

Abstract
Studies involving pharmacologic or molecular biologic manipulation of Group VIA phospholipase A2 (iPLA2β) activity in pancreatic islets and insulinoma cells suggest that iPLA2βparticipates in insulin secretion. It has also been suggested that iPLA2βis a housekeeping enzyme that regulates cell 2-lysophosphatidylcholine (LPC) levels and arachidonate incorporation into phosphatidylcholine (PC). We have generated iPLA2β-null mice by homologous recombination and have reported that they exhibit reduced male fertility and defective motility of spermatozoa. Here we report that pancreatic islets from iPLA2β-null mice have impaired insulin secretory responses to D-glucose and forskolin. Electrospray ionization mass spectrometric analyses indicate that the abundance of arachidonate-containing PC species of islets, brain, and other tissues from iPLA2β-null mice is virtually identical to that of wild-type mice, and no iPLA2β mRNA was observed in any tissue from iPLA2β-null mice at any age. Despite the insulin secretory abnormalities of isolated islets, fasting and fed blood glucose concentrations of iPLA2β-null and wild-type mice are essentially identical under normal circumstances, but iPLA2β-null mice develop more severe hyperglycemia than wild-type mice after administration of multiple low doses of the β-cell toxin streptozotocin, suggesting an impaired islet secretory reserve. A high fat diet also induces more severe glucose intolerance in iPLA2β-null mice than in wild-type mice, but PLA2β-null mice have greater responsiveness to exogenous insulin than do wild-type mice fed a high fat diet. These and previous findings thus indicate that iPLA2β-null mice exhibit phenotypic abnormalities in pancreatic islets in addition to testes and macrophages.
Phospholipases A2 (PLA2)2 catalyze hydrolysis of the sn-2 fatty acid substituent from glycerophospholipid substrates to yield a free fatty acid, e.g. arachidonic acid, and a 2-lysophospholipid (1, 2) that have intrinsic mediator functions (3, 4) and can initiate synthesis of other mediators (5). Arachidonic acid, for example, is converted to prostaglandins, leukotrienes, and epoxytrienes, and acetylation of 2-lysoplasmanylcholine yields platelet-activating factor (PAF) (5).
Of mammalian PLA2s so far cloned, the PAF-acetylhydrolase PLA2 family exhibits substrate specificity for PAF and oxidized phospholipids, and secretory PLA2 (sPLA2) are low molecular weight enzymes that require mM [Ca2+] for catalysis and affect inflammation and other processes (1). Of Group IV cytosolic PLA2 (cPLA2) family members (1), cPLA2α was the first identified and prefers substrates with sn-2 arachidonoyl residues, catalyzes arachidonate release for subsequent metabolism, associates with its substrates in membranes when cytosolic [Ca2+], rises, and is also regulated by phosphorylation (6). There are additional members of the cPLA2 family encoded by separate genes (7–10).
The Group VI PLA2 (iPLA2) enzymes (11–13) do not require Ca2+ for catalysis and are inhibited by a bromoenol lactone (BEL) suicide substrate (14) that does not inhibit sPLA2 or cPLA2 at similar concentrations (14–17). The Group VIA PLA2 (iPLA2β) resides in the cytoplasm of resting cells, but Group VIB PLA2 (iPLA2γ) contains a peroxisomal targeting sequence and is membrane-associated(18,19). These enzymes belong to a larger class of serine lipases that are encoded by multiple genes (20, 21). The iPLA2β enzymes cloned from various species are 84–88 kDa proteins that contain a GXSXG lipase consensus sequence and eight stretches of a repetitive motif homologous to that in the protein binding domain of ankyrin (11–13).
It has been proposed that iPLA2β plays housekeeping roles in phospholipid metabolism (22, 23), such as generating lysophospholipid acceptors for incorporating arachidonic acid into phosphatidylcholine (PC) of murine P388D1 macrophage-like cells, based on studies involving reducing iPLA2 activity with BEL or an antisense oligonucleotide that suppresses [3H]arachidonate incorporation into PC and reduces [3H]lysophosphatidylcholine (LPC) levels (23–25). Arachidonate incorporation involves a deacylation/reacylation cycle of phospholipid remodeling (26, 27), and the level of LPC is thought to limit the [3H]arachidonic acid incorporation rate into P388D1 cell PC (24, 25).
Another housekeeping function for iPLA2β in PC homeostasis has been proposed from studies of overexpression of CTP: phosphocholine cytidylyltransferase (CT) (28, 29), which catalyzes the rate-limiting step in PC synthesis. Cells that overexpress CT exhibit increased rates of PC biosynthesis and degradation and little net change in PC levels, suggesting that PC degradation is up-regulated to prevent excess PC accumulation. Increased PC degradation in CT-overexpressing cells is prevented by BEL, and iPLA2β protein and activity increase, suggesting that iPLA2β is up-regulated (28, 29).
Many other iPLA2β functions have been proposed (30–43), and the fact that multiple splice variants are differentially expressed among cells and form hetero-oligomers with distinct properties suggest that iPLA2β gene products might have multiple functions (40–44). Proposed functions include signaling in secretion (40, 41, 45–50), and BEL attenuates glucose-induced insulin secretion, arachidonate release, and rises in cytosolic [Ca2+] in pancreatic islet β-cells and insulinoma cells (41, 45–50).
Many cells, including β-cells, express multiple distinct PLA2 (13, 16, 17, 51–53), which might reflect redundancy or specific functions of individual PLA2. The mechanism-based iPLA2 inhibitor BEL and its enantiomers inhibit iPLA2 at concentrations lower than those required to inhibit sPLA2 or cPLA2 (14–17, 32), and this has been widely exploited to discern potential biological roles for iPLA2 (30–40, 54–56). BEL also inhibits enzymes other than iPLA2β; however, including serine proteases (57) and phosphatidate phosphohydrolase-1 (PAPH-1) (58), which accounts for some of its biological effects. In addition, BEL inhibits iPLA2γ (18) and at least four other serine lipases (20, 21).
The ambiguity of pharmacologic studies with BEL makes manipulating iPLA2β expression by molecular biologic means an attractive alternative to study iPLA2β functions, and physiological roles for PLA2s can be studied with genetic gain- or loss-of-function manipulations. Stably transfected INS-1 insulinoma cells that overexpress iPLA2β exhibit amplified insulin secretory responses to glucose, particularly in the presence of agents that elevate cAMP (59), and stable suppression of PLA2β expression in transfected insulinoma cells that express small interfering RNA (siRNA) directed against iPLA2β mRNA results in impaired insulin secretory responses to those stimuli (60).
Although these observations support pharmacologic evidence that iPLA2β participates in signaling or effector events involved in insulin secretion (45–51), genetic manipulations at the level of the whole organism sometimes provide information about physiological role(s) of specific gene products that are not readily apparent from results of experiments with cultured cells. Disruption of the gene encoding the Kir6.2 potassium channel, for example, prevents the rise in intracellular [Ca2+] ordinarily induced by D-glucose in pancreatic islet β-cells and suppresses glucose-induced insulin secretion, but, contrary to expectations, non-obese Kir6.2-null mice do not exhibit hyperglycemia because of a counterbalancing increase in insulin action (61), although glucose intolerance can be induced in these mice by dietary interventions and obesity (62, 63).
Disruption of the gene encoding cPLA2α by homologous recombination to yield cPLA2α-null mice has revealed a role for that enzyme in parturition, allergic responses, and post-ischemic brain injury (64, 65), and we have generated iPLA2β-null mice by similar methods in order to examine the physiological functions of iPLA2β (66). Among various tissues, testes of wild-type mice express the highest iPLA2β levels, and male iPLA2β−/− mice produce spermatozoa with reduced motility and impaired ability to fertilize mouse oocytes. Male iPLA2β−/− mice are much less fertile than wild-type males, but female iPLA2β−/− mouse fertility is not impaired (66).
In this report, we examine the insulin secretory responses and phospholipid composition of pancreatic islets isolated from iPLA2β-null mice and their wild-type littermates and the effects on glucose homeostasis of metabolic stresses that include administration of multiple low doses of the β-cell toxin streptozotocin and prolonged feeding of a diet with a high fat content.
EXPERIMENTAL PROCEDURES
Materials
BEL [(E)-6-(bromo-methylene)tetrahydro-3-(1-naphthalenyl)-2H-pyran-2-one] was obtained from Cayman Chemical (Ann Arbor, MI); enhanced chemiluminescence (ECL) reagents from Amersham Biosciences; standard phospholipids including 1,2-dimyristoyl-sn-glycerophosphocholine (14:0/14:0-GPC) and 18:0/22:6-GPC from Avanti Polar Lipids (Birmingham, AL); SDS-PAGE supplies from Bio-Rad; organic solvents from Fisher Scientific; Coomassie reagent from Pierce; streptozotocin, ATP, ampicillin, kanamycin, common reagents, and salts from Sigma; culture media, penicillin, streptomycin, Hanks’ balanced salt solution (HBSS), L-glutamine, agarose, molecular mass standards, and RT-PCR reagents from Invitrogen (Carlsbad, CA); fetal bovine serum from Hyclone (Logan UT); Pentex bovine serum albumin (BSA, fatty acid free, fraction V) from ICN Biomedical (Aurora, OH); and forskolin from Calbiochem (La Jolla, CA). Krebs-Ringer bicarbonate buffer (KRB) contained 25 mM HEPES (pH 7.4), 115 mM NaCl, 24 mM NaHCO3, 5 mM KCl, 1 mM MgCl2, and 2.5 mM CaCl2.
Generating iPLA2−/− Knock-out Mice
The Washington University Animal Studies Committee approved all studies described here and elsewhere in this article. The knock-out construct was prepared with a P1 clone containing an iPLA2β gene fragment obtained from screening a 129/SvJ mouse genomic DNA library with rat iPLA2β cDNA (66). The 7.8-kb EcoRV-BglII fragment containing exons 7–10 was subcloned into pBluescript SK-(pBSK). A single XhoI site mapped to exon 9. A pGK-neopoly(A) cassette with a neomycin-resistance gene (neo) was inserted at this site to disrupt iPLA2β coding sequence and provide a positive selection marker. This yielded a vector with 4.1 and 3.7 kb of 5′- and 3′-sequence, respectively, homologous to the native gene for recombination.
The targeting fragment was excised with EcoRV and BglII and introduced into 129/SvJ mouse embryonic stem (ES) cells by electroporation. Clones resistant to G418 were isolated and screened for homologous recombination by Southern blotting of genomic DNA digested with EcoRV. Six ES clones contained 6.7-kb fragments characteristic of the disrupted iPLA2β gene and 8.7-kb fragments from the wild-type allele. Clones were injected into C57BL/6 mouse blastocysts, which were implanted for gestation to yield chimeras that were mated with wild-type mice to yield heterozygotes. Mating iPLA2β +/− mice with each other yielded iPLA2β−/−, iPLA2β−/+, and iPLA2β+/+ pups in a Mendelian 1:2:1 distribution.
Mice were genotyped with Southern blots of tail clipping genomic DNA digested with EcoRV using a 32P-labeled probe (EB) prepared by PCR amplification or by restriction endonuclease digestion with EcoRV and BglII to yield an 0.95-kb fragment located downstream from the targeting sequence. This probe hybridizes with an 8.7-kb DNA fragment in iPLA2β+/+ mice, with a 6.7-kb fragment in iPLA2β−/− mice, and with both in iPLA2β+/− mice.
Harvesting Mouse Tissues, Preparing Cytosol, and Extracting Tissue Phospholipids
Mice were euthanized by CO2 inhalation, and brain, kidney, muscle, aorta, and liver were removed and weighed. Tissue samples (2 grams) were minced and rinsed twice in ice-cold phosphate-buffered saline. To prepare cytosol, three volumes of 67 mM phosphate buffer containing 1.15% KCl were added, and tissue was homogenized in a Dounce apparatus. Homogenates were transferred to centrifuge tubes and centrifuged (10,000 × g, 20 min, 5 °C). Supernatants were removed and transferred to Beckman ultracentrifuge tubes and again centrifuged (100,000 × g, 1 h, 4 °C). The cytosolic supernatant was removed and stored at −80 °C until use.
For phospholipid extractions, minced tissue (1 g wet weight) was placed in a solution (2 ml) of chloroform/methanol (1:1, v/v) and homogenized (Tissue Tearor, setting 7, 60 s, Biospec Products, Bartlesville, OK). Homogenates were sonicated on ice (20% power, 5 s bursts for 60 s, Vibra Cell probe sonicator, Sonics and Materials, Danbury, CT). Samples were centrifuged (2,800 × g, 5 min) to remove tissue debris and supernatants transferred to silanized 10-ml glass tubes and extracted by adding methanol (1 ml), chloroform (1 ml), and water (1.8 ml). Samples were vortex-mixed and centrifuged (900 × g, 5 min) and supernatants removed, concentrated, and dissolved in methanol/chloroform (9:1). Lipid phosphorus was measured as described (67).
Islet Isolation
Islets were isolated from pancreata removed from female mice 12–16 weeks of age by collagenase digestion after mincing, followed by Ficoll step density gradient separation, and manual selection under stereomicroscopic visualization to exclude contaminating tissues (68, 69). Mouse islets were counted and aliquoted for insulin secretion studies (30 islets/aliquot) or analyses of phospholipids (300 islets/aliquot). Islet lipids were extracted, and their phosphorus content was measured, as described above.
Analyses of iPLA2β mRNA in Mouse Tissues
Northern blots of iPLA2β mRNA were performed as described (66). For RT-PCR, total RNA was isolated with an RNeasy kit (Qiagen Inc.). SuperScript First Strand Synthesis System (Invitrogen) was used to synthesize cDNA in 20-μl reactions that contained DNase I-treated total RNA (2 μg). The cDNA product was treated (20 min, 37 °C) with RNase H (2 units, Invitrogen), and heat-inactivated (70 °C for 15 min). A reaction without reverse transcriptase was performed to verify absence of genomic DNA. PCR performed with the pair of primers 1 and 2 was designed to amplify a fragment that spans the neomycin cassette insertion site. PCR performed with the pair of primers 3 and 2 was designed to amplify a fragment downstream from the neomycin cassette insertion site. The sequence of primer 1 is tgtgacgtggacagcactagc; that of primer 2 is ccccagagaaacgactatgga; and that of primer 3 is tatgcgtggtgtgtacttccg.
Western Blotting Analyses and Immunoprecipitation
The proteins in tissue cytosol and membrane fractions were analyzed by SDS-PAGE (7.5%), transferred onto a polyvinylidene difluoride membrane, and probed with iPLA2β antibodies obtained from commercial sources or with antibody 506 provided to us by Dr. Richard Gross, as described in the figure legends. Protein bands were visualized by ECL, as described (13). Immunoprecipitation studies were performed with the Catch and Release® reversible immunoprecipitation system (Upstate Inc., Lake Placid, NY) using iPLA2β antibody obtained from Cayman Chemical (Ann Arbor, MI) according to the manufacturer’s instructions.
Identifying a Protein Other than iPLA2β that Cross-reacts with iPLA2β Antibody
Western blotting analyses of cytosol from liver of both wild-type and iPLA2β-null mice revealed a 100-kDa immunoreactive band recognized by iPLA2β polyclonal antibody T14 from Santa Cruz Biotechnology. This band was excised from Coomassie-stained SDS-PAGE gels and digested with trypsin, and the digest was analyzed by LC/ESI/MS/MS as described (70). Data base searching revealed a match with mouse glycogen phosphorylase with a high Mascot score of 965. Standard glycogen phosphorylase was then purchased from Sigma, analyzed by SDS-PAGE, and transferred to a nylon membrane. Probing with iPLA2β polyclonal antibody T14 indicated that the 100-kDa protein was recognized by the antibody.
Liquid Chromatographic Mass Spectrometric Analyses of Tryptic Digests of Protein
As described (70), samples (0.2–2 μl) from tryptic digests were injected into a Micromass CapLC liquid chromatography system (Micromass, Manchester, UK). Peptides were concentrated on a PepMap C18 precolumn (300 μm × 5 mm), eluted onto an analytical C18 column (75 μm × 150 mm), and analyzed with a solvent gradient from solution A (3% acetonitrile) to solution B (95% acetonitrile) containing 0.1% formic acid over 50 min at a flow rate reduced from 5 μl/min to 200 nl/min by stream splitting. LC eluant was introduced into the nanoflow source of a Micromass Q-TOF Micro mass spectrometer (Micromass, Manchester, UK). Data-dependent switching from MS to MS/MS analyses was performed, and the resultant data were searched against data bases, as described (70).
Cloning, Expression, and Purification of Polyhistidine-tagged Native iPLA2β and an iPLA2β Mutant Protein Lacking Sequence Encoded by Exon 9
An iPLA2β cDNA that lacked sequence encoded by exon 9 of the iPLA2β gene was constructed using PCR to eliminate exon 9 and to link exons 8 and 10 in a manner that maintained the reading frame. Spodoptera frugiperda (Sf9) cells were cultured as described (71). For protein expression, cDNA encoding native iPLA2β with a polyhistidine tag or a cDNA encoding a C-terminal-polyhistidine-tagged iPLA2β mutant that lacked sequence encoded by exon 9 (NE9-iPLA2β) was cloned into the EcoRI-SalI site of pFast-Bac™1 baculovirus shuttle vector (Invitrogen). Sf9 cell suspensions were infected by baculovirus, collected by centrifugation, and disrupted by sonication. His-tagged proteins were purified with a TALON metal affinity column, as described (71). Aliquots of protein solutions were analyzed by SDS-PAGE. Proteins were visualized by Coomassie staining or by immunoblotting after transfer to nylon membranes, as described (13).
Ca2+-independent Phospholipase A2 Activity Assay
Tissue Ca2+-independent PLA2 specific activity was determined as described (48) in cytosol by monitoring hydrolysis of 1-palmitoyl-2-[14C]linoleoyl-sn-glycero-3-phosphocholine in assay buffer (40 mM Tris, pH 7.5, 5 mM EGTA) to [14C]linoleate as measured by TLC and liquid scintillation spectrometry. Specific activity was calculated from released [14C] dpm and protein content.
Static Insulin Secretion
Islets were rinsed with KRB medium containing 3 mM glucose and 0.1% bovine serum albumin and placed in silanized tubes (12 × 75 mm) in the same buffer, through which 95% air/5% CO2 was bubbled before the incubation. The tubes were capped and incubated (37 °C, 30 min) in a shaking water bath, as described (68, 69). The buffer was then replaced with KRB medium containing 3 or 20 mM glucose and 0.1% BSA without or with forskolin (2.5 or 10 μM), and the samples were incubated for 30 min. Insulin secreted into the medium was measured by radioimmunoassay.
Electrospray Ionization Mass Spectrometric Analyses of Glyc-erophosphocholine Lipids
PC and LPC were analyzed as Li+ adducts by positive ion ESI/MS on a Finnigan (San Jose, CA) TSQ-7000 triple stage quadrupole mass spectrometer with an ESI source controlled by Finnigan ICIS software. Phospholipids were dissolved in methanol/chloroform (2/1, v/v) containing LiOH (10 pmol/μl), infused (1 μl/min) with a Harvard syringe pump, and analyzed as described (72–74). For tandem MS, precursor ions selected in the first quadrupole were accelerated (32–36 eV collision energy) into a chamber containing argon (2.3–2.5 mtorr) to induce collisionally activated dissociation (CAD), and product ions were analyzed in the final quadrupole. Identities of GPC species were determined from their tandem spectra (72–74), and their quantities were determined relative to the internal standards 14:0/14:0-GPC and 18:0/22:6-GPC by interpolation from a standard curve (60, 75, 76).
Multiple Low Dose Streptozotocin Administration
Wild-type mice and their iPLA2β-null littermates 18–22 weeks of age received 40 mg/kg of streptozotocin (STZ) intraperitoneally on 5 consecutive days between 1300 and 1400 h, as described (77). The mice were housed in a temperature-controlled room with 12-hour light/12-hour dark cycles. Mice were given free access to water and standard laboratory chow during these studies. Blood was obtained at various intervals from the saphenous vein and used for glucose and insulin determinations. Specimens were obtained after an overnight fast. Mice were followed for 7 weeks.
High Fat Dietary Intervention Study
Female mice were housed in a pathogen-free barrier facility with unrestricted access to water and standard mouse chow (Purina Mills Rodent Chow 5053) containing 6% fat. For dietary intervention studies, mice were fed standard chow until 8 weeks of age and thereafter were randomized into groups that were fed either standard chow or a high fat diet continuously for the next six months, as described (78). The composition of the high fat diet (Harlan Teklad catalog TD88137) was: energy from fat 42%; casein 195 g/kg; sucrose 341 g/kg; corn starch 150 g/kg; cellulose 50 g/kg, anhydrous milk fat (g/kg) 210 g/kg; cholesterol 1.5 g/kg; 18.95 kilojoules per gram.
Glucose and Insulin Tolerance Tests
Female mice in clean cages with free access to water were fasted overnight and then weighed, and baseline blood glucose was determined using Ascensia ELITE XL blood glucose meter. The animals were injected intraperitoneally with 50% (w/v) dextrose at a dose of 2 mg/g body weight, and blood glucose was measured at 30, 60, and 120 min, as described (69). Insulin tolerance tests were conducted in a similar manner, except that mice were not fasted and received an intraperitoneal injection of human regular insulin (Lilly, Indianapolis, IN) at a dose of 0.75 units/kg body weight, as described (69).
Other Analytical Procedures
As described (69), serum glucose was measured using reagents from Sigma. Serum insulin was assessed by enzyme-linked immunosorbent assay (Crystal Chem. Inc., Downer’s Grove, IL).
Statistical Methods
Results are presented as mean ± S.E. Data were evaluated by unpaired, two-tailed Student’s t test or by analysis of variance with appropriate post-hoc tests. Significance levels are described in individual figure legends.
RESULTS
Production of iPLA2β-Null Mice and Characterization of iPLA2β mRNA Expression in Mouse Pancreatic Islets, Brain, and Other Tissues
We recently generated iPLA2β-null mice via homologous recombination and observed that they exhibit a phenotype of greatly reduced male fertility associated with impaired production and motility of spermatozoa (66). Expression of iPLA2β was found to be highest in wild-type mouse testes among tissues examined, which did not include pancreatic islets in our first report on iPLA2β-null mice, and no iPLA2β mRNA or protein was observed in testes or in other examined tissues from iPLA2β-null mice (66).
High levels of iPLA2β are expressed in brain (79), and brain shares many biochemical similarities with pancreatic islets that other tissues do not (80). Another group of investigators (36) informed us that they observed iPLA2β-immunoreactive protein in brains of iPLA2β-null mice obtained from our laboratory, and this caused us to re-examine iPLA2β expression in brain of iPLA2β-null mice to determine whether functional protein might be produced from an alternatively spliced mRNA species derived from the disrupted iPLA2β gene.
The knock-out construct for disrupting the iPLA2β gene resulted in insertion of a neomycin-resistance cassette into exon 9, which precedes the GTSTG serine lipase consensus motif encoded in exon 10. This was predicted to alter the reading frame and result in premature stop codons that would cause termination of translation at a point preceding the GTSTG motif. Because we observed no iPLA2β mRNA of any size by Northern blotting or by RT-PCR in iPLA2β-null mouse tissues with various primer sets that should have recognized even truncated mRNA species, we concluded that any mRNA produced from the disrupted iPLA2β gene was rapidly degraded by non-sense-mediated decay.
Nonetheless, several isoforms of iPLA2β arise from alternatively spliced mRNA species in various cells (42–44), and we considered the possibility that an exon-skipping mechanism of splicing that resulted in exclusion of exon 9 might yield a functional protein that contained the lipase consensus sequence and that such splicing might be tissue- or age-dependent. We therefore performed Northern blotting analyses for iPLA2β mRNA in several tissues including brains from mice aged one month to two years using a probe that recognizes sequence spanning exons 7 through 12 of the iPLA2β gene. Brain and other tissues, including pancreatic islets, from wild-type mice did contain mRNA species of the expected size that were recognized by the iPLA2β probe, but none of any size was observed in tissues from iPLA2β-null mice (Fig. 1A).
FIGURE 1. Expression of iPLA2β mRNA in tissues of wild-type and iPLA2β-null mice.
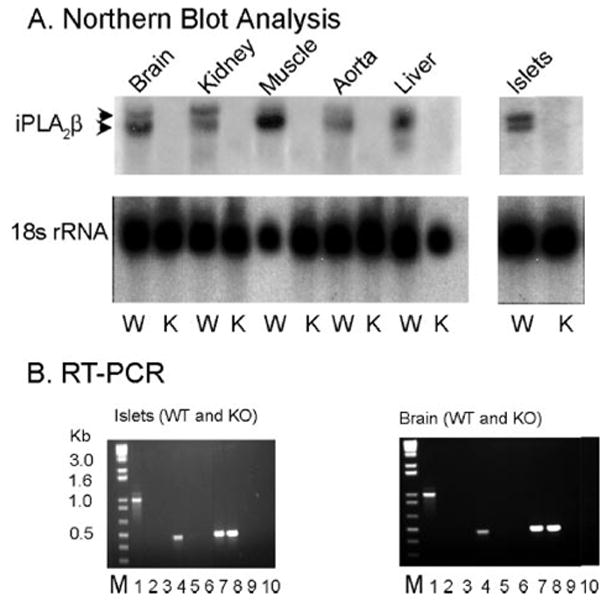
A represents Northern blot analyses of total RNA from wild-type (WT or W) and iPLA2β-null (KO or K) mouse brain, kidney, skeletal, muscle, aorta, liver, and isolated pancreatic islets. The upper panels were probed for iPLA2β mRNA, and the lower panels were probed for 18 S ribosomal RNA. B depicts analyses of products from RT-PCR amplification of iPLA2β mRNA fragments using total RNA isolated from pancreatic islets and brain as template and two different sets of primers. Lane M represents molecular weight standards. Lanes 1, 4, and 7 represent WT tissue, and lanes 2, 5, and 8 represent iPLA2β-knock-out (KO) tissues. Lanes 3, 6, 9, and 10 represent control reactions in which no template was added. In lanes 1–3, a primer set was used that amplifies an mRNA fragment that spans the site of insertion of the neomycin resistance cassette in the knock-out construct. In lanes 4 – 6, a primer set was used that amplifies a fragment downstream from the neomycin resistance cassette insertion site. In lanes 7–9, a primer set was used that amplifies a fragment of actin mRNA as a control.
RT-PCR experiments were also performed with two sets of primers, the first of which amplified sequence in iPLA2β mRNA beginning in exon 6 and extending into exon 14, which spans the insertion site for the neomycin resistance cassette. The second pair of primers amplifies sequence that starts in exon 11 at a point after the neomycin resistance cassette insertion site and ends in exon 14. Each primer set yielded a product of the expected size in wild-type brain, pancreatic islets (Fig. 1B), and other tissues, but no such products or any shorter ones were observed in tissues of iPLA2β-null mice. These Northern blotting and RT-PCR studies thus indicated that brain, pancreatic islets, and other tissues from iPLA2β-null mice contained no full-length or alternatively spliced iPLA2β mRNA species that lacked exon 9.
Expression of iPLA2β-Immunoreactive Protein in Mouse Brain
Because we could demonstrate no iPLA2β mRNA species in tissues of iPLA2β-null mice of any age, we suspected that iPLA2β immunoreactive protein observed in brain of iPLA2β-null mice was not produced from an alternatively spliced iPLA2β mRNA but rather reflected a non-iPLA2β protein that cross-reacted with iPLA2β antibodies. To examine this issue further, we performed immunoprecipitation studies using an iPLA2β antibody obtained from Cayman Chemical, analyzed the immunoprecipitates by SDS-PAGE, and then performed Western blotting with a second iPLA2β antibody obtained from Santa Cruz Biotechnology.
Fig. 2A illustrates such analyses of cytosol from stably transfected INS-1 insulinoma cells (59) that overexpress iPLA2β (lane 1); a parental INS-1 cell line that expresses lower levels of iPLA2β (lane 2); wild-type (lane 3) or iPLA2β-null (lane 4) mouse brain; and washes of immunoprecipitates from INS-1 cells (lane 5), wild-type mouse brain (lane 6), and iPLA2β-null mouse brain (lane 7). Cytosolic immunoprecipitate from the iPLA2β-overexpressing (lane 1) or parental (lane 2) INS-1 cell lines and the immunoprecipitate from wild-type mouse brain (lane 3) all contain an 84 kDa iPLA2β-immunoreactive protein that corresponds to iPLA2β, but the immunoprecipitate from iPLA2β-null mouse brain does not (lane 4). This material is also absent from washes of the immunoprecipitates from INS-1 cells or wild-type or iPLA2β-null mouse brain (lanes 5–7), indicating that the immunoprecipitating antibody effectively removed iPLA2β from solution. A number of other proteins are observed in lanes 2–7 that we believe represent cross-reacting proteins that do not arise from iPLA2β mRNA.
FIGURE 2. Immunoblotting analyses of proteins in tissues of wild-type and iPLA2β-null mice that are recognized by iPLA2β antibodies.
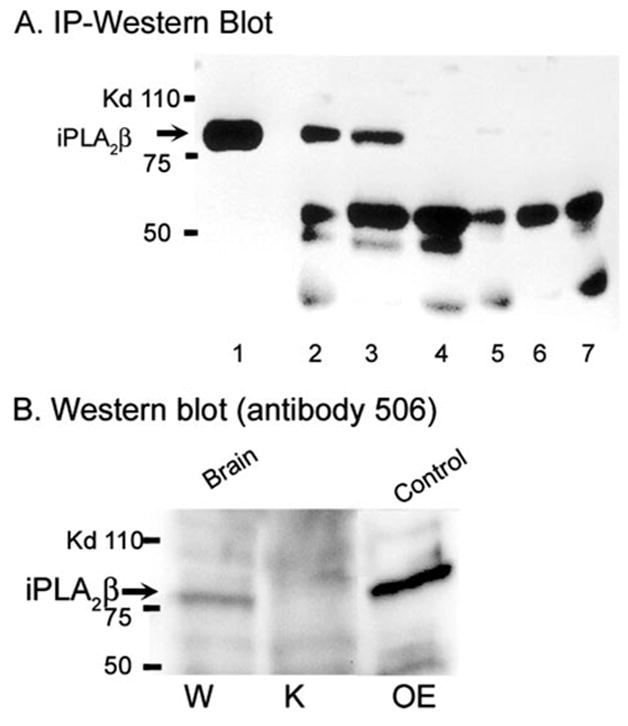
A represents immunoblots of immunoprecipitates of mouse tissues obtained with an iPLA2β antibody from Cayman Chemical. The immunoprecipitates were analyzed by SDS-PAGE, transferred to nylon membranes, and probed with iPLA2β antibody T14 from Santa Cruz Biotechnology. Lane 1 represents analysis of an immunoprecipitate from a stably transfected INS-1 insulinoma cell line that overexpresses iPLA2β by more than 10-fold. Lane 2 represents an immunoprecipitate from a parental INS-1 cell line that expresses lower levels of iPLA2β. Lanes 3 and 4 represent immunoprecipitates from brain of wild-type (W) and iPLA2β -knock-out (K) mice, respectively. Lanes 5, 6, and 7 represent analyses of washes of immunoprecipitates from INS-1 cells, wild-type brain, and iPLA2β-null brain, respectively, and indicate that the iPLA2β anti-body effectively removed the 84-kDa immunoreactive protein, although other cross-reacting proteins of various molecular masses remained. B represents immunoblots that were probed with iPLA2β antibody 506 prepared in the laboratory of Dr. Richard Gross. Lanes 1 and 2 represent analyses of brain cytosol from wild-type and iPLA2β-null mice, respectively. Lane 3 represents an analysis of cytosol from a stably transfected INS-1 cell line that overexpresses iPLA2β.
Similar experiments were performed with another iPLA2β antibody given to us by Dr. Richard Gross that was prepared in his laboratory (Fig. 2B). This antibody recognizes a protein with a molecular mass of 84 kDa in the cytosol of INS-1 cells that overexpress iPLA2β and from brain of wild-type mice, but no such protein is observed in cytosol from brain of iPLA2β-null mice (Fig. 2B).
We conclude that iPLA2β protein is expressed in brain of wild-type but not iPLA2β-null mice and that iPLA2β antibodies recognize a number of proteins other than iPLA2β. In support of this, we have identified one of the proteins that cross-reacts with iPLA2β antibodies in liver (see figure in supplemental data) by digesting the cross-reactive band with trypsin followed by LC/MS/MS analyses and data base searching, which identified mouse glycogen phosphorylase. Authentic glycogen phosphorylase was then purchased and analyzed by SDS-PAGE and Western blotting and shown to cross-react with iPLA2β antibody.
An iPLA2β Mutant Protein That Lacks Sequence Encoded by Exon 9 Is Catalytically Inactive
Although our findings indicated that no iPLA2β mRNA or protein is expressed in iPLA2β-null mouse tissues, we wondered whether a catalytically active protein could be produced from an alternatively spliced iPLA2β mRNA species that lacked sequence encoded by exon 9. We therefore prepared a cDNA construct encoding such a mutant protein (NE9-iPLA2β) with a polyhistidine tag (Fig. 3A), expressed it in a baculovirus-Sf9 cell system, purified it by immobilized metal affinity chromatography, and compared its catalytic activity to that of native iPLA2β with a polyhistidine tag.
FIGURE 3. Expression of a polyhistidine-tagged iPLA2β mutant protein that lacks sequence encoded by exon 9 of the iPLA2β gene and determination of its catalytic activity.
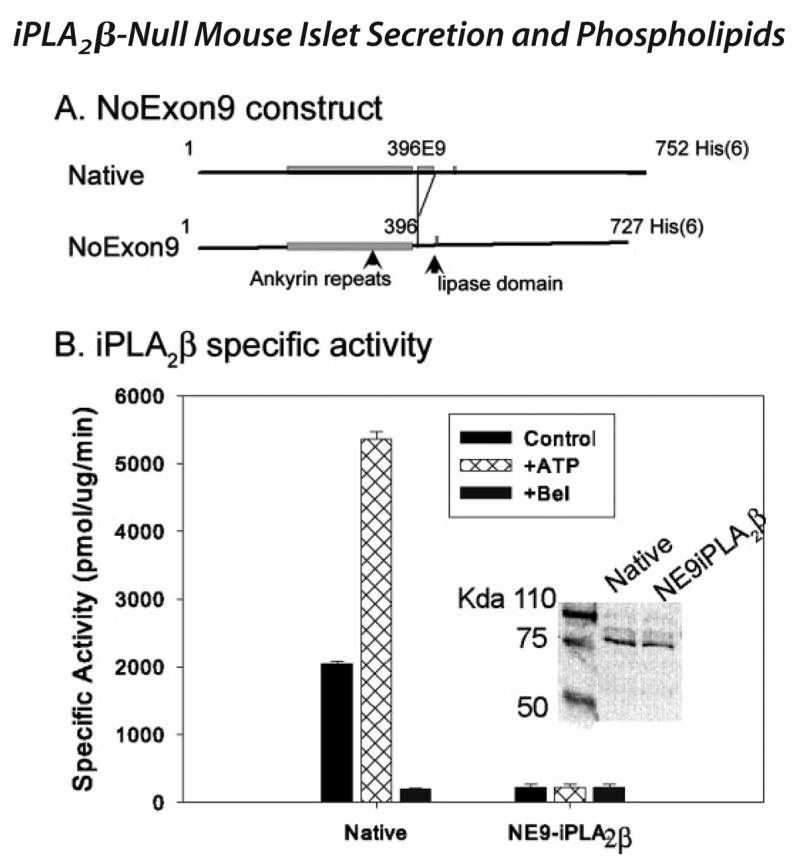
A illustrates the design of the construct used to prepare the polyhistidine-tagged iPLA2β mutant protein (NE9iPLA2β) that lacks sequence encoded by exon 9 of the iPLA2β gene. The positions of the regions of iPLA2β cDNA that encode the ankyrin repeat domain, exon 9, and the GTSTG lipase domain are indicated. The numbers indicate the amino acid residue number in the sequence of the 84-kDa isoform of rat iPLA2β. The protein was expressed in a baculovirus-Sf9 cell system, purified by immobilized metal affinity chromatography, and assayed for catalytic activity. B represents specific activity measurements for polyhistidine-tagged versions of native iPLA2β (leftmost set of bars) and of the NE9-iPLA2β mutant (rightmost set of bars). Control (black bars) assays were performed in the presence of EGTA and absence of added Ca2+, ATP, or BEL. Cross-hatched bars denote assays to which 1 mM ATP was added, and gray bars denote assays to which 10 μM BEL was added. Displayed values represent mean ± S.E. (n = 6).
Fig. 3B illustrates that native iPLA2β catalyzes hydrolysis of the phospholipid substrate 1-palmitoyl-2-[14C]linoleoyl-sn-glycero-3-phosphocholine in the absence of Ca2+ and that this activity is stimulated by ATP and inhibited by the iPLA2β suicide substrate BEL. In contrast, the mutant protein exhibits no ATP-stimulated or BEL-inhibited PLA2 activity. We conclude that any protein produced from an iPLA2β mRNA species that lacked sequence encoded by exon 9 would not be catalytically active.
Insulin Secretory Properties of Pancreatic Islets Isolated from Wild-type and iPLA2β-Null Mice
Our reports that the iPLA2β suicide substrate BEL inhibits glucose-induced insulin secretion from rat (47) and human (81) pancreatic islets and insulinoma cells (48, 49) suggest that iPLA2β might participate in insulin secretion. This is supported by findings that stably transfected INS-1 insulinoma cell lines that overexpress iPLA2β exhibit more robust insulin secretory responses to glucose and forskolin than do parental INS-1 cells (59) and that stably transfected INS-1 cells that produce siRNA that suppresses iPLA2β expression have impaired insulin secretory responses (60). This suggests that iPLA2β-null mouse pancreatic islets might exhibit impaired insulin secretory responses, although some mouse tissues express much less iPLA2β than do the corresponding rat or human tissues (82). Fig. 1, A and B provide the first evidence that mouse pancreatic islets, like those from rats and humans (13, 43), do express iPLA2β.
Fig. 4 illustrates that insulin secretion from pancreatic islets isolated from wild-type mice is stimulated by 20 mM D-glucose and that this response is amplified by the adenylyl cyclase activator forskolin (2.5 μM), as is also the case for rat and human islets. Insulin secretion from islets isolated from iPLA2β-null mice is also stimulated by 20 mM D-glucose and amplified by 2.5 μM forskolin, but these responses are significantly reduced with iPLA2β-null compared with wild-type islets. This suggests that the absence of iPLA2β in islets results in attenuation of insulin secretory responses to some stimuli.
FIGURE 4. Insulin secretion from pancreatic islets isolated from wild-type and iPLA2β-null mice.
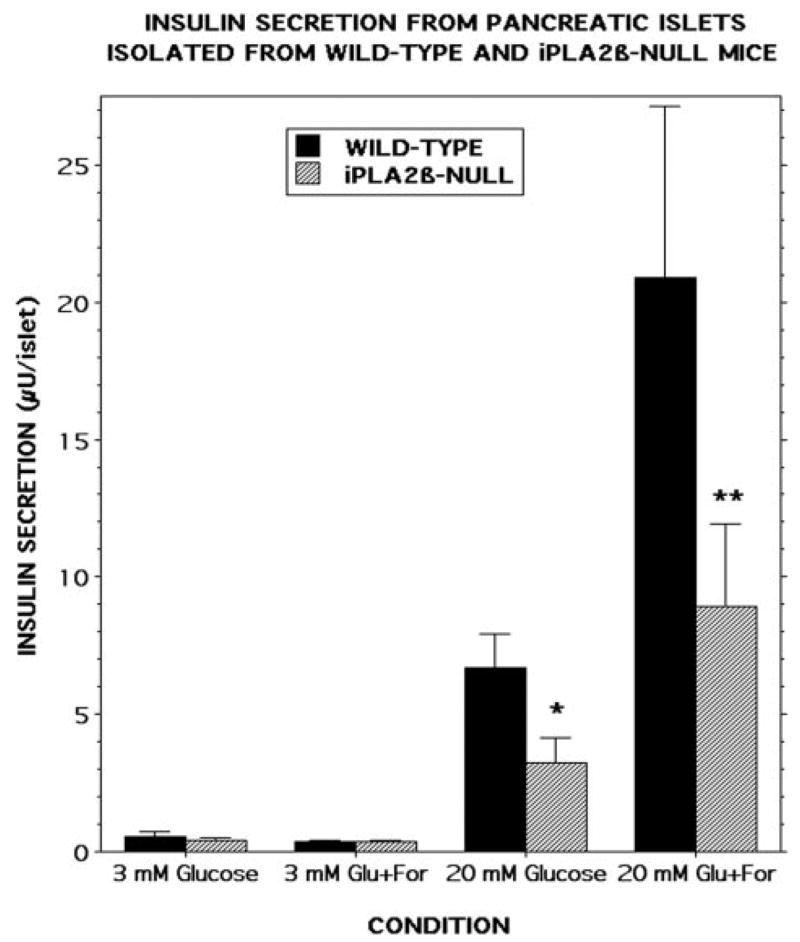
Pancreatic islets were isolated from wild-type (black bars) or iPLA2β-null (cross-hatched bars) mice and incubated (30 islets/condition, 30 min, 37 °C) at a glucose concentration of 3 mM or 20 mM in the absence or presence of 2.5 μM forskolin. Medium was then removed for measurement of insulin by radioimmunoassay. Values represent mean ± S.E. (n = 7). Single asterisk denotes p < 0.01, and double asterisk denotes p < 0.003 versus wild-type.
Nonetheless, neither the fasting nor fed blood glucose concentrations differed between wild-type and iPLA2β-null mice at any age examined, suggesting that in vivo compensation occurred for the insulin secretory impairment of isolated islets from iPLA2β-null mice. The insulin secretory impairment of iPLA2β-null islets at 2.5 μM forskolin could be overcome at 10 μM forskolin, suggesting that the secretory defect of iPLA2β-null mice can be overridden under some conditions, and this could explain the apparent normality of glucose homeostasis of iPLA2β-null mice under non-stressed conditions.
Phospholipid Composition of Pancreatic Islets and Brain of Wild-type and iPLA2β-Null Mice
It has been suggested that iPLA2β plays housekeeping functions in maintaining PC homeostasis by providing 2-lysophosphatidylcholine acceptors for arachidonic acid incorporation into PC and by cooperating with cytidylylphosphocholine transferase to maintain appropriate cellular PC levels (22, 23, 28, 29). This suggests that the PC composition of tissues from iPLA2β-null mice, particularly the abundance of arachidonate-containing PC species, might differ from that of wild-type mice.
We therefore examined the composition of glycerophosphocholine (GPC) lipids from brain and pancreatic islets of wild-type and iPLA2β-null mice as Li+ adducts by ESI/MS (Fig. 5) and by ESI/MS/MS (Fig. 6). Ions representing arachidonate (20: 4)-containing GPC lipids are prominent in the spectra in Fig. 5 and include those at m/z 788 (16:0/20:4-GPC) and m/z 816 (18:0/20:4-GPC). The identities of the parent [M + Li]+ ions were established from their tandem spectra (Fig. 6).
FIGURE 5. Electrospray ionization mass spectrometric analyses of glycerophosphocholine lipids from brain and pancreatic islets of wild-type and iPLA2β-null mice.
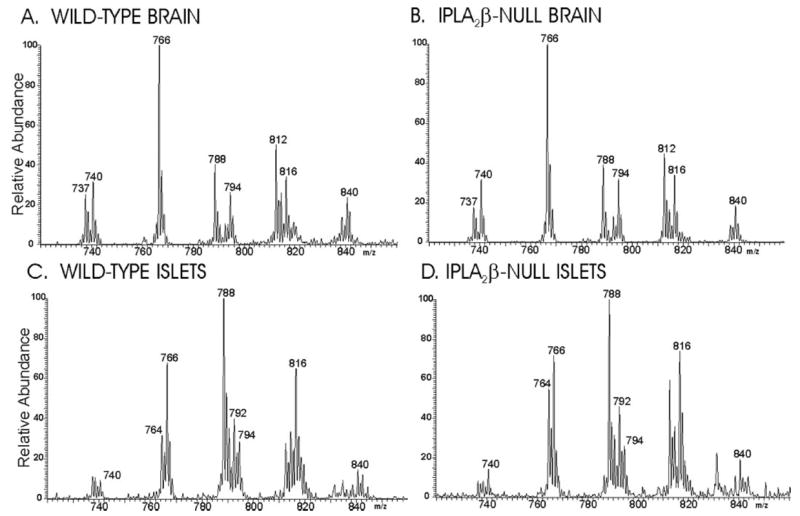
Phospholipids from wild-type (A and C ) or iPLA2β-null (B and D) mouse brain (A and C ), or pancreatic islets (B and D) were analyzed as Li+ adducts by positive ion ESI/MS, and the relative abundances of the ion currents were plotted as a function of m/z value.
FIGURE 6. Tandem mass spectra of glycerophosphocholine lipids from mouse brain and islets.
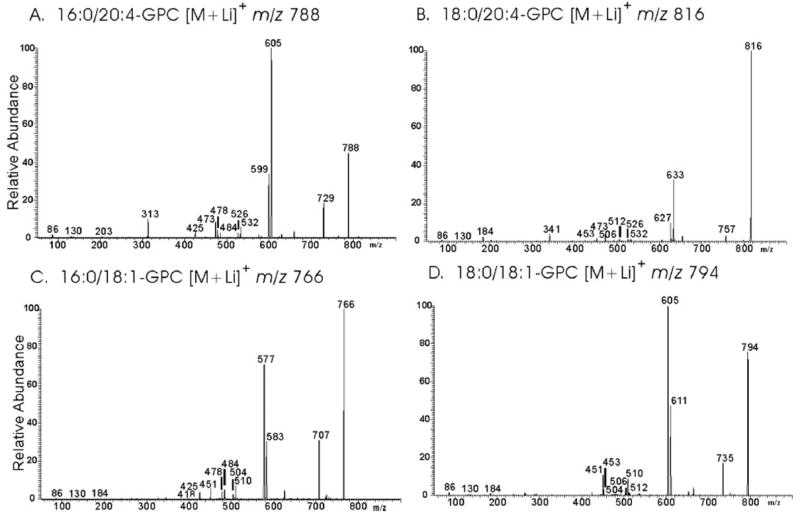
GPC lipid-Li+ adducts from wild-type or iPLA2β-null mouse brain extracts were analyzed by positive ion ESI/MS/MS. Specific ions were selected in the first mass-analyzing quadrupole and accelerated into the collision cell to induce CAD. Product ions were analyzed in the final quadrupole. Ions selected in the first quadrupole included m/z 788 (A), 816 (B), 766 (C ), and 794 (D).
CAD of m/z 788 yields a spectrum (Fig. 6A) that contains ions reflecting neutral losses of trimethylamine plus either the sn-1 substituent (MLi+-315) or the sn-2 substituent (MLi+-363) as free fatty acids at m/z 473 and m/z 425, respectively. The former is more abundant than the latter, indicating that palmitate and arachidonate are the sn-1 and sn-2 substituents, respectively (72–74). Analogous ions in Fig. 6B (m/z 473 and 453) from CAD of m/z 816 indicate that stearate and arachidonate are the sn-1 and sn-2 substituents, respectively, of the parent [M + Li]+ ion.
Fig. 6A also shows neutral losses of the sn-1 substituent as a free fatty acid (MLi+-256) or as a Li+ salt (MLi+-262) at m/z 532 and m/z 526, respectively. Ions of the same m/z values are observed in Fig. 6B and represent [MLi+-284] and [MLi+-290], respectively. Neutral losses of the sn-2 substituent as a free fatty acid (MLi+-304) or as a Li+salt (MLi+-310) are seen at m/z 484 and m/z 478, respectively, in Fig. 6A and at m/z 512 and m/z 506, respectively, in Fig. 6B. The ion m/z 313 (MLi+-475) in Fig. 6A reflects net elimination of [LiPO4(CH2)2N(CH3)3] and loss of the sn-2 substituent as a ketene (72–74). An analogous m/z 341 ion (MLi+-475) is seen in the tandem spectrum 18:0/20:4-GPC-Li+ (Fig. 6B). Other diagnostic ions in Fig. 6A include those for loss of trimethylamine (m/z 729) or net loss of phosphocholine or its Li+ salt from [M + Li]+ at m/z 605 and 599, respectively. Analogous ions in Fig. 6B are observed at m/z 757, m/z 633, and m/z 627, respectively.
Fig. 6C also contains ions m/z 484, 478, and 425 that identify oleate as a fatty acid substituent. Those ions reflect neutral losses of oleic acid (MLi-282), of Li+-oleate (MLi-288), and of trimethylamine plus oleic acid (MLi-341), respectively. Palmitate is the other fatty acid substituent, as shown by ions reflecting losses of palmitic acid (m/z 510), of Li+-palmitate (m/z 504), and of trimethylamine plus palmitic acid (m/z 451). The ion reflecting loss of trimethylamine plus the sn-1 substituent is more abundant than the ion reflecting loss of trimethylamine plus the sn-2 substituent in GPC lipid-Li+ CAD spectra (71–73), and the relative abundance of ions m/z 425 and 451 in Fig. 6C indicates that palmitate and oleate are the sn-1 and sn-2 substituents, respectively.
The most abundant ion in mass spectra of wild-type brain GPC lipids is m/z 766 (Fig. 5A) and represents 16:0/18:1-GPC [M + Li]+, as shown by its tandem spectrum (Fig. 6C). Loss of trimethylamine (MLi+-59) yields the ion m/z 707, and the ions m/z 583 (MLi+-183) and m/z 577 (MLi+-189) reflect net loss of [HPO4(CH2)2N(CH3)3] or [LiPO4(CH2)2N(CH3)3], respectively, and identify the head group (71–73). Fig. 6C also contains ions m/z 484, 478, and 425 that identify oleate as a fatty acid substituent. Those ions reflect neutral losses of oleic acid (MLi-282), of Li+-oleate (MLi-288), and of trimethylamine plus oleic acid (MLi-341), respectively. Palmitate is the other fatty acid substituent of the parent [M + Li]+ion in Fig. 6C, as shown by ions reflecting losses of palmitic acid (m/z 510), of Li+-palmitate (m/z 504), and of trimethylamine plus palmitic acid (m/z 451). The relative abundance of ions m/z 425 and 451 in Fig. 6C indicates that palmitate and oleate are the sn-1 and sn-2 substituents, respectively.
An analogous tandem spectrum in Fig. 6D identifies the ion at m/z 794 in Fig. 5A as 18:/0/18:1-GPC, as reflected by ions representing neutral losses of trimethylamine plus either the sn-1 substituent (MLi+-341) or the sn-2 substituent (MLi+-343) as free fatty acids at m/z 453 and m/z 451, respectively, and by losses of these substituents from [M + Li]+ as free fatty acids (m/z 512 and m/z 510) or as Li+ salts (m/z 506 and m/z 504), respectively. Identities of other GPC lipids represented by ions in Fig. 5A were similarly determined and include 16:0/16:0-GPC (m/z 740), 16:0/22:6-GPC (m/z 812), and 18:0/22:6-GPC (m/z 840). The tandem spectrum of the ion at m/z 737 indicates that it represents the sphingomyelin species with stearate as the fatty acid substituent.
Fig. 5B is the ESI/MS spectrum of GPC lipid Li+ adducts from iPLA2β-null brain tissue and is virtually identical to that for wild-type brain (Fig. 5A). Ions representing the 20:4-containing GPC species at m/z 788 and 816 are no less abundant in the latter spectrum than in the former, and quantitative measurements with internal standards and normalization to measured lipid phosphorus levels also indicate that 16:/20:4-GPC and 18:0/20:4-GPC are no less abundant in brain from iPLA2β-null mice than in brain from wild-type mice. This indicates that the absence of iPLA2β does not result in a deficiency of arachidonate-containing GPC lipids in mouse brain.
Fig. 5C is the ESI/MS spectrum of Li+ adducts of GPC lipids of pancreatic islets from wild-type mice. Most GPC lipid species observed in brain are also observed in islets, although the relative abundance differs between the two tissues, as has also been observed for rat brain and islets (84). Tandem spectra establish that the ion at m/z 764 represents a mixture of 16:0/18:2-GPC and 16:1/18:1-GPC, that the ion at m/z 792 represents a mixture of 18:0/18:2-GPC and 18:1/18:1-GPC, and that the ion at m/z 814 represents predominantly 18:1/20:4-GPC. The relative abundances of those components of the GPC mixture are greater in islets than brain.
In addition, the relative abundances of the arachidonate-containing species 16:0/20:4-GPC (m/z 788) and 18:0/20:4-GPC (m/z 816) is notably greater in mouse islets than in brain, as is also the case for rat islets and brain (84). Fig. 5D is the ESI/MS spectrum of Li+ adducts of GPC lipids from islets isolated from iPLA2β-null mice and is similar to that for wild-type islets, except for a slightly greater relative abundance of 16:0/22:6-GPC in the iPLA2β-null islets. The relative abundances of the ions representing 16:0/20:4-GPC (m/z 788) and 18:0/20:4-GPC (m/z 816) are similar in the two spectra and do not indicate a deficiency of 20:4-containing GPC lipids in iPLA2β-null islets.
Responses of Wild-type and iPLA2β-Null Mice to Multiple Low Dose Streptozotocin Administration
The phospholipid analyses described above do not indicate an impaired ability to achieve a normal content of arachidonate-containing GPC lipids in brain or islets of iPLA2β-null mice, but insulin secretory responses to some stimuli are reduced with islets from iPLA2β-null mice compared with those from wild-type mice. These findings support a signaling role for iPLA2β in insulin secretion rather than a housekeeping function in the maintenance of islet phosphatidylcholine composition, but the fasting and fed blood glucose concentrations of iPLA2β-null mice do not differ from those of wild-type mice.
This calls into question the importance of any signaling function of iPLA2β in insulin secretion. We wondered whether compensatory mechanisms might override any secretory defect in iPLA2β-null islets under normal conditions but imposing a metabolic stress might unmask a latent defect in glucose homeostasis. One such stress that was tested is the administration of multiple low doses of streptozotocin (77).
Streptozotocin is a β-cell toxin, and single large doses are used to induce insulinopenic diabetes in mice and rats by destroying essentially the entire β-cell mass (85). Another protocol involves administration of multiple, small doses of streptozotocin over a period of time (77). This results in a smoldering inflammatory process that reduces β-cell number but leaves a substantial functional β-cell mass. This state is somewhat analogous to type II diabetes, in which some β-cell function is preserved, but insulin secretion is sufficiently impaired to result in defects in glucose homeostasis. This model has been used to examine effects of gene disruption on sensitivity to diabetogenic stimuli (77).
Wild-type and iPLA2β-null mice were thus subjected to five daily intraperitoneal injections of streptozotocin (40 mg/kg) and then maintained on a standard laboratory chow diet, and their blood glucose concentrations were determined after an overnight fast at various intervals. Wild-type mice developed a modest increase in fasting blood glucose concentrations that rose progressively in the period 1–7 weeks after streptozotocin administration (Fig. 7A). The iPLA2β-null mice experienced a significantly greater increase in fasting blood glucose concentration than did wild-type mice at 1 week and experienced greater further increases in fasting blood glucose concentration than did wild-type mice at every tested interval up to 7 weeks after injection. This is compatible with an impaired insulin secretory reserve in iPLA2β-null islets that is latent under unstressed conditions but unmasked by reducing β-cell mass.
FIGURE 7. Fasting blood glucose and insulin concentrations as a function of time after administration of multiple low doses of streptozotocin to wild-type or iPLA2β-null mice.

Wild-type (closed squares) or iPLA2β-null (closed circles) mice 12–16 weeks of age were treated with five daily intraperitoneal injections of streptozotocin and then maintained on a normal chow diet. Fasting blood glucose (A) and insulin (B) concentrations were determined at various intervals after the first streptozotocin injection. Glucose values are expressed as percent of the time 0 value on the day before the first streptozotocin injection and represent mean ± S.E. (n = 14). Asterisk denotes p ≤ 0.05 versus wild-type. Insulin concentrations are expressed in ng/ml as mean ± S.E. (n = 14).
Fig. 7B illustrates that fasting blood insulin concentrations decline after streptozotocin administration, and the falling insulin levels indicate that streptozotocin did injure β-cells and impair insulin secretion. The insulin levels are quite variable, as is often the case with mouse blood insulin measurements (69), and do not differ significantly between wild-type and iPLA2β-null mice. Nonetheless, the significantly higher fasting glucose levels of the latter compared with the former reflect the expected impairment of insulin secretion after streptozotocin administration.
The fact that different insulin values are not observed between wild-type and iPLA2β-null mice is attributable partly to the variability in mouse blood insulin measurements, but, more importantly, to the fact that, if there were no impairment of insulin secretion in the iPLA2β-null mice, one would expect them to exhibit higher insulin values than wild-type mice because the former have higher blood glucose concentrations.
Similar observations have been made with genetically modified mice with altered islet lipoprotein lipase (LPL) expression (69). Although these mice developed hyperglycemia after challenge, there were no differences in blood insulin concentration between these animals and their normoglycemic wild-type littermates. The failure to detect higher insulin levels in the setting of hyperglycemia was taken to reflect defective insulin secretion, which was confirmed using isolated islets (69).
There are a number of other examples in which there is a discrepancy between blood insulin levels and genetic perturbations of β-cell function that disturb glucose homeostasis in mice, and some of them are summarized in “Discussion.” As discussed further below, no differences in insulin-tolerance testing responses were observed between wild-type and iPLA2β-null mice fed a regular diet.
Responses of Wild-type and iPLA2β-Null Mice Fed a High Fat Diet
Another metabolic stress that was tested is maintaining the mice on a high fat diet, which is also a known diabetogenic stimulus (86) that can result in both insulin resistance (87) and attenuation of insulin secretion (88). Wild-type and iPLA2β-null mice were thus placed on a high fat diet or regular chow at 8 weeks of age and maintained on the diet for 6 months. At the end of that period, glucose tolerance testing (GTT) and insulin tolerance testing (ITT) were performed.
Compared with mice maintained on regular chow, both wild-type (Fig. 8A) and iPLA2β-null mice (Fig. 8B) fed a high fat diet developed a significant increase in fasting blood glucose concentrations, as reflected by the time 0 points in the GTT curves in Fig. 8. In response to an intraperitoneal glucose load, wild-type mice maintained on the high fat diet were less glucose tolerant than those fed regular chow, as reflected by an increase in the area under the glucose concentration versus time curve (89) from 213 ± 4.3 area units (au) for mice fed standard chow to 318 ± 35 au for mice fed the high fat diet (p = 0.0035), which represented a difference in areas under the two GTT curves of 105 au (Fig. 9A).
FIGURE 8. Glucose tolerance testing of wild-type and iPLA2β-null mice that had been maintained on normal chow or a high fat diet.

Wild-type (A) or iPLA2β-null (B) mice were fed a normal mouse chow diet after weaning until they were 8 weeks of age. Thereafter, they were randomized to receive normal chow (closed squares) or a high fat diet (closed circles) for 6 months. Intraperitoneal glucose-tolerance testing was then performed after an overnight fast. Values represent mean ± S.E. (n = 18). A single asterisk denotes p < 0.05, and a double asterisk denotes p < 0.005 versus normal chow.
FIGURE 9. Areas under glucose concentration versus time curves and blood insulin/glucose ratios in glucose-tolerance testing of wild-type and iPLA2β-null mice that had been maintained on normal chow or a high fat diet.

Experiments were performed as described in the legends of Fig. 8 and Table 1, and the areas under the glucose concentration versus time curves (A) and the 30-min blood glucose/insulin concentration ratios (B) were determined (107) after intraperitoneal glucose administration for wild-type and iPLA2β-null mice that had been fed regular chow (lightly cross-hatched bars) or a high fat diet (dark bars). Differences between the values obtained with the regular and high fat diets were also computed (lightly shaded bars). Displayed values represent mean ± S.E. (n = 18), and asterisks denote p < 0.05 for the comparison between wild-type and iPLA2β-null mice.
The iPLA2β-null mice fed the high fat diet experienced a greater deterioration in glucose tolerance than did wild-type mice, as reflected by a significant increase in the area under the GTT curve from 200 ± 8.6 au for mice fed standard chow to 411 ± 27 au for mice fed the high fat diet (p = 0.0002). This represented a difference in areas under the two GTT curves of 211 au for iPLA2β-null mice, a value twice as great that for wild-type mice (Fig. 9A).
Although the fasting blood glucose concentrations at time 0 of the GTT did not differ between wild-type (112 ± 3.0 mg/dl) and iPLA2β-null (112 ± 3.5 mg/dl) mice that had been fed a high fat diet, the mean blood glucose values after intraperitoneal glucose administration were significantly (p = 0.022) higher for iPLA2β-null (244 ± 19 mg/dl) than for wild-type (189 ± 13 mg/dl) mice, as were the increases in glucose concentrations (132 ± 19 mg/dl for iPLA2β-null mice and 78 ± 13 mg/dl for wild-type mice, respectively, p = 0.022). This indicates that iPLA2β-null mice experience a greater deterioration in glucose tolerance when fed a high fat diet than do wild-type mice and provides another example of a metabolic stress that exposes a latent defect in glucose homeostasis in iPLA2β-null mice that is not apparent in the basal state.
For wild-type mice maintained on a regular diet, intraperitoneal glucose administration resulted in a rise in mean blood insulin concentration from 323 ± 48 pg/ml to 781 ± 129 pg/ml, and the corresponding values for iPLA2β-null mice were 296 ± 47 and 644 ± 6 pg/ml, respectively (Table 1). Wild-type mice fed a high fat diet experienced an increase in fasting blood insulin concentration (to 1080 ± 143 pg/ml) relative to those fed regular chow, and their insulin levels rose to a mean of 2600 ± 420 pg/ml after intraperitoneal glucose administration. The corresponding values for iPLA2β-null mice were 990 ± 129 and 2330 ± 227 pg/ml, respectively (Table 1).
TABLE 1. Blood insulin levels after intraperitoneal administration of glucose to wild-type and iPLA2β-null mice that had been maintained on normal chow or a high fat diet.
Wild-type or iPLA2β-knock-out mice were fed a normal mouse chow diet after weaning until they were 8 weeks of age. Thereafter, they were randomized to receive normal chow regular diet (R) or a high fat diet (HF) for 6 months. An intraperitoneal glucose load was then administered after an overnight fast, and insulin levels were measured in blood samples obtained just before and 30 min after glucose administration. Values represent mean ± S.E. (n = 18).
| Genotype | Diet | Time after glucose injection | Insulin |
|---|---|---|---|
| min | pg/ml | ||
| WT | R | 0 | 323 ± 048 |
| WT | R | 30 | 781 ± 129 |
| KO | R | 0 | 296 ± 047 |
| KO | R | 30 | 644 ± 006 |
| WT | HF | 0 | 1080 ± 143 |
| WT | HF | 30 | 2600 ± 220 |
| KO | HF | 0 | 990 ± 129 |
| KO | HF | 30 | 2330 ± 227 |
The fact that mice fed a high fat diet exhibit higher blood insulin concentrations than do those fed normal chow likely reflects an increase in insulin resistance, and the fact that blood glucose levels also rise reflects an insulin secretory defect, consistent with previously reported effects of a high fat diet on glucose homeostasis in mice (86–88).
Although there is a trend for iPLA2β-null mice in both dietary groups to exhibit lower blood insulin levels than wild-type mice before and after intraperitoneal glucose administration, this was not statistically significant because of the magnitude of the variability in measured insulin levels. Given that the iPLA2β-null mice developed higher blood glucose concentrations than wild-type mice after intraperitoneal glucose admin istration, they would be expected to have higher blood insulin concentrations if their insulin secretory defect were not more severe that that of wild-type mice, as discussed above and elsewhere (69).
Significant differences can be observed in blood insulin/glucose ratios in glucose tolerance testing in genetically modified mice with insulin secretory defects compared with wild-type mice even when absolute insulin concentrations do not differ significantly (90), and this also is the case for iPLA2β-null mice fed a high fat diet (Fig. 9B). These mice exhibit significantly lower blood insulin/glucose ratios than do wild-type mice fed a high fat diet at 30 min after intraperitoneal glucose administration (Fig. 9B). For wild-type mice, the high fat diet results in more than a doubling of the blood insulin/glucose ratio achieved in response to intraperitoneal glucose challenge compared with mice fed a regular diet, which presumably represents a compensatory response to increased insulin resistance induced by high fat feeding. An increase in the stimulated blood insulin/glucose ratio also occurs in iPLA2β-null mice fed a high fat diet, but the magnitude of the rise is significantly less than that for wild-type mice (Fig. 9B).
Interestingly, iPLA2β-null mice fed a high fat diet exhibited greater sensitivity to exogenous insulin than did wild-type mice fed a high fat diet, as reflected by significantly lower blood glucose concentrations in iPLA2β-null than in wild-type mice at 30 min (p = 0.011), 60 min (p = 0.017), and 120 min (p = 1.5 × 10−9) after insulin administration (Fig. 10). No differences in insulin tolerance testing responses were observed between wild-type and iPLA2β-null mice fed regular chow. This suggests that iPLA2β-null mice are less sensitive than wild-type mice to the high fat diet-induced increase in insulin resistance. The greater glucose intolerance of iPLA2β-null mice compared with wild-type mice fed a high fat diet is thus likely to reflect a more pronounced impairment in insulin secretory responses.
FIGURE 10. Insulin tolerance testing of wild-type and iPLA2β-null mice that had been maintained on normal chow or a high fat diet.
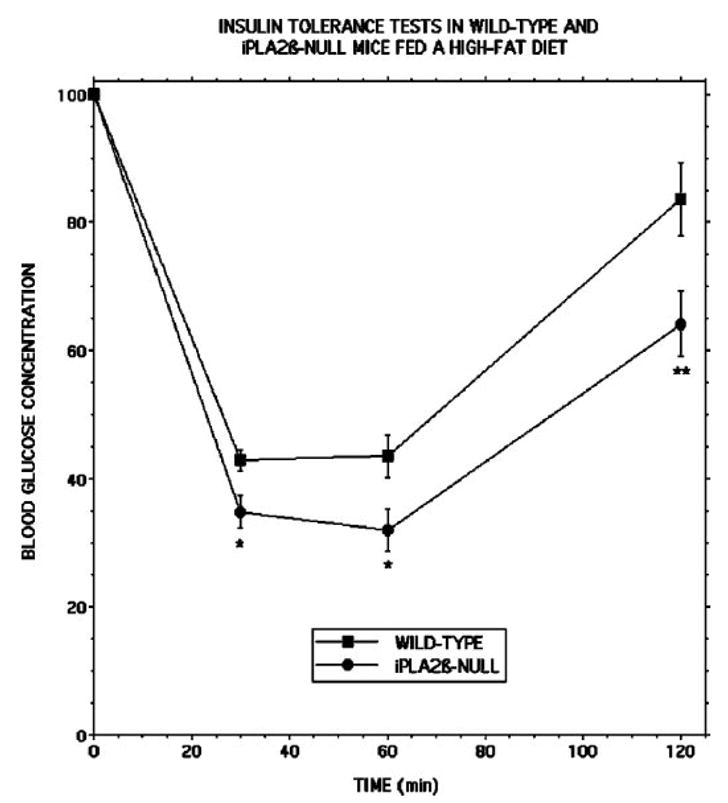
Wild-type (closed squares) or iPLA2β-null (closed circles) mice were fed a normal mouse chow diet after weaning until they were 8 weeks of age and were then fed a high fat diet for the next 6 months. Intraperitoneal insulin tolerance testing was then performed after an overnight fast. Values represent mean ± S.E. (n = 24). A single asterisk denotes p < 0.02, and a double asterisk denotes p < 0.002 versus wild-type.
To further examine this possibility, pancreatic islets were isolated from wild-type and iPLA2β-null mice that had been fed a high fat diet, and their insulin secretory responses were studied ex vivo (Fig. 11). Compared with islets isolated from wild-type mice fed a regular diet (Fig. 4), islets from wild-type mice fed a high fat diet exhibited exaggerated insulin secretory responses to 8 and 20 mM D-glucose in the absence or presence of 2.5 μM forskolin (Fig. 11), which is consistent with the higher blood insulin levels observed in the high fat fed animals that presumably reflect compensation for diet-induced insulin resistance (Table 1). The ex vivo insulin secretory responses of islets from iPLA2β-null mice fed a high fat diet were even more impaired compared with wild-type islets (Fig. 11) than were those from mice fed a regular diet (Fig. 4).
FIGURE 11. Insulin secretion from pancreatic islets isolated from wild-type and iPLA2β-null mice that had been fed a high fat diet.
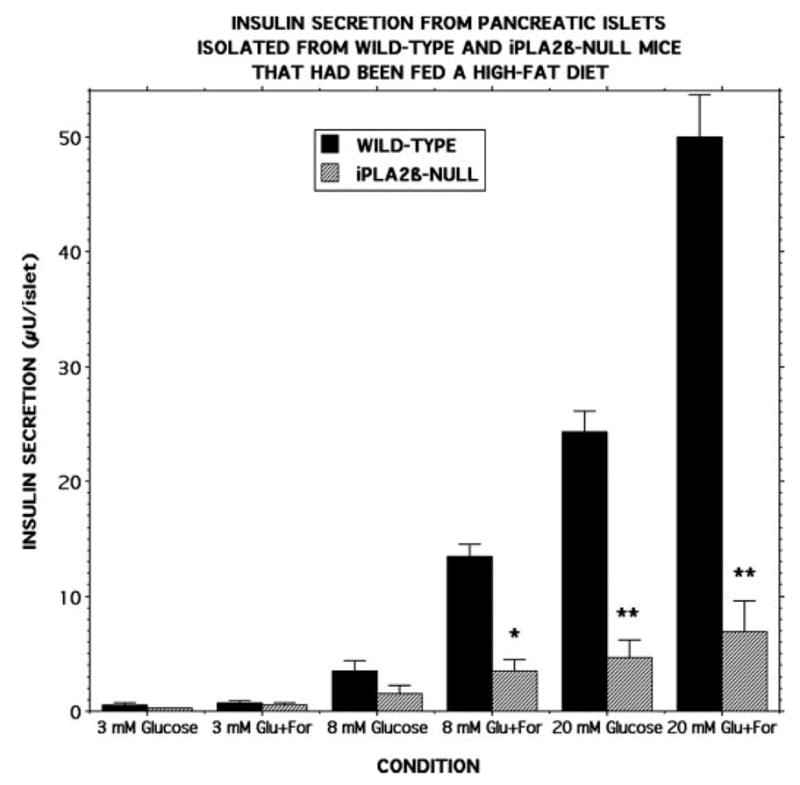
Pancreatic islets were isolated from wild-type (black bars) or iPLA2β-null (cross-hatched bars) mice that had been fed a high fat diet and incubated (30 islets/condition, 30 min, 37 °C) at a glucose concentration of 3, 8, or 20 mM in the absence or presence of 2.5 μM forskolin. Medium was then removed for measurement of insulin by radioimmunoassay. Values represent mean ± S.E. (n = 7). Single asterisk denotes p < 0.01, and double asterisk denotes p < 0.001 versus wild-type.
DISCUSSION
We have generated iPLA2β-null mice by homologous recombination that have previously been reported to exhibit the phenotypic abnormalities of reduced male fertility associated with impaired production and motility of spermatozoa (66) and defective signaling in macrophages (38). We confirm here that no iPLA2β mRNA is expressed in any tissue examined from iPLA2β-null animals of any age, including pancreatic islets and brain, although proteins that cross-react with anti-bodies to iPLA2β are frequently observed in brain and other tissues. In addition, an iPLA2β mutant protein that lacks sequence encoded by exon 9 of the iPLA2β gene has been expressed in a baculovirus-Sf9 cell system, purified by immobilized metal affinity chromatography, and demonstrated not to exhibit iPLA2β catalytic activity.
It has been proposed that iPLA2β plays the housekeeping roles of providing 2-lysophosphatidylcholine (LPC) acceptors for incorporating arachidonic acid into PC (22–25) and of maintaining membrane phospholipid homeostasis by degrading excess PC (28, 29), This could have important implications for pancreatic islet function because islets contain the highest fraction of arachidonate esterified in their phospholipids, including phosphatidylcholine, of any known tissue (81, 84), and the physical properties of such phospholipids are likely to have an important influence on fusion of secretory granule and plasma membranes (84, 92), which is the final event in insulin exocytosis.
Nonetheless, we observe a PC composition in pancreatic islets and brain of iPLA2β-null mice that is similar or identical to that of wild-type mice, as is also the case with the PC composition of testes from iPLA2β-null and wild-type mice (66). In particular, there is no deficiency in arachidonic acid-containing PC species in any of these tissues from iPLA2β-null mice. Our ESI/MS findings that the PC content and composition of pancreatic islets from iPLA2β-null and wild-type mice are virtually identical are consistent with the lack of effects of pharmacologic inhibition of iPLA2β activity (50, 76) or molecular biologic manipulation of iPLA2β expression (59, 60) on these parameters in insulinoma cells and islets from rats. Neither stable over-expression (59) nor suppression (60) of iPLA2β expression in INS-1 insulinoma cells results in a significant change in their PC content or composition or rates of arachidonic acid incorporation into PC.
Although we find no evidence that iPLA2β plays the proposed housekeeping roles in PC homeostasis (28, 29) and remodeling (23–25) in pancreatic islet β-cells, iPLA2β is widely expressed and might have multiple functions that vary among tissues and cell types, perhaps dependent in part on which splice variants (42–45) and proteolytic processing products (31, 50, 70) of iPLA2β are expressed in a given cell and on what interacting proteins (71) are present in the cell compartment (59, 93) in which the iPLA2β isoform resides.
Our findings do indicate that pancreatic islets isolated from iPLA2β-null mice exhibit impaired insulin secretory responses to stimulatory concentrations of D-glucose and forskolin compared with those from wild-type mice, and this is consistent with other evidence that iPLA2β participates in insulin secretion. Pharmacologic inhibition of iPLA2β with BEL impairs stimulated insulin secretion from rat (47) and human (81) pancreatic islets and from several clonal β-cell lines (48–50).
Because BEL affects several targets in addition to iPLA2β (17, 18, 20, 21, 57, 58, 94), it is important to use molecular biologic manipulation of iPLA2β activity to complement pharmacologic findings. Stable overexpression of iPLA2β in INS-1 insulinoma cells amplifies stimulated insulin secretion (59), and transient (41) or stable (60) suppression of iPLA2β expression impairs insulin secretion, consistent with the reduced secretory responses of iPLA2β-null islets reported here.
In glucose-induced insulin secretion, glucose enters β-cells via GLUT-II transporters and is phosphorylated by glucokinase, which results in generation of metabolic signals that include increased [ATP]/[ADP] (95). This inactivates plasma membrane (PM) ATP-sensitive K+ channels (KATP) (96), causing membrane depolarization, activation of PM voltage-operated Ca2+ channels (VOCC), Ca2+ influx, and a rise in [Ca2+] (97) that triggers insulin exocytosis through Ca2+-sensitive effectors, including Ca2+/calmodulin-dependent protein kinase IIβ (98).
Phospholipid hydrolysis catalyzed by iPLA2β and accumulation of the products nonesterified arachidonate and LPC in β-cell membranes (99–102) have been proposed to amplify the glucose-induced rise in β-cell [Ca2+] and insulin secretion by several mechanisms. These include facilitating Ca2+ entry (99, 102), perhaps by arachidonate effects on VOCC (103), effects of LPC and arachidonate on KATP (104), effects of an arachidonate 12-lipoxygenase product on a PM Na+/K+-ATPase (45), and effects of LPC (36) on PM store-operated cation channels (105).
Although abnormal insulin secretory responses are demonstrable with pancreatic islets isolated from iPLA2β-null mice, these mice do not exhibit fasting or fed hyperglycemia under ordinary circumstances and gain weight at the same rate as wild-type littermates. The failure to observe overt hyperglycemia despite defective insulin secretory responses of isolated islets is also observed in mice in which the gene encoding the Kir6.2 component of the KATP channel is disrupted by homologous recombination (61). Glucose intolerance is induced in the Kir6.2-null mice by imposing the metabolic stress of a high fat diet or the development of obesity, and this is associated with exacerbation of insulin secretory defects (62, 63).
Prolonged feeding of a high fat diet also induces glucose intolerance in iPLA2β-null mice that is more severe than that induced in wild-type mice, and this does not appear to be attributable to a more severe deterioration in insulin-sensitivity because iPLA2β-null mice are more responsive to exogenous insulin administration than are wild-type mice after 6 months of high dietary fat intake, as reflected by the magnitude of the hypoglycemic response. The observation that iPLA2β-null mice develop more severe hyperglycemia than do wild-type mice after administration of multiple low doses of streptozotocin also suggests that the islet secretory reserve is reduced in iPLA2β-null mice and that this results in an impaired ability to compensate for metabolic stresses.
Although there is a trend for blood insulin levels to be lower in iPLA2β-null mice than in wild-type mice in glucose-tolerance testing studies, the large variability in measured insulin levels prevents the difference from achieving statistical significance. We nonetheless believe that the more severe impairment of glucose tolerance of iPLA2β-null mice compared with wild-type mice fed a high fat diet is attributable to more severely impaired insulin secretion. Mouse blood insulin levels are not always a reliable indicator of β-cell function in vivo (e.g. Refs. 69, 83, 90, 91, 105), in part because of great analytical variability to which a mouse red cell insulinase is thought to contribute (69).
In addition, blood insulin levels must be interpreted in the context of blood glucose levels and cannot be compared directly in absolute concentrations without considering glucose levels. The blood insulin/glucose concentration ratio achieved 30 min after intraperitoneal glucose administration was significantly lower in iPLA2β-null than in wild-type mice that had been fed a high fat diet, and this ratio has also been observed to be lower in other genetically modified mice with an insulin secretory defect compared with wild-type mice even when the absolute blood insulin concentrations do not differ significantly between the groups (90).
Because iPLA2β-null mice fed a high fat diet develop higher blood glucose concentrations than do wild-type mice after intraperitoneal glucose administration, they would be expected to have higher blood insulin concentrations if their insulin secretory defect were not more severe that that of wild-type mice (69, 83, 90, 91, 105). The fact that the iPLA2β-null mice have blood insulin levels that are less than or equal to those of wild-type mice, despite the fact that the iPLA2β-null mice have higher glucose levels in GTT studies after high fat feeding, implies that their insulin secretory defect is more severe than that of wild-type mice (69, 83, 90, 91, 105).
There are many examples in which there are discrepancies between measured blood insulin levels and genetic perturbations of β-cell function that disturb glucose homeostasis in mice. As discussed above, β-cell-specific disruption of the lipoprotein lipase (LPL) gene results in mice that have impaired glucose tolerance, but their blood insulin levels do not differ significantly from those of wild-type mice during GTT studies (69). Nonetheless, islets isolated from β-cell-specific LPL knock-out mice exhibit impaired insulin secretory responses in vitro; the mice exhibit normal insulin sensitivity in insulin-tolerance tests; and their impaired glucose tolerance is attributable to impaired insulin secretion despite measured blood insulin levels that do not differ from those of wild-type mice (69).
Similarly, β-cell specific knock-out of the ARNT transcription factor gene results in male mice that have impaired glucose tolerance on GTT studies, but their blood insulin levels are indistinguishable from those of wild-type mice during such studies (91). Nonetheless, islets isolated from the male β-cell-specific ARNT-null mice have defective insulin secretory responses, and their impaired glucose tolerance is attributable to impaired insulin secretion, even though their measured blood insulin levels are not different from those of wild-type mice (91).
Moreover, β-cell-specific knock-out of the Hnf1β transcription factor gene results in mice that have impaired glucose tolerance on GTT studies (90). Their absolute blood insulin levels are nonetheless indistinguishable from those of wild-type mice during such studies, although islets isolated from the β-cell-specific Hnf1β-null mice have defective insulin secretory responses, and their glucose intolerance is also attributable to impaired insulin secretion (90). This is also reflected by the lower blood insulin/glucose concentration ratios during the GTT for the genetically modified compared with wild-type mice (90).
In addition, 8-month-old female transgenic mice that over-express the Sir2 protein deacetylase specifically in β-cells have significantly better glucose tolerance on GTT studies than do wild-type mice, but their glucose-induced rise in blood insulin levels is not statistically different from that of wild-type mice during such studies (83). Nonetheless, glucose-stimulated insulin release from their perfused pancreata is significantly greater than that for wild-type mice, and their improved glucose tolerance is attributable to augmented insulin secretion despite the fact that measured blood insulin levels are not elevated (83).
Our findings are thus quite similar to those of each of the four studies of glucose homeostasis in genetically modified mice just described (69, 83, 90, 91, 105). Disruption of the iPLA2β gene results in mice that exhibit impaired glucose tolerance after exposure to stressors that include administration of multiple low doses of streptozotocin or a high fat diet. There is a trend for the iPLA2β-null mice to exhibit lower blood insulin levels than do wild-type mice after intraperitoneal glucose administration, but this does not achieve statistical significance because of the large variability in measured blood insulin levels. Nonetheless, islets isolated from the iPLA2β-null mice exhibit impaired insulin secretory responses ex vivo; this insulin secretory impairment compared with wild-type islets is exaggerated with islets from iPLA2β-null mice fed a high fat diet; the results of insulin tolerance testing indicate that iPLA2β-null mice do not have reduced insulin sensitivity; and their reduced glucose tolerance thus appears to result from inadequate insulin secretion.
These findings thus contribute to a large body of evidence that iPLA2β plays a role in stimulated insulin secretion and indicate that homozygous iPLA2β gene disruption produces phenotypic abnormalities in several tissues and cells that include pancreatic islets in addition to testes (66) and macrophages (38).
Acknowledgments
We thank Sheng Zhang and Min Tan for excellent technical assistance and Dr. Richard Gross for the gift of iPLA2β antibody 506.
Footnotes
This work was supported by United States Public Health Service Grants R37-DK34388, P01-HL57278, P41-RR00954, P60-DK20579, R01-69455, and P30-DK56341.
The on-line version of this article (available at http://www.jbc.org) contains supplemental figures.
The abbreviations used are: PLA2, phospholipase A2; BEL, bromoenol lactone suicide substrate; CAD, collisionally activated dissociation; ESI, electrospray ionization; GPC, glycerophosphocholine; HBSS, Hank’s balanced salt solution; iPLA2β, Group VIA phospholipase A2; LPC, lysophosphatidylcholine; MS, mass spectrometry; MS/MS, tandem mass spectrometry; PAF, platelet-activating factor; PAPH, phosphatidate phosphohydrolase; PC, phosphatidylcholine; PM, plasma membrane; RT, reverse transcriptase; siRNA, small interfering RNA; SOC, store operated channel; TLC, thin layer chromatography; VOCC, voltage-operated Ca2+ channel; WT, wild type; KO, knock-out; au, area units; CT, CTP:phosphocholine cytidylyltransferase.
References
- 1.Six DA, Dennis EA. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 2.Ma Z, Turk J. Prog Nucleic Acids Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 3.Brash AR. J Clin Investig. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radu CG, Yang LV, Riedinger M, Au M, Witte ON. Proc Natl Acad Sci U S A. 2004;101:245–250. doi: 10.1073/pnas.2536801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy RC, Sala A. Methods Enzymol. 1990;187:90–98. doi: 10.1016/0076-6879(90)87013-s. [DOI] [PubMed] [Google Scholar]
- 6.Gijon MA, Spencer DM, Kaiser AL, Leslie CC. J Cell Biol. 1999;145:1219–1232. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. J Biol Chem. 1998;273:21926–21932. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 8.Pickard RT, Strifler BA, Kramer RM, Sharp JD. J Biol Chem. 1999;274:8823–8831. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]
- 9.Song C, Chang XJ, Bean KM, Proia MS, Knopf JL, Kriz RW. J Biol Chem. 1999;274:17063–17067. doi: 10.1074/jbc.274.24.17063. [DOI] [PubMed] [Google Scholar]
- 10.Ohto T, Uozumi N, Hirabayashi T, Shimizu T. J Biol Chem. 2005;280:24576–24583. doi: 10.1074/jbc.M413711200. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 12.Balboa MA, Balsinde J, Jones SS, Dennis EA. J Biol Chem. 1997;272:8576–8580. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 13.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. J Biol Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 14.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 15.Balsinde J, Dennis EA. J Biol Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 16.Ma Z, Ramanadham S, Hu Z, Turk J. Biochim Biophys Acta. 1998;1391:384–400. doi: 10.1016/s0005-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 17.Balsinde J, Dennis EA. J Biol Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 18.Mancuso DJ, Jenkins CM, Gross RW. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Takeya R, Sumimoto H. Biochem Biophys Res Commun. 2000;272:320–326. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 20.van Tienhoven M, Atkins J, Li Y, Glynn P. J Biol Chem. 2002;277:20942–20948. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 21.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 22.Balsinde J. Biochem J. 2002;364:695–702. doi: 10.1042/BJ20020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balsinde J, Balboa MA. Cell Signal. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Proc Natl Acad Sci U S A. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balsinde J, Balboa MA, Dennis EA. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 26.Lands WEM, Crawford CG. The Enzymes of Biological Membranes. Plenum Press; New York: 1997. [Google Scholar]
- 27.Chilton FH, Fonteh AN, Surette ME, Triggiani M, Winkler JD. Biochim Biophys Acta. 1996;1299:1–15. doi: 10.1016/0005-2760(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 28.Baburina I, Jackowski S. J Biol Chem. 1999;274:9400–9408. doi: 10.1074/jbc.274.14.9400. [DOI] [PubMed] [Google Scholar]
- 29.Barbour SE, Kapur A, Deal CL. Biochim Biophys Acta. 1999;1439:77–88. doi: 10.1016/s1388-1981(99)00078-5. [DOI] [PubMed] [Google Scholar]
- 30.Akiba S, Mizunaga S, Kume K, Hayama M, Sato T. J Biol Chem. 1999;274:19906–19912. doi: 10.1074/jbc.274.28.19906. [DOI] [PubMed] [Google Scholar]
- 31.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 32.Jenkins CM, Han X, Mancuso DJ, Gross RW. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 33.Seegers HC, Gross RW, Boyle WA. J Pharmacol Exp Ther. 2002;302:918–923. doi: 10.1124/jpet.302.3.918. [DOI] [PubMed] [Google Scholar]
- 34.Perez R, Melero R, Balboa MA, Balsinde J. J Biol Chem. 2004;279:40385–40391. doi: 10.1074/jbc.M402562200. [DOI] [PubMed] [Google Scholar]
- 35.Yellaturu CR, Rao GN. J Biol Chem. 2003;278:43831–43837. doi: 10.1074/jbc.M301472200. [DOI] [PubMed] [Google Scholar]
- 36.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 37.Martinson BD, Albert CJ, Corbett JA, Wysolmerski RB, Ford DA. J Lipid Res. 2003;44:1686–1691. doi: 10.1194/jlr.M300018-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Moran JM, Buller RM, McHowat J, Turk J, Wohltmann M, Gross RW, Corbett JA. J Biol Chem. 2005;280:28162–28168. doi: 10.1074/jbc.M500013200. [DOI] [PubMed] [Google Scholar]
- 39.Guo Z, Su W, Ma Z, Smith GM, Gong MC. J Biol Chem. 2003;278:1856–1863. doi: 10.1074/jbc.M211075200. [DOI] [PubMed] [Google Scholar]
- 40.Balboa MA, Saez Y, Balsinde J. J Immunol. 2003;170:5276–5280. doi: 10.4049/jimmunol.170.10.5276. [DOI] [PubMed] [Google Scholar]
- 41.Song K, Zhang X, Zhao C, Ang NT, Ma ZA. Mol Endocrinol. 2005;19:504–515. doi: 10.1210/me.2004-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsson Forsell PK, Kennedy BP, Claesson HE. Eur J Biochem. 1999;262:575–585. doi: 10.1046/j.1432-1327.1999.00418.x. [DOI] [PubMed] [Google Scholar]
- 43.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 1999;274:9607–9616. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Larsson PK, Claesson HE, Kennedy BP. J Biol Chem. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 45.Owada S, Larsson O, Arkhammar P, Katz AI, Chibalin AV, Berggren PO, Bertorello AM. J Biol Chem. 1999;274:2000–2008. doi: 10.1074/jbc.274.4.2000. [DOI] [PubMed] [Google Scholar]
- 46.Simonsson E, Ahren B. Int J Pancreatol. 2000;27:1–11. doi: 10.1385/IJGC:27:1:01. [DOI] [PubMed] [Google Scholar]
- 47.Ramanadham S, Gross RW, Han X, Turk J. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 48.Ramanadham S, Wolf MJ, Jett PA, Gross RW, Turk J. Biochemistry. 1994;33:7442–7452. doi: 10.1021/bi00189a052. [DOI] [PubMed] [Google Scholar]
- 49.Ramanadham S, Wolf MJ, Li B, Bohrer A, Turk J. Biochim Biophys Acta. 1997;1344:153–164. doi: 10.1016/s0005-2760(96)00139-7. [DOI] [PubMed] [Google Scholar]
- 50.Ramanadham S, Song H, Hsu FF, Zhang S, Crankshaw M, Grant GA, Newgard CB, Bao S, Ma Z, Turk J. Biochemistry. 2003;42:13929–13940. doi: 10.1021/bi034843p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramanadham S, Ma Z, Arita H, Zhang S, Turk J. Biochim Biophys Acta. 1998;1390:301–312. doi: 10.1016/s0005-2760(97)00189-6. [DOI] [PubMed] [Google Scholar]
- 52.Su X, Mancuso DJ, Bickel PE, Jenkins CM, Gross RW. J Biol Chem. 2004;279:21740–21748. doi: 10.1074/jbc.M314166200. [DOI] [PubMed] [Google Scholar]
- 53.Shirai Y, Balsinde J, Dennis EA. Biochim Biophys Acta. 2005;1735:119–129. doi: 10.1016/j.bbalip.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 54.Akiba S, Sato T. Biol Pharm Bull. 2004;27:1174–1178. doi: 10.1248/bpb.27.1174. [DOI] [PubMed] [Google Scholar]
- 55.Larsson Forsell PK, Runarsson G, Ibrahim M, Bjorkholm M, Claesson HE. FEBS Lett. 1998;434:295–299. doi: 10.1016/s0014-5793(98)00999-5. [DOI] [PubMed] [Google Scholar]
- 56.Murakami M, Kambe T, Shimbara S, Kudo I. J Biol Chem. 1999;274:3103–3115. doi: 10.1074/jbc.274.5.3103. [DOI] [PubMed] [Google Scholar]
- 57.Daniels S, Cooney E, Sofia M, Chakravarty P, Katzenellenbogen J. J Biol Chem. 1983;258:15046–15053. [PubMed] [Google Scholar]
- 58.Fuentes L, Perez R, Nieto ML, Balsinde J, Balboa MA. J Biol Chem. 2003;278:44683–44690. doi: 10.1074/jbc.M307209200. [DOI] [PubMed] [Google Scholar]
- 59.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 60.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. J Biol Chem. 2006;281:187–198. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Miki T, Nagashima K, Tashiro F, Kotake K, Yoshitomi H, Tamamoto A, Gonoi T, Iwanaga T, Myazaki J, Seino S. Proc Natl Acad Sci. 1998;95:10402–10496. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seino S, Iwanaga T, Nagashima K, Miki T. Diabetes. 2000;49:311–318. doi: 10.2337/diabetes.49.3.311. [DOI] [PubMed] [Google Scholar]
- 63.Remedi MS, Koster JC, Markova K, Seino S, Miki T, Patton BL, McDaniel ML, Nichols CG. Diabetes. 2000;53:3159–3167. doi: 10.2337/diabetes.53.12.3159. [DOI] [PubMed] [Google Scholar]
- 64.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622–625. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 65.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature. 1997;390:618–622. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 66.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hallberg A. Biochim Biophys Acta. 1984;796:328–335. doi: 10.1016/0005-2760(84)90134-6. [DOI] [PubMed] [Google Scholar]
- 68.McDaniel ML, Colca JR, Kotagal N, Lacy PE. Methods Enzymol. 1983;98:182–200. doi: 10.1016/0076-6879(83)98149-1. [DOI] [PubMed] [Google Scholar]
- 69.Pappan KL, Pan Z, Kwon G, Marshall CA, Coleman T, Goldberg IJ, McDaniel ML, Semenkovich CF. J Biol Chem. 2005;280:9023–9029. doi: 10.1074/jbc.M409706200. [DOI] [PubMed] [Google Scholar]
- 70.Song H, Hecimovic S, Goate A, Hsu FF, Bao S, Vidavsky I, Ramanadham S, Turk J. J Am Soc Mass Spectrom. 2004;12:1780–1793. doi: 10.1016/j.jasms.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Z, Ramanadham S, Ma ZA, Bao S, Mancuso DJ, Gross RW, Turk J. J Biol Chem. 2005;280:6840–6849. doi: 10.1074/jbc.M405287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hsu FF, Bohrer A, Turk J. J Am Soc Mass Spectrom. 1998;9:516–526. doi: 10.1016/S1044-0305(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 73.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2001;12:1036–1043. doi: 10.1016/S1044-0305(00)00194-X. [DOI] [PubMed] [Google Scholar]
- 74.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2003;14:352–363. doi: 10.1016/S1044-0305(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 75.Nowatzke W, Ramanadham S, Ma Z, Hsu FF, Bohrer A, Turk J. Endocrinology. 1998;139:4073–4085. doi: 10.1210/endo.139.10.6225. [DOI] [PubMed] [Google Scholar]
- 76.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 77.Bleich D, Chen S, Zipser B, Sun D, Funk CD, Nadler JL. J Clin Invest. 1999;103:1431–1436. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheverud JM, Ehrich TH, Hrbek T, Kenney JP, Pletscher LS, Semenkovich CF. Diabetes. 2004;53:3328–3336. doi: 10.2337/diabetes.53.12.3328. [DOI] [PubMed] [Google Scholar]
- 79.Yang HC, Mosior M, Ni B, Dennis EA. J Neurochem. 1999;73:1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- 80.Hsu FF, Bohrer A, Turk J. Biochim Biophys Acta. 1998;1392:202–216. doi: 10.1016/s0005-2760(98)00034-4. [DOI] [PubMed] [Google Scholar]
- 81.Ramanadham S, Bohrer A, Gross RW, Turk J. Biochemistry. 1993;32:13499–13509. doi: 10.1021/bi00212a015. [DOI] [PubMed] [Google Scholar]
- 82.Mancuso DJ, Abendschein DR, Jenkins CM, Han X, Saffitz JE, Schuessler RB, Gross RW. J Biol Chem. 2003;278:22231–22236. doi: 10.1074/jbc.C300033200. [DOI] [PubMed] [Google Scholar]
- 83.Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 84.Ramanadham S, Hsu FF, Bohrer A, Nowatzke W, Ma Z, Turk J. Biochemistry. 1998;37:4533–4567. doi: 10.1021/bi9722507. [DOI] [PubMed] [Google Scholar]
- 85.Eizrik DL, Strandell E, Bendtzen K, Sandler S. Diabetes. 1998;37:916–919. doi: 10.2337/diab.37.7.916. [DOI] [PubMed] [Google Scholar]
- 86.Surwit RS, Kuhn M, Cochrane C, McCubbin JA, Feinglos MN. Diabetes. 1988;37:1163–1167. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 87.Collins S, Martin TL, Surwit RS, Robidoux J. Physiol Behav. 2004;81:243–248. doi: 10.1016/j.physbeh.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 88.Wencel HE, Smothers C, Opara EC, Kuhn CM, Feinglos MN, Surwit RS. Physiol Behav. 1995;57:1215–1220. doi: 10.1016/0031-9384(95)00022-b. [DOI] [PubMed] [Google Scholar]
- 89.Kubota N, Terauchi Y, Kubota T, Kumagai H, Itoh S, Satoh H, Yano W, Ogata H, Tokuyama I, Mineyama T, Ishikawa M, Moroi M, Sugi K, Yamauchi T, Ueki K, Tobe K, Noda T, Nagai R, Kadowaki T. J Biol Chem. 2006;281:8748–8755. doi: 10.1074/jbc.M505649200. [DOI] [PubMed] [Google Scholar]
- 90.Wang L, Coffinier C, Thomas MK, Gresh L, Eddu G, Manor T, Levitsky LL, Yaniv M, Rhoads DB. Endocrinology. 2004;145:3941–3949. doi: 10.1210/en.2004-0281. [DOI] [PubMed] [Google Scholar]
- 91.Gunton JE, Kulkarni RN, Yim S, Okada T, Hawthorne WJ, Tseng YH, Roberson RS, Ricordi C, O’Connell PJ, Gonzalez FJ, Kahn CR. Cell. 2005;122:337–349. doi: 10.1016/j.cell.2005.05.027. [DOI] [PubMed] [Google Scholar]
- 92.Han X, Ramanadham S, Turk J, Gross RW. Biochim Biophys Acta. 1998;1414:95–107. doi: 10.1016/s0005-2736(98)00154-0. [DOI] [PubMed] [Google Scholar]
- 93.Bao S, Jin C, Zhang S, Turk J, Ma Z, Ramanadham S. Diabetes. 2004;53 (Suppl 1):S186–S189. doi: 10.2337/diabetes.53.2007.s186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Song H, Ramanadham S, Bao S, Hsu FF, Turk J. Biochemistry. 2006;45:1061–1073. doi: 10.1021/bi052065q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meglasson MD, Matschinsky FM. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 96.Cook DL, Satin LS, Ashford ML, Hales CN. Diabetes. 1988;37:495–498. doi: 10.2337/diab.37.5.495. [DOI] [PubMed] [Google Scholar]
- 97.Arkhammar P, Nilsson T, Rorsman P, Berggren PO. J Biol Chem. 1987;262:5448–5454. [PubMed] [Google Scholar]
- 98.Easom RA. Diabetes. 1999;48:675–684. doi: 10.2337/diabetes.48.4.675. [DOI] [PubMed] [Google Scholar]
- 99.Wolf BA, Pasquale SM, Turk J. Biochemistry. 1991;30:6372–6379. doi: 10.1021/bi00240a004. [DOI] [PubMed] [Google Scholar]
- 100.Wolf BA, Colca JR, Comens PG, Turk J, McDaniel ML. J Biol Chem. 1986;261:16284–16287. [PubMed] [Google Scholar]
- 101.Turk J, Mueller M, Bohrer A, Ramanadham S. Biochim Biophys Acta. 1992;1125:280–291. doi: 10.1016/0005-2760(92)90057-3. [DOI] [PubMed] [Google Scholar]
- 102.Ramanadham S, Gross R, Turk J. Biochem Biophys Res Commun. 1992;184:647–653. doi: 10.1016/0006-291x(92)90638-2. [DOI] [PubMed] [Google Scholar]
- 103.Vacher P, McKenzie J, Dufy B. Am J Physiol. 1989;257:E203–E211. doi: 10.1152/ajpendo.1989.257.2.E203. [DOI] [PubMed] [Google Scholar]
- 104.Eddlestone GT. Am J Physiol. 1995;268:C181–C190. doi: 10.1152/ajpcell.1995.268.1.C181. [DOI] [PubMed] [Google Scholar]
- 105.Roe MW, Worley JF, 3rd, Qian F, Tamarina N, Mittal AA, Dralyuk F, Blair NT, Mertz RJ, Philipson LH, Dukes ID. J Biol Chem. 1998;273:10402–10410. doi: 10.1074/jbc.273.17.10402. [DOI] [PubMed] [Google Scholar]