

Abstract
We estimated DNA sequence variation within and between four populations of Drosophila ananassae at Om(1D) and vermilion (v) by using single-strand conformation polymorphism analysis and direct DNA sequencing. Om(1D) is located on the X chromosome in a region with a normal recombination rate; v is in a region of low recombination. In each population, levels of nucleotide diversity at v are reduced 10- to 25-fold relative to those at Om(1D). Divergence between D. ananassae and its sibling species D. pallidosa, however, is comparable for both loci. This lack of correlation between levels of polymorphism and divergence led to the rejection of a constant-rate, neutral model. To distinguish among alternative models, we propose a test of the background selection hypothesis based on the observed pattern of differentiation between populations. Although the degree of differentiation (measured by FST) among all pairs of subpopulations is similar at Om(1D), we found substantial differences at v. The two northern populations from Burma and Nepal are very homogeneous, whereas comparisons between northern and southern populations (e.g., between Nepal and middle India) produced large FST values. A coalescent-based simulation of the background selection model (in a geographically structured species with a finite number of demes) showed that the observed homogeneity among the northern populations is inconsistent with the background selection hypothesis. Instead, it may have been caused by a recent hitchhiking event that was limited to the northern species range.
In the past decade, a number of studies of DNA sequence variation in Drosophila populations have focused on the detection of natural selection at the DNA level by comparing patterns of variation in gene regions of low and high recombination rates. Most of these studies have found that levels of average nucleotide diversity in low-recombination regions are reduced (1, 2). In contrast, divergence between closely related species is not affected by recombination (3–6). This lack of correlation between levels of variation and divergence led to the conclusion that a constant-rate, neutral model (7) is not compatible with the observations and opened the door to the introduction of alternative models that invoke natural selection.
The selection models that have been proposed to account for the reduction in variability in low-recombination regions, and for related phenomena, fall roughly into three categories: (i) the hitchhiking model, which considers the effect of rare, strongly advantageous mutations on linked, neutral polymorphisms (8–10), (ii) the background selection model, which, in a dual manner, assumes that the driving mutations are frequent and strongly deleterious (11–13), and (iii) models that assume intermediately strong fluctuating or directional selection pressures at linked loci (14, 15).
These models so far have been tested based on intra-populational sequence data. However, most attempts to distinguish the proposed selective forces (in particular, genetic hitchhiking and background selection) were of limited success. In this paper, we propose a test of the background selection model using data on variation and differentiation in a spatially structured species. This test is based on the idea that background selection in a geographically subdivided species is expected to increase FST, a relative measure of genetic differentiation between populations, in gene regions of low recombination because the effective size of local demes is reduced relative to that of high-recombination regions (16). In contrast, genetic hitchhiking may lead to greater homogeneity among subpopulations if a selected allele causing a hitchhiking event in one deme migrates to other demes and causes a hitchhiking event in these demes as well. In the case of local adaptation (i.e., the selected allele causing the hitchhiking event is locally adapted), however, a hitchhiking event may be restricted to a single deme or part of the species range. As a consequence, genetic differentiation between subpopulations (measured by FST) may be large and thus indistinguishable from background selection (17, 18).
We used D. ananassae to test these ideas. In contrast to other often studied Drosophila species, including D. melanogaster (18), D. ananassae shows substantial population substructuring. The picture that emerged from analyses of chromosome inversions (19), isozyme polymorphism (20, 21), and DNA polymorphism (2, 22) is that D. ananassae exists in many semi-isolated populations. It is largely tropical, but has been found on all continents (23). Its zoogeographical center is thought to be in Southeast Asia. Relative to our previous restriction fragment length polymorphism (RFLP) studies (2, 17, 24), which included samples from only two localities in Southeast Asia (Hyderabad, India and Mandalay, Burma), the number of demes was increased to obtain a more complete pattern of genetic differentiation between local populations in Southeast Asia. The gene regions surveyed are the same as in our previous studies: vermilion (v) located on the X chromosome in a region of low recombination, and Om(1D) also located on the X chromosome in a region of intermediate to high recombination (2, 24). Instead of RFLP analysis, sequence variation was measured by the use of single-strand conformation polymorphism (SSCP) and direct DNA sequencing.
MATERIALS AND METHODS
Strains and DNA Preparation.
A total of 45 D. ananassae X chromosome lines that originated from four different collections were used in this survey: nine lines from Mandalay (Burma), 10 lines from Hyderabad (India) (both samples are described in ref. 2), 12 lines from four localities in Nepal (Bharatpur, Godawari, Hetauda, and Kathmandu), and 14 from two localities in Sri Lanka (Beruwala and Colombo). The isofemale lines from Nepal were collected by N. Asada in 1990 and kindly provided by Y. N. Tobari (Kyorin University, Tokyo, Japan); those from Sri Lanka were collected by J. R. David in 1994 and kindly provided by M.-L. Cariou (Centre National de la Recherche Scientifique, Gif/Yvette, France). The X chromosomes were isolated from wild-caught males by using a compound-X chromosome stock constructed by S. Tanda (University of Maryland, College Park, MD). The v sequence of D. ananassae (STD) was determined from a clone derived from a ca;px strain (2). The D. pallidosa strain was obtained from the National Drosophila Stock Center (Bowling Green, OH) and put through four generations of brother-sister mating. Genomic DNA was purified by using CsCl-Sarkosyl gradients (25).
DNA Amplification and Sequencing of Om(1D).
For each of the 45 D. ananassae lines, a 1,762-bp fragment was amplified by PCR. The primers, constructed from the previously published D. ananassae Om(1D) sequence (26), were from 3689 to 3708 and from 5432 to 5450. The nucleotide sequence was determined for both strands of DNA by using a set of seven oligonucleotide primers spaced approximately every 225 bp, a set of seven oligonucleotide primers that were the reverse complement of the previous set, and the PCR primers. These same primers were used to amplify and sequence D. pallidosa, with the addition of one primer that had to be designed from the D. pallidosa sequence itself. PCR amplification was carried out according to Kirby and Stephan (25). Sequencing was performed by using an Applied Biosystems automated sequencer (Molecular Genetics Instrumentation Facility at the University of Georgia). The overlapping fragments were assembled by using the genejockey computer program (Biosoft, Milltown, NJ). Because of a technical difficulty, a 41-bp segment from coordinates 5027 to 5067 encompassing the end of the first intron and a small fragment of the second exon was not included in the variation study of D. ananassae.
DNA Sequencing of vermilion Clone.
The v sequence (STD) was determined by using a 12-kb ca;px clone that was in a pBlueScript vector (2). Sequencing was performed with the dsDNA Cycle Sequencing System according to manufacturer’s specifications (Life Technologies, Grand Island, NY). Both DNA strands were sequenced. The total length of the region sequenced was 3,565 bp and included the entire coding region, the entire 5′ untranslated region, part of the 3′ untranslated region, and part of the 5′ flanking region. The exon/intron structure was determined by comparison with D. melanogaster (27).
SSCP Analysis and Sequencing of the Wild-Caught vermilion Alleles.
SSCP and stratified sequencing were used instead of direct sequencing because of the greater efficiency in regions with low levels of variation (28). SSCP analysis was run according to the protocol in ref. 28. Fourteen pairs of primers were used to amplify overlapping segments of v (numbers in parentheses are distances between primers in bp); 198–217 and 436–453 (256), 415–432 and 679–697 (283), 657–674 and 924–942 (286), 902–921 and 1163–1183 (283), 1144–1161 and 1428–1446 (303), 1400–1420 and 1673–1693 (294), 1652–1670 and 1927–1944 (293), 1904–1924 and 2217–2236 (333), 2181–2199 and 2470–2490 (310), 2447–2466 and 2705–2724 (278), 2684–2702 and 2965–2983 (300), 2944–2961 and 3228–3246 (303), 3198–3218 and 3447–3467 (270), and 3391–3409 and 3549–3565 (175). After the initial SSCP scoring of the 45 wild-caught lines, the mobility of variant alleles were retested by rerunning the samples in a different order in which samples with similar mobilities were grouped together. For each primer pair two randomly chosen lines were sequenced for each mobility class (unless there was only one variant available). Sequencing was done as for Om(1D). Sequencing did not reveal any undetected polymorphism, thus we proceeded by assuming all variation was detected. Primers used for the SSCP analysis were used to sequence both v strands of D. pallidosa, with the addition of five primers that had to be designed from the D. pallidosa sequence itself.
A Test of the Background Selection Model.
Background selection generates genealogies that are approximately identical to those produced by a neutral model, except that the effective population size under neutrality has to be adjusted such that selection and the recombination rate of the locus under background selection are taken into account (12). This results in a reduced effective size, Nev, for the locus of interest (e.g., v). The fact that the background selection model predicts slight distortions of the allele frequency spectrum can be neglected because this effect cannot be observed for typical sample sizes (11). The effect of background selection on neutral variation at this locus in a subdivided species thus can be investigated by simulating a coalescent in a finite island model with k demes (ref. 29, chapter 3.4) in which the recombination rate per locus, Rv = 2Nevr, the mutation rate per locus θv = 3Nevμ (or 4Nevμ for autosomal gene regions), and the migration rate Mv = Nevm have been specified (30). The equation for Rv assumes that recombination in males is negligible. The values of k and Rv are usually unknown, and a range of reasonable numbers has to be assumed. The other two parameters, however, can be estimated from the data as follows:
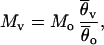 |
1 |
and
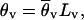 |
2 |
where Mo is the migration rate of the reference locus (located in a region of high recombination), which is estimated for the l subpopulations of interest (l ≤ k) based on Wright’s classical result (31) for the infinite island model, and θ̄v and θ̄o are the arithmetic means of the per-site nucleotide diversities in these subpopulations at the locus under consideration and at the reference locus, respectively. Lv is the number of silent sites at the locus of interest. Eq. 1 assumes that the nucleotide mutation rates of the locus of interest and of the reference locus are identical.
Each simulation run considers k subpopulations, and FST is estimated for a subset of l subpopulations (l ≤ k). We used the same coalescent simulation method as Hudson et al. (30) and ran a modified version of their code, kindly provided by R. R. Hudson (University of California, Irvine, CA). Repeating this procedure generates a probability density of FST values. The P-value of the observed FST then is estimated as the proportion of runs that produced an FST value less than or equal the observed one. A small P-value (≤ 0.05) means that the frequencies of DNA polymorphisms among the l subpopulations are more homogeneous than what would be predicted by the background selection hypothesis in conjunction with the finite island model for the given parameter values.
RESULTS
DNA Polymorphism at Om(1D).
A region of 1.8 kb spanning part of intron 1, exon 2, intron 2, and part of exon 3 was sequenced in 45 lines of D. ananassae, and the homologous segment was sequenced in one line (PAL) of D. pallidosa (Fig. 1). Polymorphism in D. ananassae was assessed based on 1,543 silent sites, and divergence was estimated from 1,735 silent sites. The results are presented in Table 1. A total of 73 silent segregating sites and two replacement polymorphisms were detected. The estimates of average nucleotide diversity, π̂ and θ̂, in the two northern populations (Burma and Nepal) are around 1% and thus approximately twice as large as those of the more southern populations from India and Sri Lanka. These results are comparable to previous RFLP studies on Om(1D) and forked (2, 24); as Om(1D), forked is located on the X chromosome in a region of intermediate to high recombination. Furthermore, π̂ and θ̂ are comparable within each of the four samples. This results in values of Tajima’s D statistic (32) that are close to zero, suggesting that at Om(1D) each population is in (or near) neutral equilibrium. Similarly, the Fu-Li test (33) did not indicate a departure from neutrality (results not shown). Thus, the pattern of sequence variation found at Om(1D) confirms our previous RFLP studies. The differences in levels of average diversity between northern and southern populations is consistent with the interpretation that the northern populations are closer to the geographic species center (2).
Figure 1.

DNA polymorphisms of the Om(1D) and vermilion genes found in 45 lines of D. ananassae. The localities of the strains are indicated on the far left side. The nucleotides within the reference sequence STD (from ref. 26 and as determined from a ca;px clone) are shown along the top. ∗ indicate insertions (I), deletions (D), or complex mutations (M). The numbers above the sequence represent the position numbers of each segregating site within the reference sequences, the nucleotide preceding an insertion, the start of a deletion or the nucleotide preceding a complex mutational event. The homologous nucleotides within the D. pallidosa sequences (PAL) are along the bottom. Dashes indicate where the homologous nucleotide of D. pallidosa could not be determined. Sequence domains of both genes are indicated at the top, E (exon) and I (intron), and F5 (5′ flanking region of v). + indicates amino acid replacement polymorphisms at sites 5395 and 5413 of Om(1D). Insertions, deletions, complex mutations, and amino acid changes in Om(1D) are as follows: a, 3694; b, 60 bp; c, 18 bp; d, 3791; e, TTACTA replaced by a 50-bp sequence; f, T; g, 141 bp; h, 17 bp; i, A; j, 4194 to 4205; k, 4218; l, AGA; m, 4276; n, 4330; o, 4331; p, 4582; q, T; r, T; s, G; t, 5390; u, Thr (ACC) to Asn (AAC); v, Met (AUG) to Lys (AAG). The deletions and the complex mutation in v are as follows: a, TTG replaced by ACC; b, 1094 to 1098; c, 1426 to 1434. The v sequences of D. ananassae and D. pallidosa have been submitted to GenBank (accession nos. AF028834 and AF028835, respectively).
Table 1.
Polymorphism at Om(1D) and vermilion
| Total | Burma | Nepal | India | Sri Lanka | |
|---|---|---|---|---|---|
| Sample size | 45 | 9 | 12 | 10 | 14 |
| Om(1D) | |||||
| Segregating sites | 73 | 46 | 47 | 24 | 27 |
| Singletons | 16 | 23 | 9 | 8 | 6 |
| Diversity θ̂ | 0.011 | 0.010 | 0.0055 | 0.0055 | |
| Diversity π̂ | 0.010 | 0.011 | 0.0056 | 0.0053 | |
| Tajima’s D | −0.41 | +0.43 | +0.13 | −0.15 | |
| vermilion | |||||
| Segregating sites | 7 | 3 | 3 | 4 | 3 |
| Singletons | 4 | 2 | 2 | 2 | 1 |
| Diversity θ̂ | 0.0004 | 0.0004 | 0.0006 | 0.0004 | |
| Diversity π̂ | 0.0003 | 0.0003 | 0.0004 | 0.0006 | |
| Tajima’s D | −0.94 | −0.83 | −0.82 | +0.43 |
DNA Polymorphism at vermilion.
A region of 3.6 kb encompassing the v gene was sequenced in a ca;px strain of D. ananassae (STD) (2) and one line of D. pallidosa (PAL) (Fig. 1). Polymorphism in the four natural D. ananassae populations was assessed by SSCP and stratified sequencing (see Materials and Methods). Seven segregating sites were detected at 2,549 silent sites surveyed (Table 1). The estimates of nucleotide diversity, π̂ and θ̂, in the two northern populations (Burma and Nepal) are 0.0003 and 0.0004, respectively, and are by a factor 25 lower than those at Om(1D). Furthermore, in contrast to Om(1D), they are slightly lower than those of the two southern populations from India and Sri Lanka. The values of Tajima’s D statistic in the samples from Burma, Nepal, and India (but not Sri Lanka) are quite negative, reflecting the fact that most of the segregating sites are singletons (i.e., occur only once in the sample); however, these D values do not deviate significantly from zero.
Polymorphism and Divergence.
Average divergence (at silent sites) between D. ananassae and D. pallidosa at Om(1D) and v is 0.032 and 0.022, respectively. This confirms that these species are very closely related. Reproductive isolation between D. ananassae and D. pallidosa was reported by Futch (34), but may not be complete under laboratory conditions (Y.N. Tobari, unpublished results). Using our data on polymorphism and divergence, we ran Hudson, Kreitman, and Aguadé (HKA) tests (35) for each population. The results are as follows: X2 = 8.29 (P < 0.005) for Burma, 9.62 (P < 0.005) for Nepal, 5.52 (P < 0.025) for India, and 7.78 (P < 0.01) for Sri Lanka. These analyses suggest that levels of polymorphism and divergence are not correlated, thus rejecting a constant-rate, neutral model.
Population Structure.
We used the statistical test of Hudson et al. (36) for detecting genetic differentiation of subpopulations. We applied this test to pairs of populations (Table 2). The test statistic used was KST. The results indicate that at Om(1D) all populations are genetically different from each other, except for the Nepal–Sri Lanka comparison, which is only marginally significant. In contrast, two of the six comparisons for the v locus have rather high P-values: {Burma, Nepal} and {India, Sri Lanka}.
Table 2.
Results of population structure analysis
| Population 1 | Population 2 |
Om(1D)
|
vermilion
|
||
|---|---|---|---|---|---|
| KST | FST | KST | FST | ||
| Burma | Nepal | 0.029 (0.05) | 0.115 | 0.012 (0.292) | 0.039 |
| Burma | India | 0.019 (0.03) | 0.241 | 0.084 (0.022) | 0.423 |
| Burma | Sri Lanka | 0.040 (<0.001) | 0.318 | 0.060 (0.043) | 0.153 |
| Napal | India | 0.026 (0.001) | 0.229 | 0.143 (<0.001) | 0.543 |
| Nepal | Sri Lanka | 0.009 (0.07) | 0.282 | 0.121 (<0.001) | 0.322 |
| India | Sri Lanka | 0.014 (0.017) | 0.088 | −0.008 (0.688) | 0.114 |
The first two columns show the pair of populations compared by the test of Hudson et al. (36). The third and fourth columns give the results of these tests for Om(1D), where the numbers in the third column are the P-values of the observed KST. The FST values (in column 4) have been estimated by using the method of Hudson et al. (30). The remaining columns contain the results for v.
We also measured the extent of genetic differentiation by estimating FST for all pairs of populations (Table 2). For both loci, the populations from Burma and Nepal and from India and Sri Lanka showed the least amount of differentiation. This suggests that polymorphism frequencies among the two northern populations and among the two southern ones are the most homogeneous. Between northern and southern populations, however, genetic differentiation is large, in particular at v. This latter result is consistent with our previous RFLP study, which also showed that the Indian and Burmese populations are genetically more different at v (and furrowed) than at Om(1D) (and forked) (17, 37). However, this does not mean that differentiation (as measured by FST) is generally stronger in regions of reduced recombination. For instance, the {Burma, Nepal} pair produced a much lower FST value at v than at Om(1D), suggesting that the two northern populations are genetically more homogeneous at v than at Om(1D).
Test of the Background Selection Model.
Background selection against deleterious alleles is expected to increase FST in regions of reduced recombination (16). This prediction can be tested by generating the probability density of FST values for a finite island model with k demes, a given migration rate Mv = Nevm, a given mutation rate per locus θv = 3Nevμ, and a given recombination rate per locus Rv = 2Nevr. Nev is the effective (local) population size for the gene region under consideration (i.e., v). We chose a range of reasonable values for the unknown parameters k and Rv; Mv and θv were estimated from the data according to Eqs. 1 and 2, respectively, and l = 2. The results are presented in Table 3. For all combinations of k and Rv values used in the simulations, the {Burma, Nepal} subsample produced the lowest probabilities (P ≤ 0.003). This suggests that the FST value observed for these two northern subpopulations is not compatible with those predicted by the background selection model for the specified migration and mutation parameters. For the two southern subpopulations we found P ≤ 0.01. Relatively low values also were obtained for the subpopulations from Burma and Sri Lanka and from Nepal and Sri Lanka. The other P-values are higher.
Table 3.
Probability of obtaining the observed FST given the background selection model
| Population 1 | Population 2 |
k = 10
|
k = 100
|
||
|---|---|---|---|---|---|
| Rv = 0 | Rv = 0.1 | Rv = 0 | Rv = 0.1 | ||
| Burma | Nepal | 0.003 | 0.001 | 0.002 | 0.001 |
| Burma | India | 0.032 | 0.028 | 0.034 | 0.010 |
| Burma | Sri Lanka | 0.008 | 0.008 | 0.012 | 0.010 |
| Nepal | India | 0.061 | 0.054 | 0.052 | 0.030 |
| Nepal | Sri Lanka | 0.011 | 0.015 | 0.012 | 0.010 |
| India | Sri Lanka | 0.010 | 0.004 | 0.007 | 0.006 |
We also studied by simulation how the P-value of the observed FST is affected by the various model parameters. The number of demes, k, has only minor impact (Table 3) even when k is increased to 1,000 (results not shown). Increasing recombination rate produces lower P-values and thus makes the test more conservative (Table 3). This trend continues to hold when Rv is raised to 1 or 10 (results not shown). The only parameter to which the model is sensitive is the migration rate. An analysis of the effect of migration is important because the estimate of the migration rate (Eq. 1) assumes that the neutral mutation rates in the two loci compared are identical, which may not be the case for v and Om(1D) indicated by the difference in their rates of divergence. Furthermore, although sequencing did not reveal any polymorphism that was not scored by SSCP, a small percentage of (rare) variants may be undetected (28). If so, migration rate Mv inferred from the data would be an underestimate (see Eq. 1). For these reasons, we varied Mv and provide in Table 4 for each parameter set k, Rv, and θv a critical value, Mvu, of the migration rate above which background selection is no longer rejected (i.e., the rate for which P = 0.05 for given values of k, Rv, and θv). We note that for the {Burma, Nepal} subsample the estimated migration rate Mv is more than 20 times lower than its critical value. This strongly suggests that our analysis of the data from these two northern populations (Table 3) is robust. For the {India, Sri Lanka} pair, the estimated migration rate is less than the critical value by a factor 5, which is relatively robust. For the other pairs, however, the results are not as robust.
Table 4.
Critical migration rates where the background selection model is no longer rejected
| Population 1 | Population 2 | Mv |
k = 10
|
k = 100
|
||
|---|---|---|---|---|---|---|
| Rv = 0 | Rv = 0.1 | Rv = 0 | Rv = 0.1 | |||
| Burma | Nepal | 0.033 | 0.66 | 0.90 | 0.66 | 1.7 |
| Burma | India | 0.052 | 0.076 | 0.08 | 0.11 | 0.13 |
| Burma | Sri Lanka | 0.042 | 0.21 | 0.26 | 0.26 | 0.50 |
| Nepal | India | 0.056 | 0.043 | 0.051 | 0.052 | 0.081 |
| Nepal | Sri Lanka | 0.044 | 0.14 | 0.15 | 0.16 | 0.23 |
| India | Sri Lanka | 0.078 | 0.41 | 0.41 | 0.41 | 0.63 |
Column 3 contains the estimated migration rates used in the simulations (Table 3). The critical migration rates (for the given values of the parameters k and Rv) are in columns 4–7.
DISCUSSION
Overview.
We measured levels of DNA polymorphism in two gene regions on the X chromosome in four D. ananassae populations by using SSCP and DNA sequencing. One gene is located in a region of low recombination near the centromere (v), the other one in a region of normal to high recombination [Om(1D)]. One population sample was from Burma, the other three were collected on the Indian subcontinent: Nepal in the north, Hyderabad in middle India, and Sri Lanka in the south. In addition, we determined the DNA sequences of v and part of Om(1D) in the sibling species D. pallidosa and measured divergence between D. ananassae and D. pallidosa. This study was designed to analyze the differential migration behavior of low- and high-recombination genes in a substructured species and to infer from this behavior the effect of natural selection on genetic variation and differentiation.
The level of average nucleotide diversity at Om(1D) is around 0.01 in the two northern populations from Burma and Nepal, and lower by a factor of about 2 in the two southern ones from India and Sri Lanka. These values are comparable with estimates obtained from RFLP analysis for Om(1D) (24) and forked (2), which maps to the same polytene band as Om(1D). At v, however, average nucleotide diversity ranges from 0.0003 to 0.0006. Thus, levels of variation at the low-recombination locus are much lower than at the high-recombination gene. Divergence levels between the two gene regions, on the other hand, are comparable [0.032 for Om(1D) vs. 0.022 for v]. This led to a rejection of a constant-rate, neutral model by the Hudson, Kreitman, and Aguadé (HKA) test. The values of Tajima’s D reported in Table 1 for v led us to conclude that this locus deviates from neutrality whereas Om(1D) does not.
Tests for detecting genetic differentiation between pairs of populations revealed that all four populations are genetically distinct at the Om(1D) locus. At v, by contrast, we found that the two northern populations were not significantly different, and the same was the case for the two southern ones. Genetic differentiation (measured by FST) between northern and southern populations, however, was generally stronger at v. Strong differentiation between the Indian and Burmese pair of populations at v, and even more so at furrowed, another locus near the centromere of the X chromosome, also was observed in a previous RFLP study (17, 37).
Distinguishing Among Selection Hypotheses.
Several alternatives to the constant-rate, neutral model, which we rejected for the v data, have been proposed: (i) the hitchhiking model that considers the effect of rare, strongly advantageous mutations on linked, neutral polymorphisms (8–10), (ii) the background selection model that assumes that the driving mutations are frequent, nearly recessive, and strongly deleterious (11–13), and (iii) “intermediate” models that assume intermediately strong fluctuating or directional selection pressures at linked loci (14, 15). All three models predict a reduction of variability within subpopulations. Our observation of low levels of variation within each subpopulation is in qualitative agreement with this prediction and therefore can not be used as a criterion for distinguishing these models.
Polymorphism data from different populations of a geographically structured species offered a unique approach to distinguishing among the above models. The background selection model predicts increased FST values in regions of reduced recombination (16). We have shown that this property can be tested because the genealogical structure of the background selection model is similar to that of the neutral model. Background selection could be ruled out as an explanation for the observed pattern of differentiation at v between the two northern populations. These two populations showed very little genetic differences at v, whereas at Om(1D) the differences between pairs of subpopulations are larger and more uniform (among all pairs).
Our results are robust in several respects. First, our assumption of low levels of recombination is conservative. Tables 3 and 4 indicate higher rates of rejection of the background selection model with increasing levels of recombination. Second, even if there were polymorphisms undetected by SSCP (for which there is no evidence), this would not affect our results much as the background selection model is rejected for the two northern populations for a wide range of migration rates (Table 4). Third, assessment of population differentiation using absolute levels of divergence as an alternative to FST (38) shows that (absolute) divergence at v between the two northern populations is more than 100 times lower than at Om(1D), which is consistent with our results.
Can the pattern of variation at v in the two northern populations be explained by genetic hitchhiking or by the models with intermediate selection coefficients? One aspect of the data seems to indicate a relatively recent selective sweep that is limited to the northern species range (including Burma and Nepal). During this process the haplotype identical to STD appears to have increased in frequency. This is suggested by the pattern of variation at nucleotide position 849, as the ancestral haplotype is likely to carry C at this site (see D. pallidosa sequence), whereas the new haplotype has a T (Fig. 1). However, another aspect of the data provides evidence that the hitchhiking event in the two northern populations is not caused by strong directional selection as described by the first model. All polymorphisms in the two northern populations are in relatively low frequency, resulting in D values of −0.83 and −0.94 for Nepal and Burma, respectively (Table 1), but these D values do not significantly deviate from zero. Braverman et al. (39) suggest that the observed Tajima’s Ds were unlikely under the hitchhiking model for the cases they examined, but the test is often said to have low power (e.g., ref. 40).
The alternative hypothesis is that the homogeneity in the northern populations may be resulting from a hitchhiking event caused by relatively weak directional selection. Because the allele (STD) that has risen in frequency in the northern populations also is found in the southern localities, this sweep still may be going on and be slowly spreading from the northern to the southern species range. Evidence for the slow dynamics of this process is that at least one recombination event (between coordinates 837 and 849) in the Indian population involving the STD haplotype can be inferred from the data (Fig. 1). This latter scenario would be more in line with the models of the third category that predict negative D values in an intermediate range (14).
Our observation of sequence homogeneity in a region of low recombination among geographically differentiated populations may extend to the species level if subpopulations go on to become separate species and divergence occurred very recently. For instance, among the simulans complex species, D. simulans, D. mauritiana, and D. sechellia, the low-recombination loci asense and ciD show less divergence than zeste, per, and yp2, which are located in regions of intermediate recombination (41). This pattern was interpreted as the result of a sweep that has occurred among these forms even after divergence into separate species had begun (41).
Acknowledgments
We thank R. Hudson for sharing his computer code, B. Charlesworth for sharing an unpublished manuscript, and M.-L. Cariou, S. Tanda, and Y. Tobari for supplying fly stocks. Useful advice was provided by two reviewers, R. Hudson, B. Charlesworth, and all members of our lab. A. Mohseni helped with the SSCP. Our research was supported by National Science Foundation Grant DEB-9407226. L.X. was supported in part by the Academica Sinica (China), and J.M.B. is a National Science Foundation/Sloan Foundation postdoctoral fellow.
ABBREVIATIONS
- SSCP
single-strand conformation polymorphism
- RFLP
restriction fragment length polymorphism
Footnotes
References
- 1.Aguadé M, Miyashita N, Langley C H. Genetics. 1989;122:607–615. doi: 10.1093/genetics/122.3.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stephan W, Langley C H. Genetics. 1989;121:89–99. doi: 10.1093/genetics/121.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Begun D J, Aquadro C F. Genetics. 1991;129:1147–1158. doi: 10.1093/genetics/129.4.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berry A J, Ajioka J W, Kreitman M. Genetics. 1991;129:1111–1117. doi: 10.1093/genetics/129.4.1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martín-Campos J M, Comerón J M, Miyashita N, Aguadé M. Genetics. 1992;130:805–816. doi: 10.1093/genetics/130.4.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langley C H, MacDonald J, Miyashita N, Aguadé M. Proc Natl Acad Sci USA. 1993;90:1800–1803. doi: 10.1073/pnas.90.5.1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kimura M. The Neutral Theory of Molecular Evolution. Cambridge: Cambridge Univ. Press; 1983. [Google Scholar]
- 8.Maynard Smith J, Haigh J. Genet Res. 1974;23:23–35. [PubMed] [Google Scholar]
- 9.Kaplan N L, Hudson R R, Langley C H. Genetics. 1989;123:887–899. doi: 10.1093/genetics/123.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephan W, Wiehe T H E, Lenz M W. Theor Popul Biol. 1992;41:237–254. [Google Scholar]
- 11.Charlesworth B, Morgan M T, Charlesworth D. Genetics. 1993;134:1289–1303. doi: 10.1093/genetics/134.4.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hudson R R, Kaplan N L. Genetics. 1995;141:1605–1617. doi: 10.1093/genetics/141.4.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charlesworth B. Genet Res. 1996;68:131–149. doi: 10.1017/s0016672300034029. [DOI] [PubMed] [Google Scholar]
- 14.Gillespie J H. In: Non-Neutral Evolution: Theories and Molecular Data. Golding B, editor. New York: Chapman and Hill; 1994. pp. 1–17. [Google Scholar]
- 15.Barton N H. Genetics. 1995;140:821–841. doi: 10.1093/genetics/140.2.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Charlesworth B, Nordborg M, Charlesworth D. Genet Res. 1997;70:155–174. doi: 10.1017/s0016672397002954. [DOI] [PubMed] [Google Scholar]
- 17.Stephan W, Mitchell S J. Genetics. 1992;132:1039–1045. doi: 10.1093/genetics/132.4.1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Begun D J, Aquadro C F. Nature (London) 1993;365:548–550. doi: 10.1038/365548a0. [DOI] [PubMed] [Google Scholar]
- 19.Tomimura Y, Matsuda M, Tobari Y N. In: Drosophila ananassae: Genetical and Biological Aspects. Tobari Y N, editor. Tokyo: Japan Scientific Societies Press; 1993. pp. 139–151. [Google Scholar]
- 20.Johnson F M. Genetics. 1971;68:77–95. doi: 10.1093/genetics/68.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cariou M-L, Da Lage J-L. In: Drosophila ananassae: Genetical and Biological Aspects. Tobari Y N, editor. Tokyo: Japan Scientific Societies Press; 1993. pp. 160–171. [Google Scholar]
- 22.Da Lage J-L, Cariou M-L, David J R. Heredity. 1989;63:67–72. doi: 10.1038/hdy.1989.76. [DOI] [PubMed] [Google Scholar]
- 23.Patterson J T, Stone W S. Evolution in the Genus Drosophila. New York: Macmillan; 1952. [Google Scholar]
- 24.Stephan W. Mol Biol Evol. 1989;6:624–635. doi: 10.1093/oxfordjournals.molbev.a040580. [DOI] [PubMed] [Google Scholar]
- 25.Kirby D A, Stephan W. Genetics. 1996;144:635–645. doi: 10.1093/genetics/144.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanda S, Corces V G. EMBO J. 1991;10:407–417. doi: 10.1002/j.1460-2075.1991.tb07962.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Searles L L, Ruth R S, Pret A-M, Fridell R A, Ali A J. Mol Cell Biol. 1990;10:1423–1431. doi: 10.1128/mcb.10.4.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aguadé M, Meyers W, Long A D, Langley C H. Proc Natl Acad Sci USA. 1994;91:4658–4662. doi: 10.1073/pnas.91.11.4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crow J F. Basic Concepts in Population, Quantitative, and Evolutionary Genetics. New York: Freeman; 1986. [Google Scholar]
- 30.Hudson R R, Slatkin M, Maddison W P. Genetics. 1992;132:583–589. doi: 10.1093/genetics/132.2.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wright S. Ann Eugen. 1951;15:323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 32.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fu Y-X, Li W-H. Genetics. 1993;133:693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Futch D G. Evolution. 1973;27:456–467. doi: 10.1111/j.1558-5646.1973.tb00692.x. [DOI] [PubMed] [Google Scholar]
- 35.Hudson R R, Kreitman M, Aguadé M. Genetics. 1987;116:153–159. doi: 10.1093/genetics/116.1.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson R R, Boos D D, Kaplan N L. Mol Biol Evol. 1992;9:138–151. doi: 10.1093/oxfordjournals.molbev.a040703. [DOI] [PubMed] [Google Scholar]
- 37.Stephan W. In: Non-Neutral Evolution: Theories and Molecular Data. Golding B, editor. New York: Chapman and Hill; 1994. pp. 57–66. [Google Scholar]
- 38.Charlesworth B. Mol Biol Evol. 1998;15:538–543. doi: 10.1093/oxfordjournals.molbev.a025953. [DOI] [PubMed] [Google Scholar]
- 39.Braverman J M, Hudson R R, Kaplan N L, Langley C H, Stephan W. Genetics. 1995;140:783–796. doi: 10.1093/genetics/140.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonsen K L, Churchill G A, Aquadro C F. Genetics. 1995;141:413–429. doi: 10.1093/genetics/141.1.413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilton H, Kliman R M, Hey J. Evolution. 1994;48:1900–1913. doi: 10.1111/j.1558-5646.1994.tb02222.x. [DOI] [PubMed] [Google Scholar]
- 42.Watterson G A. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 43.Nei M. Molecular Evolutionary Genetics. New York: Columbia Univ. Press; 1987. [Google Scholar]