

Abstract
A major challenge for kidney transplantation is balancing the need for immunosuppression to prevent rejection, while minimizing drug-induced toxicities.
We used DNA microarrays (HG-U95Av2 GeneChips, Affymetrix) to determine gene expression profiles for kidney biopsies and peripheral blood lymphocytes (PBLs) in transplant patients including normal donor kidneys, well-functioning transplants without rejection, kidneys undergoing acute rejection, and transplants with renal dysfunction without rejection. We developed a data analysis schema based on expression signal determination, class comparison and prediction, hierarchical clustering, statistical power analysis and real-time quantitative PCR validation. We identified distinct gene expression signatures for both biopsies and PBLs that correlated significantly with each of the different classes of transplant patients. This is the most complete report to date using commercial arrays to identify unique expression signatures in transplant biopsies distinguishing acute rejection, acute dysfunction without rejection and well-functioning transplants with no rejection history. We demonstrate for the first time the successful application of high density DNA chip analysis of PBL as a diagnostic tool for transplantation. The significance of these results, if validated in a multicenter prospective trial, would be the establishment of a metric based on gene expression signatures for monitoring the immune status and immunosuppression of transplanted patients.
Keywords: DNA microarrays, gene expression, kidney, rejection, transplant
Introduction
Kidney transplantation has extended and improved the quality of life for the majority of patients with end stage renal disease. Most transplants involve genetically nonidentical donor-to-recipient combinations. As a consequence the immune response is a major impediment to successful graft survival, necessitating lifelong treatment with potent immunosuppressive drugs. These drugs suppress the host immune system in a nonspecific manner and have many side-effects including, but not limited to, increased risk of life-threatening infections and cancer. Another key point is that responses of the donor organ itself are also major contributors to post transplant events. Despite recent reductions in the incidence of acute rejection, chronic allograft nephropathy and immunosuppressive drug side-effects are still major causes of graft loss and patient morbidity. In this context, it is essential to further our understanding of the immune system and the transplanted organ to both immune and non-immune mechanisms of injury.
High-density microarray technology provides one means to measure the differential expression of hundreds to thousands of genes simultaneously. While its basic applications in gene discovery are well established, high-density microarrays also have promise as a clinical tool. For example, this technology has been used with different cancers to predict prognosis and response to therapy (1-3) and in multiple sclerosis to identify inflammatory genes in brain lesions (4). Several publications have examined gene expression in kidney transplant patients using quantitative PCR (5,6), and demonstrated that for a very small set of immunologically relevant gene transcripts good correlations with acute rejection and clinical outcomes were present. Studies in small animal transplant models using DNA microarrays supported the potential use of this technology in a clinical setting (7,8). A small study of kidney transplant patients with acute rejection demonstrated the up-regulation of four genes consistently and two transcripts down-regulated (9). Recently the experience using the Stanford Lymphochip cDNA glass slide array (10) with kidney transplant biopsies of 50 pediatric patients defined three different gene expression signatures for acute rejection that correlated with graft survival (11). Finally, a study using the Hu95Av2 Affymetrix GeneChip for kidney biopsies performed 6 months post transplant identified 10 genes for which expression correlated with the risk of developing chronic rejection defined by biopsy at 12 months post transplant (12).
In the present study we extended the work previously carried out in this field. We developed a data analysis strategy based on expression signal determination, class comparison and prediction, hierarchical clustering, statistical power analysis and real-time quantitative PCR validation. We determined gene expression profiles in biopsies obtained from normal kidneys at the time of their recovery for living donor transplantation, creating a unique control population for gene expression profiling of any renal disease including transplanted kidneys. This study includes a collection of profiles for transplant patients with normal graft function on full immunosuppression compared with transplant patients with biopsy-documented acute rejection. In addition, we provide the first gene expression profile information on patients with acute kidney transplant dysfunction who did not demonstrate evidence of histological acute rejection by biopsy. Finally, this is the first report of high-density DNA array gene expression profiles of peripheral blood lymphocytes (PBLs) from each of these classes of patients.
Hierarchical clustering of samples and statistical analysis of individual gene expression signals demonstrated significant differences in the profiles of biopsies and PBLs from patients with acute rejection and acute dysfunction without rejection as compared with normal donors and well-functioning transplant patients with no history of rejection. One implication of these results is that gene profiling of PBLs could be used as a minimally invasive surrogate marker for rejection and identify patients with acute dysfunction but without rejection. These data support the hypothesis that the gene expression profiles of PBLs can be used to dynamically monitor the state of the immune response to the transplant. Thus, it may be possible to determine the impact and adequacy of immunosuppression in individual patients at any time post transplant using DNA array technology.
Methods
Patients
Patients signed Cleveland Clinic Foundation-approved IRB consent forms. Kidney biopsies were obtained from nine living donor controls, seven recipients with histologically confirmed acute rejection, five recipients with renal dysfunction without rejection on biopsy, and 10 protocol biopsies carried out more than one year post transplant in patients with good transplant function and normal histology (Table 1). Peripheral blood lymphocytes were obtained from one living kidney donor and seven healthy volunteer blood donor controls, seven recipients with biopsy-proven acute rejection, eight recipients biopsied for renal dysfunction without rejection, and from nine of the 10 recipients who had protocol biopsies carried out more than 1 year post transplant (Table 1). It is important to emphasize that all the acute rejection profiles of transplant biopsies and PBLs are matched to the same patients for all samples. For example, AR3 PBLs are from the patient of biopsy AR3. Evaluation of renal function for living donors included creatinine clearance, protein excretion and renal imaging with ultrasound and angiography. Acute rejection episodes were Banff criteria scored (13) and confirmed by response to anti-rejection therapy. Patients with clinical or laboratory evidence of CMV or other infections were excluded. Immunosuppression comprised a calcineurin inhibitor or sirolimus, with mycophenolate mofetil and steroids. Control biopsies were obtained from the cortex of diuresing kidneys during open-donor nephrectomies. Transplant biopsies were obtained under ultrasound guidance by spring-loaded 15-gauge needles (ASAP Automatic Biopsy, Microvasive, Watertown, MA). Cores went immediately into 1.5 mL of RNALater (Ambion, Austin, TX), and −80°C freezers within 4 h. Peripheral blood (20 mL) was obtained before biopsy, placed on ice and mononuclear cells were isolated within 1 h by density-gradient centrifugation and stored in RNALater at −80°C.
Table 1.
Clinical and demographic data of patients entered into the study
| Patient ID | BX | PBL | Age | Sex | Immunosuppression | Histopathology | LD/CAD | Scr (mg/dL) | Days post TX |
|---|---|---|---|---|---|---|---|---|---|
| C1 | • | 38 | Female | 0.8 | |||||
| C2 | • | 42 | Male | 0.9 | |||||
| C3 | • | 35 | Female | 0.6 | |||||
| C4 | • | 39 | Female | 0.9 | |||||
| C5 | • | • | 39 | Male | 1.2 | ||||
| C6 | • | 44 | Male | 0.8 | |||||
| C7 | • | 36 | Male | 1.2 | |||||
| C8 | • | 35 | Female | 0.8 | |||||
| C9 | • | 50 | Female | 0.6 | |||||
| AR1 | • | • | 42 | Male | CsA/MMF/P | BanffIIA | CAD | 12 | 285 |
| AR2 | • | • | 28 | Male | FK/MMF/P | BanffIIA | LD | 5.9 | 1467 |
| AR3 | • | • | 18 | Male | CsA/MMF/P | BanffIA | CAD | 2.2 | 119 |
| AR4 | • | • | 28 | Female | FK/MMF/P | BanffIIA | CAD | 1.5 | 366 |
| AR5 | • | • | 26 | Female | CsA/MMF/P | Borderline | CAD | 2 | 278 |
| AR6 | • | • | 55 | Male | SRL/MMF/P | Borderline | CAD | 2.9 | 68 |
| AR7 | • | • | 35 | Male | SRL/MMF/P | BanffIA | CAD | 2 | 184 |
| TX1 | • | 51 | Male | CsA/MMF/P | Normal | CAD | 1.5 | 932 | |
| TX2 | • | 56 | Male | CsA/MMF/P | Normal | LD | 1.3 | 911 | |
| TX3 | • | 52 | Male | CsA/MMF/P | Normal | CAD | 1.2 | 902 | |
| TX4 | • | 31 | Female | CsA/MMF/P | Normal | LD | 1.1 | 651 | |
| TX5 | • | 53 | Female | CsA/MMF/P | Normal | LD | 1.1 | 689 | |
| TX6 | • | 32 | Male | CsA/MMF/P | Normal | LD | 1.6 | 776 | |
| TX7 | • | 46 | Female | CsA/MMF/P | Normal | CAD | 1.2 | 713 | |
| TX8 | • | 61 | Male | CsA/MMF/P | Normal | CAD | 0.9 | 733 | |
| TX9 | • | 44 | Male | CsA/MMF/P | Normal | LD | 1.8 | 718 | |
| TX10 | • | 21 | Male | CsA/MMF/P | Normal | CAD | 1.5 | 674 | |
| TXPBL1 | • | 38 | Male | CsA/MMF/P | CAD | 1.4 | 461 | ||
| TXPBL2 | • | 57 | Female | FK/MMF/P | LD | 1.3 | 42 | ||
| TXPBL3 | • | 65 | Male | CsA/MMF/P | CAD | 1.5 | 213 | ||
| TXPBL4 | • | 65 | Female | FK/MMF/P | CAD | 0.8 | 246 | ||
| TXPBL5 | • | 36 | Female | CsA/MMF/P | CAD | 1.1 | 1278 | ||
| TXPBL6 | • | 68 | Male | CsA/MMF/ | CAD | 1.7 | 376 | ||
| TXPBL7 | • | 39 | Male | SRL/MMF/P | CAD | 0.9 | 36 | ||
| TXPBL8 | • | 61 | Female | CsA/MMF/P | CAD | 0.9 | 1491 | ||
| TXPBL9 | • | 46 | Male | SRL/MMF/P | LD | 1.2 | 81 | ||
| NR1 | • | • | 55 | Male | CsA/MMF/P | CNI toxicity | LD | 1.7 | 456 |
| NR2 | • | • | 38 | Male | FK/MMF/P | CNI toxicity | LD | 2.3 | 155 |
| NR3 | • | • | 61 | Male | SRL/MMF/P | ATN | LD | 5.2 | 11 |
| NR4 | • | • | 43 | Male | CsA/MMF/P | CNI toxicity | CAD | 3.8 | 262 |
| NR5 | • | • | 35 | Male | CsA/MMF/P | ATN | CAD | 6.3 | 16 |
| NR6 | • | 35 | Female | FK/MMF/P | CNI toxicity | CAD | 2.6 | 37 | |
| NR7 | • | 44 | Male | SRL/MMF/P | ATN | CAD | 6.3 | 40 | |
| NR8 | • | • | 22 | Female | FK/MMF/P | FSGS | LD | 3.3 | 78 |
| NR9 | • | 58 | Male | CsA/MMF/P | ATN | CAD | 5 | 47 |
BX, biopsy; PBL, peripheral blood lymphocytes; CsA, cyclosporine; MMF, mycophenolate mofetil; P, prednisone; FK, tacrolimus; SRL, sirolimus; CAD, cadaveric; LD, live donor; Scr, serum creatinine; ATN, actute tubular necrosis; CNI, calcineurin inhibitor; FSGS, focal segmental glomerulosclerosis.
RNA isolation
Frozen biopsy specimens were thawed, poured into 2-mL tissue grinders with 1 mL of Trizol (Invitrogen, Carlsbad, CA) and manually homogenized. Frozen PBLs were thawed and disrupted in 1 mL of Trizol using a 21-gauge needle. Total RNA was isolated from homogenates using chloroform: isopropanol and further purified using an RNeasy column (Qiagen, Valencia, CA) and DNAse-treated (DNA-free, Ambion) to remove genomic DNA. RNA quality was confirmed by electropherograms using an Agilent 2100 BioAnalyzer (Palo Alto, CA). Total RNA yields from 14 consecutive 15-gauge needle biopsies were 14.9 ± 3.9 μG.
Microarray analysis
For tissue biopsies, standard Affymetrix GeneChip (Santa Clara, CA) protocols were used [affymetrix.com (14)]. RNA extracted from PBLs underwent one additional round of RNA amplification owing to limited RNA yields in the early samples of the study. Amplification was carried out starting with 100 nG of total RNA using the Ambion MEGAscript™ aRNA Amplification Kit following the manufacturer's protocols. All labeled samples were hybridized to HG-U95Av2 GeneChip arrays. GeneChip data were analyzed using Microarray Suite 5.0 (MAS 5.0, Affymetrix) and DNA Chip Analyzer (dChip) (15,16) software using the PM only model. ‘Present’ and ‘Absent’ calls were determined with MAS 5.0. The dChip software used all the Affymetrix.CEL files generated in this study as a training set. BRB Array-Tools (http://linus.nci.nih.gov/BRB-ArrayTools.html) was used to perform hierarchical clustering and class prediction. Statistically significant changes in gene expression were measured with Significance Analysis of MicroArrays (SAM v1.3; 17). Delta values were chosen to minimize the median false discovery rate (FDR) at a level less than one false discovery per gene list. Two additional methods were used to filter the gene list. First, we applied the limit fold change model, which systematically bins genes by signal intensity; those genes within the top 10% of the highest fold changes for each bin were selected (18). Second, MAS 5.0 Present/Absent calls were used to filter the list; we required the majority of calls in the up-regulated group to be ‘Present’.
Real-time quantitative PCR (Q-PCR)
Q-PCR was performed on 15 genes selected for relatively large fold-changes from the list of 65 genes shown in Figure 3B using predesigned primer and probe sets from the Assays-on-Demand Genomic Assays (12 genes) and Assays-by-Design service (three genes) (Applied Biosystems, Foster City, CA). Each assay consisted of two unlabeled PCR primers and a FAM™ dye-labeled TaqMan® MGB probe. The endogenous control, 18S rRNA, was detected with a VIC™ dye-labeled TaqMan® MGB probe. Briefly, cDNA was transcribed from 100 nG total RNA using the ABI cDNA Archive kit (Applied Biosystems). Nine μL of the cDNA reaction was added to 11 μL of Platinum® Quantitative PCR SuperMix-UDG PCR reagent (Invitrogen, Carlsbad, CA) and PCR performed on an ABI Prism 7900HT (Applied Biosystems). All amplifications were carried out in triplicate and threshold cycle (Ct) scores were averaged for calculations of relative expression values. The Ct scores for genes of interest were normalized against Ct scores for the corresponding 18S rRNA control. Relative expression was determined by the following calculation where the amount of target is normalized to an endogenous reference (18S rRNA) and relative to an arbitrary calibrator (the reference class of patients used in the comparison):
Figure 3.

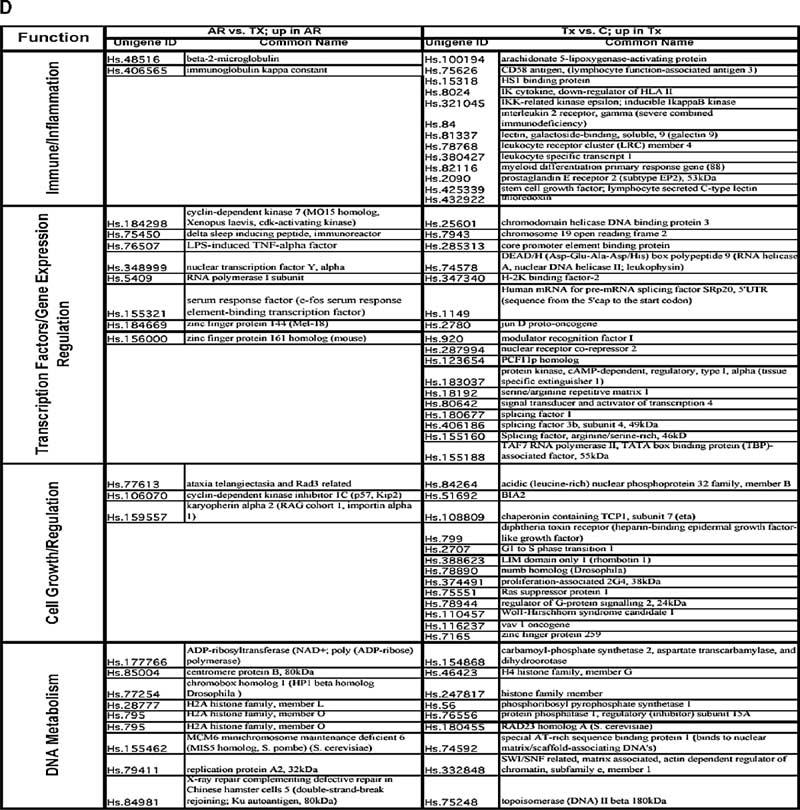
Gene expression profiles generated from peripheral blood lymphocytes (PBLs) according to different clinical classes. Classes comprised PBLs from patients with transplanted kidneys and stable renal function on full immunosuppression (TX), patients undergoing acute rejection (AR), patients with acute dysfunction but where rejection was not found on histological examination (NR), and PBLs from healthy blood donors (C). (A) Hierarchical clustering (unsupervised) of gene expression profiles from PBLs comparing the three transplant patient classes (TX, AR, NR). (B) Hierarchical clustering (unsupervised) of healthy donor PBLs (C) demonstrated distinct separation from the gene profiles generated from stable, well-functioning transplant recipient PBLs (TX). (C) Functional categories for the genes up or down-regulated according to different clinical classes. The PBLs from patients undergoing acute rejection were compared with stable immunosuppressed recipients (AR vs. TX); stable immunosuppressed recipients were compared with healthy blood donor controls (TX vs. C). (D) Up-regulated genes identified in PBLs from different clinical classes. The PBLs from recipients undergoing acute rejection were compared with stable immunosuppressed recipients (AR vs. TX); and stable immunosuppressed recipients were compared with healthy blood donor controls (TX vs. C).
Power calculations
Power calculations for application to microarray experiments has been attempted by several research groups (Simon, 2003; Zien, 2003). The basic premise is to determine the variability for measurements of gene expression by standard deviation of the results of multiple samples. While there is not general agreement on a single best method to perform these calculations, the data we had collected to date provided us with real data upon which to make estimates of variability. Variability for a measurement is described in terms of the standard deviation and is the key experimental metric for sample size calculations. In this context, the measurement is the mean signal intensity measured for each gene's probe set on the GeneChip. The variance value (s) was based on the median log2 transformed signal intensities derived from our data on more than 30 experiments using the GeneChips on either transplant biopsy or PBL samples. The next step is to set values for an acceptable alpha error (false-positive rate), beta error (false-negative) and the delta (minimal detectable change that will be confidently determined). We used values of 0.001 (alpha; a), beta (b) of 0.8 and a minimal detectable fold change of 2 (delta; d).
Calculations were performed using the following equation:
Web site data
All the.cel files for the Affymetrix GeneChips used in these studies are available to the public at our TSRI DNA Array Core in MIAME compliant format (URL: www.scripps.edu/services/dna_array/). We also provide at this site a series of annotated gene lists including literature references.
Results
Gene expression analysis of kidney transplant biopsies
To define gene expression profiles in kidney transplant patients we assembled a series of biopsy samples from normal living kidney donors (C) and several classes of patients including well-functioning kidneys more than 1-year post transplant (TX), biopsy-confirmed acute rejection (AR), and acute renal dysfunction without rejection (NR) (Table 1). After signal expression determination using dChip, we used hierarchical clustering of samples based on their individual gene expression profiles as a tool to examine the relationships between experimental groups. Clustering of (C), (TX), and (AR) indicates that each group is distinct with respect to their gene expression profiles (Figure 1A). It is important to note that this cluster analysis was performed using an unsupervised data set, essentially all genes called as Present on at least one chip (8320 genes; 66% of the probe sets on the chip). The purpose of an unsupervised clustering is to avoid introduction of bias based on preclassification of gene expression by sample type.
Figure 1.


Gene expression profiles generated from kidney biopsies according to different clinical classes. Classes represented were histologically confirmed and comprised healthy kidney donors (C), transplanted kidneys with stable function on full immunosuppression (TX), and kidneys undergoing acute rejection (AR). (A) Hierarchical clustering (unsupervised) of gene expression profiles from kidney biopsies. (B) Functional categories of genes up- or down-regulated from kidney biopsies. Functional gene categories were defined using gene names and annotations available from public domain databases. (C) Up-regulated immune/inflammation response genes identified in kidney biopsies from different clinical classes. The kidneys undergoing acute rejection were compared with stable immunosuppressed recipients (AR vs. TX); the 44 genes shown are up-regulated in AR (see Figure 2B). Stable immunosuppressed recipients were compared with healthy donor kidney controls (TX vs. C); the 45 genes shown are up-regulated in TX (see Figure 2B).
This clustering pattern demonstrates that gene expression defines distinct groups of transplant patient biopsies, specifically separating acute rejection from well-functioning transplants and from normal kidneys unex-posed to immunosuppression. Therefore, we performed a class comparison analysis between acute rejection and biopsies from fully immunosuppressed patients with good graft function (AR vs. TX) (Figure 1B). This comparison identified the subset of differentially expressed genes, up-and down-regulated, that define acute rejection. We also compared gene expression in healthy donor kidneys with that of transplant recipients with good graft function and full immunosuppression (TX vs. C). This comparison identifies gene expression profiles that define the impact of transplantation and immunosuppression on a normal donor kidney.
We determined significant changes in gene expression comparing biopsies of acute rejection to those of the stable transplants (AR vs. TX). Using SAM we identified 96 up-regulated and 619 down-regulated genes (median FDR < 0.14% per comparison). We created an annotated gene list based on a literature search (Figure 1B). These results show that genes involved in immune and inflammatory responses represent the dominant category of up-regulated genes in acute rejection (44 of 96 genes; 46%). Interestingly, a large number of the genes down-regulated in acute rejection are involved in different categories of basic cellular metabolism that might reflect the impact of rejection and immunosuppressive drug-mediated tissue injury on the kidney.
Next we compared the biopsies of the fully immunosuppressed recipients with normal graft function with those from normal living donors (TX vs. C). We identified and classified 612 up-regulated and 28 down-regulated genes (median FDR < 0.16%; Figure 1B and supplemental data). Even a year or more post transplant, well-functioning kidneys had a distinct gene profile compared with the normal donor controls. Possible explanations for these differences in gene expression include an underlying subclinical immune response, the impact of post transplant drug therapies, compensatory physiological changes in a single kidney, and tissue responses by the transplanted kidney to these injury pathways. For example, genes that are up-regulated and define the differences between the transplanted and normal donor kidneys include 45 genes classified with cell growth and regulation, 47 with protein metabolism, 35 as structural and 66 as transcription factors or other gene expression regulators.
An important question is the nature of the immune response in well-functioning kidney transplants without clinical or biopsy evidence of rejection. There are 45 up-regulated genes classified as immune/inflammatory in well-functioning transplants (Figure 1B) compared with the normal donor control kidneys (7.3% of 619). Interestingly, this gene set does not overlap with the list of immune/inflammatory genes significantly increased when acute rejection biopsies are compared with the well-functioning transplants (AR vs. TX; Figure 1C). The largest group within the immune/inflammatory genes up-regulated in the well-functioning transplants is histocompatibility antigens consistent with the hypothesis of an ongoing but low-grade immune response or some form of tissue injury resulting in cytokine-mediated induction of MHC molecule expression.
A common clinical problem is acute renal dysfunction resulting from nonimmune-mediated injury of the transplant (i.e. drug toxicity and ischemic injury). Roughly 50% of the biopsies carried out during this study for acute renal dysfunction did not reveal acute rejection by histology. Therefore, we examined the differential gene expression profiles of patients with acute renal transplant dysfunction in which the biopsy histology did not demonstrate rejection (NR). Unsupervised hierarchical clustering demonstrated a good separation of the well-functioning transplants (TX) from the profiles of kidneys with acute dysfunction (AR and NR; Figure 2A). However, it was not possible to distinguish the AR and NR biopsy groups.
Figure 2.


Unsupervised and supervised hierarchical clustering of gene expression profiles generated from kidney biopsies according to different clinical classes. Classes were histologically confirmed and comprised transplanted kidneys with stable renal function on full immunosuppression (TX), kidneys undergoing acute rejection (AR), and kidneys with acute dysfunction but where rejection was not found on histological examination (NR). (A) Unsupervised hierarchical clustering of gene expression profiles. (B) Supervised clustering was performed using the 65 genes identified by a class comparison analysis using BRB ArrayTools as distinguishing AR from NR. C. The common names, Unigene numbers and functional categories of the 65 genes that distinguished the AR from the NR clinical classes by kidney biopsy profiles.
We hypothesized that there were at least two predominant gene groups within the expression profiles of the AR and NR biopsies, one comprised of genes related directly to the acute immune-mediated rejection and another representing genes common to tissue injury and kidney dysfunction. If the second group of injury-associated genes was much larger, then it could explain the inability of unsupervised cluster analysis to separate the AR from NR biopsies. Therefore, we performed a two-class comparison analysis in BRB ArrayTools of the gene expression profiles comparing AR with NR. This gave us 65 genes at a 0.001 significance level. The results of a three-class comparison analysis comparing AR with NR with TX was 3550 genes at the 0.001 significance level; consistent with our hypothesis that the set of genes associated with kidney injury/dysfunction is indeed larger then the gene list associated with acute rejection. Thus, we performed a supervised hierarchical clustering using just the 65 genes identified as distinguishing AR from NR (Figure 2B). The supervised approach gives a clear separation of all three clinical groups. By functional class, the 65 genes identified as distinguishing AR from NR contain 12 genes associated with immune/inflammation responses (17%), seven of which are also in the immune/inflammation group of 44 genes up-regulated in the profiles of acute rejection biopsies compared with well-functioning transplants (AR vs. TX; Figure 1C).
Gene expression analysis of peripheral blood lymphocytes
To assess the impact of immunosuppression and acute rejection, PBLs were collected from: a control group of healthy, nonimmunosuppressed blood donors (C), immunosuppressed kidney transplant recipients with well-functioning kidneys and no history of rejection (TX), and immunosuppressed kidney transplant recipients with acute renal dysfunction documented by biopsy to be owing to either rejection (AR), or non-immune-mediated pathology (NR). Unsupervised hierarchical clustering analysis of the array data was performed (Figure 3A,B).
These data show that PBLs from immunosuppressed transplant patients with well-functioning kidneys (TX) cluster separately (Figure 3A). Peripheral blood lymphocytes from transplant patients with renal dysfunction owing to either AR or without biopsy evidence of rejection (NR) cluster predominantly into separate groups. However, AR5 clusters with the NR PBLs, and NR2 clusters with the AR PBLs. These exceptions suggest the possibility that some level of acute renal dysfunction can be immune-mediated and yet fall below the level detected by the biopsy. However, a much larger data set will be required to test this hypothesis. Unsupervised clustering of the healthy donor PBLs (C) and PBLs from immunosuppressed kidney transplant recipients with well-functioning kidneys (TX) demonstrates distinct separation of the PBL profiles from nonimmunosuppressed donors (Figure 3B). Nonetheless, for reasons that are unclear, samples C5 and C1 cluster independently from the other control PBL samples with a low correlation branch to the cluster of PBL from immunosuppressed patients with well-functioning transplants.
We created an annotated gene list based on a literature search (Figure 3C). One striking difference for the PBLs, in comparison with the transplant biopsies (Figure 1B), is that genes classified as immune/inflammatory are not a dominant category, particularly in patients with biopsy-proven AR. However, the PBL profiles for patients with well-functioning transplants on full immunosuppression compared with normal blood donors (TX vs. C) reveal a significant up-regulation of genes classified as immune/inflammatory (13; 8%), cell growth and regulation (13; 8%), protein metabolism (24; 15%) and transcription factors/regulators of gene expression (17; 11%). Interestingly, none of the 13 immune/inflammatory genes up-regulated in the profiles of PBL from patients with well-functioning transplants (TX vs. C) are identified in the list of 45 such genes identified in the same comparison based on the biopsy data (Figure 1B). Analysis of the specific genes in the four functional classes (Figure 3D) up-regulated in PBLs from patients with acute rejection compared with well-functioning transplants (AR vs. TX) and PBLs from patients with well-functioning transplants compared with normal blood donors (TX vs. C) reveals that there are only three genes that overlap with the genes up-regulated in the biopsies (Unigene #: Hs. 183037, Hs. 18192, Hs. 75248). Thus, it is evident that the gene expression profiles of PBLs are very different than those of the biopsies in the various classes of transplant patients.
Predicting clinical status of kidney transplants from gene expression profiles
A test of our hypothesis that distinct gene expression profiles correlate with clinically and biopsy-defined phenotypes in kidney transplantation is to demonstrate successful use of class prediction tools to correctly separate the phenotypes. We used six class predictors implemented in BRB ArrayTools for determining to which of two or more predefined groups an unknown sample belongs. If class prediction results of PBLs gene expression profiles correlate with clinical phenotypes, then monitoring of patient status would be possible with blood sampling. Thus, we tested all of the six class prediction algorithms currently available in BRB Array Tools for both biopsy and PBLs profiles (Table 2).
Table 2.
Class prediction analysis of kidney biopsy and PBL samples using multiple statistical alogorithms
| Classifier | Sample type |
Phenotype | Number of samples |
Compound covariate predictor |
Linear discriminant analysis |
1-nearest neighbor |
3-nearest neighbor |
Nearest centroid |
Support vecor machines |
Classifier p-value |
Average number of genes in classifier |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Tx vs. AR | Biopsy | Tx | 10 | 100% | 100% | 100% | 100% | 94% | 100% | 0.005– <0.0005 |
2837 |
| AR | 7 | ||||||||||
| PBL | Tx | 9 | 93% | 93% | 93% | 93% | 93% | 93% | 0.009–0.005 | 743 | |
| AR | 6 | ||||||||||
| AR vs. NR | Biopsy | AR | 7 | 83% | 75% | 67% | 92% | 83% | 67% | 0.247–0.017 | 73 |
| NR | 5 | ||||||||||
| PBL | AR | 6 | 79% | 79% | 79% | 79% | 79% | 79% | 0.121–0.083 | 77 | |
| NR | 8 | ||||||||||
| Tx vs. NR | Biopsy | Tx | 10 | 100% | 100% | 100% | 100% | 100% | 100% | 0.002– <0.0005 |
3792 |
| NR | 5 | ||||||||||
| PBL | Tx | 9 | 94% | 94% | 94% | 94% | 94% | 94% | 0.003– <0.0005 |
1812 | |
| NR | 8 | ||||||||||
| Tx vs. AR/NR | Biopsy | Tx | 10 | 100% | 100% | 100% | 100% | 100% | 100% | <0.0005 | 3728 |
| AR/NR | 12 | ||||||||||
| PBL | Tx | 9 | 100% | 100% | 100% | 100% | 100% | 100% | <0.0005 | 1017 | |
| AR/NR | 14 | ||||||||||
| C vs. Tx | Biopsy | C | 9 | 100% | 100% | 100% | 100% | 100% | 100% | <0.0005 | 5165 |
| Tx | 10 | ||||||||||
| PBL | C | 8 | 88% | 88% | 88% | 88% | 88% | 94% | 0.02–0.001 | 1910 | |
| Tx | 9 |
In the comparison of TX vs. AR, the performance of a ‘leave-one-out’ cross-validation correctly classified from 94 to 100% of the biopsies and 93% of the PBL gene expression profiles. Class prediction results for the comparison of PBLs and biopsy profiles of acute rejection with non-rejection patients (AR vs. NR) were generally unsatisfactory. These results match the problems we encountered in the unsupervised clustering of these data (Figure 2A). In contrast, comparison of the samples from well-functioning transplants with those from the non-rejection patients (TX vs. NR) correctly classified 100% of the biopsies and 94% of the PBL profiles. Moreover, class prediction comparing well-functioning transplants with the combined AR/NR group resulted in 100% correct classifications for both biopsy and PBL data. These results are consistent with the hierarchical clustering shown above (Figure 2). Finally, class prediction for normal donor kidneys compared with well-functioning kidney transplants (C vs. TX) was successful in classifying 100% of the biopsies and 88–94% of the PBL gene expression profiles. Therefore, it is evident that gene expression in well-functioning kidney transplants is not the same as normal kidneys and these differences may help to identify the impacts of immunosuppressive drugs, immunity and transplant surgery. These results also support the hypothesis that the impact of immunosuppression and transplantation may be profiled successfully in the peripheral blood compartment.
Validation of GeneChip data
One method of data validation is quantitative PCR. We chose 15 genes from the list of 62 classifying AR vs. NR biopsies (Figure 2C) for validation by quantitative PCR (Table 3). Three biopsies from each of the four clinical classes were chosen based on adequate material for analysis. We compared all the clinical classes for all the comparisons involving these 15 genes where SAM analysis indicated a significant change was present. These results demonstrated agreement in 20 of 21 comparisons with respect to the direction of gene expression change at a highly significant level (p = 0.0001). In general, fold changes determined by quantitative PCR were greater than those detected by GeneChip data analysis. These results suggest that the dynamic range of current GeneChip technology is relatively low, though the direction of expression changes are accurate. Thus, Q-PCR and similar quantitative measures of RNA expression are important and complementary tools.
Table 3.
Real time PCR analysis of selected gene transcripts
| Unigene ID | Gene name | Gene list | Upregulated chip in |
Fold difference by chip |
Upregulated Q-PCR in |
Fold difference by Q-PCR |
Agreement |
|---|---|---|---|---|---|---|---|
| Hs. 73946 | Enothelial cell growth factor (platelet-derived) | TX vs. AR | AR | 2.6 | AR | 218 | Yes |
| Hs. 155597 | D component of complement (adipsin) | TX vs. AR | AR | 1.9 | AR | 982 | Yes |
| Hs. 76364 | Allograft inflammatory factor 1 | TX vs. AR | AR | 1.6 | AR | 529 | Yes |
| Hs.73885 | HLA-G histocompatibility antigen, class I, G | C vs. TX | TX | 3.2 | TX | >105 | Yes |
| Hs. 6150 | Rho-specific guanine nucleotide exchange factor p114 | C vs. TX | TX | 2.5 | C | 7 | No |
| Hs. 79356 | Lysosomal-associated multispanning membrane protein-5 | TX vs. AR | AR | 2.7 | AR | 6 | Yes |
| Hs. 21486 | Signal transducer and activator of transcription 1, 91 kDa | AR vs. NR | AR | 3.98 | AR | 6105 | Yes |
| TX vs. AR | AR | 3.99 | AR | 6 | Yes | ||
| Hs. 327 | Interleukin 10 receptor, alpha | AR vs. NR | AR | 2.16 | AR | 121 | Yes |
| TX vs. AR | AR | 1.92 | AR | 266 | Yes | ||
| Hs. 202 | Benzodiazapine receptor (peripheral) | AR vs. NR | AR | 2.14 | AR | 4 | Yes |
| Hs. 75367 | Src-like-adaptor | TX vs. AR | AR | 3.65 | AR | 72 | Yes |
| Hs. 425777 | Ubiquitin-conjugating enzyme E2L 6 | AR vs. NR | AR | 2.47 | AR | 2 | Yes |
| Hs. 193852 | ATP-binding cassette, sub-family C (CFTR/MRP), member 2 | AR vs. NR | NR | 2.26 | NR | 72 | Yes |
| TX vs. AR | TX | 3.66 | TX | 4 | Yes | ||
| Hs. 83968 | Integrin, beta 2 (antigen CD18 (p95), lymphocyte function-associated antigen 1; macrophage antigen 1 (mac-1) beta subunit) | AR vs. NR | AR | 2.27 | AR | 161 | Yes |
| TX vs. AR | AR | 2.41 | AR | 7 | Yes | ||
| Hs. 184411 | Human serum albumin (ALB) gene | AR vs. NR | NR | 3.83 | NR | 143 | Yes |
| TX vs. AR | TX | 4.77 | TX | 7 | Yes | ||
| Hs. 1765 | Lymphocyte-specific protein tyrosine kinase | AR vs. NR | AR | 1.97 | AR | 36 | Yes |
| TX vs. AR | AR | 1.66 | AR | 2 | Yes |
Another important aspect of validating data for gene expression signatures correlating with specific patient groups is the appropriateness of the sample sizes studied. While there is not general agreement on a single best method for statistical power calculations in microarray experiments the development of formulas has been attempted by several research groups (19). We performed a power analysis of this study using our sample sizes and variance based on the median standard deviation of gene expression measurements. Our power to compare the expression profiles of acute rejection (AR) with the nonrejection and well-functioning patient groups (NR, TX) is 86% and 99%, respectively, for the biopsy data and 97% and 99%, for the PBLs. Thus, these data do reveal that our gene expression signatures correlate significantly with specific patient groups.
Discussion
The ability to measure gene expression profiles in kidney transplantation allows us to test several hypotheses that will directly impact on clinical practice. Currently, there is no objective measure for determining the adequacy of immunosuppression, and no objective way of predicting an individual patient's response to therapy. Clinical practice is based on experience with large populations of patients that are empirically individualized by transplant physicians to take into account factors identified as unique to a given patient such as an early acute rejection episode, evidence of drug toxicity, and serial measurements of renal function. There is also a constant pressure to reduce or eliminate drugs to avoid long-term toxicity and cost. Therefore, if gene expression profiling identifies a signature for acute rejection, then a patient on any given immunosuppressive regime could be monitored for that signature as a measure of the adequacy of immunosuppression. In turn, decisions to reduce or eliminate immunosuppressive drugs could be made with a strategy to safely monitor the results before clinically apparent changes in kidney function occur. It may also be possible to improve the safety of new immunosuppressive drugs, particularly in establishing dose responses, and testing the efficacy of combining new agents with existing drug regimes.
The data presented in this study reporting an acute rejection signature for both PBLs and transplant biopsies supports the hypothesis that a prospective approach to monitoring molecular changes in transplant patients could also be used to predict acute rejection. If determining the adequacy of immunosuppression and predicting rejection could be carried out with PBLs alone, then the potential for a minimally invasive monitoring strategy would be realized. Moreover, an important goal of molecular medicine is to develop tools that effectively allow physicians to individualize therapy. However, we understand that an adequately powered prospective clinical trial would be required to test this hypothesis developed with our data and validate such a diagnostic strategy.
Another hypothesis that should be tested is that gene expression signatures can be used to predict chronic allograft nephropathy early enough to alter therapy. In this context, subclinical rejection identified in early protocol biopsies supports the hypothesis that rejection can be present long before evidence of clinical kidney dysfunction emerges (20-23). The results of Scherer et al. support this hypothesis, indicating that gene expression profiles of protocol biopsies at 6 months could predict biopsy changes of chronic rejection at 12 months (12). Therefore, a major question is whether there is a continuum between subclinical acute rejection and chronic allograft nephropathy that represents the mechanistic link between the events determining rejection, tissue injury, and repair. If such a continuum can be defined in molecular terms, then the potential of therapeutic interventions can be tested.
There remain a number of problems with the present approach that must be considered. The heterogeneity of our patient populations, differences in immunosuppressive therapy, and different degrees of rejection all contribute to biological variability in gene expression profiles that will reduce the number of statistically significant genes we have identified. Thus, while our statistical power analysis demonstrates that our group sizes are sufficient to support the conclusions we have made regarding the significance of expression signatures, it does not mean that all the genes that play a significant role in transplantation have been identified. Moreover, much larger sample sizes of patients are required to draw conclusions regarding the correlations between these gene expression signatures and clinical outcomes such as response to antirejection therapy, long-term graft function and survival. In addition, a limitation of the current microarray technology is that the sensitivity and specificity of gene expression profiling is difficult to determine objectively when thousands of genes are studied simultaneously. Of course, the HG-U95Av2 GeneChip used here represents conservatively one-third of what is now considered the full human genome and the technology has already advanced to the latest version, the HG-U133 chip set. Thus, for all these reasons it is certain that many important genes are not included in our lists. One way to address these limitations would be to design the large and prospective trial discussed above and use the latest microarrays with a more complete representation of the human transcriptosome as well as other technologies such as quantitative PCR to validate and extend these studies.
While the clinical impact of gene expression signatures that can predict rejection and monitor immunosuppression is clear, the potential contributions to our basic understanding of transplantation biology are also important to consider. Thus, the ultimate objective of gene expression profiling is to identify specific genes and associate these with specific pathways mediating cellular mechanisms of rejection, tissue injury and repair, immunosuppression and tolerance. Therefore, we have taken care to provide lists organized by both function and specific gene names for all our significant group comparisons. We also have placed all our data files in MIAME format at our web site for public access. However, a key point is that the fields of bioinformatics and systems biology are still in their infancy with respect to taking specific gene sets and reliably establishing biological pathways. Therefore, we have concentrated on establishing the validity of our first hypothesis that gene signatures can be correlated with well characterized clinical phenotypes all established by the current gold standard of a transplant biopsy. Of course, in all these sets there are genes that we recognize and can find literature regarding their biological function and correlation with immune responses and transplantation models of various types. These are provided with annotations at our web site. But there are also many genes and pathways that are presently not fully understood or characterized and some are likely to be misunderstood at the current time.
How are lymphocytes in the peripheral lymphoid compartment influenced by events that occur within the kidney transplant such as antigen recognition and the signaling events responsible for allo-immune activation? Our results demonstrate that PBL gene expression profiles in acute rejection are distinctly different from those of normal controls and from patients with well-functioning transplants. Therefore, acute rejection does influence the gene expression profile of the circulating lymphocyte pool. Moreover, despite the fact that surprisingly we found very little common gene expression between PBLs and kidney biopsies, we did identify a large number of lymphocyte-specific genes in the kidney tissue. One interpretation is that there are compartment-specific differences between the PBLs in the circulation and the subset of lymphocytes that are activated and recruited to the transplant kidney during acute rejection. The significance of these results in the context of monitoring patients after transplantation is that they may explain the failure of more than a decade of work testing PBLs for an array of activation antigens based on findings in rejecting allografts and other immune models. In other words, the activated lymphocytes infiltrating the rejecting allograft are a distinct population compared with the circulating PBL pool. It is possible that the gene expression profile of the PBLs represents the adequacy of immunosuppression such that the rejecting patients reflect the profile of inadequate immunosuppression as compared with the PBLs sampled from patients with well-functioning transplants. Perhaps future drug therapies could be advanced by targeting the genes that are up-regulated in these PBL profiles. Nonetheless, our results do demonstrate that there is a distinct gene expression profile in the PBL pool that correlates with acute rejection and immunosuppression. If these results can be confirmed in a large, prospective trial it would support the use of such profiles as a minimally invasive monitoring strategy for the immunological status of the graft and support the potential of using them to monitor the adequacy of immunosuppression.
One limitation to consider is that we purified PBLs for analysis using a density gradient and performed one round of amplification of the mRNA before the standard labeling procedure. It is known that such physical handling of PBLs can result in ex vivo cell activation and gene induction. Secondly, amplification of RNA transcripts can also bias gene expression measurements. We were consistent in using the same protocol for all PBLs samples studied, both for amplification and processing, such that there should be no class-specific bias in the expression profiles obtained. However, recently several new technologies have been developed that will eliminate this issue by allowing investigators to draw peripheral blood samples directly into preservation solutions that instantly capture the transcriptosome at that time of the draw. Finally with respect to the possibility of RNA amplification introducing bias, it is important to note that a number of studies have been carried out demonstrating consistent gene expression profiles carried out with two and in some instances three rounds of amplification (24,25).
Given that chronic allograft nephropathy is a major cause of transplant dysfunction and loss, another question is the status of the well-functioning kidney transplant. Our results demonstrate that despite good graft function in this group there is a distinct up-regulation of inflammatory/immune response genes in both biopsies and PBLs. One possibility is that there is a continuum of immune activation that defines the status of a transplant at any given time. This activation state is influenced by factors such as the adequacy of immunosuppression, genetics, and environment. We believe that the long-term function of the transplanted kidney is determined by the intersecting effects of both recipient and donor genetics. Namely, the nature of the recipient's immune response integrated with the donor organ's response to tissue injury, including the impact of nephrotoxic drugs. Theoretically, it should be possible to distinguish genes expressed by the donor organ from genes expressed by the host's infiltrating cells using techniques such as laser capture microdissection.
In conclusion, we have developed a strategy for integrating a number of gene expression profiling and supervised and unsupervised statistical tools to generate lists of genes that represent at least parts of the complex biological pathways involved in transplantation biology. In this context, we acknowledge the fact that at the present time the function of only a minority of the human genome is documented. As the knowledge base that can be accessed through bioinformatics grows to better define cellular pathways and regulatory networks, these gene lists linked to well-defined clinical events in transplantation will provide additional opportunities to advance our understanding of the basic biology of transplantation and identify new targets for therapeutics.
Acknowledgments
It should be noted that the efforts of the first two authors were complementary and contributed equally to the development of the manuscript. We wish to acknowledge the work of Barbara Mastroianni, RN, and Kathy Savas, RN, in obtaining the different patient samples at the Cleveland Clinic. We also acknowledge the critical support of The Scripps Research Institute's General Clinical Research Center (M01 RR00833-28).
References
- 1.Jenssen TK, Kuo WP, Stokke T, Hovig E. Associations between gene expressions in breast cancer and patient survival. Hum Genet. 2002;111:411–20. doi: 10.1007/s00439-002-0804-5. [DOI] [PubMed] [Google Scholar]
- 2.Moos PJ, Raetz EA, Carlson MA, et al. Identification of gene expression profiles that segregate patients with childhood leukemia. Clin Cancer Res. 2002;8:3118–3130. [PubMed] [Google Scholar]
- 3.Gordon GJ, Jensen RV, Hsiao LL, et al. Translation of microarray data into clinically relevant cancer diagnostic tests using gene expression ratios in lung cancer and mesothelioma. Cancer Res. 2002;62:4963–4967. [PubMed] [Google Scholar]
- 4.Lock C, Hermans G, Pedotti R, et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med. 2002;8:500–508. doi: 10.1038/nm0502-500. [DOI] [PubMed] [Google Scholar]
- 5.Suthanthiran M. Molecular analyses of human renal allografts: differential intragraft gene expression during rejection. Kidney Int. 1997;58:S15–S21. [PubMed] [Google Scholar]
- 6.Strehlau J, Pavlakis M, Lipman M, Maslinski W, Shapiro M, Strom TB. The intragraft gene activation of markers reflecting T-cell-activation and – cytotoxicity analyzed by quantitative RT-PCR in renal transplantation. Clin Nephrol. 1996;46:30–33. [PubMed] [Google Scholar]
- 7.Stegall MD, Park WD, Kim DY, Covarrubias M, Khair A, Kremers WK. Changes in intragraft gene expression secondary to ischemia reperfusion after cardiac transplantation. Transplantation. 2002;74:924–930. doi: 10.1097/00007890-200210150-00004. [DOI] [PubMed] [Google Scholar]
- 8.Stegall M, Park W, Kim D, Kremers W. Gene expression during acute allograft rejection: novel statistical analysis of microarray data. Am J Transplant. 2002;2:913–925. doi: 10.1034/j.1600-6143.2002.21007.x. [DOI] [PubMed] [Google Scholar]
- 9.Akalin E, Hendrix RC, Polavarapu RG, et al. Gene expression analysis in human renal allograft biopsy samples using high-density oligoarray technology. Transplantation. 2001;72:948–953. doi: 10.1097/00007890-200109150-00034. [DOI] [PubMed] [Google Scholar]
- 10.Alizadeh A, Eisen M, Davis RE, et al. The lymphochip: a specialized cDNA microarray for the genomic-scale analysis of gene expression in normal and malignant lymphocytes. Cold Spring Harbor Symposia on Quantitative Biology. 1999;64:71–78. doi: 10.1101/sqb.1999.64.71. [DOI] [PubMed] [Google Scholar]
- 11.Sarwal M, Chua MS, Kambham N, et al. Molecular heterogeneity in acute renal allograft rejection identified by DNA microarray profiling.[comment] N Engl J Med. 2003;349:125–138. doi: 10.1056/NEJMoa035588. [DOI] [PubMed] [Google Scholar]
- 12.Scherer A, Krause A, Walker JR, Korn A, Niese D, Raulf F. Early prognosis of the development of renal chronic allograft rejection by gene expression profiling of human protocol biopsies. Transplantation. 2003;75:1323–1330. doi: 10.1097/01.TP.0000068481.98801.10. [DOI] [PubMed] [Google Scholar]
- 13.Racusen L, Rayner D, Trpkov K, Olsen S, Solez K. The Banff classification of renal allograft pathology: where do we go from here? Transplant Proc. 1996;28:486–488. [PubMed] [Google Scholar]
- 14.Lockhart DJ, Dong H, Byrne MC, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 15.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2 doi: 10.1186/gb-2001-2-8-research0032. RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li C, Wong WH. Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc Natl Acad Sci U S A. 2001;98:31–36. doi: 10.1073/pnas.011404098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mutch DM, Berger A, Mansourian R, Rytz A, Roberts MA. The limit fold change model: a practical approach for selecting differentially expressed genes from microarray data. BMC Bioinformatics. 2002;3:17. doi: 10.1186/1471-2105-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simon R, Radmacher MD, Dobbin K, McShane LM. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Nat Cancer Inst. 2003;95:14–18. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 20.Rush D. Protocol biopsies should be part of the routine management of kidney transplant recipients. Pro.[comment] Am J Kid Dis. 2002;40:671–673. doi: 10.1053/ajkd.2002.36427. [DOI] [PubMed] [Google Scholar]
- 21.Rush D, Nickerson P, Gough J, et al. Beneficial effects of treatment of early subclinical rejection: a randomized study. J Am Soc Nephrol. 1998;9:2129–2134. doi: 10.1681/ASN.V9112129. [DOI] [PubMed] [Google Scholar]
- 22.Rush DN, Nickerson P, Jeffery JR, McKenna RM, Grimm PC, Gough J. Protocol biopsies in renal transplantation: research tool or clinically useful? Curr Opin Nephrol Hyperten. 1998;7:691–694. doi: 10.1097/00041552-199811000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Lipman ML, Shen Y, Jeffery JR, et al. Immune-activation gene expression in clinically stable renal allograft biopsies: molecular evidence for subclinical rejection. Transplantation. 1998;66:1673–1681. doi: 10.1097/00007890-199812270-00018. [DOI] [PubMed] [Google Scholar]
- 24.Baugh LR, Hill AA, Brown EL, Hunter CP. Quantitative analysis of mRNA amplification by in vitro transcription. Nucleic Acids Res. 2001;29:E29. doi: 10.1093/nar/29.5.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polacek DC, Passerini AG, Shi C, et al. Fidelity and enhanced sensitivity of differential transcription profiles following linear amplification of nanogram amounts of endothelial mRNA. Physiol Genomics. 2003;13:147–156. doi: 10.1152/physiolgenomics.00173.2002. [DOI] [PubMed] [Google Scholar]
- 26.Zien A, Fluck J, Zimmer R, Lengauer T. Microarrays: how many do you need? J Comput Biol. 2003;10:653–667. doi: 10.1089/10665270360688246. [DOI] [PubMed] [Google Scholar]