

Abstract
Activin A is a member of the transforming growth factor-β superfamily, which we have identified as having a role in inflammatory responses. We show that circulating levels of activin increase rapidly after LPS-induced challenge through activation of Toll-like receptor 4 and the key adaptor protein, MyD88. Treatment with the activin-binding protein, follistatin, alters the profiles of TNF, IL-1β, and IL-6 after LPS stimulation, indicating that activin modulates the release of several key proinflammatory cytokines. Further, mice administered one 10-μg dose of follistatin to block activin effects have increased survival after a lethal dose of LPS, and the circulating levels of activin correlate with survival outcome. These findings demonstrate activin A's crucial role in the inflammatory response and show that blocking its actions by the use of follistatin has significant therapeutic potential to reduce the severity of inflammatory diseases.
Keywords: lipopolysaccharide, cytokines, Toll-like receptor, MyD88, innate immunity
One of the most potent pathogen-derived inflammatory signals is the Gram-negative bacterial cell wall component, LPS, which induces a massive release of cytokines and other inflammatory mediators in the infected host. TNF, IL-1β, and IL-6 are key proinflammatory cytokines induced by LPS. In particular, TNF rapidly increases in the circulation after exposure to LPS and is regarded as one of the earliest cytokines released. This release is critical to the orchestration of the innate immune response, but requires tight regulation to limit the resulting inflammation, which can result in endotoxic shock and, at worst, multiple organ failure (1). Endotoxic shock continues to be one of the most frequent causes of death in the modern hospital intensive care unit. It is now understood that LPS mediates its effects through a member of the highly conserved Toll-like receptor (TLR) family, TLR4, leading to the induction of numerous proinflammatory genes in most cell types, particularly macrophages (2).
Activin A, isolated initially as a stimulator of pituitary follicle-stimulating hormone (FSH) secretion, also influences a number of other processes such as embryogenesis, neuroprotection, apoptosis, and fibrosis (3). These activin A actions can be blocked by its high-affinity-binding protein, follistatin (4). Activin A levels and expression are increased in several inflammatory diseases such as septicemia, inflammatory bowel disease, and rheumatoid arthritis (5–7), which raises the possibility that it plays a significant role in the acute inflammatory response.
In this study, we report the response of activin A in mice given LPS, determine its mode of stimulation, and define its importance in the inflammatory cascade by blocking its bioactivity by using follistatin (4). Our data implicate activin A as a component of the innate immune response, initiated by TLR4 activation through the MyD88 pathway, and show that it modulates the release of key proinflammatory cytokines. Blockade of its actions with follistatin alters the cytokine response to LPS and reduces mortality during lethal endotoxemia in mice.
Results
Circulating Activin A Is Acutely Elevated After LPS Challenge.
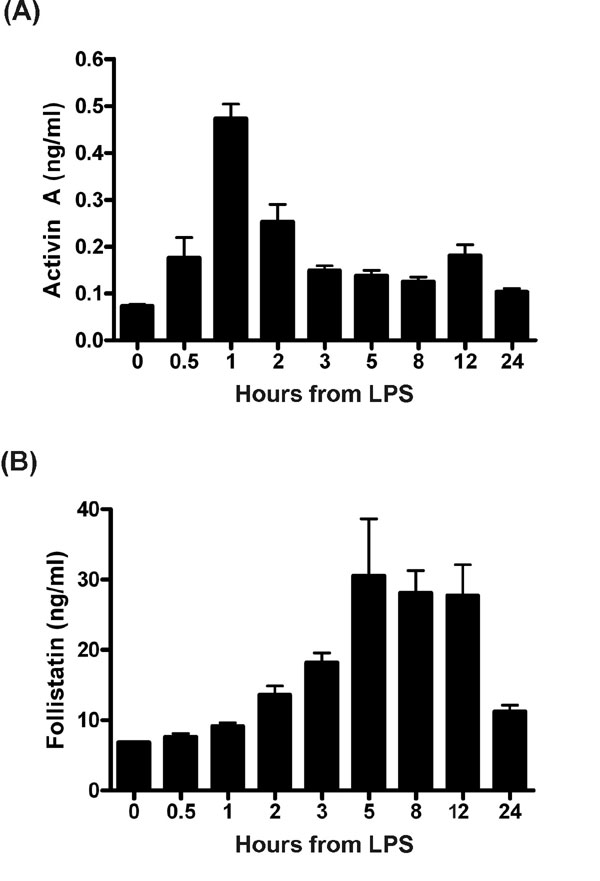
We first established whether activin A was released rapidly into the circulation of mice in response to LPS challenge, suggestive of being a core component of the innate immune response. In C57BL/6 male mice injected with 100 μg of LPS from Escherichia coli (serotype O111:B4), serum activin A concentrations showed a rapid and significant rise, peaking within 1 h and returning to basal levels within 5–8 h [supporting information (SI) Fig. 6]. These animals also had elevated concentrations of circulating follistatin between 5 and 12 h after LPS administration. To remove the possibility that the rise in activin was caused by contaminating toxins other than LPS, such as bacterial lipoproteins, usually present in commercially available LPS preparations, a modified phenol extraction technique was used to remove potential contaminants (8). The injection of purified LPS produced a similar peak in serum activin A when compared with mice injected with nonpurified LPS (Fig. 1A), indicating that this activin A release was triggered by LPS. Comparison of key proinflammatory cytokines revealed that activin A release was concurrent with TNF but significantly earlier than either IL-6 or IL-1β (Fig. 1 C–E). Importantly, activin A release occurred 2 h earlier than the release of its binding protein, follistatin (Fig. 1B), which inhibits activin's actions in vivo and in vitro (4).
Fig. 1.

Release of activin A, follistatin, and other cytokines in mice challenged with phenol-purified LPS. (A) Activin A showed a typical rapid but transient increase within 1 h of LPS. (B) Follistatin was significantly elevated 3 h after LPS. (C) TNF concentrations peaked sharply and were significantly elevated 30 min after LPS injection, peaking at 1 h (P < 0.01). (D) IL-6 peaked at 2 h after LPS (P < 0.01) and returned to basal levels between 5 and 8 h. (E) IL-1β slowly increased, peaked at 5 h, and then returned to basal levels. All data are mean ± SE.
Release of Activin Is Directly Downstream of TLR4 Activation.
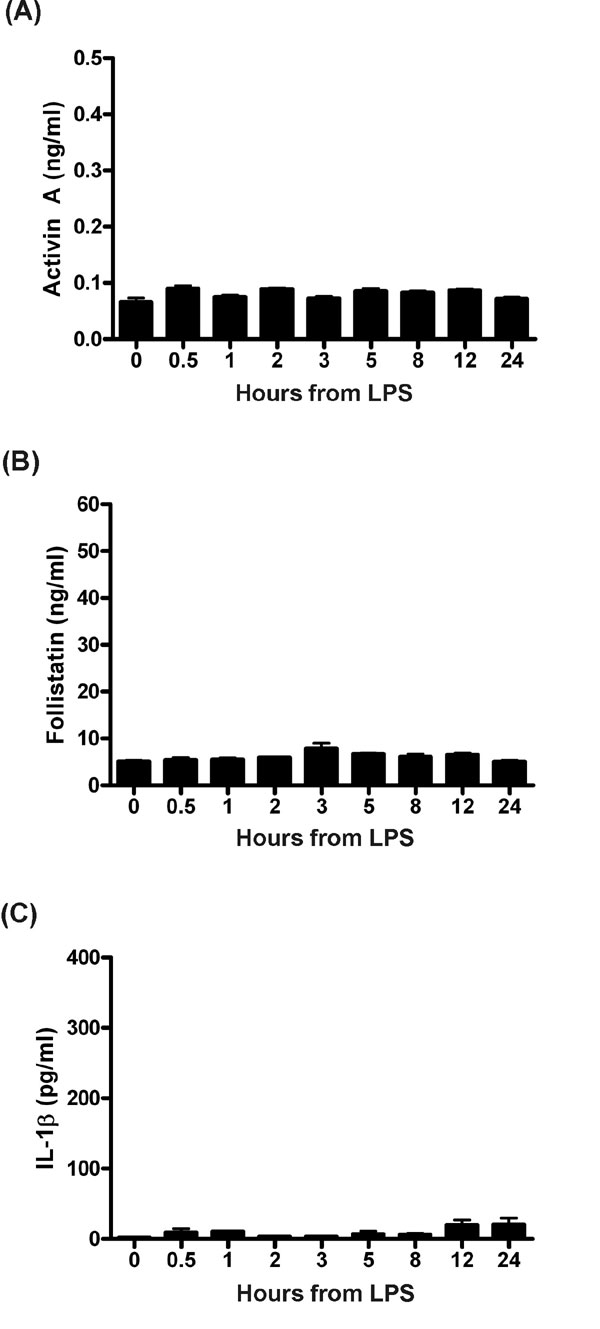
Because our initial findings suggested that activin A release depended on an LPS-driven TLR4 response, we tested this finding directly in C3H/HeJ mice, a strain with a spontaneous inactivating mutation in TLR4 (9, 10). Significantly, following phenol-purified LPS administration, there was no activin A response observed in these mice (SI Fig. 7), and circulating follistatin levels only increased transiently to ≈20% of the response observed in normal mice. These mice also displayed a subdued cytokine response to LPS, with circulating TNF and IL-6 being ≈1% and IL-1β 10% of normal levels in response to LPS. Therefore, these data indicate that activin A release depends on a TLR4 signal, highlighting a mechanism whereby activin A is directly downstream of LPS signaling. Furthermore, the absence of the activin A response and the markedly subdued rise of the other cytokines (TNF and IL-1β) indicate that activin A release is a critical step in the inflammatory response initiated by LPS. To address the question of where in the TLR4 pathway activin is released, we tested the response to LPS in mice lacking MyD88 (11), a critical adaptor protein in TLR4 signaling. LPS exposure of MyD88−/− mice did not cause a notable release of activin A or follistatin (data not shown). We confirmed this obligate requirement of MyD88 by using a dominant-negative mutant in vitro approach. The TM4 epithelial cell line is sensitive to activation by TLR pathways (12) and releases activin A after LPS stimulation. Transient transfection of a dominant-negative MyD88 P-H mutant (13) dose dependently blocked the LPS-driven release of activin in TM4 cells (Fig. 2). Thus, these separate outcomes implicate activin A as an early component in the TLR4 signaling cascade dependent on MyD88 recruitment to the TLR4 signaling complex (2).
Fig. 2.

Release of activin A by LPS is downstream of MyD88. Treatment of the TM4 cell line with LPS for 5 h caused significant release of activin A. Transient transfection of increasing amounts of a dominant-negative MyD88 construct with a nonfunctional TIR domain progressively blocked this activin release, confirming that the LPS stimulation occurs in the TLR4 pathway after binding to MyD88. Data are presented as mean ± SE.
Blockade of Activin Action Modulates Proinflammatory Cytokine Profiles.
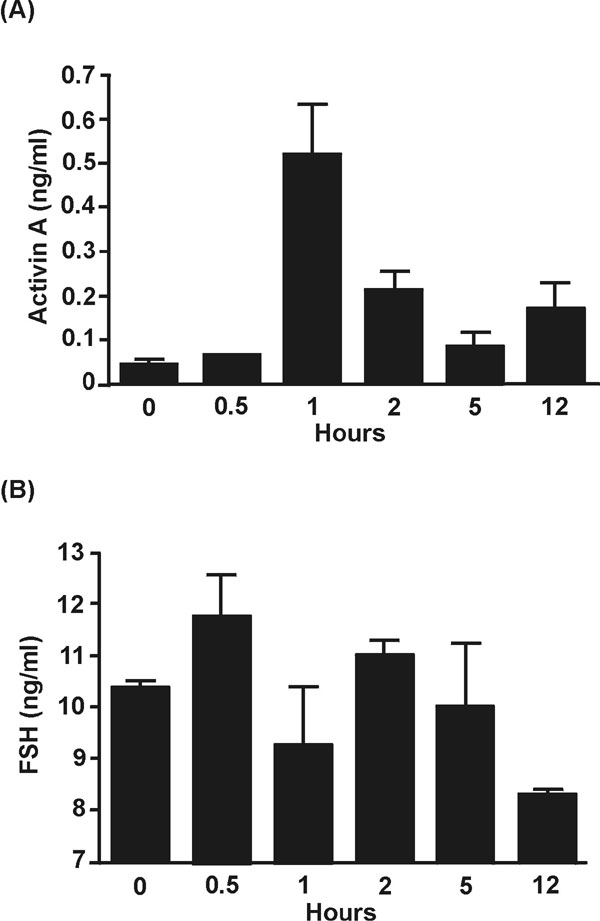
The previous studies clearly established activin A as an early component of innate immunity and the LPS–TLR4 signaling cascade. To further investigate the role of activin in the modulation of the levels of the critical proinflammatory cytokines, namely TNF, IL-1β, and IL-6 during the LPS-induced inflammatory response, we used follistatin (4) to block the actions of activin in vivo. We administered follistatin i.p. to the mice 30 min before LPS challenge. Several pilot studies were conducted to confirm the ability of follistatin to block activin bioactivity as assessed by the decrease of FSH release (14). Administration of 1 μg of human recombinant follistatin per mouse injected i.p. 30 min before LPS effectively blocked the FSH-stimulating actions of activin A (SI Fig. 8). Treatment with follistatin did not alter the initial release of activin A to LPS (Fig. 3A), but interestingly the rise of endogenous follistatin was suppressed (Fig. 3B) presumably because of neutralization of the activin-based stimulation of follistatin secretion (3). Importantly, by blocking the actions of the activin release, follistatin administration altered the profiles of proinflammatory cytokines in response to LPS. TNF and IL-1β release were decreased ≈50% (Fig. 3C) and ≈65% (Fig. 3E), respectively, whereas IL-6 secretion increased ≈250% (Fig. 3D). Furthermore, the temporal release profile of both IL-6 and IL-1β were shifted forward by 1 and 2 h, respectively.
Fig. 3.

Cytokine responses in mice administered 1 μg of follistatin to block the effects of activin after LPS challenge. (A) Activin A after LPS was not affected by follistatin treatment. (B) Follistatin in plasma was significantly suppressed at 5–8 h. (C) Peak plasma concentrations of TNF were significantly suppressed by blockade of activin. (D) The IL-6 response after LPS and follistatin treatment was both elevated and earlier. (E) Peak concentrations of IL-1β were ≈35% of LPS-administered animals, and the peak occurred earlier, shifting from 5 to 2 h. The equivalent response to LPS alone is depicted by a dotted line for comparison. All data are mean ± SE.
This modulation of the release of proinflammatory cytokines was further explored by altering the dose of follistatin used to block the activin bioactivity. In mice given half the amount of follistatin (0.5 μg), peak IL-6 levels were about half those when using the 1-μg dose but double those when LPS alone was used (Table 1), confirming a dose-dependent stimulation of IL-6 by activin. There were similar trends apparent when the amount of IL-6 released was calculated by using area-under-curve estimations. Intriguingly, neither the amount of TNF released nor the peak concentrations was altered by the dose of follistatin used, but in every case these were less than half the values when LPS was administered without follistatin pretreatment (Table 1). Peak levels of IL-1β showed a biphasic response to the dose of follistatin, with the 2-μg dose inducing a 3-fold increase in peak concentrations, and the amount of IL-1β released (area under curve) compared with lower doses of follistatin.
Table 1.
Effect of blocking activin activity with differing doses of follistatin on the release of LPS-stimulated proinflammatory cytokines
| Dose of follistatin, μg | TNF |
IL-6 |
IL-1β |
|||
|---|---|---|---|---|---|---|
| Peak concentration, pg/ml | Area under curve, pg/ml/h | Peak concentration, pg/ml | Area under curve, pg/ml/h | Peak concentration, pg/ml | Area under curve, pg/ml/h | |
| 0 | 7,426 ± 1,811a | 9,047 ± 1,226 | 18,776 ± 1,123a | 59,204 ± 4,476ab | 283 ± 96b | 887 ± 258a |
| 0.5 | 2,827 ± 452b | 5,041 ± 1,362 | 35,748 ± 6,086b | 86,750 ± 14,398b | 203 ± 37b | 393 ± 94a |
| 1 | 3,154 ± 344b | 5,224 ± 685 | 50,455 ± 5,507c | 83,586 ± 10,989b | 116 ± 51a | 452 ± 132a |
| 2 | 3,214 ± 490b | 5,123 ± 642 | 16,043 ± 1,113a | 47,028 ± 3,787a | 914 ± 93c | 3,520 ± 416b |
The peak concentration and the amount of cytokine released (area under the curve) are given for TNF, IL-6, and IL-1β. Data are mean ± SE; differing superscripts within a column indicate significant (P < 0.05) differences between doses.
To directly address the effect of activin A on cytokine release, we also injected human recombinant activin A into normal mice based on the 100% sequence homology in the amino acid sequences of human and mouse activin A. Administration of 1 μg of activin A induced release of IL-6 within 30 min (Fig. 4), but did not cause release of TNF or IL-1β (data not shown). Taken together, these findings point to a complex interaction in the activin A modulation of the proinflammatory cytokine responses during acute inflammation.
Fig. 4.

Activin A induces IL-6 release into the circulation. Injection of 1 μg of human recombinant activin A into mice results in an increase in IL-6 concentrations in the circulation within 30 min. Data are mean ± SE.
Increased Survival in Lethal Endotoxemia When Activin Action Is Blocked.
To address the biological outcome of blocking activin A effects during acute endotoxemia, mice were administered 35 μg/g body weight of phenol-purified LPS with or without 10 μg of follistatin given 30 min before the LPS. We found that this dose of LPS was sufficient to cause septic shock and death in 50–65% of mice by 24 h. Significantly, mice that received this single dose of follistatin before LPS had prolonged survival, and the overall number of animals surviving was greater than in mice injected with LPS alone (Fig. 5A). Serum activin A in those mice that succumbed or who had severe symptoms and were killed for ethical reasons had significantly (P < 0.0001) higher circulating activin A concentrations than those that survived for 48 h (Fig. 5B). Overall, this latter study demonstrates that blocking the actions of activin A during lethal endotoxemia increases survival, and that under these circumstances circulating activin is predictive of outcome.
Fig. 5.

Survival of mice was increased in mice administered follistatin before a dose of LPS that causes significant shock and death. (A) Mice administered follistatin before LPS (FS, open circles) displayed an increased survival compared with LPS alone (filled circles). (B) Activin A levels in the circulation of mice treated with the lethal dose of LPS, irrespective of follistatin administration, correlate with outcome, with significantly (P < 0.001) higher concentrations in those animals that died compared with those that survived the endotoxemia. The dotted line shows that there were no overlapping values between the two outcomes.
Discussion
This study demonstrates that LPS challenge results in the release of activin A through mechanisms downstream of TLR4 and the key adaptor protein, MyD88. Additionally, activin A also appears to be responsive to activation of other Toll-like pathways, such as a TLR2 challenge, as demonstrated by studies in a rabbit model of Gram-positive meningitis (15). We confirmed that activin A is responsive to a TLR2 agonist, Pam3Cys, given to mice in vivo (data not shown). These data, together with some earlier studies in a sheep model, suggest that the response of activin A to inflammatory stimuli is representative of all mammals (6, 16, 17).
A 10-μg dose of follistatin markedly decreased the mortality of mice in response to lethal endotoxemia, strongly arguing that activin A plays an important role in the LPS acute inflammatory event. The profoundly elevated activin A concentrations in the circulation of mice that died, compared with those that survived endotoxemia, are consistent with clinical septicemia, where we have shown that activin A is elevated in those patients with a poor prognosis (6). Furthermore, the current findings illustrate the potential of follistatin as a therapeutic agent in both acute and chronic inflammatory disorders, particularly as the 10-μg dose of follistatin used equates to a molar equivalent of only 2.5 μM. Follistatin has been recently used in a therapeutic setting in mouse models of inflammatory bowel disease with significant efficacy (18).
The observed decrease in serum TNF levels after blockade of activin bioactivity by follistatin suggests that activin modulates the production of this cytokine in vivo. This finding is in agreement with previously published in vitro studies using rat bone marrow-derived macrophages (BMMs) (19). However, in a noninflammatory setting, activin A did not elicit release of TNF when injected directly, suggesting that activin promotes additional release of TNF, rather than initiating de novo secretion. Conversely, the release of activin A in response to LPS in vivo is not dependent on the action of TNF because the use of a specific TNF receptor antagonist did not affect the normal release profile of activin A after LPS administration to sheep (17). Further support for a link between high levels of activin A and TNF bioactivity is the profound cachexia and gonadal tumor development noted in mice homozygous for the targeted disruption of the inhibin α-subunit gene (20). The inhibin α-subunit dimerizes with the subunits of activin, and its absence leads to considerably elevated endogenous levels of activin A. The cachexia associated with these tumors fails to occur when the actions of activin A are blocked in mice, where the activin type II receptor and the inhibin α-subunit genes are disrupted (21). Unfortunately, it is not possible to test mice with targeted disruption of the activin subunit gene because these mice die within hours of birth because of feeding difficulties associated with a cleft palate (22).
In contrast to TNF, activin A was able to elicit secretion of IL-6. This result suggests a dual role of activin A in reducing the amount of IL-6 stimulated through the classical NF-κB pathway, but conversely being able to instigate de novo release of this cytokine.
Despite the complexity of the modulation of the LPS-induced inflammatory cytokine response to different doses of follistatin, this finding was consistent with existing knowledge of the effects of activin on cytokine production by various cell types in vitro. Activin stimulates I-κB activation, nuclear translocation of NF-κB, and phosphorylation of ERK1/2 and p38 MAP kinase in murine BMMs under basal conditions and when stimulated with the receptor activator of NF-κB ligand and/or colony-stimulating factor-1 (23, 24). Accordingly, activin has been found to stimulate the production of IL-1β and TNF by rat BMMs (19) and IL-1β, IL-6, and TNF by human monocytes and peripheral blood mononuclear cells (25). However, activin also has been shown to inhibit the conversion of the IL-1β precursor to its secreted bioactive form, presumably by inhibiting caspase-1 activity (26), and to inhibit secretion of IL-6 by human endometrial epithelial cells (27) and rat thymocytes (28). Notably, activin has stimulatory effects on IL-6 production by human amnion cells at lower doses, but is inhibitory at higher doses (29). Altogether, these data indicate that activin can exert either stimulatory or inhibitory effects on IL-1β and IL-6 in a dose-dependent manner, and that modulating activin activity through follistatin leads to biphasic effects on the production of these two cytokines, as was observed in the present study. The effects of activin on TNF, by contrast, appear to be exclusively stimulatory.
We suggest that the effects of activin on key proinflammatory cytokines during acute inflammation may occur by cross-talk between the cytokine and activin signaling pathways. This theory is supported by the recent demonstration that the activated Smad pathway, specifically Smad2 that both activin and TGF-β use, down-regulates IL-6-induced STAT signaling, highlighting a mechanism by which activin can interact with IL-6 (30). The identification of structural homology between IFN regulatory factor-3 and Smads provides further possibilities for cross-talk between these apparently diverse cytokine signaling pathways (31).
The rapidity and time frame of the activin response in the current study, as well as our previous findings in a sheep model (17), suggest that this release is secretion of prestored activin A, rather than synthesis of a new dimer (see also SI Fig. 9). The source of this release is currently unknown, although we believe it is not coming from the vascular endothelium (32); monocytes, for instance, although responsive to LPS, have little stored activin A under basal conditions (33). Similarly, whether such an inflammatory release extends to other activins, such as activin B, is not yet known because there is currently no validated immunoassay specific for this form. Nevertheless, there is a dramatic increase in activin βB-subunit mRNA after LPS administration (SI Fig. 9).
The in vivo studies presented here reveal the pathophysiological relevance of activin A release to inflammatory pathways by clearly demonstrating that modulation or blockade of activin A during an inflammatory challenge profoundly alters the magnitude and kinetics of proinflammatory cytokine release and ultimately affects the degree of mortality to severe inflammatory insults. The data presented here suggest that modulation of cytokine release by activin A may favor the pro- as opposed to the antiinflammatory side of inflammation and highlights the therapeutic potential of follistatin in treatment modalities for inflammatory disorders, such as in sepsis and the development of septic shock. This regulatory network lies immediately downstream of the presumptive LPS receptor and the key adaptor protein, MyD88. Therefore, activin A is a clearly distinct factor critical to a number of aspects of the innate immune response in mammalian organisms.
Methods
Reagents.
LPS (E. coli serotype 0127:B8) was obtained from Sigma–Aldrich (St. Louis, MO). LPS was purified by using a modified phenol-water extraction method as previously described (8). Recombinant human follistatin 288 was obtained from Biotech Pty Ltd. (Sydney, Australia), or purified bovine follistatin isolated from ovarian follicular fluid was used (14). Human recombinant activin A was a generous gift from Teresa Woodruff (Northwestern University, Evanston, IL).
Mice.
Male C57BL/6, C3H/HeJ, or MyD88−/− (11) mice used in experiments were 4–8 weeks old. The mice were weighed before experimentation began and had ad libitum access to mouse chow and tap water. All of the animal studies were conducted in accordance with the National Health and Medical Research Council Australian Code of Practice for the Care of Animals for Scientific Purposes (1997) and were approved by the Monash Medical Centre Animal Ethics Committee.
In Vivo Experiments.
In general, groups of six mice were randomly allocated to individual groups or time points. One group of mice was killed to provide basal cytokine levels. All other groups received a 100-μg injection of LPS in 100 μl of nonpyrogenic saline. All injections were given i.p. In experiments where the effect of follistatin was assessed, animals were given an i.p. injection of follistatin (at the dose specified) in 100 μl of isotonic, nonpyrogenic saline solution 30 min before an injection of LPS. Animals were killed at the time points shown in the various figures, and blood was collected into a 1.5-ml Eppendorf (Boulder, CO) tube and stored overnight at 4°C. The blood was then centrifuged at 250 × g for 20 min at room temperature, with the resultant serum removed, and stored at −20°C until assayed for activin A, follistatin, TNF, IL-6, and IL-1β. In the study assessing the effect of follistatin when a lethal dose of LPS was used, 45 mice were randomly allocated into a control group of 25 mice and a group of 20 mice for follistatin administration. The control group received LPS alone (35 μg/g body weight), and the second group also received a 10-μg injection of of follistatin 30 min before the LPS injection. Mice were monitored every 20 min, and the time of death was recorded or individuals were killed for ethical reasons. Any mice alive 48 h after the LPS injection were deemed to have survived the event and were killed. In all circumstances, blood was collected and serum derived as detailed above.
In Vitro Experiments.
The TM4 mouse epithelial Sertoli cell line was cultured in DMEM/F12 medium supplemented with antibiotics and 5% FCS. Cells were plated overnight and then transfected with designated amounts of MyD88 dominant-negative construct, a NF-κB reporter construct, TK Renilla, and PEFBOS filler plasmid (34). This transfection occurred overnight, followed by a medium change and a 5-h exposure to 10 ng/ml LPS. Cell lysates and conditioned media samples were collected at the conclusion of this LPS treatment. Lysates were measured for transfection efficiency, and media samples were assayed for activin A concentrations by using the specific ELISA detailed next.
Immunoassays.
Activin A was measured by ELISA as previously described by using human recombinant activin A as standard (35). This ELISA measures both free and follistatin-bound activin A and does not cross-react significantly with other isoforms of activin or with TGF-β. The mean sensitivity was 0.01 ng/ml, and the mean intra- and interassay coefficients of variation (CVs) were 3.9% and 5.1%, respectively. Follistatin was measured by RIA as described in ref. 36. As with the activin A ELISA, this RIA measures both free and bound forms. The assay uses human recombinant follistatin 288 as both standard and tracer. The mean assay sensitivity was 2.7 ng/ml, ED50 was 13.3 ng/ml, and the intra- and interassay CVs were 6.4% and 10.2%, respectively. Mouse TNF, IL-6, and IL-1β were measured by ELISA (R&D Systems, Minneapolis, MN). These assays use mouse recombinant proteins as standards and monoclonal antibodies for detection. The sensitivity of the TNF assay was 0.5 pg/ml, and the intra- and interassay CVs were both <10%. The sensitivity of the IL-6 assay was 0.2 pg/ml, and the intra- and interassay CVs were <10% and <12%, respectively. The sensitivity of the IL-1β was 3 pg/ml, and the intra- and interassay CVs were <10% and <11%, respectively (see SI Methods).
Statistical Analyses.
One-way ANOVA was used to determine significant effects, followed by the Student–Newman–Keuls post hoc test. Where appropriate, data were log n-transformed before analysis to correct for heterogeneity of variance. Area-under-curve derivations were made by using the GraphPad Prism 4.03 package (San Diego, CA). Data are presented as mean ± SE unless otherwise specified, with P < 0.05 considered significant.
Supplementary Material
Acknowledgments
We thank Teresa Woodruff for the generous gift of human recombinant activin A and Anne O'Connor, Sue Hayward, Kathy Wilson, and Kim Sebire for excellent technical assistance. This work was supported in part by National Health and Medical Research Council of Australia Program Grant 334011.
Abbreviations
- BMM
bone marrow-derived macrophages
- FSH
follicle-stimulating hormone
- TLR
Toll-like receptor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0705971104/DC1.
References
- 1.Ulevitch RJ, Tobias PS. Curr Opin Immunol. 1999;11:19–22. doi: 10.1016/s0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K, Kaisho T. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 3.Phillips DJ. In: The Cytokine Handbook. 4th Ed. Thomson A, Lotze MT, editors. Vol 2. London: Academic; 2003. pp. 1153–1177. [Google Scholar]
- 4.Phillips DJ, de Kretser DM. Front Neuroendocrinol. 1998;19:287–322. doi: 10.1006/frne.1998.0169. [DOI] [PubMed] [Google Scholar]
- 5.Hübner G, Brauchle M, Gregor M, Werner S. Lab Invest. 1997;77:311–318. [PubMed] [Google Scholar]
- 6.Michel U, Ebert S, Phillips D, Nau R. Eur J Endocrinol. 2003;148:559–564. doi: 10.1530/eje.0.1480559. [DOI] [PubMed] [Google Scholar]
- 7.Yu EW, Dolter KE, Shao L-E, Yu J. Clin Exp Immunol. 1998;112:126–132. doi: 10.1046/j.1365-2249.1998.00522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hirschfeld M, Ma Y, Weis JH, Vogel SN, Weis JJ. J Immunol. 2000;165:618–622. doi: 10.4049/jimmunol.165.2.618. [DOI] [PubMed] [Google Scholar]
- 9.Poltorak A, He X, Smirnova I, Liu M-Y, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, et al. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 10.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, Malo D. J Exp Med. 1999;189:615–625. doi: 10.1084/jem.189.4.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- 12.Riccioli A, Starace D, Galli R, Fuso A, Scarpa S, Palombi F, De Cesaris P, Ziparo E, Filippini A. J Immunol. 2006;177:7122–7130. doi: 10.4049/jimmunol.177.10.7122. [DOI] [PubMed] [Google Scholar]
- 13.Dunne A, Ejdebäck M, Ludidi PL, O'Neill LAJ, Gay NJ. J Biol Chem. 2003;278:41443–41451. doi: 10.1074/jbc.M301742200. [DOI] [PubMed] [Google Scholar]
- 14.Robertson DM, Klein R, de Vos FL, McLachlan RI, Wettenhall REH, Hearn MTW, Burger HG, de Kretser DM. Biochem Biophys Res Commun. 1987;149:744–749. doi: 10.1016/0006-291x(87)90430-x. [DOI] [PubMed] [Google Scholar]
- 15.Michel U, Gerber J, O'Connor AE, Bunkowski S, Brück W, Nau R, Phillips DJ. J Neurochem. 2003;86:238–245. doi: 10.1046/j.1471-4159.2003.01834.x. [DOI] [PubMed] [Google Scholar]
- 16.Jones KL, Brauman JN, Groome NP, de Kretser DM, Phillips DJ. Endocrinology. 2000;141:1905–1908. doi: 10.1210/endo.141.5.7531. [DOI] [PubMed] [Google Scholar]
- 17.Jones KL, de Kretser DM, Clarke IJ, Scheerlinck J-PY, Phillips DJ. J Endocrinol. 2004;182:69–80. doi: 10.1677/joe.0.1820069. [DOI] [PubMed] [Google Scholar]
- 18.Dohi T, Ejima C, Kato R, Kawamura YI, Kawashima R, Mizutani N, Tabuchi Y, Kojima I. Gastroenterology. 2005;128:411–423. doi: 10.1053/j.gastro.2004.11.063. [DOI] [PubMed] [Google Scholar]
- 19.Nüsing RM, Barsig J. Br J Pharmacol. 1999;127:919–926. doi: 10.1038/sj.bjp.0702626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Proc Natl Acad Sci USA. 1994;91:8817–8821. doi: 10.1073/pnas.91.19.8817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coerver KA, Woodruff TK, Finegold MJ, Mather J, Bradley A, Matzuk MM. Mol Endocrinol. 1996;10:534–543. doi: 10.1210/mend.10.5.8732684. [DOI] [PubMed] [Google Scholar]
- 22.Matzuk MM, Kumar TR, Vassalli A, Bickenbach JR, Roop DR, Jaenisch R, Bradley A. Nature. 1995;374:354–356. doi: 10.1038/374354a0. [DOI] [PubMed] [Google Scholar]
- 23.Murase Y, Okahashi N, Koseki T, Itoh K, Udagawa N, Hashimoto O, Sugino H, Noguchi T, Nishihara T. J Cell Physiol. 2001;188:236–242. doi: 10.1002/jcp.1113. [DOI] [PubMed] [Google Scholar]
- 24.Sugatani T, Alvarez UM, Hruska KA. J Cell Biochem. 2003;90:59–67. doi: 10.1002/jcb.10613. [DOI] [PubMed] [Google Scholar]
- 25.Yamashita N, Nakajima T, Takahashi H, Kaneoka H, Mizushima Y, Sakane T. Clin Exp Immunol. 1993;94:214–219. doi: 10.1111/j.1365-2249.1993.tb06003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ohguchi M, Yamato K, Ishihara Y, Koide M, Ueda N, Okahashi N, Noguchi T, Kizaki M, Ikeda Y, Sugino H, Nisihara T. J Interferon Cytokine Res. 1998;18:491–498. doi: 10.1089/jir.1998.18.491. [DOI] [PubMed] [Google Scholar]
- 27.Perrier d'Hauterive S, Charlet-Renard C, Dubois M, Berndt S, Goffin F, Foidart J-M, Geenen V. Neuroimmunomodulation. 2005;12:157–163. doi: 10.1159/000084848. [DOI] [PubMed] [Google Scholar]
- 28.Hedger MP, Phillips DJ, de Kretser DM. Cytokine. 2000;12:595–602. doi: 10.1006/cyto.1999.0597. [DOI] [PubMed] [Google Scholar]
- 29.Keelan JA, Zhou R-L, Mitchell MD. Placenta. 2000;21:38–43. doi: 10.1053/plac.1999.0451. [DOI] [PubMed] [Google Scholar]
- 30.Walia B, Wang L, Merlin D, Sitaraman SV. FASEB J. 2003;17:2130–2132. doi: 10.1096/fj.02-1211fje. [DOI] [PubMed] [Google Scholar]
- 31.Moustakas A, Heldin C-H. Nat Struct Biol. 2003;10:874–876. doi: 10.1038/nsb1103-874. [DOI] [PubMed] [Google Scholar]
- 32.Wilson KM, Smith AI, Phillips DJ. Mol Cell Endocrinol. 2006;253:30–35. doi: 10.1016/j.mce.2006.03.041. [DOI] [PubMed] [Google Scholar]
- 33.Erämaa M, Hurme M, Stenman U-H, Ritvos O. J Exp Med. 1992;176:1449–1452. doi: 10.1084/jem.176.5.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mansell A, Brint E, Gould JA, O'Niell LA, Hertzog PJ. J Biol Chem. 2004;279:37227–37230. doi: 10.1074/jbc.C400289200. [DOI] [PubMed] [Google Scholar]
- 35.Knight PG, Muttukrishna S, Groome NP. J Endocrinol. 1996;148:267–279. doi: 10.1677/joe.0.1480267. [DOI] [PubMed] [Google Scholar]
- 36.O'Connor AE, McFarlane JR, Hayward S, Yohkaichiya T, Groome NP, de Kretser DM. Hum Reprod. 1999;14:827–832. doi: 10.1093/humrep/14.3.827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}