereby providing a powerful method for specific and quantitative labeling of the SH2 domain at a single site with a wide variety of probes (e.g., biotin, fluorescein, and Texas Red).
Fig. 2.
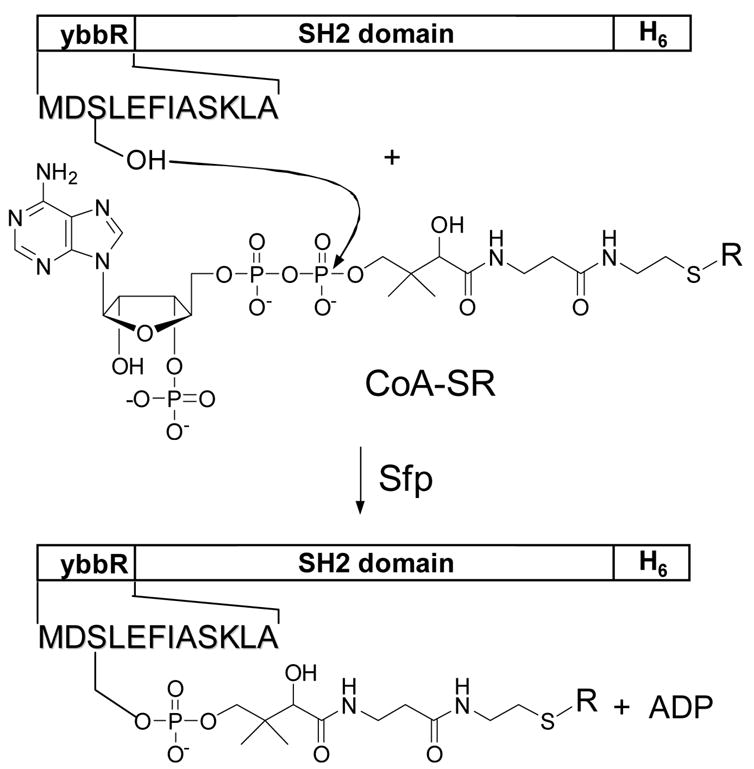
Specific labeling of a ybbR-tagged protein by Sfp-mediated transfer of an S-alkylated phosphopantetheinyl group from CoA-SR to the ybbR tag. CoA-SR, S-alkylated coenzyme A where R is biotin or a fluorescent group.
2.2.1 Materials
The Marathon-Ready™ human cDNA library and Talon resin were purchased from Clontech (Mountain View, CA). The pMAL-c2 vector, all DNA modifying enzymes, and amylose resin were from New England Biolabs (Ipswich, MA). Prokaryotic expression vector pET-28a was purchased from Novagen (San Diego, CA). The expression vector for the addition of the ybbR tag (pET22-ybbR) and the Sfp-overproducing plasmid (pET29-Sfp) were generous gifts from Dr. C. T. Walsh (Harvard Medical School) [39]. Antibiotics, N-hydroxysuccinimido-biotin, Sephadex G-25 resin, and other reagents were obtained from Sigma-Aldrich.
2.2.2 Protocols
2.2.2.1 Cloning and protein expression
The SH2 domains were constructed as three different fusion proteins: MBP fusion, ybbR tagged, and six-histidine tagged. The coding sequences of the SH2 domains were amplified from the Marathon-Ready™ cDNA library by polymerase chain reaction using primers containing appropriate restriction sites on their 5′ and 3′ termini. For MBP-SH2 fusions, the PCR product was digested with EcoRI and HindIII and cloned into the corresponding site of pMAL-c2. The same restriction sites were used for cloning into plasmid pET-28a, which produces proteins with an N-terminal six-histidine tag. For cloning into plasmid pET22-ybbR [39], HindIII and XhoI restriction sites were utilized. The authenticity of all DNA constructs was confirmed by dideoxy sequencing.
Escherichia coli DH5α cells harboring the proper pMAL-c2-SH2 plasmid or E. coli BL21 (DE3) cells harboring the proper pET22-SH2-ybbR or pET28a-SH2 plasmid were grown in LB medium to the mid-log phase and induced by the addition of 0.2 mM isopropyl β-D-thiogalactoside (IPTG) for 3 h at 30 °C. The cells were harvested by centrifugation and lysed in the presence of protease inhibitors by passing through a French pressure cell. The MBP-SH2 protein and the histidine-tagged SH2 domains were purified from the crude lysate on amylose and Talon cobalt affinity columns, respectively, according to the manufacturer’s instructions. The histidine-tagged SH2 proteins were concentrated in an Amicon concentrator, before being passed through a Sephadex G-25 column. The elution buffer contained 10 mM HEPES (pH 7.4), 150 mM NaCl, 3 mM EDTA, and 1 mM tris(carboxyethyl)phosphine (TCEP). The protein was flash frozen in liquid nitrogen and stored at −80 °C.
2.2.2.2 Protein labeling
The MBP-SH2 protein was concentrated in an Amicon concentrator to approximately 4 mg/mL (in 20 mM HEPES, pH 8.0, 150 mM NaCl, 2 mM 2-mercaptoethanol, and 10 mM maltose) and treated with 2 molar equivalents of N-hydroxysuccinimido-biotin at room temperature for 30 min (total volume of 1 mL). The reaction was quenched by the addition of 10 μL of a 2 M Tris-HCl solution (pH 8.0). The biotinylated MBP-SH2 protein was passed through a Sephadex G-25 column to remove any free biotin and concentrated again. Glycerol was added to a final concentration of 40% and the protein was quickly frozen in liquid nitrogen or a dry ice/isopropanol bath, before being stored at −80 °C. Biotinylated MBP was similarly prepared as a negative control.
The Sfp protein was expressed and purified from E. coli BL21 (DE3) Rosetta cells using a Talon cobalt affinity column as previously described [39]. Texas Red-CoA conjugate was prepared following a modified procedure of Yin et al [39]. Briefly, 10 μmol of CoA lithium salt (Sigma) was dissolved in 1 mL of 100 mM sodium phosphate buffer (pH 7.0), whereas Texas Red C2 maleimide (Molecular Probes; 11 μmol) was dissolved in 0.2 mL of DMSO. The two solutions were mixed and stirred in the dark and at room temperature for 2 h. After that, 11 μmol of 2-mercaptoethanol was added to quench the reaction and the solution was stirred for 10 additional min. The mixture was divided into 20-μL aliquots and stored at −80 °C. Other CoA-small molecule conjugates were prepared by the same procedure.
Protein labeling reaction followed the procedure of Yin et al [39]. The reaction mixture (total volume of 10 mL) contained 50 mM HEPES (pH 7.5), 10 mM MgCl2, 0.5 μM Sfp, 10 μM CoA-small molecule conjugate, and 5 μM ybbR-tagged protein and was incubated at 37 °C for 15 min. The resulting labeled protein was purified on a Talon cobalt affinity column to remove any remaining free dye molecules (or biotin). The protein was quickly frozen in a dry ice/isopropanol bath and stored at −80 °C.
2.3 Library screening
2.3.1 Materials
Micro-BioSpin columns (0.8 mL) were obtained from BioRad (Hercules, CA). Streptavidin-alkaline phosphatase conjugate (SA-AP) was from Prozyme (San Leandro, CA). 5-Bromo-4-chloro-3-indolyl phosphate (BCIP) was from Sigma-Aldrich.
2.3.2 Protocols
2.3.2.1 Colorimetric screening
In a typical screening experiment (Figure 3), 30–50 mg of the peptide library was placed in a micro-BioSpin column (0.8 mL) and washed exhaustively with dichloromethane, methanol, ddH2O, and HBST buffer (30 mM HEPES, pH 7.4, 150 mM NaCl, and 0.01% Tween 20). The resin was blocked for 1 h using HBST buffer containing 0.1% (w/v) gelatin. After blocking, the solution was drained and the library was resuspended in fresh HBST buffer containing 0.1% gelatin and varying concentrations of the biotinylated MBP-SH2 protein (typically 10–50 nM). The solution was incubated at 4 °C for 4–16 h with gentle mixing, during which the MBP-SH2 protein is recruited to beads that carry peptides of high affinity to the SH2 domain. Next, the protein solution was drained, and 0.8 mL of SA-AP buffer (30 mM Tris, pH 7.6, 1 M NaCl, and 20 mM K/PO4) containing 1 μL of SA-AP (1 mg/mL) was added to the column. The mixture was incubated for 10 min at 4 °C with gentle mixing. This allowed SA-AP to bind to the resin-bound biotinylated SH2 domain (Figure 3). The solution was again drained and the resin was quickly washed with 400 μL of SA-AP buffer, 400 μL of HBST buffer, and 400 μL of staining buffer (30 mM Tris, pH 8.5, 100 mM NaCl, 5 mM MgCl2, and 20 μM ZnCl2). The washing steps are necessary to remove any excess or nonspecifically bound protein. Finally, the resin was transferred into a 35-mm Petri dish with 3 × 300 μL of the staining buffer and 100 μL of BCIP (5 mg/mL in the staining buffer) was added. The resin-bound alkaline phosphatase hydrolyzes BCIP into an indole, which instantaneously dimerizes in air into indigo, resulting in a turquoise colored precipitate deposited on the bead surface. Therefore, as a result of the reaction cascade, a bead that carries a high-affinity ligand for the SH2 domain becomes intensely colored in 30–45 min, while the rest of the beads remain colorless (Figure 3). The staining reaction was quenched by the addition of 2 mL of 0.1 M HCl. The resin was transferred back into the Bio-Spin column and washed extensively with water. The beads were replaced in a Petri dish, from which positive beads (turquoise colored) were manually picked with a micropipette under a dissecting microscope. The number of colored beads depends on the affinity and specificity of the protein domain as well as the stringency of screening conditions (e.g., SH2 domain concentration, number of washings, and length of staining time). The screening reactions are controlled so that typically 10–100 colored beads are obtained from 100 mg of resin (~286,000 beads). The number of positive beads is generally very reproducible when multiple screenings are performed against the same SH2 domain. Control experiments conducted with biotinylated MBP produced no colored beads under identical conditions.
Fig. 3.
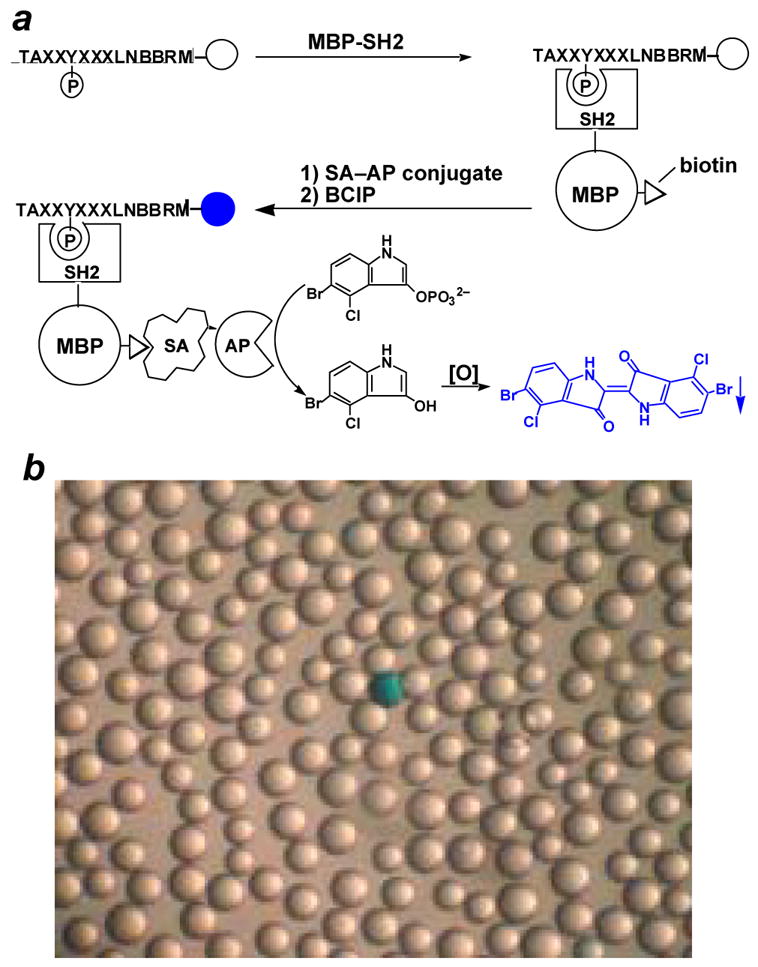
Screening of the pY peptide library against an SH2 domain. (a) Scheme showing the steps involved in colorimetric screening of the pY peptide library against a biotinylated MBP-SH2 domain. Key: MBP, maltose binding protein; SA, streptavidin; AP, alkaline phosphatase. (b) A photograph of a portion of the screened library (~50x magnification). A positive bead (turquoise colored) is shown.
2.3.2.2 Fluorescent screening
The fluorescent screening assay employed a slightly modified procedure. Briefly, 30–50 mg of the peptide library was placed in a micro-BioSpin column (0.8 mL), washed with solvents, and blocked with 0.1% gelatin as described above. The blocking solution was drained and the resin was resuspended in HBST buffer containing 0.1% gelatin and 0.1–0.5 μM fluorescently labeled SH2 domain and transferred into a 35 mm Petri dish. The dish was gently shaken at 4 °C for 4–16 h. The dish was then viewed under a fluorescent microscope (Olympus, SZX12) and the most fluorescent beads were removed using a micropipette. It should be noted that some TentaGel beads exhibit strong background fluorescence in the green and shorter wavelength regions. We found that by labeling the protein with dyes of longer excitation and emission wavelengths (e.g., Texas Red), one can eliminate most of the interference from background fluorescence. Also, we routinely wash the colored beads (from either calorimetric or fluorescent screening) with aqueous as well as organic solvents to remove the bound proteins and dyes, and subject the beads to further rounds of screening, to eliminate any false positive beads.
2.4 Sequence determination of positive beads
Screening against an SH2 domain typically results in ~100 colored beads, which must be sequenced individually. The availability of a relatively large number of individual binding sequences is critical for statistical analysis in order to draw definitive conclusions about the specificity of a given domain, especially when the domain recognizes multiple consensus sequences. Peptide sequencing on this scale used to be an insurmountable challenge, as the Edman method is both expensive and time consuming. The advent of tandem MS techniques has not adequately addressed this problem either. Therefore, we have developed an inexpensive, high-throughput peptide sequencing technique, termed “PED/MS”, which is ideally suited for sequencing resin-bound peptides. This technique was initially developed in 2001 [40] and has subsequently been improved [35, 36]. The latest version (Version 2006) is described here. Briefly, the selected beads (1 to 106 beads) are treated with a 10–30:1 (mol/mol) mixture of phenyl isothiocyanate (PITC) and N-(9-fluorenylmethoxycarbonyloxy)succinimide (Fmoc-OSU), followed by TFA (Figure 4a). This cleaves the N-terminal amino acid from 90–95% of the peptides (Edman degradation); for the other 5–10% peptides on each bead, reaction with Fmoc-OSU causes N-terminal blocking and retention of the N-terminal residue. Repetition of the above procedure for n-1 cycles (n = No. of residues to be sequenced) produces a peptide ladder. After removal of the N-terminal Fmoc group by piperidine, the treated beads are individually picked, placed in separate microcentrifuge tubes (one bead/tube), and treated with CNBr. The sample from each tube is then mixed with matrix material and spotted on 96-well sample plates and analyzed by MALDI-TOF MS in an automated format. Figure 4b shows the mass spectrum of the degradation products derived from a single 90-μm bead carrying the sequence TAFIpYDNVLNBBRM. The peptide sequence is read from the mass spectrum by inspecting the mass differences between adjacent peaks. For example, the mass difference of 101 between peaks of the largest (m/z 1735.0) and second largest m/z values (m/z 1633.8) indicates a Thr at the N-terminus. Mass degenerate amino acids (e.g., Nle, Leu, and Ile) are unambiguously resolved in the spectra by the addition of a small amount (5%) of a capping agent to peptide coupling reactions during library synthesis. We chose CD3CO2D (for Leu and Lys) and CH3CD2CO2D (for Nle) as the capping agents [36]. Thus, a singlet peak 113 mass units from the preceding peak indicates an Ile residue; a set of doublet peaks 45 (m/z + CD3CO) and 113 mass units (m/z + Leu) from the preceding peak indicates a Leu; whereas doublet peaks 58 (m/z + CH3CD2CO) and 113 mass units (m/z + Nle) from the preceding peak indicate a Nle residue. Lysine and glutamine, which have the same nominal residue mass (128), are similarly differentiated. The PED/MS method is sensitive, reliable, inexpensive, and high-throughput. On a Bruker Reflex III MALDI-TOF instrument, 20–30 beads can be processed in an hour at a cost of $0.5 per bead (reagents and instrument time). Only 10% of the sample from a single 90-μm bead is needed for each MS experiment. This permits re-runs (if necessary) to ensure correct sequence determination. The success rate is typically ~95%.
Fig. 4.
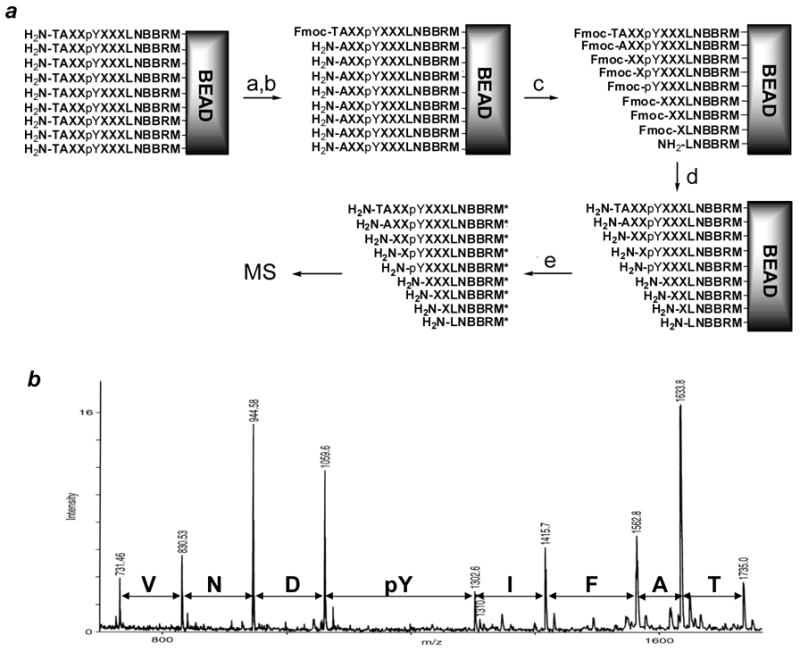
Peptide sequencing by PED/MS. (a) Scheme showing the reactions involved in partial Edman degradation. Reagents and conditions: (a) 10–30:1 PITC:Fmoc-OSU; (b) TFA; (c) Repeat steps a and b 7 times; (d) 20% piperidine in DMF; and (e) CNBr in 70% TFA. (b) The MALDI mass spectrum of peptide TAFIpYDNVLNBBRM and its degradation products (from a single 90 μm bead). Key: M*, homoserine lactone.
2.4.1 Materials
Fmoc-OSU was from Advanced ChemTech. PITC was purchased in 1 mL sealed ampoules from Sigma-Aldrich. 4-Hydroxy-α-cyanocinnamic acid and other organic solvents were also from Sigma-Aldrich.
2.4.2 Protocols
Positive beads (typically 10–150 beads) from the same category (i.e., screened against the same protein receptor and having similar color intensity) were placed in a custom-designed glass vessel (12-mm i.d., 20-mm height) fitted with a Luer tip (1-mm i.d.) at one end and a fine-porosity frit (10–20-μm pore size) just above the Luer tip. The beads were exhaustively washed with water, pyridine, and a 2:1 (v/v) pyridine/water solution containing 0.1% triethylamine. The solutions were removed by vacuum suction, with the Luer tip attached to a vacuum manifold (Figure 1). The Luer tip was then capped with a plastic cap, and the beads were suspended in 160 μL of 2:1 (v/v) pyridine/water solution containing 0.1% triethylamine. A stock solution of Fmoc-OSU (31 mM) was freshly prepared in pyridine. An aliquot of this solution (160 μL) was mixed with PITC (6–18 μL, 0.05–0.15 mmol) to give a degradation reagent mixture containing 290–870 mM PITC and 29 mM Fmoc-OSU (PITC/Fmoc-OSU ratio of 10–30:1). An equal volume of this reagent mixture (160 μL) was added to the suspended beads and the degradation was allowed to proceed at room temperature for 6 min (with gentle shaking on a rotary mixer). The solution was drained, and the beads were washed with pyridine (1.5 mL), dichloromethane (3 × 1.5 mL), and dried by suction. The beads were washed with 1 mL of anhydrous TFA and treated twice with 1 mL of anhydrous TFA at room temperature (6 min each time). The beads were washed with dichloromethane (3 × 1.5 mL), dried by suction, washed again with 1.5 mL of pyridine, and ready for the next reaction cycle. After the final round of PED, the beads were treated twice with 1 mL of 20% piperidine in DMF at room temperature (5 min each time), to remove the Fmoc group from the N-terminus and any capped amino acid side chains. The beads were suspended in 1 mL of TFA containing 20 μL (0.27 mmol) of dimethyl sulfide and 25 mg (0.18 mmol) of ammonium iodide and incubated on ice for 30 min to reduce any oxidized methionine. The beads were washed exhaustively with water and transferred to a Petri dish, where they were picked under a microscope and placed into separate microcentrifuge tubes.
Individual beads in microcentrifuge tubes were spun under vacuum in a SpeedVac concentrator to evaporate any residual water. Each bead was treated overnight in the dark with 20μL of 70% TFA containing CNBr (40 mg/mL), to cleave the peptides from the bead. The excess CNBr and TFA were removed under vacuum in a SpeedVac concentrator. The released peptides were dissolved in 10 μL of 0.1% TFA in ddH2O. The tube was vortexed and centrifuged three times to dissolve any peptides that may be on the walls of the tube. For MS analysis, a matrix solution was prepared by saturating a 50:50:0.1 (v/v) acetonitrile/water/TFA solution with 4-hydroxy-α-cyanocinnamic acid. One μL of the peptide solution was mixed with 2–5 μL of the matrix solution, and 1 μL of this mixture was spotted onto a 96-well sample plate. Mass spectrometry analysis was performed on a Bruker Reflex III MALDI-TOF instrument in an automated manner. Sequence determination from the mass spectra was performed manually.
2.5 Individual peptides synthesis
2.5.1 Materials
CLEAR-amide resin (0.46 mmol/g) and Fmoc-8-amino-3,6-dioxooctanoic acid (miniPEG) were from Peptides International. Benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP) were from NovaBiochem.
2.5.2 Protocols
Peptides were synthesized on 50–100 mg of CLEAR-amide resin, using standard Fmoc/HBTU/HOBt chemistry. Fmoc-8-amino-3,6-dioxooctanoic acid (miniPEG) was added to the N-terminus of the peptides to provide a flexible linker, before the addition of biotin. D-Biotin was coupled with 4 equivalents of biotin and PyBOP and 8 equivalents of DIPEA. The reaction was allowed to proceed for 2 h at room temperature. A ninhydrin test confirmed the completion of the reaction. Peptide cleavage from the resin and side-chain deprotection were carried out with modified reagent K as previously described. The crude peptides were precipitated in cold diethyl ether, washed with diethyl ether, dried under vacuum, and purified by reversed-phase HPLC on a C18 column (Varian Dynamax 300 Å, 250 × 10 mm). The identity of each peptide was confirmed by MALDI-TOF mass analysis.
2.6 Determination of dissociation constants by surface plasmon resonance (SPR)
2.6.1 Materials
SPR analysis was performed on a BIAcore 3000 instrument. Streptavidin-coated SA biosensor chips and HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% polysorbate 20) were purchased from BIAcore (Piscataway, NJ).
2.6.2 Protocols
The binding affinity of the selected peptides to an SH2 domain of interest was determined by SPR analysis, using N-terminally histidine-tagged domain (not MBP fusion) to validate the screening results. Assays were performed in HBS-EP buffer at room temperature. The sensorchip was conditioned using a solution of 1 M NaCl and 50 mM NaOH (aqueous) according to the manufacturer’s instructions. Each biotinylated peptide (~0.05 nmol) was injected at a flow rate of 15 μL/min until a constant level of response unit (400–500 RU) was obtained. Varying concentrations of the SH2 domain (0.05–5.0 μM) were passed over the chip for 2 min at a flow rate of 15 μL/min. A blank flow cell (no immobilized peptide) was used as a control. In between two runs, the sensorchip was regenerated by injecting a strip buffer (10 mM NaOH, 200 mM NaCl, 0.05% SDS) for 5–10 s at a flow rate of 100 μL/min. The equilibrium response unit (RUeq) at a given protein concentration was obtained by subtracting the response of the blank flow cell from the flow cells containing the peptide. The dissociation constant (KD) was obtained by nonlinear regression fitting of the data to the equation:
where RUeq is the measured response unit at a given domain concentration and RUmax is the maximum response unit.
3. Applications
Over the past few years, the Pei laboratory has applied the above method to successfully determine the sequence specificity of a dozen different SH2 domains [40–44, unpublished results], 3 BIR domains [45], and several chromodomains and WW domains) [unpublished results]. Below we describe our results on Src, Grb2, Csk, SHP-1, SHP-2, and SHIP1 SH2 domains and compare them with those reported earlier by Cantley and coworkers. One example is given on its application to the determination of PTP substrate specificity [46].
3.1. SHP-1, SHP-2, and SHIP1 SH2 domains
The four SH2 domains of SHP-1 and SHP-2 and the single SH2 domain of SHIP1 exhibit overlapping specificities, as these proteins often compete for binding to the same pY protein receptors [47]. This has generated much confusion with regard to which phosphatase is the physiologically relevant regulator in various signaling pathways. To help alleviate this problem, we determined the sequence specificity of all 5 SH2 domains [40–42]. Several important findings resulted from this study. First, we found that a single modular domain can recognize more than one type of peptide motifs. For example, the N-terminal SH2 domain of SHP-2 binds with high affinity to four different classes of ligands of the consensus sequences (I/L/V/m)XpY(T/V/A)X(I/V/L/f) (class I), W(M/T/v)pY(y/r)(I/L)X (class II), (I/V)XpY(L/M/T)Y(A/P/T/S/g) (class III), and (I/V/L)XpY(F/M)XP (class IV) [41], where the lower case letters represent less frequently selected residues and X is any amino acid. The class I ligands correspond to the pITIM motifs found on many cell surface receptors, which are well known ligands of SHP-1, SHP-2, and SHIP1 SH2 domains in vivo [47]. Several proteins have also been reported to bind SHP-1/2 through class III and IV motifs, including c-Kit (YVpYIDP as binding motif), CTLA-4 (QPpYFIP), E-selectin (GSpYQKP), prolactin receptor (LDpYLDP), and STAT3 (putatively LVpYLYP) [48–52]. Interestingly, previous investigators proposed that these receptors might bind to SHP-1/2 indirectly [49]. Our data show for the first time that these motifs bind directly to the N-SH2 domain of SHP-1/2. No class II motif has so far been found to mediate SHP-2 binding in vivo. This property is not unique to SHP-2 SH2 domain, as tensin SH2 domain [44] and XIAP BIR3 domain [45] have been reported to bind to three and two different types of ligands, respectively. This emphasizes the importance of obtaining individual binding sequences from library screening. In fact, when we intentionally mixed all four classes of SHP-2-binding sequences and examined the amino acids enriched at each position, the pattern was dominated by the class I sequences and we were not able to tease out the class III or IV sequences [41]. Another observation was that the most frequently selected sequences do not necessarily bind to the target protein with the highest affinity, especially when comparing two different classes of peptides. In fact, the opposite trend is often true. For example, the class I sequences for SHP-2 N-SH2 domain were most frequently selected (84 out of 150 total sequences), whereas only 12 class IV sequences were selected from the library [41]. Yet, the class IV peptides tested bound to SHP-2 N-SH2 domain with an order of magnitude higher affinity than class I peptides (KD = 0.044–0.11 μM for class IV peptides vs 1.4–9.7 μM for class I peptides). Presumably, a high-affinity interaction requires a better fit between the protein and peptide structures, necessarily limiting the number of possible choices in the library.
SHP-2 C-SH2 domain has a single consensus sequence (T/V/I/y)XpY(A/s/t/v)X(I/v/l) [41]. The C-terminal SH2 domain of SHP-1 also recognizes a single class of peptides with a very similar consensus sequence, (T/v/i)XpY(Abu/A/t)X(L/m/v) [40, 41], whereas SHIP1 SH2 domain recognizes the consensus of pY(Y/S/T/v)(L/y/nle/f)(L/Nle/i/v) [41]. Thus, the five SH2 domains have overlapping sequence specificities but also significant differences. SHP-1/2 SH2 domains all require a hydrophobic residue at pY-2 position (L, I, V, or T), whereas SHIP1 SH2 domain does not. On the other hand, SHIP1 SH2 domain requires a hydrophobic residue (especially L or Y) at pY+2 position but SHP SH2 domains do not. There are also differences between the SH2 domains of SHP-1 and SHP-2 [detailed in ref 41]. For example, SHP-1 SH2 domains prefer Leu at pY+3 position, whereas SHP-2 SH2 domains favor Ile. The high-resolution specificity data can now rationalize many of the previously observed cellular phenomena on a molecular basis and predict new protein-protein interactions [41, 42].
3.2. c-Src SH2 domain
A total of ~200 mg of the pY peptide library was screened against Src SH2 domain, resulting in 130 complete sequences (Table 1). Analysis of these sequences reveals that Src SH2 recognizes a single consensus motif, pY(T/A/s/e/n)X(I/M/l/v) (Figure 5). The most critical specificity determinant is the pY+3 position, where isoleucine and norleucine (methionine) are most preferred, although valine and leucine are also tolerated. In addition, there is relatively strong preference for a small residue (e.g., Thr, Ala, and Ser) at the pY+1 position. A wide variety of residues were found at pY−2, pY−1, and pY+2 positions, although there is a significant overrepresentation of histidine at the pY−2 position. To confirm the screening results, 8 representative peptides were individually synthesized and their dissociation constants toward Src SH2 domain were determined by SPR (Table 2). The pYEEI peptide, a known Src SH2 domain ligand [4], was used as a positive control. All of the pY peptides bound to the SH2 domain with submicromolar KD values (0.11–0.71 μM). Consistent with the selection for His at position pY−2, mutation of this histidine of peptide HYpYEMM into an alanine resulted in a small (1.3-fold) but reproducible decrease in affinity (compare peptides 1 and 2 in Table 2). Note that substitution of norleucine for methionine has minimal effect on the binding affinity (compare peptides 1 vs 4 and 3 vs 5 in Table 2). Cantley and coworkers previously determined the binding sequences of Src SH2 domain by pooled sequencing and reported a consensus of pY(E/d/t)(E/n/d)(I/v/m/l) [4].
Table 1.
Peptide sequences selected from the pY library against Src, Grb2, and Csk SH2 domainsa
| Src | Grb2 | Csk | |||||
|---|---|---|---|---|---|---|---|
| RMpYAII | QRpYSKI | NFpYYTL | FRpYTSM | HHpYENA | TYpYVNM | LYpYRNV | FCpYASA |
| FGpYAMI | WQpYVQI | FMpYAQV | VRpYTVM | HHpYENA | VTpYVNM | MYpYRNV | QKpYARI |
| ERpYAMI | HLpYDFI | HIpYSFV | HMpYDMM | VLpYQNA | LQpYVNM | VYpYTNV | ATpYAKI |
| AIpYAQI | HMpYDKI | VRpYSIV | FLpYEAM | SYpYVNA | NRpYVNM | HQpYVNV | CRpYAHI |
| RYpYAQI | HHpYDQI | HYpYSMV | HHpYEDM | SYpYVND | VYpYYNM | DYpYVNV | AGpYASM |
| NMpYAQI | TYpYDQI | YGpYTLV | HLpYEKM | VRpYVNF | GYpYMNQ | ILpYYNV | PRpYACP |
| RApYAQI | VYpYDVI | TWpYTQV | HVpYENM | HFpYQNH | PYpYMNQ | LYpYINY | VPpYSFP |
| HHpYARI | HMpYNEI | DGpYTVV | MLpYEMM | AKpYENI | QRpYQNQ | HGpYKNY | AApYSVV |
| TGpYARI | HYpYNII | LSpYVYV | IVpYEMM | QRpYKNI | VRpYQNQ | YYpYRNY | ARpYSMV |
| QVpYATI | AHpYNKI | EWpYMLV | HYpYEMM | LRpYENL | QVpYVNR | WMpYVNY | FHpYSRV |
| QNpYATI | FWpYNVI | HRpYDFV | HLpYEVM | NRpYMNL | VYpYVNT | SVpYSRV | |
| RGpYAVI | EFpYNVI | GYpYQVV | TSpYEVM | NRpYMNL | HVpYANV | HRpYTHC | |
| VYpYAVI | HApYNVI | KYpYAFM | PFpYNEM | VSpYQNL | YMpYANV | PYpYTQP | |
| IVpYAVI | YYpYQTI | HDpYAIM | IFpYNEM | VSpYQNL | FQpYENV | SEpYTRV | |
| SVpYAYI | WMpYQVI | NHpYAIM | PYpYNMM | WHpYVNL | HFpYENV | ALpYTRV | |
| RFpYTAI | HMpYQKI | RGpYAIM | YMpYNMM | NMpYANM | IFpYENV | KGpYTIL | |
| HTpYTFI | RGpYQLI | VVpYAIM | VFpYNSM | NRpYANM | ATpYENV | RApYRSH | |
| YGpYTII | HLpYFHI | GGpYAIM | TLpYNYM | ARpYENM | HYpYENV | ARpYRCI | |
| HHpYTII | FApYFYI | AYpYALM | HVpYMNM | HTpYMNM | LRpYENV | EYpYRRI | |
| RKpYTKI | RLpYHAI | IApYAYM | VVpYYYM | QHpYMNM | LRpYENV | RCpYRSI | |
| RApYTKI | ALpYHQI | VFpYSMM | SNpYYMM | NDpYMNM | FYpYHNV | RMpYRFP | |
| TFpYTLI | HIpYHKI | HApYSPM | EMpYYQM | TGpYMNM | HSpYKNV | YPpYRTP | |
| HQpYTLI | GVpYYAI | HLpYTAM | MYpYAEA | TGpYMNM | VFpYNNV | QLpYRPP | |
| PVpYTQI | GGpYYFI | HMpYTAM | VTpYAFA | AIpYQNM | VYpYNNV | HMpYRKV | |
| HTpYTQI | YDpYYLI | HMpYTEM | KFpYRTF | HYpYQNM | LYpYQNV | ||
| HMpYTRI | RLpYYQI | WHpYTEM | FSpYMYF | HLpYQNM | FNpYQNV | ||
| SYpYTRI | YGpYYVI | GYpYTFM | IGpYVLH | IRpYQNM | FVpYQNV | ||
| DMpYTSI | VFpYYYI | MYpYTIM | MFpYSMR | EMpYQNM | FVpYQNV | ||
| HYpYTTI | HHpYEEL | DYpYTLM | MYpYAMR | EMpYQNM | GWpYQNV | ||
| YRpYTYI | ARpYKKL | HFpYTMM | FHpYGRR | YGpYRNM | GWpYQNV | ||
| HEpYSII | RHpYQKL | SYpYTMM | KYpYKER | QLpYRNM | ILpYQNV | ||
| KApYSVI | SYpYTTL | HLpYTQM | HHpYVNM | VFpYQNV | |||
| MFpYSFI | HYpYTYL | IYpYTSM | RApYVNM | IYpYRNV | |||
Boldfaced sequences were resynthesized and tested for binding by SPR. M, norleucine; C, α-aminobutyric acid. The pY library screened against c-Src and Grb2 SH2 domains did not contain α-aminobutyric acid.
Fig. 5.
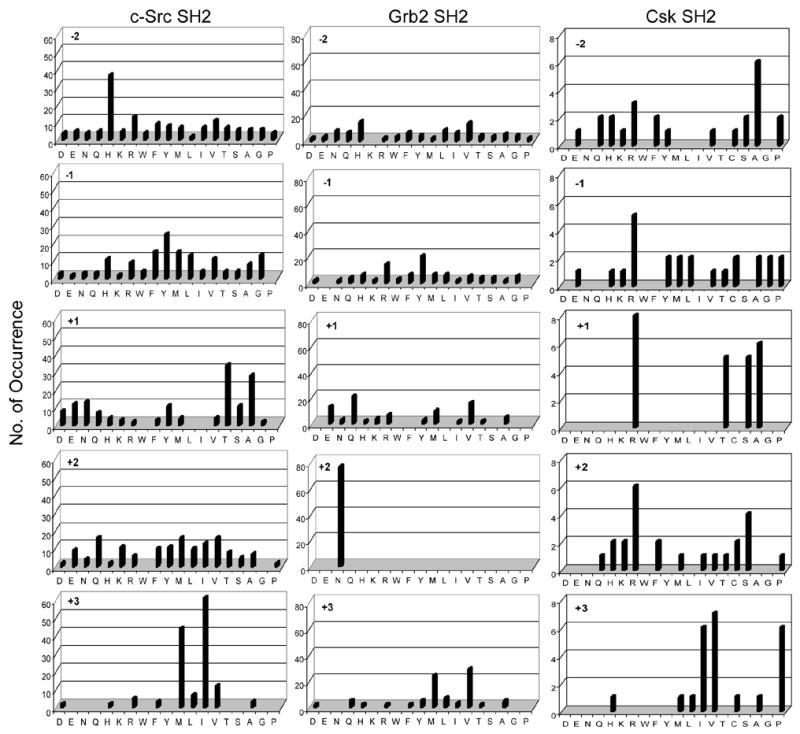
Sequence specificity of c-Src, Grb2 and Csk SH2 domains. Displayed are the amino acids identified at each position from −2 to +3 relative to pY (position 0). Number of occurrence on the y axis represents the number of selected sequences that contain a particular amino acid at a certain position. Key: M, norleucine; C, α-aminobutyric acid. The pY library screened against c-Src and Grb2 SH2 domains did not contain α-aminobutyric acid.
Table 2.
Dissociation constants (KD) of selected pY peptides toward Src and Csk SH2 domainsa
| Peptide | Peptide sequence | KD (μM) | |
|---|---|---|---|
| Src SH2 | Csk SH2 | ||
| 1 | HYpYEMM | 0.26 ± 0.03 | ND |
| 2 | AYpYEMM | 0.34 ± 0.03 | ND |
| 3 | HYpYTTM | 0.71 ± 0.05 | ND |
| 4 | HypYE(Nle)(Nle)LN | 0.19 ± 0.03 | ND |
| 5 | HYpYTT(Nle)LN | 0.44 ± 0.04 | ND |
| 6 | HYpYEEILN | 0.11 ± 0.01 | ND |
| 7 | SVpYAYILN | 0.67 ± 0.07 | ND |
| 8 | HHpYTIILN | 0.50 ± 0.05 | ND |
| 9 | ALpYHQILN | 0.47 ± 0.05 | ND |
| 10 | RCpYRSILN | ND | 4.1 ± 0.2 |
All peptides (except for peptide 10) contained an N-terminally acetyl group and a C-terminal BK-NH2 linker. The lysine side chain was modified with a (PEG)4-biotin moiety for immobilization. Peptide 10 contained biotin, an 8-atom linker (miniPEG), and two β-alanine residues added at its N-terminus. M, methionine; C, α-aminobutyric acid; ND, not determined.
3.3. Grb2 SH2 domain
The selected sequences (Table 1) indicate that Grb2 SH2 also recognizes a single consensus motif: pY(Q/V/e/m)N(V/M/l) (Figure 5). It shows little selectivity on the N-terminal side of pY. The most striking feature is that each of the 76 selected sequences has an asparagine at the pY+2 position, confirming the absolute requirement for an asparagine at pY+2 that had previously been reported by others [4, 22, 23, 53]. Selectivity at pY+3 positions is less stringent, preferring hydrophobic amino acids such as valine and methionine. Still less stringent selectivity was found at pY+1, where both hydrophobic (Val and Met) and hydrophilic residues (Gln and Glu, and Arg) were selected. Interestingly, the selected sequences exhibit significant sequence covariance. When the pY+3 residue is Val, the pY+1 residue is predominantly hydrophilic; however, when Met is at pY+3 the majority of the selected sequences contain a hydrophobic residue at pY+1 (Table 1). Overall, our results are in good agreement with the previous library screening data [4, 22, 23, 53] as well as the identified pY motifs in Grb2-binding proteins, including Shc (binding motif: pYVNV) [54], Bcr-Abl (pYVNV) [55], HGFR/SF (pYVNV) [56], and FAK (pYENV) [57]. The requirement for an asparagine residue at pY+2 has been explained by the crystal structure of this domain complexed with pY peptides [58, 59]. The pY peptide assumes a β turn conformation, in contrast to other SH2-pY peptide structures where the pY peptide adopts an extended conformation. At position pY+2, the asparagine residue defines the specificity pocket because its carboxamide side chain forms three critical hydrogen bonds with the protein. At position pY+3, the peptide side chain makes contacts with an extended hydrophobic surface of the protein, justifying the preference for a hydrophobic amino acid at this position.
3.4. Csk SH2 domain
For Csk SH2 domain, library screening was carried out by the fluorescent method, using Texas Red-labeled SH2 domain. A total of 24 sequences were obtained (Table 1) and suggest a consensus sequence of pY(R/A/S/T)(R/x)(V/I/P) (Figure 5). Cantley and coworkers reported a consensus sequence of pY(T/A/s)(K/R/Q/N)(M/I/V/R) for Csk SH2 domain [60]. Because positively charged amino acids especially Arg can sometimes cause nonspecific binding to proteins during library screening, one of the selecetd Arg-containing peptide, R(Abu)pYRSILN, was synthesized and tested for binding to Csk SH2 domain by SPR. A KD value of 4.1 μM was obtained (Table 2 peptide 10). Thus, preference for positively charged residues is a genuine feature of Csk SH2 domain.
3.5 Substrate specificity of PTP1B
The above library method cannot be directly applied to determine the substrate specificity of PTPs, because there is no stable association between a resin-bound pY peptide and the PTP during a catalytic process. Previous library screening experiments employed catalytically inactive PTP mutants or non-hydrolyzable pY analogues [28–31]. However, a high-affinity binding sequence may not act as a good substrate for the active enzyme. In order to apply our library method to PTPs, one must first find a reliable method to selectively derivatize the PTP reaction product (i.e., tyrosine) with a specific probe such as biotin or a fluorescent group, without affecting any of the other functional groups in the library. We have recently developed such a methodology, which takes advantage of the exquisite substrate specificity of tyrosinase to selectively oxidize the tyrosine side chain into dopaquinone in the presence of O2 (Figure 6) [46]. The orthoquinone is then derivatized with a nucleophilic probe. To test this strategy, a pY library containing five random residues N-terminal to pY, Fmoc-XXXXXpYAALNBBRM-resin (where X is norleucine or any of the 17 proteinogenic amino acids except for Met, Cys, and Tyr; B = β-alanine; theoretical diversity = 1.89 × 106), was synthesized on 90-μm TentaGel resin as described above. Limited treatment of the library with a small amount of PTP1B resulted in dephosphorylation of beads that carry the best substrates of PTP1B. The exposed Tyr side chain was oxidized into orthoquinone by treatment with an excess amount of mushroom tyrosinase in the presence of atmospheric O2 (Note that Tyr is excluded from the randomized region). The resulting orthoquinone was selectively derivatized with biotin hydrazide. Subsequently, the library was incubated with SA-AP and BCIP and the dephosphorylated (and thus biotinylated) beads became turquoise colored. After removal of the N-terminal Fmoc group with piperidine, the positive beads were sequenced by PED/MS [35, 36].
Fig. 6.
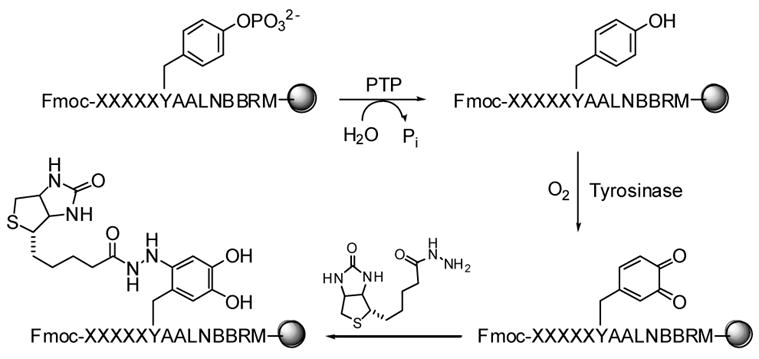
Reactions involved in pY library screening against a PTP.
A preliminary screening of ~10 mg of the above pY peptide library against PTP1B resulted in 13 intensely colored beads and their peptide sequences are listed in Table 3. These pY peptides can be classified into at least 3 classes. The most abundant class (peptides 11–17) has a consensus sequence of (K/R)(A/s/t)(I/V/l/m)XXpYAA, where X is any amino acid. Peptides in the second class (peptides 18–21) all contain a glutamate at pY−2 position and frequently a tryptophan at pY−1 position. The class II substrates have previously been reported by other investigators [28–34]. The third class (peptides 22 and 23) contains an acidic residue at pY−5 position. Four representative peptides were individually synthesized and assayed against PTP1B in the solution phase. All of them are excellent substrates of PTP1B, having kcat/KM values in the range of 3–7 × 105 M−1 s−1 (Table 3). Although additional work is needed to completely define the substrate specificity of PTP1B (in progress), the preliminary data clearly demonstrate that our combinatorial library method is readily applicable to PTPs.
Table 3.
Sequences and kinetic constants of peptides selected against PTP1Ba
| Peptide | Peptide Sequence | kcat (s−1) | KM (μM) | kcat/KM (× 105 M−1 s−1) |
|---|---|---|---|---|
| 11 | KAVFIpYAA | 16 ± 2 | 23 ± 6 | 7.0 |
| 12 | KAIFLpYAA | |||
| 13 | KALHGpYAA | |||
| 14 | KSVENpYAA | |||
| 15 | RAMLMpYAA | |||
| 16 | RTINEpYAA | 12 ± 1 | 18 ± 1 | 6.9 |
| 17 | RTIEWpYAA | 7.8 ± 0.1 | 26 ± 1 | 3.0 |
| 18 | VPGEWpYAA | |||
| 19 | TMHEWpYAA | |||
| 20 | MGDEQpYAA | |||
| 21 | QEDEPpYAA | 8.3 ± 0.6 | 20 ± 5 | 4.1 |
| 22 | DRRVApYAA | |||
| 23 | DLVSApYAA |
M, norleucine. PTP assay was performed in 100 mM Hepes (pH 7.4), 100 mM NaCl, 2 mM EDTA, and 5 mM TCEP.
4. Concluding remarks
The combinatorial library method described above is ideally suited for identifying peptide ligands for protein or non-protein receptors. Compared to the previously reported methods, our method has many advantages. First, our method identifies individual binding sequences; this feature is crucial for receptors that recognize multiple consensus sequences. Second, our method allows for “fair” competition among all library peptides, as each bead contains roughly the same amount of peptide molecules (~100 pmol). This ensures that library screening is based on affinity only, so that an underrepresented high-affinity ligand is identified. Third, because our method employs chemically synthesized libraries, modified (e.g., pY) and/or unnatural amino acids (e.g., D-amino acids) can be easily incorporated into the libraries. Fourth, our method is high-throughput, cost effective, and highly reproducible. A typical pY peptide library (5 g) can be synthesized in a week and is sufficient for screening against 20 different SH2 domains. For each SH2 domain, library screening and peptide sequencing can be easily completed in another week (assume that ~100 positive beads are identified for each SH2 domain). As demonstrated with PTP1B, the library method can also be adapted to screen for optimal substrates of enzymes. Finally, the sequence motifs identified from combinatorial libraries can be used to search protein and genomic databases for potential protein partners of SH2 domain-containing proteins [41, 42]. For example, two novel tensin-binding pY proteins have been discovered by this chemical approach [44]. The library method can also employed to identify potent and specific peptidyl inhibitors against modular domains and PTPs.
Acknowledgments
The authors thank other past and present members of the Pei research group for their contributions to the development of the methodologies described in this manuscript. This work was supported by National Institutes of Health (GM062820).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Venter JC, et al. Science. 2001;291:1304–1351. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- 2.Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, Cowburn D, Hanafusa H, Mayer BJ, Overduin M, Resh MD, Rios CB, Silverman L, Kuriyan J. Nature. 1992;358:646–653. doi: 10.1038/358646a0. [DOI] [PubMed] [Google Scholar]
- 3.Blaikie P, Immanuel D, Wu J, Li N, Yajnik V. B Margolis J Biol Chem. 1994;269:32031–3203. [PubMed] [Google Scholar]
- 4.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B. L C Cantley Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 5.Burshtyn DN, Yang W, Yi T, Long EO. J Biol Chem. 1997;272:13066–13072. doi: 10.1074/jbc.272.20.13066. [DOI] [PubMed] [Google Scholar]
- 6.Machida K. B J Mayer Biochim Biophys Acta – Proteins and Proteomics. 2005;1747:1–25. doi: 10.1016/j.bbapap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 7.Alonso A, Sasin J, Bottini N, Friedberg Ilan, Friedberg Iddo, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 8.Zhang ZY, Dixon JE. Advance in Enzymology. 1994;68:1–36. doi: 10.1002/9780470123140.ch1. [DOI] [PubMed] [Google Scholar]
- 9.Tonks NK, Neel BG. Curr Opin Cell Biol. 2001;13:182–195. doi: 10.1016/s0955-0674(00)00196-4. [DOI] [PubMed] [Google Scholar]
- 10.Hubbard MJ, Cohen P. TIBS. 1993;18:172–177. doi: 10.1016/0968-0004(93)90109-z. [DOI] [PubMed] [Google Scholar]
- 11.Mauro LJ, Dixon JE. Trends Biochem Sci. 1994;19:151–155. doi: 10.1016/0968-0004(94)90274-7. [DOI] [PubMed] [Google Scholar]
- 12.Cho H, et al. Biochemistry. 1991;30:6210–6216. doi: 10.1021/bi00239a019. [DOI] [PubMed] [Google Scholar]
- 13.Zhang ZY, Maclean D, McNamara DJ, Sawyer TK, Dixon JE. Biochemistry. 1994;33:2285–2290. doi: 10.1021/bi00174a040. [DOI] [PubMed] [Google Scholar]
- 14.Harder KW, Owen P, Wong LKH, Aebersold R, Clark-Lewis I, Jirik FR. Biochem J. 1993;298:395–401. doi: 10.1042/bj2980395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bobko M, Wolfe HR, Saha A, Dolle RE, Fisher DK, Higgins TJ. Bioorg Med Chem Lett. 1995;5:353–356. [Google Scholar]
- 16.Zhang ZY, et al. Proc Natl Acad Sci USA. 1993;90:4446–4450. doi: 10.1073/pnas.90.10.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, et al. Protein Sci. 1993;2:977–984. doi: 10.1002/pro.5560020611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dechert U, et al. Eur J Biochem. 1995;231:673–681. doi: 10.1111/j.1432-1033.1995.0673d.x. [DOI] [PubMed] [Google Scholar]
- 19.Flint AJ, Tiganis T, Barford D, Tonks NK. Proc Natl Acad Sci USA. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Blanchetot C, Chagnon M, Dube N, Halle M, Tremblay ML. Methods. 2005;35:44–53. doi: 10.1016/j.ymeth.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 21.Tenev T, Keilhack H, Tomic S, Stoyanov B, Stein-Gerlach M, Lammer R, Krivtsov AV, Ullrich A, Bohmer FD. J Biol Chem. 1997;272:5866–5973. doi: 10.1074/jbc.272.9.5966. [DOI] [PubMed] [Google Scholar]
- 22.Müller K, Gombert FO, Manning U, Grossmüller F, Graff P, Zaegel H, Zuber JF, Freuler F, Tschopp C, Baumann GJ. J Biol Chem. 1996;271:16500–16505. [PubMed] [Google Scholar]
- 23.Gram H, Schmitz SR, Zuber JF, Baumann G. Eur J Biochem. 1997;246:633–637. doi: 10.1111/j.1432-1033.1997.00633.x. [DOI] [PubMed] [Google Scholar]
- 24.King TR, Fang Y, Mahon ES, Anderson DH. J Biol Chem. 2000;275:36450–36456. doi: 10.1074/jbc.M004720200. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez M, Li SS-C, Harper JW, Songyang Z. J Biol Chem. 2004;279:8802–8807. doi: 10.1074/jbc.M311886200. [DOI] [PubMed] [Google Scholar]
- 26.Jia Z, Barford D, Flint AJ, Tonks NK. Science. 1995;268:1754–1758. doi: 10.1126/science.7540771. [DOI] [PubMed] [Google Scholar]
- 27.Yang J, Cheng Z, Niu T, Liang X, Zhao ZJ, Zhou GW. J Biol Chem. 2000;275:4066–4071. doi: 10.1074/jbc.275.6.4066. [DOI] [PubMed] [Google Scholar]
- 28.Pellegrini MC, Liang H, Mandiyan S, Wang K, Yuryev A, Vlattas I, Sytwu T, Li YC, Wennogle LP. Biochemistry. 1998;37:15598–15606. doi: 10.1021/bi981427+. [DOI] [PubMed] [Google Scholar]
- 29.Huyer G, Kelly J, Moffat J, Zamboni R, Jia Z, Gresser MJ, Ramachandra C. Anal Biochem. 1998;258:19–30. doi: 10.1006/abio.1997.2541. [DOI] [PubMed] [Google Scholar]
- 30.Espanel X, van Huijsduijnen RH. Methods. 2005;35:64–72. doi: 10.1016/j.ymeth.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 31.Walchli S, Espanel X, Harrenga A, Rossi M, Cesareni G, van Huijsduiinen RH. J Biol Chem. 2004;279:311–318. doi: 10.1074/jbc.M307617200. [DOI] [PubMed] [Google Scholar]
- 32.Vetter SW, Keng YF, Lawrence DS, Zhang ZY. J Biol Chem. 2000;275:2265–2268. doi: 10.1074/jbc.275.4.2265. [DOI] [PubMed] [Google Scholar]
- 33.Wang P, Fu H, Snavley DF, Freitas MA, Pei D. Biochemistry. 2002;41:10700–10709. [Google Scholar]
- 34.Cheung YW, Abell C, Balasubramanian S. J Am Chem Soc. 1997;119:9568–9569. [Google Scholar]
- 35.Sweeney MC, Pei D. J Comb Chem. 2003;5:218–222. doi: 10.1021/cc020113+. [DOI] [PubMed] [Google Scholar]
- 36.Thakkar A, Wavreille A-S, Pei D. Anal Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 37.Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 38.Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 39.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Proc Natl Acad Sci U S A. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beebe KD, Wang P, Arabaci G, Pei D. Biochemistry. 2000;39:13251–13260. doi: 10.1021/bi0014397. [DOI] [PubMed] [Google Scholar]
- 41.Sweeney MC, Wavreille AS, Park J, Butchar J, Tridandapani S, Pei D. Biochemistry. 2005;44:14932–14947. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- 42.Imhof D, Wavreille AS, May A, Zacharias M, Tridandapani S, Pei D. J Biol Chem. 2006;281:20271–20282. doi: 10.1074/jbc.M601047200. [DOI] [PubMed] [Google Scholar]
- 43.Qin C, Wavreille A-S, Pei D. Biochemistry. 2005;44:12196–12202. doi: 10.1021/bi050669o. [DOI] [PubMed] [Google Scholar]
- 44.Wavreille AS, Pei D. ACS Chem Biol. 2007;2 doi: 10.1021/cb600433g. in press. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney MC, Wang X, Park J, Liu Y, Pei D. Biochemistry. 2006;45:14740–14748. doi: 10.1021/bi061782x. [DOI] [PubMed] [Google Scholar]
- 46.M. Garaud, D. Pei, manuscript in preparation.
- 47.Ravetch JF, Lanier LL. Science. 2000;290:84–89. doi: 10.1126/science.290.5489.84. [DOI] [PubMed] [Google Scholar]
- 48.Kozlowski M, Larose L, Lee F, Le DM, Rottapel R, Siminovitch KA. Mol Cell Biol. 1998;18:2089–2099. doi: 10.1128/mcb.18.4.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guntermann C, Alexander DR. J Immunol. 2002;168:4420–4429. doi: 10.4049/jimmunol.168.9.4420. [DOI] [PubMed] [Google Scholar]
- 50.Hu Y, Szente B, Kiely JM, Gimbrone MA., Jr J Biol Chem. 2001;276:48549–48553. doi: 10.1074/jbc.M105513200. [DOI] [PubMed] [Google Scholar]
- 51.Ali S, Ali S. J Biol Chem. 2000;275:39073–39080. doi: 10.1074/jbc.M007478200. [DOI] [PubMed] [Google Scholar]
- 52.Gunaje JJ, Bhat GJ. Biochem Biophys Res Commun. 2001;288:252–257. doi: 10.1006/bbrc.2001.5759. [DOI] [PubMed] [Google Scholar]
- 53.Kessels HWHG, Ward AC, Schumacher TNM. Proc Natl Acad Sci USA. 2002;99:8524–8529. doi: 10.1073/pnas.142224499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelicci G, Lanfrancone L, Grignani F, McGlade J, Cavallo F, Forni G, Nicoletti I, Grignani F, Pawson T, Pelicci PG. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell. 1992;70:93–104. doi: 10.1016/0092-8674(92)90536-l. [DOI] [PubMed] [Google Scholar]
- 55.Pendergast AM, Quilliam LA, Cripe LD, Bassing CH, Dai Z, Li N, Batzer A, Rabun KM, Der CJ, Schlessinger J, et al. Cell. 1993;75:175–185. [PubMed] [Google Scholar]
- 56.Fixman ED, Naujokas MA, Rodrigues GA, Moran MF, Park M. Oncogene. 1995;10:237–249. [PubMed] [Google Scholar]
- 57.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 58.Ogura K, Tsuchiya S, Terasawa H, Yuzawa S, Hatanaka H, Mandiyan V, Schlessinger J, Inagaki F. J Mol Biol. 1999;289:439–445. doi: 10.1006/jmbi.1999.2792. [DOI] [PubMed] [Google Scholar]
- 59.Rahuel J, Gay B, Erdmann D, Strauss A, Garcia-Echeverria C, Furet P, Caravatti G, Fretz H, Schoepfer J, Grutter MG. Nat Struct Biol. 1996;3:586–589. doi: 10.1038/nsb0796-586. [DOI] [PubMed] [Google Scholar]
- 60.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, et al. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]