

Abstract
We studied the relationship between recombination and the fixation time of multiple drug-resistance mutations after HIV-1 drug therapy, under a set of different realistic scenarios. We have generalized a previous model by Bretscher and coworkers in order to explore different implementations of phenotypic mixing and more realistic demographic and selective regimes. Using computer simulations we show that the effect of recombination on the evolution of drug resistance depends strongly on the intensity of selection, as well as on the viral population size. Under the high selection pressure expected during antiretroviral therapy, the strength of the Hill-Robertson effect increases and recombination favors the evolution of resistance under a wide range of population sizes, independently of the sign of the epistatic interaction. Our results suggest that recombination plays an important role in the evolution of drug resistance in HIV-1 under various realistic scenarios. These findings could be taken into account in order to develop optimal HIV-1 drug treatments.
Keywords: recombination, drug resistance, Hill-Robertson effect, epistasis, HIV-1
INTRODUCTION
Several empirical studies have related recombination with the development of HIV-1 resistance to drug therapy (Gu et al., 1995; Kellam and Larder, 1995; McDowell et al., 1998; Moutouh, Corbeil, and Richman, 1996). Indeed, population genetics theory predicts that recombination can favor adaptation in finite populations. The Hill-Robertson effect (HRE) (Hill and Robertson, 1966) implies that in finite populations negative disequilibrium accumulates on average (i.e., beneficial and deleterious alleles are found together on the same chromosome more often than expected given their frequencies) because of drift and mutation, reducing the probability of fixation of beneficial alleles, and therefore, the efficacy of selection (Felsenstein, 1974; Fisher, 1930; Muller, 1964). Indeed, the accumulation of negative disequilibrium not only occurs because of the stochastic nature of mutation and sampling in finite populations, but can also take place when fully susceptible genotypes are less fit than expected from the product of the fitness at each locus (i.e., negative epistasis). Importantly, genetic recombination reduces the strength of the HRE, increasing the rate of fixation of beneficial alleles (McVean and Charlesworth, 2000; Roze and Barton, 2006). In a multilocus system under negative linkage disequilibrium, recombination breaks up the unpaired combinations of resistant and susceptible alleles, generating both fully resistant genotypes, that will spread rapidly within the population, and fully susceptible genotypes, that will be eliminated fairly quickly. Therefore, recombination should favor the emergence of multidrug resistance, that is, the accumulation of multiple mutations that lead to resistance to one or several drugs.
While specific mathematical models have suggested that recombination is an important mechanism for the generation of HIV-1 multidrug resistance (Rouzine and Coffin, 2005), recently, Bretscher et al. (2004) and Althaus et al. (2005) disputed the generality of this result. These authors used computer simulations to study the interaction of mutation, selection, effective population size (the number of individuals in a population who contribute offspring to the next generation) and epistasis on the evolution of multidrug resistance in HIV-1. They concluded that while recombination favors the acquisition of multidrug resistance for low and (especially for) intermediate effective population sizes regardless of epistasis, and for large effective population sizes (>105) when epistasis is negative, recombination severely impairs the development of multidrug resistance for large effective population sizes (>105) when epistasis is positive.
This last result might be very relevant because estimates of HIV-1 effective population size range from 500 to 105 (Althaus and Bonhoeffer, 2005; Rouzine and Coffin, 1999), and epistasis in HIV-1 might be positive, at least in the absence of drug therapy (Bonhoeffer et al., 2004; but see Wang, Mittler, and Samudrala, 2006). However, we believe that these results depend on the particular model implemented, which we can relax in order to provide a more realistic view of HIV-1 evolution. First of all, the selection coefficient used by Althaus et al. (2005) with positive epistasis was very low (s = 0.1), considering that under drug therapy we expect a high selection pressure (Finzi et al., 1999; Ribeiro, Bonhoeffer, and Nowak, 1998; Rouzine and Coffin, 2005). Second, they used a simple function for phenotype mixing (when two proviruses are transcribed simultaneously within the same infected cell and the newly assembled virions contain mixed proteins from both parental proviruses), assuming that the fitness of a virion in a double-infected cell is always the arithmetic mean of the fitness of both parental proviruses. However, it seems more realistic to incorporate some uncertainty in this process. Third, they used a constant effective population size, while the effective population size of HIV-1 varies throughout infection, and especially under highly active antiretroviral therapy, where the population size can drop more than one order of magnitude (Rodrigo, 1999; Rouzine and Coffin, 1999; Rouzine, Rodrigo, and Coffin, 2001).
In addition, Fraser (2005) has recently extended the deterministic model of Bretscher et al. (2004) to incorporate viral dynamics, concluding that the impact of recombination on the evolution depends on the frequency of coinfection: if coinfection falls rapidly with viral load, then recombination should have no effect, whereas if the frequency of coinfection is constant (as in Bretscher et al. (2004)), then recombination slows down the emergence of drug resistance. Note that these results contradict those by Bretscher et al. (2004).
Here, and in order to better understand the role of recombination on the evolution of HIV-1 drug resistance, we have generalized the deterministic and stochastic models in (Althaus and Bonhoeffer, 2005; Bretscher et al., 2004) to include more realistic conditions. We explore different implementations of phenotypic mixing and higher selection coefficients, under infinite and finite population sizes, with constant or variable demographics. We demonstrate that the accumulation of negative linkage disequilibrium due to a strong HRE explains how recombination facilitates the evolution of drug resistance during drug therapy in HIV-1 regardless epistasis. Finally, we also study the role of pre-existent resistant mutants, which should be an important factor for the acquisition of drug resistance (Ribeiro and Bonhoeffer, 2000).
MATERIALS AND METHODS
The models implemented in this study are based on a two-locus two-allele deterministic model in Bretscher (2004) We will first describe the basic features of this model, and then explain in detail our extensions.
Deterministic model
We have four types of proviruses, ab, aB, Ab and AB, where lowercase denotes drug-sensitive wild-type alleles and uppercase indicates drug-resistant mutant alleles. Under drug therapy, provirus type ab is fully sensitive, aB and Ab are partially drug resistant, and AB is fully resistant. The change in provirus frequencies during the replication cycle is best described by dividing this cycle in three steps: cell infection, virion release, and provirus transcription.
1) Infection of cells
It is assumed that all cells will be infected by either one or two proviruses. Let f be the frequency of cells infected with two proviruses (i.e., f is the frequency of coinfection). The frequency (C) of a cell infected with a single provirus type is the product of the frequency (P) of the corresponding provirus type and the probability of single infection (1-f). For example, Cab = (1 – f) × Pab. Similarly, the frequency of double-infected cells is given by the product of the frequencies of both proviruses and the probability of double infection (f). For example, CabAB = 2 × f × Pab × PAB With four provirus types, there are four types of single-infected and ten types of double-infected cells.
2) Release of virions
The frequency of homozygous and heterozygous virions released by the cell population is determined from the cell frequencies. It is assumed that both single- and double-infected cells release the same amount of virions, and that released virions infect target cells depending on their fitness. For example, the frequency of the infecting ABAB virion is given by VABAB = [1 / V] [WAB ( CAB + CABAB) + 0.25 [g(WAB, WAb) CABAb + g( WAB, WaB) CABaB + g(WAB, Wab) CABab)], where WAB, WAb, WaB, Wab are the fitness values of the corresponding proviruses, CAB, CABAB, CABAb, CABaB and CABab are the corresponding single or double infected cell frequencies, g is a function that will depend on the phenotypic mixing implementation (see below), and V is a normalization factor to ensure that all infecting virion frequencies sum to 1. Provirus fitness during therapy is computed under a multiplicative model in which the wild type has fitness 1 – s. Genotypes carrying one mutant allele have fitness (1 – s – E)1/2 and the double mutant has fitness 1. E is the epistatic value.
3) Transcription of inserted virions to proviruses
After all released virions have infected a target cell, the frequencies of proviruses in the next generation are computed from that of virions as a function of the mutation rate μ and the recombination rate r. Mutation is considered only during the reverse transcription process.
Extensions to the basic model: Phenotype mixing
The fitness of emerging virions depends on both the parental provirus fitness and the cellular type. This separation of phenotype and genotype is known as phenotypic mixing (Brenner, 1957; Novick and Szilard, 1951). In real life, phenotypic mixing occurs when two proviruses that are being transcribed simultaneously within the same infected cell mix their corresponding viral proteins in the assembly of new virions. It is possible then that the fitness of a released virion does not necessarily reflect the genomic RNA that it carries, but that it is rather somewhere in between that of both parental proviruses. The specific implementation of the phenotypic mixing in our simulations is described by the function g. In the Arithmetic model, which is exactly the same as the one described by Bretscher and co-workers (2004), g is the arithmetic mean. In the Geometric model virion fitness is the geometric mean of the fitness of both parental proviruses, implying that, in the absence of epistasis, the fitness of virions emerging from double infected cells with provirus AB and ab will be the same as the fitness of virions emerging from double infected cells with provirus Ab and aB. Therefore, this model is unique in that there are no fitness differences between the two fully resistant double mutants AB/ab and Ab/aB. In the Minimum and Maximum models, virion fitness is computed simply as the minimum or the maximum, respectively, of the two parental proviruses. In the Random model, the most realistic model considered here, we assume that phenotypic mixing does not always occur. Instead, we consider four types of possible events, that the virion inherits the viral proteins and the genomic RNA from either parent, or that the virion inherits the viral proteins from one parent and the genomic RNA from the other, with equal probability for each type of event. Finally, if there is no phenotypic mixing then the fitness of the virion is computed assuming a multiplicative model. In this case, the fitness of the double homozygote virion ABAB is the product WAB × WAB. The same scheme applies to all provirus genotypes.
Stochastic implementation
We introduce stochasticity by considering that different cell types can contribute a variable amount of virions in each generation, and that there is a finite amount of infecting virions (N, the effective population size). First, we select a cell type according to its frequency. Second, a virion is released by the selected cell type contingent on its probability of being produced. Third, this virion will survive and become infecting depending on its fitness. For example, the probability of selecting a cell coinfected by proviruses AB and ab is CABab. From this cell we release a virion ABAB with probability ¼ that it will survive and become infecting with probability g(WAB, Wab). We repeat this process until N released virions have survived until infection.
In addition, because drug therapy affects viral load, we also consider a model with variable effective population size. In this case, we repeat the first two steps above until N0 virions have been released. From these, Nt (≤ N0) virions will survive and become infecting depending on their fitness. The expected number of infecting virions at a given generation will be simply the sum of the fitnesses of the N0 previously released virions. As a consequence, Nt will vary across generations, but always below N0. N0 is a constant specified at the beginning of the simulation that represents the maximum number of virions released by the infected cells at any generation.
Computer Simulations
To determine how recombination affects the rate at which multidrug resistance evolves during drug therapy, we started the simulations from a population consisting only of wild-type proviruses and measured the number of generations until 90% of the provirus population was fully resistant (fixation time). In the deterministic simulations, the double infection rate per cell per generation (f) was 0.3 or 0.75 (Jung et al., 2002), the mutation rate (μ) was 3 × 10−5 mutations per locus per generation (Mansky and Temin, 1995), the selection coefficient (s) was 0.1, 0.5 or 0.9 (Finzi et al., 1999; Ribeiro, Bonhoeffer, and Nowak, 1998; Rouzine and Coffin, 2005), epistasis values (E) were −0.05, 0.0, or 0.05 (Althaus and Bonhoeffer, 2005; Bretscher et al., 2004), and the recombination rate (r) was 0.0, 0.01, 0.1, 0.2, 0.3, 0.4, or 0.5 recombination events per locus per generation (Shriner et al., 2004). In addition, we ran some cases with s = 0.01, 0.2, 0.3, 0.4, 0.6, 0.7 and 0.8. We also studied the effect of recombination on the fixation time when single resistant mutations pre-exist at a frequency of 0.1%. In the stochastic simulations, we used the same parameter values as above, except that we only explored r = 0.0 and 0.1, plus a set of distinct effective population sizes (N or N0): 102, 103, 104, 105 and 106. We carried out 1000 replicates for each set of conditions, but for N = 106 we only ran 100. The number of replicates was always enough to maintain standard errors below 1%. To test our stochastic implementation, we ran an asymptotic case with N = 108, which should mimic the behavior of the deterministic model. We measured the recombination effect on the evolution of drug resistance as the percent reduction on fixation time for a specific recombination rate compared to the case of no recombination , or Re = 100 × (1 – Tr/T0), where Tr is the fixation time with recombination rate r and T0 is the fixation time without recombination. Thus, Re will be positive when recombination reduces fixation time and negative when recombination increases it. Simulations were implemented with a computer program written in C. A copy of the source code is freely available upon request from the authors.
RESULTS
Deterministic evolution of multidrug resistance
Without epistasis, s = 0.1, and with 75% of cells doubly infected, the effect of recombination during therapy varied depending on the phenotypic mixing implementation (Figure 1). Under the Minimum model recombination reduced the fixation time, while for the Maximum model the effect was the opposite. When epistasis was negative, recombination significantly reduced the fixation time of the double-resistant proviruses regardless the implementation of phenotypic mixing, especially for the Minimum model. When epistasis was positive, recombination significantly increased the fixation time for all models, although this effect was smaller for the Minimum model or in the absence of phenotypic mixing. Other values of the selection coefficient resulted in the same trends (not shown), although smaller selection coefficient values implied longer times to fixation and vice versa). Importantly, when the frequency of double infected cell was low (f = 0.3), the results obtained were similar (not shown), although the differences between the distinct phenotypic mixing implementations were weaker.
Figure 1.
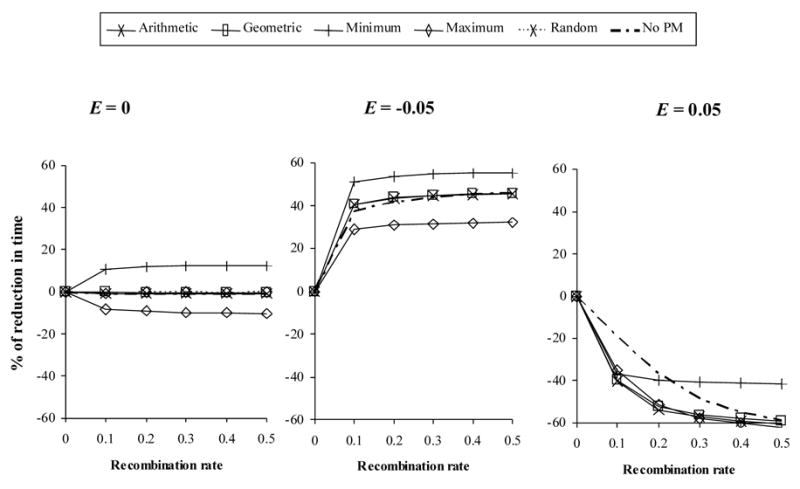
Effect of recombination on the evolution of multidrug resistance in the deterministic case. The y-axis indicates the % of reduction of fixation time produced by a given recombination rate with respect to the case of no recombination. Here f = 0.75 and s = 0.1. E: epistasis. PM: phenotypic mixing.
Stochastic evolution of multidrug resistance
The introduction of a finite effective population size had a remarkable effect. When population size was constant and selection pressure was low (s = 0.1) (Figure 2, first row) with zero or negative epistasis, recombination decreased the fixation time for a wide range of population sizes (N = 102 –106) with a maximum at intermediate sizes. When epistasis was positive, recombination slightly decreased the fixation time for small and intermediate population sizes, but increased it when N > 104. Remarkably, with higher selection pressures (s = 0.5 or 0.9) (Figure 2, second and last rows), recombination always decreased the fixation time regardless of the sign of the epistatic interaction, especially when N = 104. Phenotypic mixing had practically no effect. When we used a very large effective population size (N = 108), results matched exactly those obtained under the deterministic model (not shown), therefore validating the implementation of the stochastic model.
Figure 2.
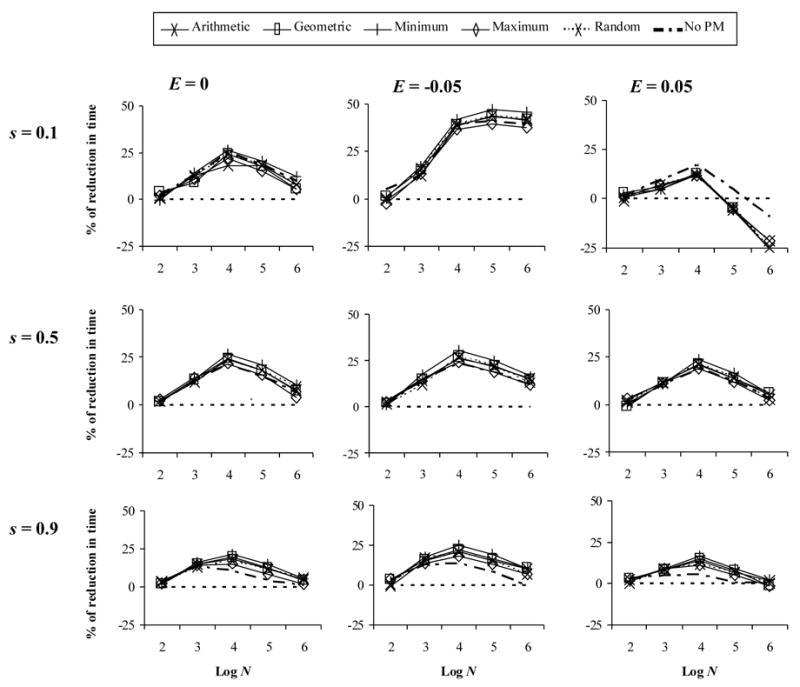
Effect of recombination on the evolution multidrug resistance with constant population size. The y-axis indicates the % of reduction onto fixation time under a recombination rate of 0.1 with respect to the case of no recombination. Here f = 0.75. E: epistasis. s: selection coefficient. Log N: logarithm of the effective population size.
With variable population size (Figure 3), results were very similar to the constant case, although it seemed that the largest decreases in fixation time were obtained under slightly larger population sizes, especially for high selective pressures. Again, phenotypic mixing had practically no effect.
Figure 3.
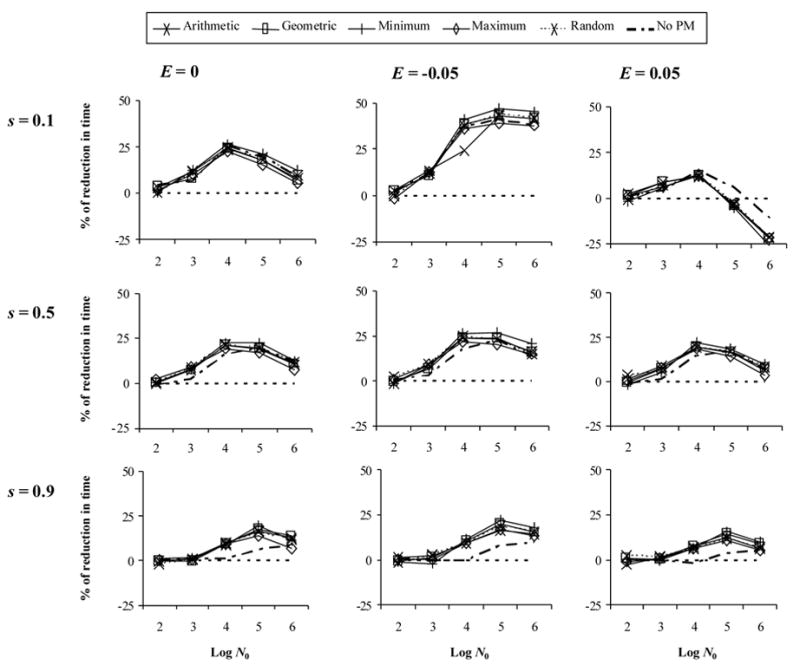
Effect of recombination on the evolution multidrug resistance with variable population size. The y-axis indicates the % of reduction onto fixation time under a recombination rate of 0.1 with respect to the case of no recombination. Here f = 0.75. E: epistasis. s: selection coefficient. Log N0: logarithm of the initial effective population size.
Pre-existence of resistant mutations
In the deterministic case, the presence of resistant mutations (0.1%) before drug therapy had no effect compared to the non-preexistence case (results not shown). In contrast, with finite population sizes the effect of recombination was magnified with respect to non-preexistence (Figure 4), but with similar trends. Under strong selection, demographics seemed to play a significant role.
Figure 4.
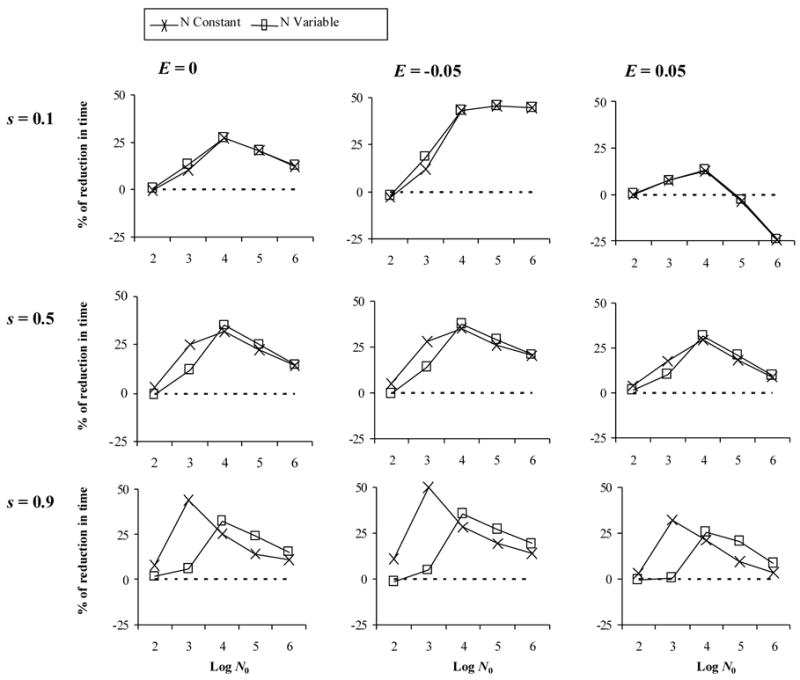
Effect of recombination on the evolution multidrug resistance when resistant mutants exist before drug therapy (pre-existence). The y-axis indicates the % of reduction onto fixation time under a recombination rate of 0.1 with respect to the case of no recombination. Here the initial frequency of mutants is 0.1%, phenotypic mixing is Random, and f = 0.75. E: epistasis. s: selection coefficient. Log N0: logarithm of the initial effective population size.
DISCUSSION
Deterministic versus stochastic models
Only under a deterministic model is negative epistasis needed for recombination to accelerate fixation time, as previously shown by Bretscher et al. (2004). This is true unless phenotypic mixing always confers the virion the fitness of the weakest parent, which does not seem very plausible. Although in the study of HIV-1 dynamics a general controversy exists regarding whether deterministic or stochastic models should be considered (Garnett, 2002; Leigh Brown, 1997; Rouzine and Coffin, 1999; Tan and Wu, 1998), we consider that the evolution of HIV-1 is better described by a stochastic process because low effective population sizes are expected due to compartmentalization (Nickle et al., 2003), to variability in progeny number between infected cells, to population expansion from a small initial inoculum, and from variable selective pressures (Leigh Brown, 1997). Furthermore, stochasticity is especially important under antiretroviral therapy, when the number of infected cells can drop by one or more orders of magnitude (Ho et al., 1995; Rodrigo, 1999; Wei et al., 1995).
From a theoretical point of view, the effect of recombination on the evolution of drug resistance can be formally explained by analyzing the models in terms of linkage disequilibrium (D) and the phenotypic mixing effect (see Appendix for details). A key difference between the deterministic and the stochastic situation is that in the former the frequency of a mutant will never go to zero. Therefore, an insignificant fraction of double resistant provirus could persist and subsequently be selected until fixation. However, in the stochastic models new mutations are often lost quickly by chance. This will generate random deviations, positive or negative, from linkage equilibrium. In the absence of selection, these deviations cancel out and D averaged across generations (or loci) equals 0. However, in the presence of selection, positive deviations from linkage equilibrium will be rapidly fixed or eliminated. On the other hand, negative D deviations are not so quickly solved by selection, because of the intermediate fitness of single mutants (a fitter allele in a less fit background). This is known as the Hill-Robertson effect (HRE) (Barton and Otto, 2005; Hill and Robertson, 1966). The accumulation of negative disequilibrium can be significant even in fairly large populations as long as the allele frequencies are initially low and subject to drift (Barton and Otto, 2005). Thus, the HRE predicts that genetic drift will interact with directional selection to produce negative D. Such negative D is all that recombination needs to favor beneficial mutations, and, as predicted by equation A1 (see Appendix), it will allow recombination to speed up the emergence of multidrug resistance. Furthermore, the form of epistasis is not critical to the advantage of recombination in finite populations, as recently suggested (Keightley and Otto, 2006),if selection is strong.
Phenotypic mixing
Bretscher et al. (2004) used the average of the parental fitnesses to include phenotypic mixing in their model. It seemed reasonable to think the outcome of phenotypic mixing might not always be the same due to the stochasticity of this process. However, in our simulations the particular implementation of phenotypic mixing had only a clear effect in the deterministic case, and even in this case only the two extreme models (Minimum and Maximum) departed from the average behavior. In fact, results under the most realistic Random model were very similar to those obtained under the Arithmetic model of Bretscher et al. (2004). That the particular implementation of phenotypic mixing did not influence the results of the stochastic simulations is important, and makes the biological interpretation of the simulations mores straightforward.
The frequency of coinfection
Importantly, the specific frequency f of double infected cells did not change the results. Although a smaller f resulted in a reduction of the effective recombination rate, recombination still favored drug resistance. It is very important to note that a reduction of the recombination rate does not necessarily reduce its effect on the fixation time, unless this rate is 0 (i.e., the effect of r = 0.5 is very similar to that of r = 0.1). Our results contrast to those obtained by Fraser (2005), who claimed that with low coinfection rates recombination does not have an effect on drug resistance. However, such results were obtained using a deterministic model under low selective pressures, which is not a realistic scenario under drug therapy.
Pre-existence of resistant mutations
Pre-existence of mutations had an effect on the role of recombination independently of epistasis, especially with the stochastic models. In particular, pre-existent mutations magnified the effect of recombination reducing the fixation time of double mutants, and therefore favor the evolution of drug resistance. This is because the pre-existence of single resistant mutants generates an initial negative linkage disequilibrium that adds to that already generated by the HRE. Under strong selection, the different meaning of N0 under constant and variable population sizes becomes evident. In the constant case N0 = 103 represents the optimal population size for negative disequilibrum to accumulate in the population under this model. In the variable case N0 = 104 represents the optimal maximum population size, which under high selective pressure will be seldom reached.
These results agree well with a previous study that showed that even a very modest amount of recombination is more efficient at generating highly fit viral genomes than mutation alone, provided the pre-existence of the more fit alleles (Rouzine and Coffin, 2005). This result is important because it suggests that if resistant mutations appear before drug therapy the fixation of resistance in the viral population could be very rapid. In addition, if transmission of drug resistance mutations increases in a population, the effect of pre-existant mutations will be even more relevant.
The effect of recombination on the evolution of drug resistance
We have shown that the effect of recombination on the evolution of multidrug resistance depends strongly not only on the viral population size but also on the intensity of selection. With higher selection pressure, the importance of the HRE prevails over epistasis, and recombination always diminishes the fixation time of resistance under a wide range of population sizes. Indeed, we expect selection to be very strong during antiretroviral therapy (Finzi et al., 1999; Ribeiro, Bonhoeffer, and Nowak, 1998; Rouzine and Coffin, 2005). In this way, we reconcile HIV-1 evolution with previous evidence (Gu et al., 1995; Kellam and Larder, 1995; McDowell et al., 1998; Moutouh, Corbeil, and Richman, 1996; Rouzine and Coffin, 2005) and with the common belief regarding the adaptive role of recombination (Felsenstein, 1974; Fisher, 1930; Hill and Robertson, 1966; Keightley and Otto, 2006; McVean and Charlesworth, 2000; Muller, 1964; Roze and Barton, 2006). Our results suggest that, everything else being equal, a combination of drugs that target close regions of the HIV-1 genome will be less susceptible to failure due to the emergence of drug resistance because of a reduced recombination rate between target loci.
Acknowledgments
We thank Armando Caballero for very useful comments on a previous version of this manuscript. This work was supported by grant R01-GM66276 from the US National Institutes of Health (AC-R, KAC, DP), grant BFU2004-02700 of the Spanish Ministry of Education and Science (DP) and by the “Ramón y Cajal” programme of the Spanish government (DP). AC-R is currently funded by an Isidro Parga Pondal research fellowship from Xunta de Galicia (Spain).
APPENDIX
Results here and from previous work (Althaus and Bonhoeffer, 2005; Bretscher et al., 2004) can be explained by analyzing the models in terms of linkage disequilibrium (D) and the phenotypic mixing effect (δ).
Linkage disequilibrium in generation t, D (t), can be defined as the difference between the product of frequencies of coupled and uncoupled provirus genotypes: D(t) = PAB(t)Pab(t) - PAb(t)PAb(t). After equilibrium is reached, the final D value accumulated through generations is Deq.
The fitness of each virion is computed assuming a phenotypic mixing function g such that g( WAB, Wab) is the fitness of virions coming from a cell coinfected with genotypes AB and ab, and g( WAb, WaB) is the fitness of virions coming from a cell coinfected with genotypes Ab and aB. In the absence of phenotypic mixing, this function represents directly the fitness of a double heterozygous virion.
The effect of recombination at generation t, on the frequency of double mutant provirus can be expressed as:
| (A1) |
where X(t-1) is a long term that includes cell frequencies, provirus fitnesses and mutation rate at generation t-1 (we will ignore it because it is independent of the recombination rate r), f is the frequency of double infected cells, and Y = (1–2μ)2 r/2, and δ = g(WAB, Wab) - g( WAb, WaB).
Results obtained in the simulations can be explained upon inspection of equation A1. Whenever Deq < 0 or δ < 0, recombination will increase the double mutant frequency, decreasing fixation time (the time until 90% of the provirus population is fully resistant). The opposite will be true when Deq > 0 or δ > 0. Moreover, negative epistasis will decrease δ and positive epistasis will increase it.
In the deterministic case, and in the absence of epistasis, Deq = 0 (Felsenstein, 1965; Lewontin, 1974), and the only possible effect is associated with δ. If δ <0 (Minimum model), recombination will reduce fixation time, while the opposite will be true if δ >0 (Maximum model). If there is epistasis, Deq will have the same sign as the epistatic interaction (Eshel and Feldman, 1970; Felsenstein, 1965). Because of this, negative epistasis will reduce fixation time, while positive epistasis will increase it.
When population size is finite, or when mutations pre-exist, Deq is negative under strong selection because of the HRE, even with positive epistasis. Note that the effect of δ will be less important because the magnitude of the multiplying factor in equation A1 is much smaller for δ than for D. Thus, under the simulated conditions of antiretroviral therapy, i.e. finite population size, strong selection pressure and null or low initial frequency of beneficial alleles, negative linkage disequilibrium will accumulate, providing the context for recombination to favor the emergence of drug resistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- Althaus CL, Bonhoeffer S. Stochastic interplay between mutation and recombination during the acquisition of drug resistance mutations in human immunodeficiency virus type 1. Journal of Virology. 2005;79(21):13572–13578. doi: 10.1128/JVI.79.21.13572-13578.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton NH, Otto SP. Evolution of recombination due to random dirift. Genetics. 2005;169(4):2353–2370. doi: 10.1534/genetics.104.032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhoeffer S, Chappey C, Parkin NT, Whitcomb JM, Petropoulos CJ. Evidence for positive epistasis in HIV-1. Science. 2004;306(5701):1547–50. doi: 10.1126/science.1101786. [DOI] [PubMed] [Google Scholar]
- Brenner S. Genetic Control and Phenotypic Mixing of the Adsorption Cofactor Requirement in Bacteriophage-T2 and Bacteriophage-T4. Virology. 1957;3(3):560–574. doi: 10.1016/0042-6822(57)90010-7. [DOI] [PubMed] [Google Scholar]
- Bretscher MT, Althaus CL, Muller V, Bonhoeffer S. Recombination in HIV and the evolution of drug resistance: for better or for worse? Bioessays. 2004;26(2):180–8. doi: 10.1002/bies.10386. [DOI] [PubMed] [Google Scholar]
- Eshel I, Feldman MW. On the evolutionary effect of recombination. Theor Popul Biol. 1970;1(1):88–100. doi: 10.1016/0040-5809(70)90043-2. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Effect of Linkage on Directional Selection. Genetics. 1965;52(2P1):349–363. doi: 10.1093/genetics/52.2.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. The evolutionary advantage of recombination. Genetics. 1974;78(2):737–56. doi: 10.1093/genetics/78.2.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. Latent infection of CD4(+) T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nature Medicine. 1999;5(5):512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The Genetical Theory of Natural Selection. Oxford University Press; Oxford: 1930. [Google Scholar]
- Fraser C. HIV recombination: what is the impact on antiretroviral therapy? J R Soc Interface. 2005;2(5):489–503. doi: 10.1098/rsif.2005.0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett GP. An introduction to mathematical models in sexually transmitted disease epidemiology. Sexually Transmitted Infections. 2002;78(1):7–12. doi: 10.1136/sti.78.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Gao Q, Faust EA, Wainberg MA. Possible involvement of cell fusion and viral recombination in generation of human immunodeficiency virus variants that display dual resistance to AZT and 3TC. Journal of General Virology. 1995;76 (10):2601–2605. doi: 10.1099/0022-1317-76-10-2601. [DOI] [PubMed] [Google Scholar]
- Hill WG, Robertson A. The effect of linkage on limits to artificial selection. Genet Res. 1966;8(3):269–94. [PubMed] [Google Scholar]
- Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S, Meyerhans A. Multiply infected spleen cells in HIV patients. Nature. 2002;418(6894):144. doi: 10.1038/418144a. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Otto SP. Interference among deleterious mutations favours sex and recombination in finite populations. Nature. 2006;443(7107):89–92. doi: 10.1038/nature05049. [DOI] [PubMed] [Google Scholar]
- Kellam P, Larder BA. Retroviral recombination can lead to linkage of reverse transcriptase mutations that confer increased zidovudine resistance. Journal of Virology. 1995;69(2):669–674. doi: 10.1128/jvi.69.2.669-674.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh Brown AJ. Analysis of HIV-1 env gene sequences reveals evidence for a low effective number in the viral population. Proceedings of the National Academy of Sciences, USA. 1997;94:1862–1865. doi: 10.1073/pnas.94.5.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewontin RC. The genetic basis of evolutionary change. Columbia University Press; New York: 1974. [Google Scholar]
- Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69(8):5087–94. doi: 10.1128/jvi.69.8.5087-5094.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell JM, Dhandaydham M, Long TA, Aarts MG, Goff S, Holub EB, Dangl JL. Intragenic recombination and diversifying selection contribute to the evolution of downy mildew resistance at the RPP8 locus of Arabidopsis. Plant Cell. 1998;10(11):1861–74. doi: 10.1105/tpc.10.11.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean GA, Charlesworth B. The effects of Hill-Robertson interference between weakly selected mutations on patterns of molecular evolution and variation. Genetics. 2000;155(2):929–44. doi: 10.1093/genetics/155.2.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moutouh L, Corbeil J, Richman DD. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proc Natl Acad Sci U S A. 1996;93(12):6106–11. doi: 10.1073/pnas.93.12.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ. The Relation Of Recombination To Mutational Advance. Mutat Res. 1964;106:2–9. doi: 10.1016/0027-5107(64)90047-8. [DOI] [PubMed] [Google Scholar]
- Nickle DC, Jensen MA, Shriner D, Brodie SJ, Frenkel LM, Mittler JE, Mullins JI. Evolutionary indicators of human immunodeficiency virus type 1 reservoirs and compartments. Journal of Virology. 2003;77(9):5540–5546. doi: 10.1128/JVI.77.9.5540-5546.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick A, Szilard L. Virus Strains of Identical Phenotype but Different Genotype. Science. 1951;113(2924):34–35. doi: 10.1126/science.113.2924.34. [DOI] [PubMed] [Google Scholar]
- Ribeiro RM, Bonhoeffer S. Production of resistant HIV mutants during antiretroviral therapy. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(14):7681–7686. doi: 10.1073/pnas.97.14.7681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro RM, Bonhoeffer S, Nowak MA. The frequency of resistant mutant virus before antiviral therapy. Aids. 1998;12(5):461–465. doi: 10.1097/00002030-199805000-00006. [DOI] [PubMed] [Google Scholar]
- Rodrigo A. HIV evolutionary genetics. Proceedings of the National Academy of Sciences. 1999;96:10559–10561. doi: 10.1073/pnas.96.19.10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine IM, Coffin JM. Linkage disequilibrium test implies a large effective population number for HIV in vivo. Proceedings of the National Academy of Sciences. 1999;96:10758–10763. doi: 10.1073/pnas.96.19.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine IM, Coffin JM. Evolution of human immunodeficiency virus under selection and weak recombination. Genetics. 2005;170(1):7–18. doi: 10.1534/genetics.104.029926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzine IM, Rodrigo A, Coffin JM. Transition between stochastic evolution and deterministic evolution in the presence of selection: General theory and application to virology. Microbiology and Molecular Biology Reviews. 2001;65(1):151–184. doi: 10.1128/MMBR.65.1.151-185.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roze D, Barton NH. The Hill-Robertson effect and the evolution of recombination. Genetics. 2006;173(3):1793–811. doi: 10.1534/genetics.106.058586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriner D, Rodrigo AG, Nickle DC, Mullins JI. Pervasive genomic recombination of HIV-1 in vivo. Genetics. 2004;167(4):1573–83. doi: 10.1534/genetics.103.023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan WY, Wu HL. Stochastic modeling of the dynamics of CD4(+) T-cell infection by HIV and some Monte Carlo studies. Mathematical Biosciences. 1998;147(2):173–205. doi: 10.1016/s0025-5564(97)00094-1. [DOI] [PubMed] [Google Scholar]
- Wang K, Mittler JE, Samudrala R. Comment on “Evidence for positive epistasis in HIV-1”. Science. 2006;312(5775):848. doi: 10.1126/science.1109904. author reply 848. [DOI] [PubMed] [Google Scholar]
- Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, Lifson J, Bonhoeffer S, Nowak MA, Hahn BH, Saag MS, Shaw GM. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]