

Abstract
Mosquitoes are vectors of parasitic and viral diseases of immense importance for public health. The acquisition of the genome sequence of the yellow fever and Dengue vector, Aedes aegypti (Aa), has enabled a comparative phylogenomic analysis of the insect immune repertoire: in Aa, the malaria vector Anopheles gambiae (Ag), and the fruit fly Drosophila melanogaster (Dm). Analysis of immune signaling pathways and response modules reveals both conservative and rapidly evolving features associated with different functional gene categories and particular aspects of immune reactions. These dynamics reflect in part continuous readjustment between accommodation and rejection of pathogens and suggest how innate immunity may have evolved.
Repeatedly during evolution, mosquitoes and other insects have adopted hematophagy to sustain abundant progeny production. In turn, blood feeding provided a new point of entry for pathogens. To counter assaults, innate immunity has evolved to recognize and respond to numerous pathogens, in a dynamic playoff where either host or pathogen may win. Although fundamental concepts mostly derive from Dm, Ag is now an important model for studies of innate immunity. A previous comparative analysis of Ag and Dm immune-related gene families (1) highlighted their diversification and pointed toward an expanded conceptual framework of insect innate immunity. The sequencing of the Aa genome (2) permitted deeper understanding of insect immune systems, as displayed by two quite different mosquito species that diverged ∼150 million years ago (Ma) and Dm, which separated from them ∼250 Ma. This three-way comparison is considerably more powerful than the previous Dm-Ag study, because it allows measuring true genetic distances rather than unrooted sequence similarities. Taking advantage of the added value from multiple species comparisons, we explore the evolutionary dynamics of innate immunity in insects and how they can address both common and species-specific immune challenges.
Multiple large-scale bioinformatic methods, manual curation, and phylogenetic analyses (3) identified 285 Dm, 338 Ag, and 353 Aa genes from 31 gene families and functional groups implicated in classical innate immunity or defense functions such as apoptosis and response to oxidative stress (table S1). Additional limited analysis of nine sequenced genomes from four holometabolous insect orders, spanning 350 million years of evolution, further defined conserved family features and assisted manual gene model curation by gene family experts. The detailed core analysis (Aa/Ag/Dm) is presented in the supporting online material (SOM) text and in figs. S1 to S22, and the total data set is organized into a web-accessible resource (http://cegg.unige.ch/Insecta/immunodb/), offering a comparative perspective across higher insects. All but 24 previously named Aa genes, as well as 79 previously unnamed Ag genes, were named in accordance with the nomenclature scheme devised for the Ag genome (1) with the use of additional guidelines as described in the SOM; this information will be incorporated in the forthcoming manual annotations of the VectorBase resource (www.vectorbase.org).
Our conservative bioinformatic analysis of the complete genomes identified 4951 orthologous trios (1:1:1 orthologs in the three species) and 886 mosquito-specific orthologous pairs (absent from both Dm and the honeybee, Apis mellifera). Combined bioinformatic analysis and manual curation of the immune repertoire identified 91 trios and 57 pairs, plus a combined total of 589 paralogous genes in the three species. Paralogs derive from family expansions and gene losses, or cases of exceptionally high sequence divergence obscuring phylogenetic relationships. Orthologs most likely serve corresponding functions in respective organisms, whereas paralogs may have acquired different functions.
By definition, orthologous trios represent a numerically conserved subset of genes. Nevertheless, a plot of Dm-Aa and Dm-Ag phylogenetic distances, measured in terms of amino acid substitutions, revealed that, on average, immunity trio orthologs are significantly more divergent (∼20%) than the totality of trios in the genomes (Fig. 1A). Indeed, the immune repertoire is one of the most divergent functional groups as defined by Gene Ontology classifications (fig. S1A). Furthermore, with Dm as reference, several Ag immunity genes are considerably more divergent than their Aa orthologs. A similar trend among all 1:1:1 orthologs was detected, implying greater accumulation of amino acid substitutions in Anopheles. One hypothesis that merits detailed testing is whether this reflects a higher speciation rate and diverse habitat colonization by Anopheles as opposed to the more cosmopolitan Aedes.
Fig. 1.
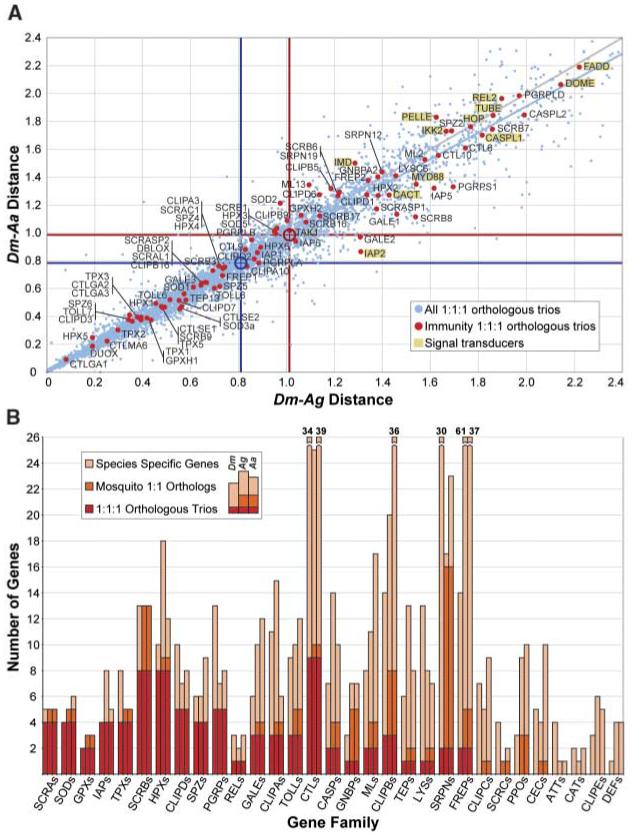
(A) Divergence of orthologous trios. Immunity single-copy trios are compared with all single-copy trios in terms of genetic distances of each mosquito species (Ag or Aa) protein to the corresponding Dm ortholog (3) (fig. S1B). Signal transducers are highlighted. Red and blue lines indicate distance means for immunity (red dots) and all trios (blue dots), respectively. (B) The repertoire of putative immune-related gene families. The numbers of 1:1:1 orthologous trios (red), mosquito-specific 1:1 orthologs (orange), and species-specific genes (light brown) are summed to give the total number of genes identified in Dm (first bar), Ag (second bar), and Aa (third bar) for each gene (sub)family. Families are arranged from left to right, according to the decreasing proportion of 1:1:1 orthologous trios within the family. Family acronyms that are not defined in the text include: CASPs, caspases; CATs, catalases; FREPs, fibrinogen-related proteins; GALEs, galectins; MLs, MD2-like receptors.
Large variation exists in different immune families in their proportions of orthologous trios, mosquito pairs, and species-specific genes (Fig. 1B). Some families display exclusively species-specific genes, some mostly trios, and others intermediate variation. At one extreme are apoptosis inhibitors (IAPs), oxidative defense enzymes [superoxide dismutases (SODs), glutathione peroxidases (GPXs), thioredoxin peroxidases (TPXs), and heme-containing peroxidases (HPXs)], and class A and B scavenger receptors (SCRs), all of which show predominantly trio orthologs. At the opposite extreme are highly diverse immune effector gene families, including three shared antimicrobial peptide (AMP) families that collectively exhibit no orthologous trio and only one confident mosquito orthologous pair. The C-type lectins (CTLs), which have been implicated in immunity as opsonins and modulators of melanization (see below), are intermediate, exhibiting large expansions while retaining nine trios and one pair. The present study reaffirms the family diversity observed in our previous Dm-Ag comparison and further reveals substantial diversity between the two mosquito species, at just over half the evolutionary distance.
A fascinating picture emerged when we disarticulated the immune responses into sequential phases (Figs. 2 and 3). Immune responses begin with molecular recognition of microbial patterns, producing immune signals. Some signals are modulated and/or transduced before activating effector mechanisms. We observed that each of the phases is characterized by different evolutionary dynamics, which may collectively account for the flexibility of the innate immune system that enables adaptation to new challenges.
Fig. 2.
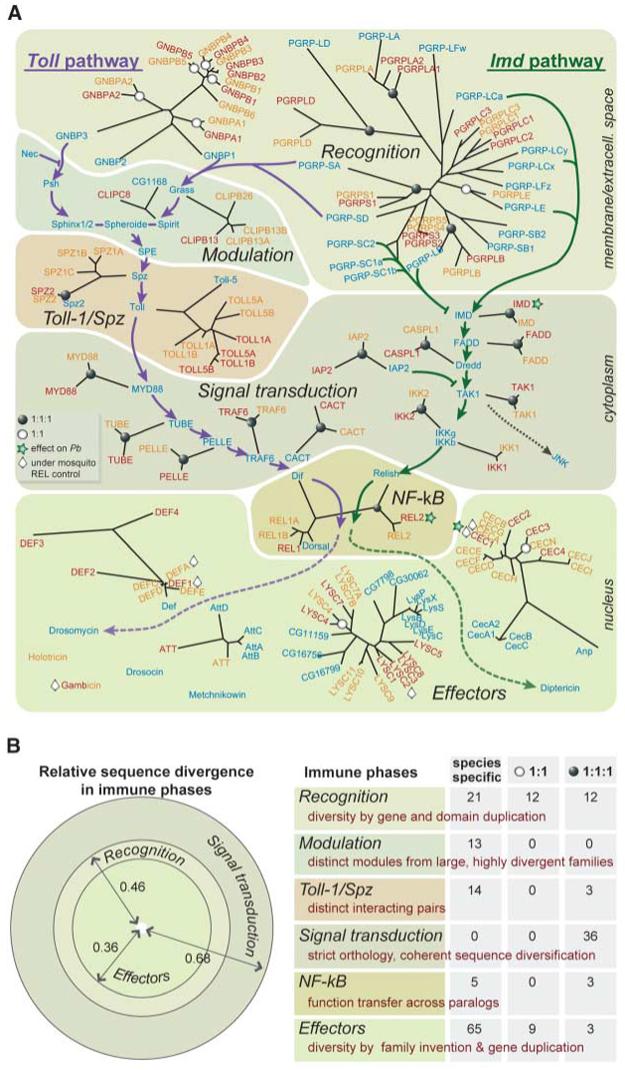
Evolution of immune signaling phases in insects. (A) Genes and gene families implicated in two immune signaling pathways, Toll and Imd (green and purple, respectively). The well-recognized phases of signaling, from recognition to effector production, are outlined. Genes known to be part of these pathways in Dm are indicated in blue, with their closest phylogenetic relatives in Ag in red and Aa in yellow (based on the analysis presented in the SOM). Single-copy orthologs (1:1:1) in all three genomes are indicated with solid circles at the branching node and mosquito 1:1 orthologs are indicated with open circles, respectively. Ag genes affecting survival of the malaria parasite P. berghei are marked with stars, and mosquito genes transcriptionally regulated by NF-κB-like mosquito REL factors are marked with diamonds; Aa CECA and Aa DEFA effectors are controlled by both REL1A and REL2 (33, 39); similarly, Ag REL2 controls expression of immune effectors, including CEC1/3 and GAM (40). Dm LYSs show little response to bacterial infection, but several are up-regulated after infection by microsporidia (41). The mosquito Ag LYSC1/2 and Aa LYSC11 (LysA) genes are up-regulated after bacterial challenge (42, 43), and Ag LYSC2 is controlled by REL1. We constructed radial trees using similarity distances of the conserved sequence cores computed by maximum likelihood. Branch-length scaling is preserved within, but not between, trees. (B) Gene families implicated in the three major immune phases (recognition, signal transduction, and effector production) are clearly different in relative sequence divergence (left panel; sum of branch lengths divided by number of members). Quantitative analysis of evolutionary divergence modes in all six phases defined in (A) is based on gene numbers: trios, mosquito pairs, and genes found in only one species (right panel). All signal transduction genes form trios but are maximally divergent in sequence. In contrast, effector families diversify not by sequence divergence but by gene duplication and creation of new families (e.g., Gambicin in mosquitoes and Diptericin, Drosocin, and others in Dm). This mode results in numerous species-specific effectors but very few trios, contrasting with the pattern seen in signal transduction. The species-specific modulators are selected separately in each species, from very large, divergent families such as SRPNs and CLIPs. Although the Toll and SPZ families are rich in trios, the mosquito genes most closely related to the Dm Toll-1/Spz interaction module are largely species-specific. Finally, the recognition phase shows an intermediate level of diversification, with species-specific genes approximately equal in number to the gene sum of trios and mosquito pairs; in this case, diversification arises by duplication of both genes and domains within genes [see (A)].
Fig. 3.
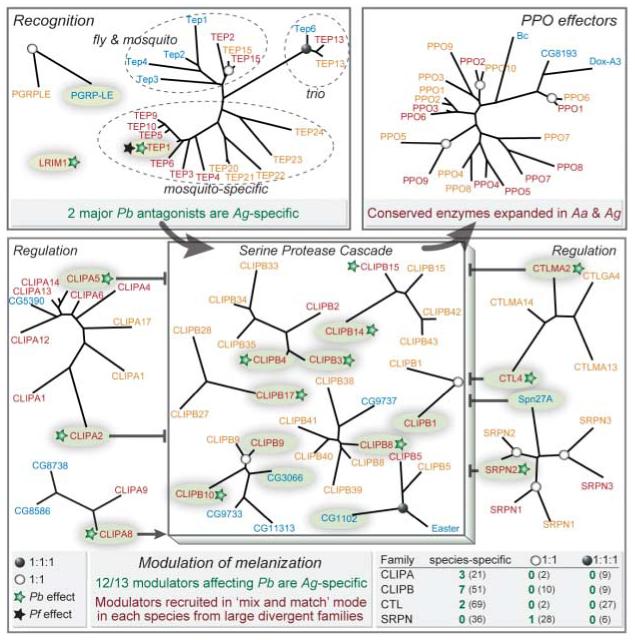
The melanization immune response evolves by convergence and is based on pathogen-related, species-specific regulatory modules. Components are highlighted and shown in relation to their closest phylogenetic relatives in Dm (blue), Ag (red), and Aa (yellow). They are grouped in three phases: recognition, signal modulation, and effectors. TEPs exhibit only one orthologous trio and otherwise form two groups: one with both Dm and mosquito genes and another with species-specific mosquito clades. Recognition genes affecting P. berghei (Pb) melanization (green stars) are Ag-specific. Similarly, among modulators, those affecting Pb melanization (numbers in green in the bottom right box) are almost exclusively specific for Ag and are recruited from large divergent families (numbers in parentheses). In the modulation phase, CLIPB cascades are regulated positively and/or negatively by serine protease homologs (CLIPAs), CTLs, and SRPNs. Among those, CLIPB1, 4, 8, 9, and 10 are involved in melanization of Sephadex beads. The PPO effectors remain conserved in sequence to preserve their enzymatic function, but the family is expanded in mosquitoes. Ag genes marked with black stars affect survival of P. falciparum (Pf). Single-copy orthologs (1:1:1) in all three genomes are indicated with solid circles, and mosquito 1:1 orthologs are indicated with open circles on respective nodes. We constructed radial trees using similarity distances of the conserved sequence cores computed by maximum likelihood, with branch-length scaling preserved within but not between trees.
The immune recognition phase seems to achieve flexibility through divergent evolution: Gene duplications result in species- or lineage-specific expansions and generation of novel genes, whereas domain duplications lead to new gene architectures. Consequently, fruit fly and mosquito recognition proteins mostly form distinct clades within each family (see SOM). Nevertheless, sequence divergence between reduplicated recognition genes or domains remains limited, possibly reflecting the relatively limited diversity of microbial molecular patterns that are known to trigger immune responses. The peptidoglycan recognition proteins (PGRPs) and the Gram-negative binding proteins (GNBPs) are recognition receptor families that trigger signaling through Toll or Imd pathways as indicated in Fig. 2 (4). The Gram-negative recognition protein Dm PGRP-LC, which functions in the Imd pathway, and its Anopheles ortholog each have three functional PGRP domains; however, these are more similar within species than between species, indicating phylogenetically separate domain reduplications. A sequence gap obscures the full structure of the Aedes PGRP-LC ortholog, which apparently derives from the same domain reduplication events that created Ag PGRP-LC. Separate reduplication of two adjacent PGRP-LC domains in Drosophila generated a novel gene, PGRP-LF, which is absent from mosquitoes.
The function of PGRP-LC in Dm is antagonized by catalytic PGRPs that cleave and inactivate peptidoglycan (5, 6). Mosquitoes also possess catalytic PGRPs, but most have emerged as species-specific paralogs (Ag PGRPS2/3 and Aa PGRPS4/5). The fruit fly recognizes Gram-positive bacteria activating Toll using the species-specific Dm PGRP-SD, as well as Dm PGRP-SA, which belongs to a trio and functions in conjunction with GNBP1, a recognition protein that processes polymeric peptidoglycan (7). The two additional Dm GNBPs are also fruit fly-specific; one of them, GNBP3, recognizes fungi, possibly through binding β1,3-glucans (8). A large expansion has generated five mosquito-specific B-type GNBPs, distinct from the two A-type orthologous pairs that resemble fruit fly GNBPs.
Recent studies in Ag identified two types of putative malaria parasite recognition receptors belonging to distinct structural classes: thioester-containing proteins (TEPs) and leucine-rich repeat (LRR) proteins. Members of each class have been associated with the killing and disposal of parasites by lysis or melanization. The TEP family is related to the vertebrate complement factors C3/C4/C5 and pan-protease inhibitors α2-macroglobulins. Ag TEP1 binds to the surface of Plasmodium berghei and mediates parasite killing (9); it also binds to bacteria and promotes phagocytosis (10, 11). TEPs exhibit only one orthologous trio and otherwise form two groups: one with both Dm and mosquito TEPs and another with only mosquito species-specific clades (the latter group includes Ag TEP1) (Fig. 3). The second class of putative receptors include LRR immune gene 1, the pioneer P. berghei LRR antagonist (12); others of similar function are Anopheles Plasmodium-responsive LRR 1 and LRR domain 7, which have been additionally implicated in resistance to P. falciparum, the human malaria parasite (13, 14). Like TEP1, none of the three has identifiable orthologs in Aa or Dm.
Immune modulation is an important process that regulates both the immediate aftermath of recognition and subsequent effector functions and evolves in a “mix and match” mode. Examples are modulation of Toll pathway activation and the melanization reaction, respectively. In both contexts, modulation uses a vast reservoir of serine proteases and their inhibitors [serpins or serine protease inhibitors (SRPNs)] or other regulators, from which particular components are picked to constitute species-specific regulatory modules.
Successful triggering of the Dm Toll pathway after fungal and Gram-positive recognition engages a dedicated proteolytic activation cascade of serine proteases and SRPNs, of which several have been identified recently (15). None of these proteins exhibit mosquito orthologs, and only Spirit and Grass have recognizable paralogs (Fig. 2). The cascade culminates in cleavage of Spaetzle by the Spaetzle proteolytic enzyme (SPE), releasing a cytokine that binds to Toll. Mosquitoes have several genes encoding Spaetzle-like proteins (SPZs), but their SPE has not been recognized. Suggestively, the short and very specific SPE cleavage site (16) recurs in Ag CLIP-domain serine protease B5 (Ag CLIPB5) and Aa CLIPB38, which are otherwise phylogenetically unrelated.
Similarly, activation of prophenoloxidases (PPOs) to phenoloxidases (POs), the executors of melanization, is induced by a protease cascade (mostly CLIPBs). The cascade is positively and negatively regulated by a network of inactive protease homologs (CLIPAs), CTLs, and SRPNs (Fig. 3). This melanization module is tightly controlled, because it generates toxic byproducts including reactive oxygen species. Reverse genetic analyses have identified a large set of Ag regulators for melanization of P. berghei (17-19) or Sephadex beads (20, 21): one SRPN, two CTLs, eight CLIPBs, and three CLIPAs (Fig. 3). Notably, all are members of mosquito-specific expansions, none has a definitive 1:1:1 ortholog, and only SRPN2 has a clear Aa ortholog. The reservoir of Aa proteases shows an underrepresentation of CLIPAs and massive expansions of CLIPBs as compared with both Ag and Dm. Finally, the melanization module may encompass additional regulators, because the genetic background determines which components are important in specific Ag strains (19).
The observed diversity of modulation components suggests that related but distinct regulatory modules may evolve in different species and even in subspecific taxa. Recruitment of individual members from very large multigene families may be followed by modulatory fine-tuning through selection imposed by particular microbes. For example, several of the genes that negatively control P. berghei melanization in Ag [CTL4, CTL mannose-binding 2 (CTLMA2), and SRPN2] do not affect P. falciparum (22, 23). Because Ag is a natural vector of P. falciparum but not of P. berghei, it is appealing to speculate that the sets of regulators of the melanization module evolve with and are manipulated by parasites. This modular mix and match evolution hinders detailed knowledge transfer between vector species but reinforces its importance in shaping the immune response. Future experimental studies of the melanization module in Aa, which can melanize bacteria and filarial worms, as well as sporozoites of the avian parasite P. gallinaceum (24, 25), will be fruitful in further exploring this fascinating mode of immune evolution.
Although Toll-like receptors (TLRs) are found throughout the animal kingdom, phylogenetic and functional studies have suggested that insect Tolls and mammalian TLRs evolved independently (26). Most Dm Tolls serve developmental functions, and the recruitment of the Toll (Toll-1) receptor to immune signaling has been ascribed to convergent evolution. Even within insects, our analysis detects diversity: species-specific Toll expansions and only three trios. Dm Toll-1 has no clear orthologs; reduplications have created a clade of four Ag and four Aa genes, all related to both Dm Toll-1 and Dm Toll-5 (Fig. 2). In addition to its role in antifungal and antibacterial responses, Dm Toll-1 has been implicated in cellular antiviral responses (27). Thus, the possibility that the expanded Toll-1/Toll-5 clade in mosquitoes is related to their interactions with viruses merits detailed functional investigation. An unexpected evolutionary pattern was also observed for Spaetzle, the cytokine partner of Dm Toll-1, which shows three Aa paralogs and no identifiable Ag ortholog. Aa SPZ1C acts together with Aa TOLL5A to activate antifungal responses (28); however, the absence of an Ag Spaetzle ortholog raises questions about the evolution of this pair of molecules as an immune module, especially because the cytokine-Toll interaction is not required for mammalian TLR signaling. The only insect Tolls that cluster with TLRs are Dm Toll-9, Ag TOLL9, and Aa TOLL9A/9B. Because Dm Toll-9 is the only other Toll linked to Drosophila immunity (29), it is possible that this clade represents the most ancient immune-related insect Tolls. Whether these receptors can directly recognize microbial or viral immune inducers remains to be seen; it is worth noting that they are more similar to lipid-binding TLRs rather than to nucleic acid-binding TLRs.
Signal transduction components exhibit an unexpected mode of evolution. Rather than duplicating to create novel cascades responding to distinct challenges, or picking up members of multiprotein families to promote adaptive interactions, these components show robustness, maintaining their distinctive identity and functionality in the face of sequence evolution. The cytoplasmic signal transduction of the Toll pathway includes a chain of interacting partners, almost invariably encoded by orthologous trios: myeloid differentiation factor 88 (MYD88), TUBE, PELLE, tumor necrosis factor receptor-associated factor 6 (TRAF6), and CACT (Fig. 2). The same is true for the components of the IMD pathway: IMD, Fas-associated death domain protein (FADD), Dredd (CASPL1), IAP2, transforming growth factor β-activated kinase (TAK1), and inhibitor of nuclear factor κB kinase subunits γ and β (IKKγ and IKKβ). Despite persistent orthology, these components show marked divergence in sequence (Fig. 1A). A similar pattern is observed in the signal transducers Dome and Hop of the immune signaling Janus kinase-signal transducers and activators of transcription (JAK-STAT) pathway, which is activated in Dm by virus infections (30). We hypothesize that the requirement for these factors to interact productively with others in the same chain causes escalating sequence divergence: A mutation in one may enhance the acceptability of certain mutations in its interacting partner, maintaining pathway function through coherent evolution rather than stasis. Consistent with this interpretation, evidence has been reported for an association between natural sequence variation of core signaling pathway components and immune competence in Drosophila (31). Similar evolutionary patterns are detected among members of the RNA interference antiviral pathway, Dicer-2 and Ago-2 (32), which also form highly divergent trios.
Signal transduction culminates in the next phase: nuclear translocation of transcription factors. The cytoplasmic nuclear factor κB (NF-κB) transcription factors remain inactive until a processed immune signal frees them from inhibitors, permitting their entry into the nucleus and transcription of effector genes. The evolutionary pattern in this phase combines aspects observed in other phases. The NF-κB s of the Imd pathway [Relish in Dm and Rel-like NF-κB protein 2 (REL2) in mosquitoes] form an orthologous trio that displays high sequence divergence, as in signal transducer trios (Figs. 1A and 2). A recent duplication in Aa has resulted in an orthologous quartet (Ag REL1, Dm Dorsal, Aa REL1A, and Aa REL1B). In contrast, Dif is absent from both mosquito species, although the intronless Aa REL1B gene may have originated by retrotransposition. Transgenic analysis has shown that REL1A controls Aedes antifungal responses, as does Dif in Dm (33); this represents an interesting case of functional transfer between paralogs. STAT, the transcription factor of the JAK-STAT pathway, shows high sequence divergence like REL2 and has been duplicated in Ag.
Immune effectors are required to target and neutralize the microbial source of the immune signal. We observed varied evolutionary dynamics for different categories of effectors, reflecting their modes of action. Those acting directly on microbes diversify rapidly or are species-specific, whereas effector enzymes that produce chemical cues to attack invaders remain conserved but independently expand in each species.
The production of AMPs, which act on bacterial membranes causing lysis, is a classic immune-inducible effector response (Fig. 2). Seven AMP families exist in Dm, but only three of them were detected in mosquitoes: Defensins (DEFs), cecropins (CECs), and attacins (ATTs) are highly diverse, together displaying no orthologous trio and only one confident 1:1 orthologous pair. Conversely, gambicins are only encountered in mosquitoes. The apparent paucity of mosquito AMPs in contrast to Dm may be attributable to different prevalence of bacteria in their respective environments.
As diverse as AMPs, the large family of antibacterial peptidoglycan-hydrolyzing lysozymes (LYSs) shows only one identifiable trio and one mosquito pair among 28 members (Fig. 2). A marked expansion in Dm is ascribable to the use of LYSs for digestion of bacteria as a food resource: These peptides are atypically acidic and are expressed in the midgut but not in other immune tissues (34). Apart from these digestive Dm LYSs, the family forms two groups: one with both Dm and mosquito LYSs and the other with only species-specific clades of mosquito LYSs—a very similar pattern to that observed for TEPs, which are also thought to function both as recognition receptors and as complement effectors.
The family of PPO melanization effectors has expanded greatly in mosquitoes as compared with Dm and larger model insects. Ag PPO1/Aa PPO6 is the only orthologous pair that clusters with Dm PPOs; the remaining 17 mosquito PPOs form a distinct clade, created by reduplication events both before and since Ag-Aa diverged (Fig. 3). The invariable catalytic activity of POs (conversion of tyrosine to melanin) is likely to restrict their functional diversification, suggesting that observed expansions may reflect diversification to accommodate differential developmental, topological, or temporal activation. Indeed, several Aa and Ag PPOs show developmental or physiological specificity (35, 36).
In Ag, increased systemic levels of hydrogen peroxide (H2O2) have been associated with Plasmodium melanization (37). H2O2 is used as an electron acceptor by HPXs that catalyze various oxidative reactions. This effector family shows a small expansion in Aa and a large one in Ag, while retaining a set of eight orthologous trios including DUOX (dual HPX and NADPH-oxidase, where NADPH is the reduced form of nicotinamide adenine dinucleotide phosphate). The latter is associated with peroxidase-mediated nitration during the apoptotic response of midgut cells to Plasmodium invasion (38). Numerous trio orthologs of HPXs and other enzyme families implicated in oxidative defense show low sequence divergence, suggestive of constraints to preserve ubiquitous catalytic activities.
The availability of the genome sequences of distantly related insects has allowed us to apply comparative genomic methods to analyze the evolutionary dynamics of the insect innate immune repertoires. Notably, we identified distinct and seemingly contrasting evolutionary modes characterizing different immune modules, which together serve to provide a flexible system capable of adapting to new challenges. The repertoire of recognition receptors of microbial groups such as bacteria and fungi, which are encountered by all species, is achieved through expansion and fine-tuning of model genes. New functions (e.g., recognition of malaria parasites) are acquired from genes bearing powerful and ancient recognition domains such as LRRs. Protein networks modulating immune signals are assembled independently in each species, in the mix and match mode of evolution described as “bricolage” by François Jacob; they therefore coevolve with pathogens and may be subject to evasion. Pathways of signal transduction, on the other hand, remain highly conserved, and their constituent genes seem to evolve always in concert. Finally, effector mechanisms follow evolutionary patterns that depend on their mode of action; most are highly divergent or even species-specific, in contrast to the ancient, conserved oxidative defense mechanisms.
Recognition of the role of Toll in Drosophila immunity led directly to the identification of TLRs as a fundamental aspect of mammalian innate immunity. Similarly, the diverse evolutionary modes of insect immunity that we detected in the present study can guide future studies on the evolution of innate immune mechanisms in vertebrates and other animals. They can also facilitate targeted studies of immunity in the two mosquito species, which together transmit some of the most devastating infectious diseases of humankind.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/316/5832/1738/DC1
Materials and Methods
SOM Text
Figs.S1 to S22
Tables S1 and S2
References
References and Notes
- 1.Christophides GK, et al. Science. 2002;298:159. doi: 10.1126/science.1077136. [DOI] [PubMed] [Google Scholar]
- 2.Nene V, et al. Science. 2007;316:1718. doi: 10.1126/science.1138878. published online 17 May 2007 (10.1126/science.1138878) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Materials and methods are available as supporting material on Science Online
- 4.Wang L, Ligoxygakis P. Immunobiology. 2006;211:251. doi: 10.1016/j.imbio.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Zaidman-Remy A, et al. Immunity. 2006;24:463. doi: 10.1016/j.immuni.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 6.Bischoff V, et al. PLoS Pathog. 2006;2:e14. doi: 10.1371/journal.ppat.0020014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang L, et al. EMBO J. 2006;25:5005. doi: 10.1038/sj.emboj.7601363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gottar M, et al. Cell. 2006;127:1425. doi: 10.1016/j.cell.2006.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blandin S, Levashina EA. Mol. Immunol. 2004;40:903. doi: 10.1016/j.molimm.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 10.Levashina EA, et al. Cell. 2001;104:709. doi: 10.1016/s0092-8674(01)00267-7. [DOI] [PubMed] [Google Scholar]
- 11.Moita LF, et al. Immunity. 2005;23:65. doi: 10.1016/j.immuni.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 12.Osta MA, Christophides GK, Kafatos FC. Science. 2004;303:2030. doi: 10.1126/science.1091789. [DOI] [PubMed] [Google Scholar]
- 13.Riehle MM, et al. Science. 2006;312:577. [Google Scholar]
- 14.Dong Y, et al. PLoS Pathog. 2006;2:e52. doi: 10.1371/journal.ppat.0020052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kambris Z, et al. Curr. Biol. 2006;16:808. doi: 10.1016/j.cub.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 16.Jang IH, et al. Dev. Cell. 2006;10:45. doi: 10.1016/j.devcel.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 17.Volz J, et al. J. Biol. Chem. 2005;280:40161. doi: 10.1074/jbc.M506191200. [DOI] [PubMed] [Google Scholar]
- 18.Michel K, et al. EMBO Rep. 2005;6:891. doi: 10.1038/sj.embor.7400478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Volz J, et al. Cell. Microbiol. 2006;8:1392. doi: 10.1111/j.1462-5822.2006.00718.x. [DOI] [PubMed] [Google Scholar]
- 20.Warr E, et al. Insect Biochem. Mol. Biol. 2006;36:769. doi: 10.1016/j.ibmb.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Paskewitz SM, Andreev O, Shi L. Insect Biochem. Mol. Biol. 2006;36:701. doi: 10.1016/j.ibmb.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 22.Cohuet A, et al. EMBO Rep. 2006;7:1285. doi: 10.1038/sj.embor.7400831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michel K, et al. Proc. Natl. Acad. Sci. U.S.A. 2006;103:16858. doi: 10.1073/pnas.0608033103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hillyer JF, Schmidt SL, Christensen BM. J. Parasitol. 2003;89:62. doi: 10.1645/0022-3395(2003)089[0062:RPAMOB]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 25.Christensen BM, et al. Trends Parasitol. 2005;21:192. doi: 10.1016/j.pt.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Imler JL, Zheng L. J. Leukocyte Biol. 2004;75:18. doi: 10.1189/jlb.0403160. [DOI] [PubMed] [Google Scholar]
- 27.Zambon RA, et al. Proc. Natl. Acad. Sci. U.S.A. 2005;102:7257. doi: 10.1073/pnas.0409181102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shin SW, Bian G, Raikhel AS. J. Biol. Chem. 2006;281:39388. doi: 10.1074/jbc.M608912200. [DOI] [PubMed] [Google Scholar]
- 29.Ooi JY, et al. EMBO Rep. 2002;3:82. doi: 10.1093/embo-reports/kvf004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dostert C, et al. Nat. Immunol. 2005;6:946. doi: 10.1038/ni1237. [DOI] [PubMed] [Google Scholar]
- 31.Lazzaro BP, Sceurman BK, Clark AG. Science. 2004;303:1873. doi: 10.1126/science.1092447. [DOI] [PubMed] [Google Scholar]
- 32.van Rij RP, et al. Genes Dev. 2006;20:2985. doi: 10.1101/gad.1482006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bian G, et al. Proc. Natl. Acad. Sci. U.S.A. 2005;102:13568. doi: 10.1073/pnas.0502815102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daffre S, et al. Mol. Gen. Genet. 1994;242:152. doi: 10.1007/BF00391008. [DOI] [PubMed] [Google Scholar]
- 35.Muller HM, et al. J. Biol. Chem. 1999;274:11727. doi: 10.1074/jbc.274.17.11727. [DOI] [PubMed] [Google Scholar]
- 36.Li JS, et al. Insect Biochem. Mol. Biol. 2005;35:1269. doi: 10.1016/j.ibmb.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Kumar S, et al. Proc. Natl. Acad. Sci. U.S.A. 2003;100:14139. doi: 10.1073/pnas.2036262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar S, et al. J. Biol. Chem. 2004;279:53475. doi: 10.1074/jbc.M409905200. [DOI] [PubMed] [Google Scholar]
- 39.Shin SW, et al. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2616. doi: 10.1073/pnas.0537347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meister S, et al. Proc. Natl. Acad. Sci. U.S.A. 2005;102:11420. doi: 10.1073/pnas.0504950102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roxstrom-Lindquist K, Terenius O, Faye I. EMBO Rep. 2004;5:207. doi: 10.1038/sj.embor.7400073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ursic Bedoya RJ, et al. Insect Mol. Biol. 2005;14:89. doi: 10.1111/j.1365-2583.2004.00534.x. [DOI] [PubMed] [Google Scholar]
- 43.Li B, et al. Gene. 2005;360:131. doi: 10.1016/j.gene.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 44.We thank M. De Iorio for help with statistics, S. Redmond for bioinformatics support, and W. M. Gelbart and J. A. Hoffmann for critical reading of the manuscript and suggestions. This work was supported by the NIH/NIAID grants P01 AI044220-06A1 to F.C.K., 5 R01 AI61576-2 to G.D., and 1 R01 AI059492-01A1 to A.S.R., the Wellcome Trust Programme grant GR077229MA to F.C.K. and G.K.C., the NIH grant GM41247 to M.R.K., and the Swiss National Science Foundation grant SNF 3100A0-112588/1 to E.M.Z. R.M.W. was supported by a Wellcome Trust Ph.D. fellowship, and A.M. was supported by a Ph.D. fellowship from the European Commission Network of Excellence grant (BioMalPar LSHP-CT-2004-553578). S.B. is a European Molecular Biology Organization Long-Term Fellow, and E.A.L. is an International Research Scholar of the Howard Hughes Medical Institute.