

Abstract
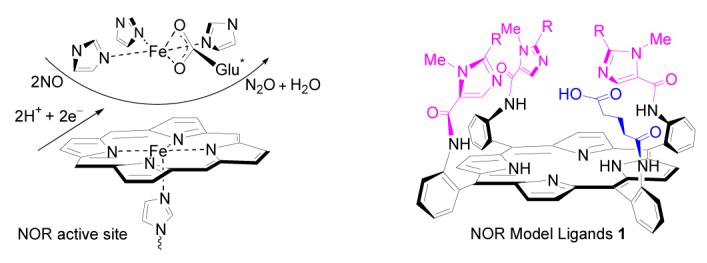 Ligands (1) for active-site models of bacterial nitric oxide reductase (NOR) have been efficiently synthesized. These compounds (1) feature three imidazolyl moieties and one carboxylic acid residue at the FeB site, which represent the closest available synthetic model ligands of NOR active center. The stereo conformations of these ligands are established based on steric effects and 1H NMR chemical shifts under the ring current effect of the porphyrin.
Ligands (1) for active-site models of bacterial nitric oxide reductase (NOR) have been efficiently synthesized. These compounds (1) feature three imidazolyl moieties and one carboxylic acid residue at the FeB site, which represent the closest available synthetic model ligands of NOR active center. The stereo conformations of these ligands are established based on steric effects and 1H NMR chemical shifts under the ring current effect of the porphyrin.
Biological denitrification is a four-step process that reduces nitrate to dinitrogen (eq 1). It represents an alternative to aerobic respiration where the nitric oxides are used as terminal electron acceptors. 1 Denitrification is not only an important process for a variety of bacteria in anaerobic energy harvest, but is also the only process that returns huge amount of fixed nitrogen to atmosphere, thereby finishing the inorganic terrestrial nitrogen cycle. Nitric oxide reductase (NOR) is a membrane-bound enzyme that catalyzes the third step of denitrification that reduces nitric oxide to nitrous oxide (eq 2).
| (eq 1) |
| (eq 2) |
The active site of NORs is bimetallic with an imidazole ligated heme iron and a non-heme iron (FeB) (Figure 1).1 The imidazole coordinated to heme iron on the proximal side is reported to disassociate from heme iron after binding of NO.2 The FeB site is coordinated by three histidyl nitrogen atoms and a postulated ligated glutamic acid residue.3 Comparing NORs with their evolutionary genetic relative Cytochrome c oxidases (CcOs), the dinuclear catalytic center in NORs is FeB-heme-Fe rather than CuB-heme-Fe in CcOs.1 The glutamic acid residues are conserved in NORs, whereas they are absent in CcOs. Additionally, in most CcOs, a redox active phenol group from a tyrosine is coupled to one of the three CuB coordinating histidine moieties.3 The glutamic acid residues have been reported to be essential for normal levels of NOR activity.3a Possible functions of these glutamic acids include increased selective binding of the distal non-heme FeB over Cu, charge regulation directing to the modulation of redox potential of the catalytic center, and mediation of the proton uptake for NO reduction.
Figure 1.
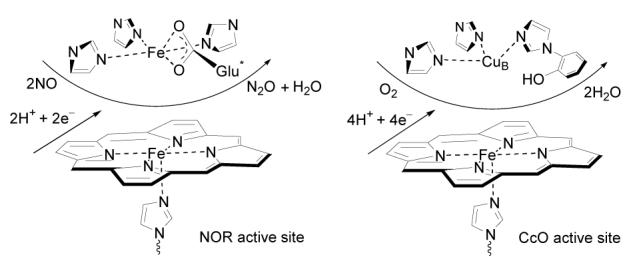
Schematic presentation of NOR and CcO active sites
The biomimetic approach to studying the structural-functional relationships of metalloenzyme activities has proven to be quite successful. 3c,4 Simulation and variation of synthetic models allows for the easy and rapid comparison of coordination environments, spectroscopic properties, and functional capabilities of the metalloenzyme active site. Such information may be difficult to obtain from wild type enzymes or their mutants due to their restricted availability and difficulty in handling. Previously, only a few synthetic models have been developed to study NOR.1a,5 Recently Karlin and co-workers reported structural models of NOR active site featuring a porphyrin covalently linked to a tetradentate chelate (TMPA, tris(2-pyridylmethyl)amine).5 In these models, the TMPA moiety was intended to mimic the trisimidazole ligand environment of FeB site in NORs. A carboxylic group that mimics the glutamic acid residue was not included in these models. A more faithful ligand environment is desirable for modeling of metalloenzymes, as a more exact mimic should shed light on metal binding affinity, spectroscopic properties, and redox potential of the active site of the native enzyme. Our interest on NOR modeling originates from our long standing effort in CcO mimicry and investigation of the chemistry occurring at the bimetallic Fe/Cu active site of CcO using synthetic bioinorganic models.6 Herein we report the syntheses of NOR model ligands 1 (1a, 1b) featuring a porphyrin bearing trisimidazole pickets and a carboxylic acid residue (Scheme 1). These ligands represent the best available NOR model ligands closely replicating the ligand environment of the active site of native NORs.
Scheme 1.
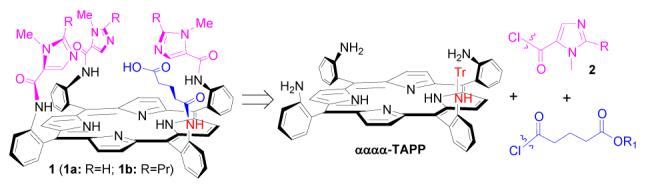
Retro-synthetic scheme of NOR model compound 1
The retro-synthetic scheme of compounds 1 is shown in Scheme 1. The four amino groups of αααα-TAPP (tetrakis-5,10,15,20-(o-aminophenyl)porphyrin) provide excellent linkages for installation of imidazole and carboxylic acid moieties by forming amide bonds with corresponding imidazolecarboxylic acid chloride and glutaric acid chloride.7 One amino group for introduction of the carboxylic acid residue is selectively discriminated from other three amino groups for imidazole moieties by protection with a trityl group.
Preparation of imidazolecarboxylic acid chloride 2 is shown in Scheme 2. Synthesis of 5-imidazolecarboxylic acids 8 followed a literature procedure incorporated with our own modification.8 Diaminomaleonitrile 3 was condensed with trimethyl orthoformate and trimethyl orthobutyrate to give dicyanoimidazole 4a and 4b in yields 83–88%, respectively. 4a was methylated with dimethyl sulfate in NaHCO3 aqueous solution providing 5a in 90% yield. Due to the lower solubility of 4b in water, an alternative method was employed for methylation of 4b. Refluxing 4b in trimethyl orthoacetate provided 1-methylated 4,5-dicyanoimidazole 5b in 84% yield. The N-methyl group in intermediates 5–8 not only results in higher solubility of this series of imidazole intermediates in organic solvents, but also leads to stereo conformational control of model compounds 1 as discussed later. Hydrolysis of 5a and 5b in refluxing sodium hydroxide following by acidification with hydrochloric acid gives imidazoledicarboxylic acids 6a and 6b in 75–90% yield, respectively. Decarboxylation of imidazoledicarboxylic acids 6 (6a, 6b) at 95–100 °C in acetic anhydride provides 5-imidazoleacrboxylic acids 8 (8a, 8b) as the major product. In order to remove side product 4-imidazoledicarboxylic acid (5–25%), the decarboxylated reaction mixture was transformed to methyl esters 7 (7a, 7b). 5-Esters 7a, 7b were isolated in 60–69% yield by column chromatography on silica gel. Pure 5-imidazoledicarboxylic acids 8a and 8b were obtained in 80–93% yield by refluxing their ester precursors in concentrated hydrochloric acid. Treatment of imidazole acids 8 (8a, 8b) with oxalyl chloride in acetonitrile give imidazole acid chlorides 2 (2a, 2b) in 72–85% yield.
Scheme 2.
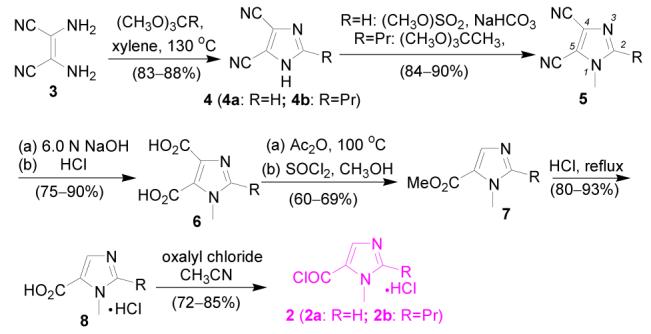
Synthetic scheme of acid chlorides 2
Introduction of the three imidazole pickets to αααα-TAPP is shown in Scheme 3. αααα-TAPP was prepared and enriched following literature procedures.7a,9 After treatment of αααα-TAPP with 1.1 equiv of trityl bromide in CH2Cl2 at room temperature, intermediate 9 with one selectively protected amino group was obtained in around 60% yield. Compound 9 is prone to atropisomerize when stored at room temperature for a long time. Reaction of both freshly prepared intermediate 9 and imidazole acid chloride 2 (2a, 2b) provided trisimidazole compounds 10 in 85–90% yields. Using an excess amount of imidazole acid chloride is important for achieving a high yield of the trisimidazole picketed compound 10. Traces of mono- and bisimidazole side products were removed during column purification. Intermediate 10 is stable; no atropisomerization was observed after storage in a refrigerator for months. Simple treatment of 10 with trifluoroacetic acid provided compound 11 in around 95% yield. This frees the trityl-protected amino group for subsequent introduction of the glutaric acid residue.
Scheme 3.
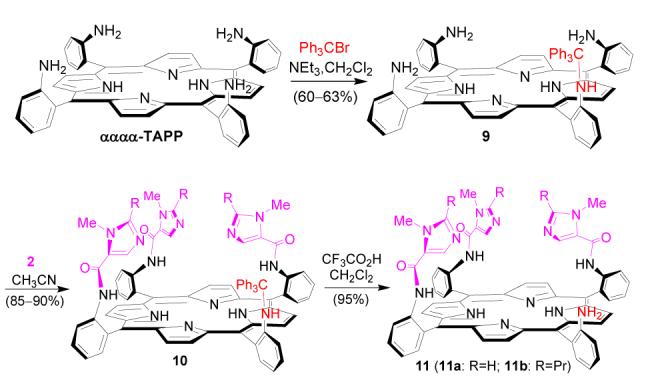
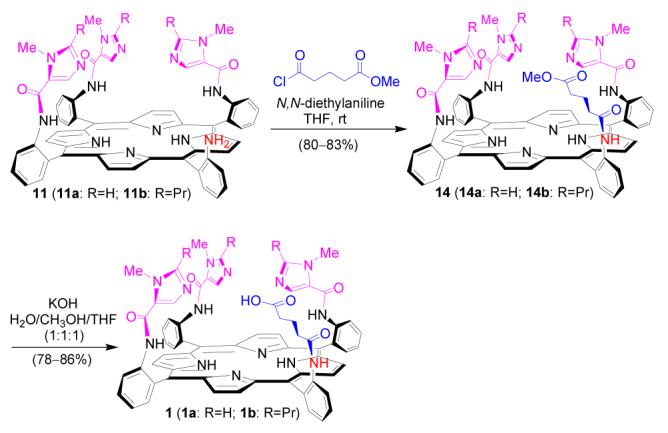
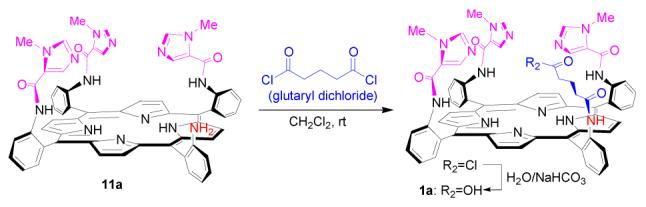
Several strategies were examined in order to find the most efficient method to introduce the glutaric acid residue (Scheme 4-6). Reaction of 11a with glutaric anhydride was first tested (Scheme 4). The reaction was found to be very sluggish and the product 1a difficult to isolate from the reaction mixture. The outcome is tentatively explained by the unfavorable steric effect from bulky trisimidazole-bearing porphyrin substrate 11a, as in a model reaction, phenylamine 12 was found to react with glutaric anhydride readily to provide corresponding amidoglutaranilic acids 13 in good yield under similar conditions (eq. 3). Reaction of 11a with an excess of glutaryl dichloride followed by hydrolysis of the acid chloride intermediate with water was also examined (Scheme 5). The reaction provided the expected compound 1a, but some side products with polarity similar to that of 1a were difficult to remove by column purification.
Scheme 4.
Scheme 6.
Scheme 5.
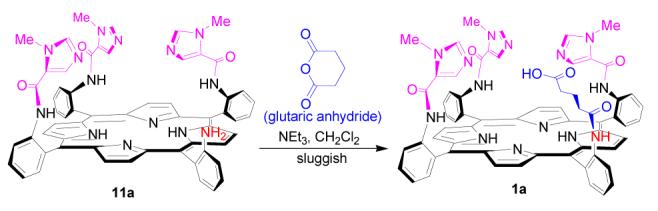
A more practical method for introducing the glutaric acid residue is shown in Scheme 6. Intermediate 11a was treated with methyl 5-chloro-5-oxoverate to provide glutaric ester 14a in 83% isolated yield. Compared with its glutaric acid counterpart 1a, ester 14a is less polar and can be easily purified by column chromatography on silica gel. Hydrolysis of methyl ester group of 14a is quite selective and efficient in a KOH solution of H2O/CH3OH/THF (v/v:1/1/1), providing compound 1a in 86% yield. An analogous model compound 1b was synthesized similarly following Scheme 6. Compared with 1a, due to the propyl groups in imidazole moieties, 1b is less polar during column purification on silica gel. The identities of NOR model compounds 1a and 1b were characterized by 1H NMR, 13C NMR, and high resolution mass spectroscopy (HRMS).10
The proposed geometry of the trisimidazole pickets and the glutaric acid residue over the αααα-TAPP platform is shown in Figure 2. The glutaric residue leans inward to the porphyrin center. The 1-NMe groups on imidazole pickets point outward leaving the 3-imidazoly nitrogen atoms closer to the porphyrin center (conformation A). A reversed stereo orientation of the 1-NMe groups directed towards the porphyrin center is unfavorable due to a strong steric repulsion among the appended groups (conformation B). Such spacial orientation is consistent with the 1H NMR spectra of the product (Figure 3 and 4).
Figure 2.
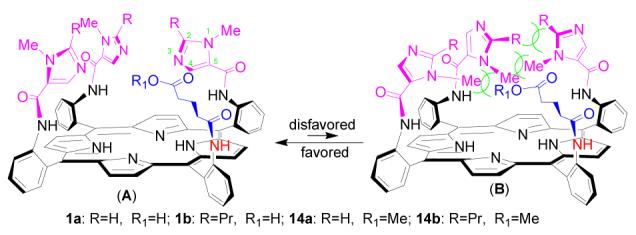
Stereo conformations of compounds 1 and 14
Figure 3.
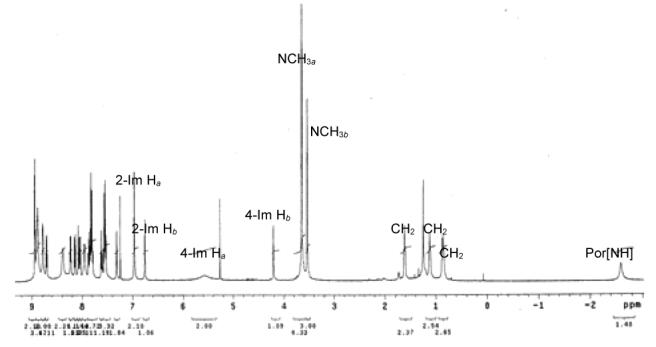
1H NMR spectrum of compound 1a in CDCl3
Figure 4.
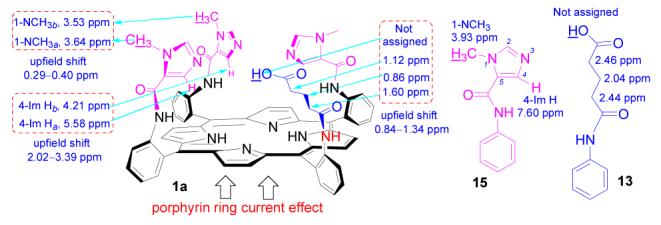
1H NMR chemical shifts of 1a, 13, and 15 in CDCl3
1H NMR spectra is a characteristic tool to probe the stereo relation between the porphyrin ring and appended substituents. Compared to those of non-porphyrin analogues, the chemical shifts of substituents closer to the porphyrin ring center are strongly shifted upfield due to the ring current effect of the porphyrin ring. The influence of the porphyrin current effect on the relation between 1H NMR chemical shifts of the substituents and their stereo orientation is demonstrated with the 1H NMR spectrum of compound 1a (Figure 3-4).11 Compared with that of its non-porphyrin analogue 15,12 the diagnostic imidazole proton 4-Im H of 1a strongly shifts upfield 2.02–3.39 ppm, which is much more than that of imidazole 1-NCH3 (0.29–0.40 ppm), suggesting that 4-Im H is close to the porphyrin center while 1-NMe is located at the porphyrin periphery. The upfield shift (0.84–1.34 ppm) of glutaric acid group on compound 1a compared to its analogue 13 implies that this glutaric acid moiety is suspended over the porphyrin plane. A similar 1H NMR shift pattern is also observed in compounds 14a, 14b, and 1b implying a similar stereo orientation of imidazole and glutaric groups in these molecules.10
In summary, two porphyrin compounds 1a and 1b with three imidazoles and one glutaric acid residue suspended over the porphyrin plane have been prepared. These compounds closely resemble the ligand environment of the diiron center of NORs. Metalation of these model compounds, spectroscopic characterization of their metal complexes, and investigation on interaction between the metal complex and NO and O2 are currently in progress. These studies should provide insightful understanding about the structure features and mechanism of NORs.
Supplementary Material
Acknowledgement
This material is based upon work supported by NIH Grants No. GM-69568-01A1 and GM-017880-35.
Footnotes
Supporting Information Available: Synthetic procedures, characterization, and 1H NMR, 13C NMR spectra, and high resolution mass spectra (HRMS) of all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
Reference
- 1.(a) Wasser IM, Vries SD, Moenne-Loccoz P, Schroder I, Karlin KD. Chem. Rev. 2002;102:1201. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]; (b) Zumft WG. Microbiology and Molecular Biology Rev. 1997;61:533. doi: 10.1128/mmbr.61.4.533-616.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Averill BA. Chem. Rev. 1996;96:2951. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]; (d) Hendriks J, Gohlke U, Saraste M. J. Bioenergetics and Biomembranes. 1998;30:15. doi: 10.1023/a:1020547225398. [DOI] [PubMed] [Google Scholar]
- 2.(a) Brudvig GW, Stevens TH, Chan SI. Biochemistry. 1980;19:5275. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]; (b) Moenne-Loccoz P, Vries SD. J. Am. Chem. Soc. 1998;120:5147. [Google Scholar]
- 3.(a) Butland G, Spiro S, Watmough NJ, Richardson DJ. J. Bacteriology. 2001;183:189. doi: 10.1128/JB.183.1.189-199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Silaghi-Dumitrescu R, Kurtz DM, Jr., Ljungdahl LG, Lanzilotta WN. Biochemistry. 2005;44:6492. doi: 10.1021/bi0473049. [DOI] [PubMed] [Google Scholar]; (c) Tshuva EY, Lippard SJ. Chem. Rev. 2004;104:987. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 4.(a) Holm RH, Solomon EI. Chem. Rev. 2004;104:347. doi: 10.1021/cr0206364. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Boulatov R, Sunderland CJ, Fu L. Chem. Rev. 2004;104:561. doi: 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]; (c) Solomon EI, Szilagyi RK, George SD, Basumallick L. Chem. Rev. 2004;104:419. doi: 10.1021/cr0206317. [DOI] [PubMed] [Google Scholar]; (d) Rao PV, Holm RH. Chem. Rev. 2004;104:527. doi: 10.1021/cr020615+. [DOI] [PubMed] [Google Scholar]; (e) Mirica LM, Ottenwaelder X, Stack TDP. Chem. Rev. 2004;104:1013. doi: 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- 5.(a) Wasser IM, Huang H-W, Moenne-Loccoz P, Karlin KD. J. Am. Chem. Soc. 2005;127:3310. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]; (b) Wasser IM, Martens CF, Verani CN, Rentschler E, Huang H-W, Moenne-Loccoz P, Zakharov LN, Rheingold AL, Karlin KD. Inorg. Chem. 2004;43:651. doi: 10.1021/ic0348143. [DOI] [PubMed] [Google Scholar]
- 6.(a) Boulatov R, Collman JP, Shiryaeva IM, Sunderland CJ. J. Am. Chem. Soc. 2002;124:11923. doi: 10.1021/ja026179q. [DOI] [PubMed] [Google Scholar]; (b) Collman JP, Boulatov R. Angew. Chem. Int. Ed. 2002;41:3487. doi: 10.1002/1521-3773(20020916)41:18<3487::AID-ANIE3487>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]; (c) Collman JP, Sunderland CJ, Boulatov R. Inorg. Chem. 2002;41:2282. doi: 10.1021/ic011191p. [DOI] [PubMed] [Google Scholar]; (d) Collman JP, Boulatov R, Shiryaeva IM, Sunderland CJ. Angew. Chem. Int. Ed. 2002;41:4139. doi: 10.1002/1521-3773(20021104)41:21<4139::AID-ANIE4139>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 7.For some examples of αααα-TAPP as a platform in synthesis of biomimetic models, see: Collman JP, Gagne RR, Reed CA, Halbert TR, Lang G, Robinson WT. J. Am. Chem. Soc. 1975;97:1427. doi: 10.1021/ja00839a026. Wuenschell GE, Tetreau C, Lavalette D, Reed CA. J. Am. Chem. Soc. 1992;114:3346. Collman JP, Yan Y-L, Eberspacher T, Xie X, Solomon EI. Inorg. Chem. 2005;44:9628. doi: 10.1021/ic0516717.
- 8.O'Connell JF, Parquette J, Yelle WE, Wang W, Rapoport H. Synthesis. 1988:767. [Google Scholar]
- 9.Lindsey J. J. Org. Chem. 1980;45:5215. [Google Scholar]
- 10.See Supporting Information for details.
- 11.The Chemical shifts of the carboxylic acid protons of 1a and 1b can not be assigned due to overlap of 1H NMR spectra.
- 12.1H NMR spectra of non-porphyrin analogues 13 and 15 are provided in Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.