

Abstract
Previous studies suggest that group VIA phospholipase A2 (iPLA2β) participates in endoplasmic reticulum (ER) stress-induced apoptosis, and mouse macrophages undergo ER stress and apoptosis upon free cholesterol loading (FCL). We recently generated iPLA2β-null mice, and here we demonstrate that iPLA2β-null macrophages have reduced sensitivity to FCL-induced apoptosis, although they and wild-type (WT) cells exhibit similar increases in the transcriptional regulator CHOP. iPLA2β-null macrophages are also less sensitive to apoptosis induced by the sarcoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin and the scavenger receptor A ligand fucoidan, and restoring iPLA2β expression with recombinant adenovirus increases apoptosis toward WT levels. WT and iPLA2β-null macrophages incorporate [3H]arachidonic acid ([3H]AA]) into glycerophosphocholine lipids equally rapidly and exhibit identical zymosan-induced, cPLA2α-catalyzed [3H]AA release. In contrast, although WT macrophages exhibit robust [3H]AA release upon FCL, this is attenuated in iPLA2β-null macrophages and increases toward WT levels upon restoring iPLA2β expression. Recent reports indicate that iPLA2β modulates mitochondrial cytochrome c release, and we find that thapsigargin and fucoidan induce mitochondrial phospholipid loss and cytochrome c release into WT macrophage cytosol and that these events are blunted in iPLA2β-null cells. Immunoblotting studies of mitochondria isolated from macrophages indicate that iPLA2β associates with mitochondria in macrophages subjected to ER stress. AA incorporation into glycerophosphocholine lipids is unimpaired in iPLA2β-null macrophages upon electrospray ionization-tandem mass spectrometry analyses, and their complex lipid composition is similar to WT cells. These findings suggest that iPLA2β participates in ER stress-induced macrophage apoptosis caused by FCL or thapsigargin but that deletion of iPLA2β does not impair macrophage arachi-donate incorporation or phospholipid composition.
Phospholipases A2 (PLA2)2 catalyze hydrolysis of the sn-2 fatty acid substituent from glycerophospholipid substrates to yield a free fatty acid, e.g. arachidonic acid, and a 2-lysophospholipid (1, 2) that have intrinsic mediator functions (3, 4) and can initiate synthesis of other mediators, such as prostaglandins, leukotrienes, epoxy-eicosatrienoates, and platelet-activating factor (PAF) (5). Mammalian PLA2s include the PAF-acetylhydrolase PLA2 family, which exhibits substrate specificity for PAF and oxidized phospholipids, and the secretory PLA2 (sPLA2), which are low molecular weight enzymes that require millimolar concentrations of [Ca2+] for catalysis and affect inflammation and other processes (1). Of the group IV cytosolic PLA2 (cPLA2) family members (1), cPLA2α was the first identified and prefers substrates with sn-2 arachidonoyl residues, associates with its substrates in membranes when cytosolic [Ca2+] rises, and is also regulated by phosphorylation (6). Additional cPLA2 family members are encoded by separate genes (7–10).
Group VI PLA2 (iPLA2) enzymes (11–13) do not require Ca2+ for catalysis and are inhibited by a bromoenol lactone (BEL) suicide substrate (14) that does not inhibit sPLA2 or cPLA2 at similar concentrations (14–17). Group VIA PLA2 (iPLA2β) resides in the cytoplasm of resting cells and undergoes subcellular redistribution upon cellular stimulation (2). Group VIB PLA2 (iPLA2γ) contains a peroxisomal targeting sequence and is membrane-associated (18, 19). These enzymes belong to a larger class of serine lipases encoded by several genes (20, 21). The iPLA2β enzymes are 84–88-kDa proteins that contain a GXSXG lipase consensus sequence and 7 or 8 stretches of a repetitive motif homologous to that of the ankyrin protein-binding domain (11–13).
In murine P388D1 macrophage-like cells iPLA2β is proposed to generate lysophosphatidylcholine (LPC) acceptors for arachidonic acid incorporation into phosphatidylcholine (PC), based on studies with BEL or an antisense oligonucleotide that reduces iPLA2 expression (22–25). It has also been proposed that iPLA2β cooperates with CTP:phosphocholine cytidylyl-transferase (26, 27), which catalyzes the rate-limiting step in PC synthesis, to maintain PC homeostasis by degrading excess PC, based on studies with BEL in cells that overexpress CTP:phosphocholine cytidylyltransferase (26, 27).
Many other iPLA2β functions have been proposed (28–41), including a role for apoptosis in human U937 promonocytes induced by anti-Fas antibody (42). Hydrolysis of arachidonic acid from U937 phospholipids induced by such antibodies is inhibited by BEL (42), and U937 cell apoptosis is associated with cleavage of iPLA2β by caspase-3 (29), a central protease in executing apoptosis. Overexpressing this iPLA2β cleavage product amplifies both thapsigargin-induced arachidonate release and cell death (29).
Thapsigargin is a SERCA inhibitor (43) and induces loss of ER Ca2+ stores and apoptosis in many cells by ER stress pathways. Thapsigargin-induced apoptosis of pancreatic islet β-cells and insulinoma cells requires hydrolysis of arachidonic acid from membrane phospholipids and generation of arachi-donate metabolites (44) by a Ca2+-independent mechanism that is suppressed by BEL (45). Overexpressing iPLA2β also amplifies thapsigargin-induced apoptosis of insulinoma cells by a BEL-sensitive mechanism (46), and these observations suggest that iPLA2β participates in ER stress-induced β-cell apoptosis.
Death of cholesterol-laden macrophages in atherosclerotic lesions (47) is thought to contribute to plaque rupture and thrombosis (48), and mouse peritoneal macrophages undergo apoptosis by a mechanism that involves ER stress when loaded with free cholesterol by incubation with acetylated low density lipoprotein (Ac-LDL) and an inhibitor of the cholesterol esterification enzyme ACAT (49–52). Free cholesterol accumulates in macrophage ER membranes, which results in SERCA-2b inhibition (51), and the macrophages subsequently undergo apoptosis, which might be attributable to ER Ca2+ loss and induction of ER stress in a manner analogous to events induced by thapsigargin (51, 53).
We generated iPLA2β-null mice by homologous recombination (54), and their peritoneal macrophages exhibit defective transcriptional regulation of the inducible nitric-oxide synthase gene in response to viral infection (36). Because iPLA2β appears to participate in ER stress-induced apoptosis in U937 promonocytes (29, 42) and β-cells (44–46) and because iPLA2β-null mouse macrophages exhibit signaling defects (36), we examined the possibility that iPLA2β might be involved in free cholesterol loading-induced apoptosis by comparing responses of wild-type and iPLA2β-null macrophages. We also compared the phospholipid composition of wild-type and iPLA2β-null macrophages to examine the proposed house-keeping functions of iPLA2β in phospholipid homeostasis (22–27).
EXPERIMENTAL PROCEDURES
Materials
[5,6,8,9,11,12,14,15-3H]Arachidonic acid (217 Ci/mmol) was obtained from Amersham Biosciences; enhanced chemiluminescence (ECL) reagents from were Amersham Biosciences; standard phospholipids were from Avanti Polar Lipids (Birmingham, AL); SDS-PAGE supplies were from Bio-Rad; organic solvents were from Fisher; Coo-massie reagent was from Pierce; ampicillin, kanamycin, zymosan, fucoidan, common reagents, and salts were from Sigma; culture media, penicillin, streptomycin, Hanks’ balanced salt solution, L-glutamine, agarose, molecular mass standards, and RT-PCR reagents were from Invitrogen; Pentex bovine serum albumin (BSA, fatty acid-free, fraction V) was from ICN Biomedical (Aurora, OH); thapsigargin was from Calbiochem. Heat-inactivated (1 h, 65 °C) fetal bovine serum was obtained from Hyclone (Logan UT). Krebs-Ringer bicarbonate buffer contained 25 mM HEPES (pH 7.4), 115 mM NaCl, 24 mM NaHCO3, 5 mM KCl, 1 mM MgCl2, and 2.5 mM CaCl2.
ACAT inhibitor 58035 (3-[decyldimethyl-silyl]-N-[2-(4methyl-phenyl)-1-phenylethyl]propanamide (55) was provided by J. Heider, formerly of Sandoz (East Hanover, NJ); a 10 mg ml−1 stock solution was prepared in dimethyl sulfoxide (Me2SO), and final Me2SO concentration for treated and control cells was 0.05%. Cholesterol (>99% pure) was obtained from Nu Chek Prep. (Elysian, MN). LDL (d, 1.020–1.063 g ml−1) from fresh human plasma was isolated by preparative ultracentrifugation. Acetyl-LDL was prepared by reaction with acetic anhydride (56). Anti-GADD153 (CHOP) antibody was obtained from Santa Cruz Biotechnology. Horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were from Bio-Rad.
Generating iPLA2β−/− Knock-out Mice
The Washington University Animal Studies Committee approved all studies described in this study. Preparation of the iPLA2β knock-out construct, its introduction into 129/SvJ mouse embryonic stem cells, their selection with G418, characterization by Southern blotting, injection into C57BL/6 mouse blastocysts, production of chimeras and then heterozygotes, and mating of heterozygotes to yield wild-type, heterozygous, and iPLA2β-null mice in a Mendelian distribution are described elsewhere, as is their genotyping by Southern blotting of tail genomic DNA (54, 57).
Isolation of Peritoneal Macrophages
As described previously (49–52), peritoneal macrophages from adult female C57BL/6 mice and mutant mice were harvested 3 days after intraperitoneal injection of 40 μg of concanavalin A in PBS (0.2 ml). Cells were incubated in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 20% L-cell-conditioned medium. Medium was replaced every 24 h until macrophages were confluent. On days of experiments, cells were washed three times with warm PBS and incubated as indicated in the figures.
Analyses of iPLA2β mRNA in Mouse Peritoneal Macrophages and Brain
Northern blots of iPLA2β mRNA were performed as described (54). For RT-PCR, total RNA was isolated with an RNeasy kit (Qiagen Inc.). SuperScript First Strand Synthesis System (Invitrogen) was used to synthesize cDNA in 20-μl reactions that contained DNase I-treated total RNA (2 μg). The cDNA product was treated (20 min, 37 °C) with RNase H (2 units; Invitrogen) and heat-inactivated (70 °C, 15 min). Reactions without reverse transcriptase were performed to verify the absence of genomic DNA. PCR with the pair of primers 1 and 2 amplifies a fragment that spans the neomycin resistance cassette insertion site. PCR with the pair of primers 3 and 2 amplifies a fragment downstream from that site. The sequence of primer 1 is tgtgacgtggacagcactagc, that of primer 2 is ccccagagaaacgactatgga, and that of primer 3 is tatgcgtggtgtgtacttccg.
Free Cholesterol Loading of Mouse Peritoneal Macrophages, Incubation with Thapsigargin and Fucoidan, Cell Death Assays, Caspase Activation, and Analyses of Internucleosomal DNA Cleavages
Macrophages were loaded with free cholesterol by incubation with 100 μg ml−1 acetyl-LDL in the presence of 10 μg ml−1 58035, which inhibits ACAT-mediated cholesterol esterification (49). Alternatively, macrophages were incubated with the SERCA inhibitor thapsigargin (0.5 μM), the scavenger receptor A ligand fucoidan (25 μg/ml), or both agents for 24 h as described (52).
At the end of the incubation, macrophages were assayed for early-to-mid stage apoptosis (i.e. externalization of phosphatidylserine) by staining with Alexa-488-labeled annexin V and for late stage apoptosis (i.e. increased membrane permeability) by staining with propidium iodide, as described (49). Cells were viewed immediately with an Olympus IX-70 inverted fluorescence microscope, and six representative fields (~1000 cells) for each condition were counted for the number of annexin-positive cells, PI-positive cells, and total cells. In other experiments, cell or nuclear preparations were subjected to immunoblot analyses, as described below. Activated caspase-3 in macrophages was detected with an EnzCheck caspase-3 assay kit (Invitrogen). DNA laddering kits (Roche Diagnostics) were used to analyze internucleosomal cleavages characteristic of apoptosis as described (46).
Immunoblotting of the Endoplasmic Reticulum Stress Marker CHOP
Immunoblotting of CHOP was performed as described (58, 59) with minor modifications. Briefly, cells were lysed in RIPA buffer to prepare whole-cell lysates, which were resuspended in 2× SDS-PAGE loading buffer and incubated (95 °C, 10 min). After SDS-PAGE analyses, samples were electrotransfered to 0.22-μm nitrocellulose membranes using a Bio-Rad mini-transfer tank and then incubated with primary antibodies. Protein bands were detected with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad) by ECL (Amersham Biosciences). Membranes were also probed with an anti-β-actin monoclonal antibody to assess loading.
Preparation of Recombinant Adenovirus to Restore iPLA2β Expression to iPLA2β-Null Cells
An adenovirus that caused expression of iPLA2β was prepared with a ViraPower adenovirus expression system (Invitrogen) according to the manufacturer’s instructions. Briefly, cDNA that encodes the 84-kDa iPLA2β (13) was subcloned into the pENTR directional TOPO cloning vector. After sequence verification, the iPLA2β cDNA was transferred into pAd/CMV/V5-DEST vector with the Gateway system using LR clonase (Invitrogen). Positive clones were confirmed by sequencing. The clones were linearized using PacI (New England Biolabs) and then transfected into 293A cells with Lipofectamine 2000 using Opti-MEM medium. Virus was prepared and amplified with the ViraPower adenoviral expression system (Invitrogen), and viral titers were determined by plaque-forming assays with 293A cells. An aliquot of viral suspension was used to infect mouse peritoneal macrophages, and iPLA2β activity was assayed 3 days after infection. As a control, pAd/CMV/V5-GW/lacZ vector (Invitrogen) was transfected into 293A cells to produce lacZ-bearing adenovirus that did not contain the iPLA2β coding sequence.
[3H]Arachidonic Acid Incorporation into and Release from Mouse Peritoneal Macrophages
Isolated macrophages were cultured in RPMI 1640 medium containing 10% heat-inactivated fetal bovine serum, 2% glutamine, and 1% nonessential amino acids (w/v). Incorporation studies involved [3H]arachidonic acid addition (final concentration 0.5 μCi/ml, 5 nM) to medium and incubation (10–60 min, 37 °C). Cells were washed three times in Krebs-Ringer bicarbonate buffer containing 5.5 mM glucose and 0.1% BSA to remove unincorporated [3H]arachidonate. Cell viability exceeded 98% by trypan blue exclusion (45). [3H]Arachidonate incorporation into phospholipid extracts was then determined by TLC and liquid scintillation spectrometry (60).
[3H]Arachidonic acid release experiments were performed at a density of 1.2 × 106 cells/ml and initiated by incubation with zymosan or free cholesterol loading at 37 °C. Incubations with zymosan were performed essentially as described (61, 62). Before use, zymosan was boiled in PBS (10 min), collected by centrifugation on a tabletop centrifuge, and resuspended in PBS. The boiling procedure was then repeated twice. One mg of zymosan consisted of about 1.3 ×107 particles, and 20 particles of zymosan per cell were used during 1-h incubations. Free cholesterol loading with Ac-LDL and ACAT inhibitor 58035 was performed as described above for various intervals. At the end of the incubation interval, cells were collected by centrifugation, and supernatant 3H content was determined by liquid scintillation spectrometry and normalized to the amount initially incorporated. The phospholipid content of [3H]arachidonic acid was determined as described above. Cell number was measured with a hemocytometer and protein with Coomassie Blue (Pierce).
Subcellular Fractionation to Prepare Mitochondria and Cytosol, Cytochrome c Release, and iPLA2β Association with Mitochondria
Macrophages (2 × 106 per 10-cm plate) were incubated (2 h) in culture medium, washed twice in PBS, and then incubated without or with thapsigargin (0.5 μM) and fucoidan (25 μg/ml) for various intervals, as described above. At the end of the incubation, cytosol was separated from mitochondria as described (63) with minor modifications. Briefly, 5 volumes of isolation buffer (20 mmol/liter HEPES-KOH, 100 mmol/liter KCl, 1.5 mmol/liter MgCl2, 1 mmol/liter EGTA, 250 mmol/liter sucrose plus the protease inhibitors phenylmethyl-sulfonyl fluoride (1 mmol/liter) and protease inhibitor mixture (50 μl/ml)) were added to the cell pellet and left on ice for 20 min. Cells were then homogenized (Dounce apparatus, 20 strokes), and the homogenate was centrifuged (750 × g, 5 min). The pellet containing any remaining intact cells and nuclei was washed once with isolation buffer and discarded. Pooled supernatant was centrifuged (105 × g, 15 min) to remove mitochondria (pellet), and the supernatant was subjected to ultracentrifugation (106 × g, 1 h). Protein content of the resultant cytosolic supernatant was measured. Alternatively, in some experiments mitochondria were prepared with the Pierce isolation kit (64, 65), and their protein content was determined. Aliquots of mitochondrial and cytosolic protein were analyzed by SDS-PAGE and immunoblotted with antibodies to cytochrome c (Santa Cruz Biotechnology, Santa Cruz, CA), tubulin, iPLA2β (antibody 509 from Dr. Richard Gross, Washington University), and the mitochondrial marker cytochrome c oxidase complex IV-subunit II (Molecular Probes, Eugene, OR) (64). Densitometric ratios of bands from immunoblots were determined with AlphaEaseFC software.
Incubating Macrophages with Exogenous Arachidonic Acid and Examining Its Incorporation into Phospholipids
Macrophages were cultured as described above in supplemented RPMI 1640 medium. Incorporation studies involved adding arachidonic acid (final concentration 10 μM) to medium and incubating (2–6 h, 37 °C). At the end of the incubation, cells were washed as above, and phospholipids were extracted and analyzed by ESI/MS/MS as described below.
Phospholipid Extraction
Macrophages collected by centrifugation and rinsed twice in PBS were sonicated on ice (20% power, 5 s bursts for 60 s, Vibra Cell probe sonicator, Sonics and Materials, Danbury, CT). Samples were centrifuged (2,800 × g, 5 min) to remove cellular debris and supernatants transferred to silanized 10-ml glass tubes and extracted by adding methanol (1 ml), chloroform (1 ml), and water (1.8 ml). Samples were vortex-mixed and centrifuged (900 × g, 5 min), and supernatants were removed, concentrated, and dissolved in methanol/chloroform (9:1). Lipid phosphorus was measured as described (66).
Positive Ion Electrospray Ionization Mass Spectrometric Analyses of Choline-containing Lipids
PC and LPC were analyzed as Li+ adducts by positive ion ESI/MS on a Finnigan (San Jose, CA) TSQ-7000 triple stage quadrupole mass spectrometer with an ESI source controlled by Finnigan ICIS software. Phospholipids were dissolved in methanol/chloroform (2:1, v/v) containing LiOH (10 pmol/μl), infused (1 μl/min) with a Harvard syringe pump, and analyzed as described (67–69). For tandem MS, precursor ions selected in the first quadrupole were accelerated (32–36 eV collision energy) into a chamber containing argon (2.3–2.5 mtorr) to induce collisionally activated dissociation (CAD), and product ions were analyzed in the final quadrupole. Identities of GPC species were determined from their tandem spectra (67–69), and their quantities were determined relative to internal standards 14:0/14:0-GPC and 18:0/22:6-GPC by interpolation from a standard curve (45, 60, 70, 71).
Statistical Methods
Results are presented as mean ± S.E. Data were evaluated by unpaired, two-tailed Student’s t test or by analysis of variance with appropriate post hoc tests. Significance levels are described in the figure legends.
RESULTS
Production of iPLA2β-Null Mice and Characterization of iPLA2β mRNA Expression in Peritoneal Macrophages and Brain
To examine expression of iPLA2β mRNA, we compared brain, which expresses high levels of iPLA2β (72), and peritoneal macrophages, which have not to our knowledge previously been demonstrated formally to express iPLA2β mRNA, from wild-type and iPLA2β-null mice generated by homologous recombination (54). Northern blotting analyses were performed with a probe that recognizes sequence spanning exons 7–12 of the iPLA2β gene. Brain and peritoneal macrophages from wild-type mice contained mRNA species of the expected size recognized by the iPLA2β probe, but none of any size was observed in brain or macrophages from iPLA2β-null mice (Fig. 1A).
FIGURE 1. Examination of expression of iPLA2β mRNA in peritoneal macrophages and brain of wild-type and iPLA2β-null mice.
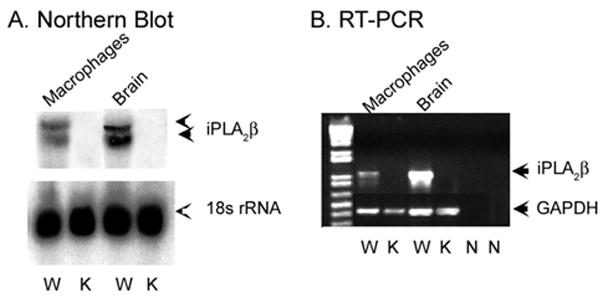
A contains Northern blots of total RNA from wild-type (W) and iPLA2β-knock-out (K) mouse peritoneal macrophages and brain. Upper panels reflect iPLA2β mRNA and lower panels 18 S rRNA. B depicts RT-PCR using total RNA from macrophages and brain as template and primers that amplify an iPLA2β mRNA fragment. The leftmost (1st) lane displays molecular weight standards. Other lanes represent wild-type (W) or knock-out (K) mice, as indicated. N lanes are controls without template. In 2nd to 5th lanes, primers that amplify a glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA fragment were included as controls.
RT-PCR was also performed with a primer pair that amplifies sequence in iPLA2β mRNA beginning in exon 6 and extending into exon 14, which spans the neomycin resistance cassette insertion site. This primer set yielded a product of the expected size in brain and peritoneal macrophages from wild-type but not iPLA2β-null mice. Northern blotting and RT-PCR studies thus indicate that brain and peritoneal macrophages from iPLA2β-null mice contained no iPLA2β mRNA species.
Induction of Macrophage Apoptosis by Free Cholesterol Loading
Mouse peritoneal macrophages undergo apoptosis when loaded with free cholesterol by incubation with acetylated low density lipoprotein (Ac-LDL) and ACAT inhibitor 58035 (49–52), as illustrated by wild-type macrophage binding of fluorophore-labeled annexin V to phosphatidylserine externalized by cells undergoing apoptosis (Fig. 2A). The fraction of macrophages from iPLA2β-null mice in which apoptosis occurred in response to free cholesterol loading was less than half that for wild-type macrophages (Fig. 2B), and the increase is caspase-3 activity upon free cholesterol loading was higher for macrophages from wild-type mice compared with those from iPLA2β-null mice (Fig. 2C). The extent of apoptosis-associated internucleosomal DNA cleavage was also greater in free cholesterol-loaded peritoneal macrophages from wild-type compared with iPLA2β-null mice (Fig. 3A).
FIGURE 2. Apoptosis and caspase-3 activation induced by free cholesterol loading of peritoneal macrophages from wild-type and iPLA2β-null mice.
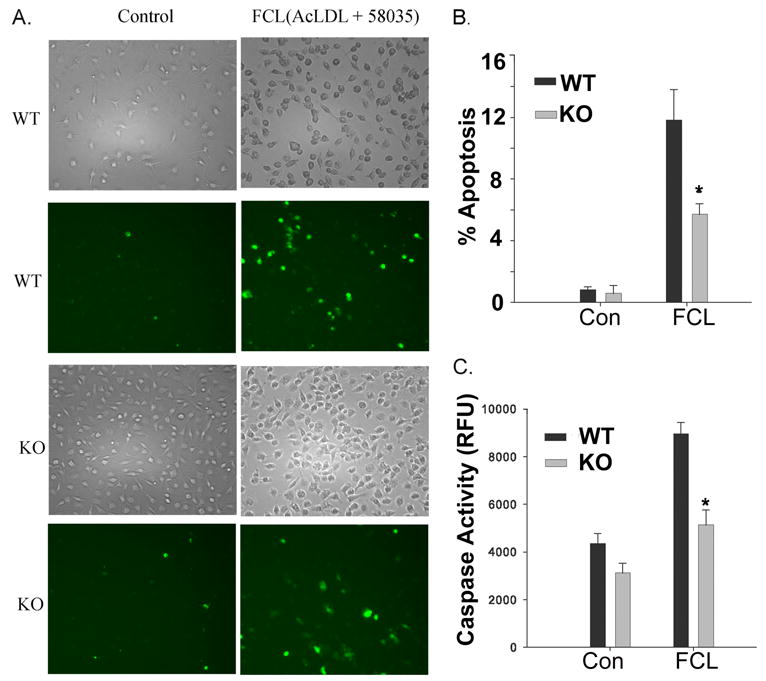
A, left column represents control macrophages from wild-type (WT) or iPLA2β-knock-out (KO) mice, and the right column represents macrophages subjected to FCL. In the 2nd and 4th rows, externalization of phosphatidylserine was visualized with Alexa-488-labeled annexin V. Total cells were determined from the light micrographs in the 1st and 3rd rows. B, percent of apoptotic cells was determined from annexin-positive and total cells in six fields of 1000 cells each. C displays macrophage caspase-3 activity. B and C, CON denotes control macrophages, and FCL denotes free cholesterol-loaded macrophages. Mean values ± S.E. are displayed (n = 6). Asterisk denotes p < 0.05 for WT versus KO.
FIGURE 3. Analyses of internucleosomal DNA cleavages and expression of the transcription factor CHOP in peritoneal macrophages from wild-type or iPLA2β-null mice incubated under control conditions, loaded with free cholesterol, or incubated with thapsigargin and fucoidan.
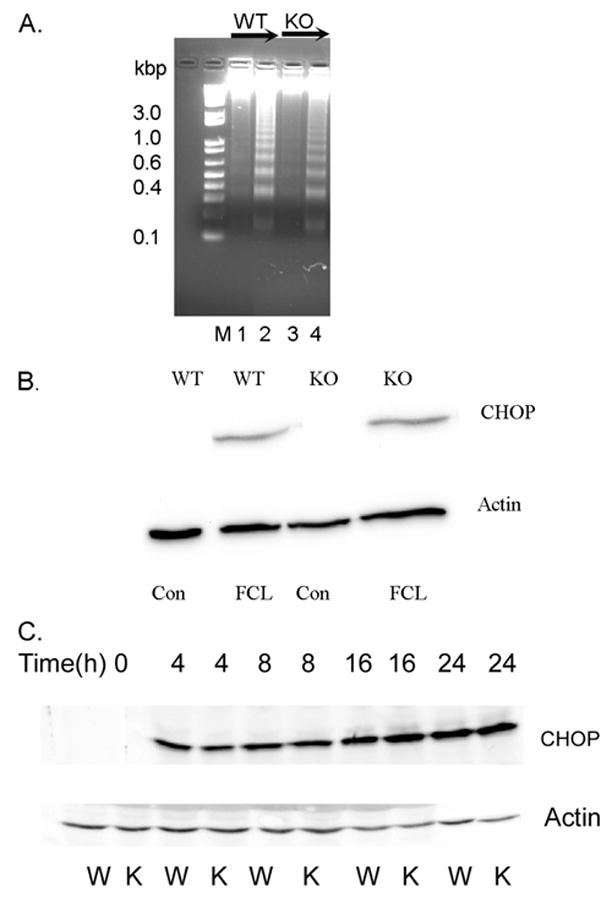
Wild-type (WT) or iPLA2β-knock-out (KO) macrophages were incubated under control conditions (CON) or subjected to FCL. A depicts DNA laddering analyses. Lane M has molecular weight markers. Lanes 1 and 3 represent DNA from untreated control macrophages from WT and KO mice, respectively, and lanes 2 and 4 represent DNA from FCL macrophages from WT and KO mice, respectively. B, immunoblotting for CHOP (upper panel) was performed with lysates from WT and KO macrophages incubated without (CON) or with (FCL) Ac-LDL and ACAT inhibitor 58035. In the lower parts of B and C, immunoblotting with an actin antibody was performed. C, immunoblotting for CHOP was performed with lysates from wild-type (W) or iPLA2β-knock-out (K) macrophages incubated without (1st two lanes from left) or with thapsigargin and fucoidan for 4 (3rd and 4th lanes), 8 (5th and 6th lanes), 16 (7th and 8th lanes), or 24 h (9th and 10th lanes).
Free cholesterol loading induces apoptosis and ER stress in mouse peritoneal macrophages by a process of accumulating in ER membrane, increasing membrane order, reducing SERCA-2b activity, and depleting ER Ca2+ (49–52), and ER stress causes expression of the transcriptional regulator CHOP (73–75). CHOP expression is induced in mouse peritoneal macrophages from wild-type mice by free cholesterol loading (50, 52) or by incubation with thapsigargin and fucoidan (52), as illustrated in Fig. 3, B and C, respectively, and a similar degree and time course of CHOP expression occurs in iPLA2β-null macrophages, although this results in apoptosis less frequently in iPLA2β-null macrophages.
Induction of Apoptosis by the SERCA Inhibitor Thapsigargin and the Scavenger Receptor A Ligand Fucoidan
Macrophage apoptosis induced by incubation with Ac-LDL and ACAT inhibitor 58035 reflects convergence of signals from multiple pathways (52, 53). One is ER stress from free cholesterol accumulation in the ER membrane and consequent SERCA inhibition. Another is SRA engagement by Ac-LDL. Either signal can be provided by alternate means. Low concentrations of thapsigargin induce ER stress, and the SRA can be engaged by fucoidan (a polymer of predominantly sulfated L-fucose) (52).
Low concentrations of thapsigargin plus fucoidan did induce apoptosis of wild-type mouse macrophages, although neither agent alone strongly induced this response (Fig. 4, upper two rows of panels, and Fig. 5A), confirming earlier reports (52). The response to the combined stimulus was attenuated with iPLA2β-null macrophages (Fig. 4, lower two rows of panels, and Fig. 5A) to a degree similar to that observed after free cholesterol loading (Figs. 2B and 5B). Apoptosis of iPLA2β-null macrophages subjected either to free cholesterol loading (Fig. 5B) or incubation with thapsigargin and fucoidan (not shown) increased toward wild-type levels upon restoring iPLA2β expression (Fig. 5C) with a recombinant adenovirus vector construct.
FIGURE 4. Fluorescence microscopic images of apoptosis induced by incubating peritoneal macrophages from wild-type and iPLA2β-null mice with thapsigargin and fucoidan.
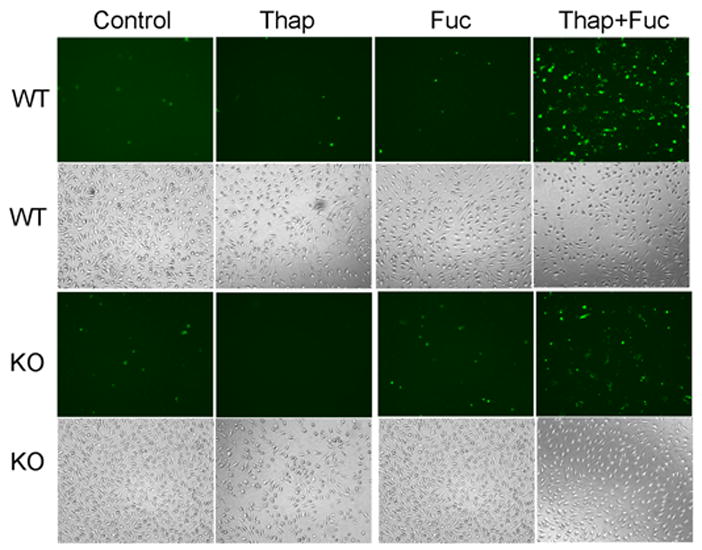
The leftmost column (1st) represents control, untreated peritoneal macrophages from wild-type (WT) or iPLA2β-knock-out (KO) mice, and the other columns represent macrophages incubated (24 h) with thapsigargin alone (Thap), fucoidan alone (Fuc), or both (Thap + Fuc). In the fluorescence micrographs in the 1st (topmost) and 3rd rows, externalization of phosphatidylserine was visualized with Alexa-488-labeled annexin V. Light micrographs in the 2nd and 4rth rows display total cells.
FIGURE 5. Apoptosis induced by incubating wild-type and iPLA2β-null mouse peritoneal macrophages with thapsigargin and fucoidan and effects of restoring iPLA2β expression to iPLA2β-null macrophages with a recombinant adenovirus.
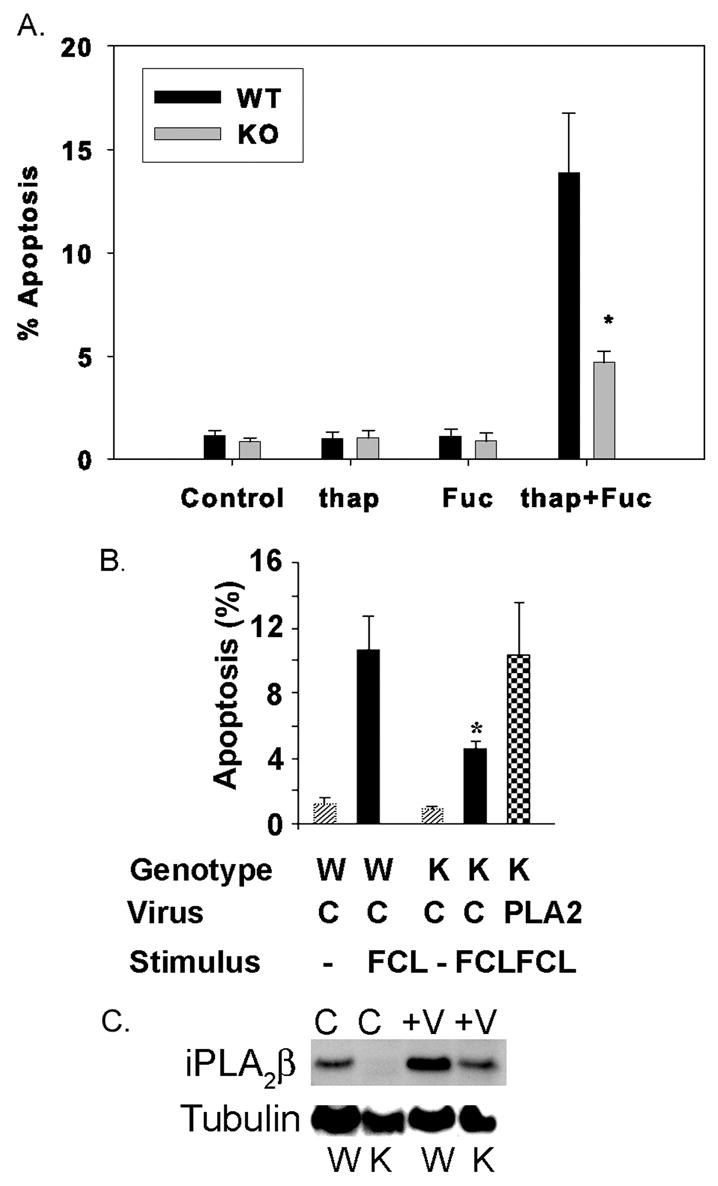
A, wild-type (WT) and iPLA2β-knock-out (KO) macrophages were incubated without (Control) or with thapsigargin (thap) alone, fucoidan (Fuc) alone, or both (thap + Fuc), and percent apoptotic cells was determined as in Fig. 2. B, WT or KO macrophages were incubated (24 h) without stimuli (lightly cross-hatched bars) or were subjected to FCL (solid black bars) with Ac-LDL and ACAT inhibitor 58035, and percent apoptotic cells was determined. Checkered bar represents experiments in which iPLA2β expression was restored recombinant adenovirus encoding iPLA2β (PLA2). Other cells were infected with control (C) virus without iPLA2β coding sequence. Immunoblots for iPLA2β and tubulin (C) illustrate iPLA2β expression in macrophages infected with virus (V) versus control (C), wild-type (W), or iPLA2β-knock-out (K) cells. Mean values ± S.E. are displayed (n = 6). Asterisk denotes p < 0.05 for WT versus KO.
Induction of ER stress in macrophages with thapsigargin or free cholesterol loading causes activation of c-Jun NH2-terminal kinase (JNK) that is required for apoptosis under these conditions (52). We confirmed that JNK is activated in wild-type macrophages by free cholesterol loading, as reflected by JNK phosphorylation visualized by immunoblotting with antibodies to phospho-JNK, but there was no obvious difference between wild-type and iPLA2β-null macrophages (not shown), suggesting that failure to activate JNK does not account for attenuated apoptosis of iPLA2β-null macrophages.
[3H8]Arachidonic Acid Incorporation into and Release from Mouse Peritoneal Macrophages
The facts that iPLA2β-null macrophages have reduced sensitivity to apoptosis induced by free cholesterol loading or by incubation with thapsigargin and fucoidan suggest that iPLA2β activation might be involved in the response of wild-type macrophages to these stimuli. To test this possibility, we determined whether free fatty acid products of iPLA2β action on phospholipids were generated by macrophages in response to free cholesterol loading.
A facile means to examine hydrolytic release of arachidonic acid from membrane phospholipids is to prelabel cellular phospholipids by incubation with [3H8]arachidonic acid ([3H]AA), remove unincorporated radiolabel by washing with albumin-containing buffers, stimulate the cells, and measure [3H]AA released into the medium (32, 76). Because iPLA2β has been proposed to generate LPC acceptors for incorporating arachidonic acid into membrane phospholipids in P388D1 macrophage-like cells (24, 25), the ability to achieve phospholipid labeling with [3H]AA in iPLA2β-null macrophages was examined first.
Wild-type and iPLA2β-null mouse peritoneal macrophages incorporated [3H]AA acid into glycerophosphocholine (GPC) lipids at essentially identical rates (Fig. 6A), suggesting that iPLA2β is not required for this process in these cells. Stimulating the cells with zymosan (an insoluble yeast cell wall carbohydrate) induced a robust release of [3H]AA of similar magnitude from both wild-type and iPLA2β-null macrophages (Fig. 6B). This is consistent with the fact that zymosan strongly stimulates arachidonic acid release from mouse peritoneal macrophages (61) catalyzed by the group IVA cPLA2α (62). That the response to zymosan is preserved in iPLA2β-null macrophages indicates they retain the ability to activate some phospholipases in response to some stimuli and do not have a general defect in phospholipid hydrolysis.
FIGURE 6. [3H8]Arachidonic acid incorporation into glycerophosphocholine lipids from peritoneal macrophages of wild-type and iPLA2β-null mice, [3H]AA release from the cells induced by incubation with zymosan or by free cholesterol loading, and effects of restoring iPLA2β expression to iPLA2β-null macrophages with recombinant adenovirus.

Wild-type (WT) and iPLA2β-knock-out (KO) macrophages were incubated with [3H8]arachidonic acid for various intervals and washed with BSA-containing buffer to remove unincorporated label. A, lipids were extracted after labeling, and their phosphorus content was measured. GPC lipids were then isolated by TLC, and their [3H]AA content was determined by liquid scintillation spectrometry. Values are expressed as dpm/nmol lipid phosphorus. B, [3H]AA-labeled macrophages were incubated (1 h) without (Control) or with zymosan. After incubation, medium [3H]AA content was determined and normalized to that initially incorporated. C, [3H]AA-labeled wild-type (squares) or iPLA2β-null (triangles) macrophages were incubated without (control, open symbols) or with Ac-LDL and ACAT inhibitor 58035 to induce free cholesterol loading (closed symbols). After various incubation intervals, medium [3H]AA content was determined and normalized. D, [3H]AA-labeled wild-type (WT) or iPLA2β-knock-out (KO) macrophages were incubated (24 h) without (−, gray bars) or with Ac-LDL and ACAT inhibitor 58035 (FCL, solid black bars). At the end of incubations, [3H]AA release was determined and normalized. The checkered bar depicts experiments in which iPLA2β expression was restored by recombinant adenovirus (PLA2). Other cells were infected with control (C) virus without iPLA2β coding sequence. Mean values ±S.E. are displayed (n = 6). Asterisk denotes p < 0.05 for WT versus KO, and double asterisk p < 0.01.
Free cholesterol loading caused [3H]AA-labeled macrophages from wild-type mice to increase release of [3H]AA in a time-dependent manner, and the magnitude of this response was greatly attenuated with iPLA2β-null macrophages (Fig. 6C). This suggests that iPLA2β catalyzes a component of cholesterol-loading induced [3H]AA hydrolysis from macrophage membrane phospholipids, which is consistent with the finding that restoring iPLA2β expression to iPLA2β-null macrophages with a recombinant adenovirus increased free cholesterol loading-induced [3H]AA release toward wild-type levels (Fig. 6D). [3H]AA release from wild-type macrophages was also induced by thapsigargin and fucoidan, and that response was attenuated with iPLA2β-null macrophages (not shown).
Cytochrome c Release and iPLA2β Subcellular Distribution
Involvement of iPLA2β in apoptosis in other cells via actions on mitochondria has been reported (78–81). The permeability transition of and cytochrome c release from hepatocyte mitochondria are modulated by iPLA2β (81), and free cholesterol loading induces both events in mouse peritoneal macrophages (82). This suggests a potential mechanism for iPLA2β participation in macrophage apoptosis induced by ER stress and SRA engagement. Mitochondrial cytochrome c was released into cytosol of wild-type macrophages incubated with thapsigargin and fucoidan, and this response was attenuated with iPLA2β-null macrophages (Fig. 7).
FIGURE 7. Cytochrome c release into cytosol of wild-type and iPLA2β-null mouse macrophages incubated with thapsigargin and fucoidan.
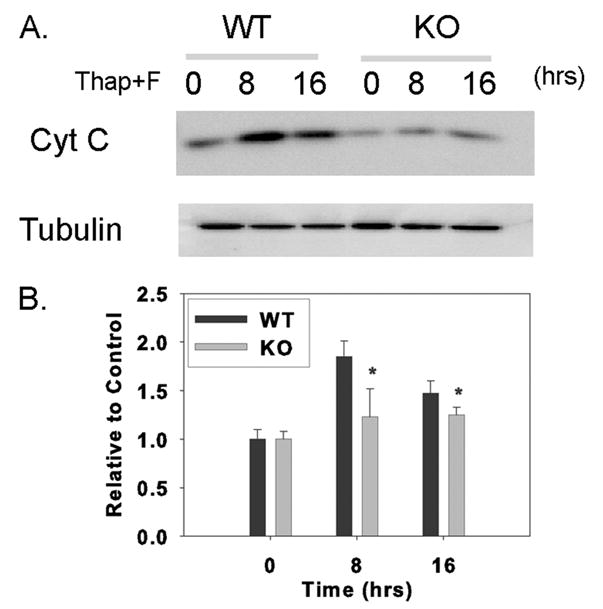
Wild-type (WT) or iPLA2β-knock-out (KO) macrophages were incubated without or with thapsigargin and fucoidan (Thap + F) for various intervals. At the end of the incubations, cytosol was prepared, analyzed by SDS-PAGE, electrotransferred to nylon membranes, and probed with cytochrome c and tubulin antibodies, as illustrated in A. B represents densitometric ratios of iPLA2β and tubulin immunoblot signals determined with AlphaEaseFC software. Mean values ± S.E. are displayed (n = 4). Asterisk denotes p < 0.05 for WT versus KO.
That cytochrome c release into wild-type macrophage cytosol induced by incubation with thapsigargin and fucoidan is blunted in iPLA2β-null macrophages suggests that an interaction between iPLA2β and mitochondria might occur during the apoptotic response. Fig. 8 illustrates experiments examining the distribution of iPLA2β in mitochondrial and cytosolic fractions of disrupted cells using antibodies to iPLA2β and to the mitochondrial marker protein cytochrome c oxidase-complex IV. Immunoreactive iPLA2β signal associated with isolated mitochondria relative to that for cytochrome c oxidase-complex IV is significantly greater for macrophages loaded with free cholesterol compared with control cells.
FIGURE 8. Subcellular redistribution of iPLA2β in macrophages subjected to free cholesterol loading.
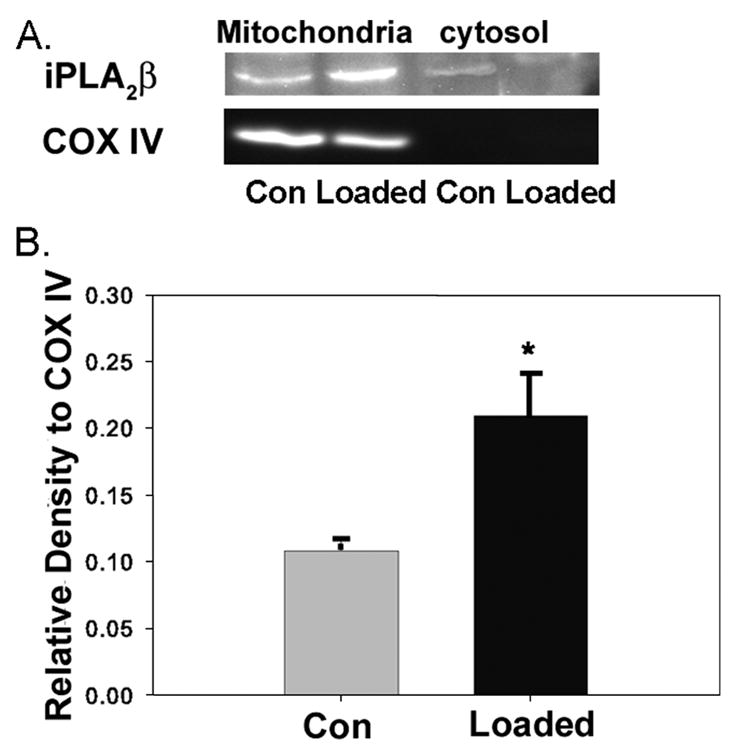
Wild-type macrophages were incubated (24 h) without (Con) or with (loaded) Ac-LDL and ACAT inhibitor 58035. At the end of incubations, mitochondrial and cytosolic fractions were prepared and analyzed by SDS-PAGE. A shows immunoblots for iPLA2β and the mitochondrial marker cytochrome c oxidase-complex IV subunit II (COX IV). B shows densitometric ratios of iPLA2β immunoblot signal divided by that for cytochrome c oxidase-complex IV in mitochondrial fractions determined by AlphaEaseFC software. Mean values ± S.E. are displayed (n = 4). Asterisk denotes p < 0.05 (WT versus KO).
Glycerophosphocholine Lipid Composition of Peritoneal Macrophages from Wild-type and iPLA2β-null Mice
The content and composition of mouse peritoneal macrophage GPC lipids are important determinants of the ability of a cell to survive free cholesterol loading (83), and iPLA2β is proposed to play housekeeping functions in maintaining GPC lipid homeostasis by providing 2-LPC acceptors for arachidonate incorporation into the PC and by cooperating with cytidylylphosphocholine transferase to maintain appropriate cellular PC levels (24–27). This suggests that the iPLA2β-null macrophage PC composition, particularly the abundance of arachidonate-containing PC species, might differ from wild-type macrophages, and this could affect their susceptibility to apoptosis induced by cholesterol loading (83).
We therefore examined the composition of GPC lipids from wild-type and iPLA2β-null peritoneal macrophages as Li+ adducts by ESI/MS (Fig. 9) and by ESI/MS/MS (Fig. 10). Ions representing arachidonate (20:4)-containing GPC lipids are prominent in the spectra in Fig. 9 and include those at m/z 788 (16:0/20:4-GPC) and m/z 816 (18:0/20:4-GPC). The identities of the parent [M + Li] + ions were established from their tandem spectra (Fig. 10). Because arachidonate-containing GPC-Li+ species tend to eliminate phosphocholine (183 Da) more readily than do GPC lipid species that contain more saturated fatty acids (67), ESI/MS/MS scanning for constant neutral loss of 183 Da accentuates the prominence of the arachidonate-containing species represented by the ions at m/z 788 and 816 (Fig. 9, C and D).
FIGURE 9. Electrospray ionization mass spectrometric analyses of glycerophosphocholine lipids from peritoneal macrophages of wild-type and iPLA2β-null mice.

Phospholipids from wild-type (A and C) or iPLA2β-null (B and D) macrophages were analyzed as Li+ adducts by positive ion ESI/MS, and relative abundances of ion currents plotted versus m/z value. A and B represent total ion current, and C and D represent ESI/MS/MS scans for constant neutral loss of 183.
FIGURE 10. Tandem mass spectra of glycerophosphocholine lipids from mouse peritoneal macrophages.

GPC lipid-Li+ adducts from wild-type or iPLA2β-null macrophage lipid extracts were analyzed by positive ion ESI/MS/MS. Specific ions selected in the first mass-analyzing quadrupole were accelerated into the collision cell to induce CAD. Product ions were analyzed in the final quadrupole. Ions selected in the first quadrupole included m/z 788 (A), 816 (B), 740 (C), and 766 (D).
CAD of m/z 788 yields a spectrum (Fig. 10A) that contains ions reflecting neutral losses of trimethylamine plus either the sn-1 substituent (M + Li+ − 315) or the sn-2 substituent (M + Li+ − 363) as free fatty acids at m/z 473 and m/z 425, respectively. The former is more abundant than the latter, which conforms to published rules established from reference spectra that palmitate and arachidonate are the sn-1 and sn-2 substituents, respectively (67–69). Analogous ions in Fig. 10B (m/z 473 and 453) from CAD of m/z 816 indicate that stearate and arachidonate are the sn-1 and sn-2 substituents, respectively.
Fig. 10A also shows neutral losses of the sn-1 substituent as a free fatty acid (M + Li+ − 256) or as a Li+ salt (M + Li+ − 262) at m/z 532 and m/z 526, respectively. Ions of the same m/z values are observed in Fig. 10B and represent [MLi+ − 284] and [MLi+ − 290], respectively. Neutral losses of the sn-2 substituent as a free fatty acid (MLi+ − 304) or as a Li+ salt (MLi+ − 310) are seen at m/z 484 and m/z 478, respectively, in Fig. 10C, and at m/z 512 and m/z 506, respectively, in Fig. 10B. The ion m/z 313 (MLi+ − 475) in Fig. 10A reflects net elimination of [LiPO4(CH2)2N(CH3)3] and loss of the sn-2 substituent as a ketene (80–82). An analogous m/z 341 ion (MLi+ − 475) is seen in the tandem spectrum 18:0/20:4-GPC-Li+ (Fig. 10B). Other diagnostic ions in Fig. 10A include those for loss of trim-ethylamine (m/z 729) or net loss of phosphocholine or its Li+ salt from [M + Li]+ at m/z 605 and 599, respectively. Analogous ions in Fig. 10B are observed at m/z 757, m/z 633, and m/z 627, respectively.
The two most abundant ions in the mass spectra of GPC lipids from peritoneal macrophages of wild-type or iPLA2β-null mice occur at m/z 740 and m/z 766 (Fig. 9, A and B). The tandem spectra in Fig. 10, C and D, identify these two ions as the Li+ adducts of 16:/0/16:0-GPC and 16:0/18:1-GPC, respectively, as described in the rationalization of these spectra in the Supplemental Material. Identities of other GPC lipids represented by ions in Fig. 9, A and B, were similarly determined and include 16:0/16:1-GPC (m/z 738), 18:1/18:1-GPC (m/z 792), and 18:0/22:6-GPC (m/z 840). The tandem spectra of the ions at m/z 726, 752, and 772 indicates that they represent the Li+ adducts of the alkyl ether (plasmanylcholine) species 16:0e/16: 0-GPC, 16:0e/18:1-GPC, 16:0e/20:4-GPC, respectively.
Fig. 9B is the ESI/MS spectrum of GPC lipid Li+ adducts from iPLA2β-null peritoneal macrophages and is virtually identical to that for wild-type peritoneal macrophages (Fig. 9A). Ions representing the 20:4-containing GPC species at m/z 788 and 816 are no less abundant in the latter spectrum than in the former, and quantitative measurements with internal standards and normalization to lipid phosphorus levels also indicate that 16:0/20:4-GPC and 18:0/20:4-GPC are no less abundant in macrophages from iPLA2β-null mice than in macrophages from wild-type mice, as summarized in supplemental Table S1. This indicates that the absence of iPLA2β does not result in a deficiency of arachidonate-containing GPC lipids in mouse peritoneal macrophages or in other changes in GPC lipid composition.
ESI/MS/MS Analyses of Incorporation of Extracellular Arachidonic Acid into Glycerophosphocholine Lipids
The proposal that iPLA2β is a housekeeping enzyme that generates LPC acceptors for arachidonate incorporation into GPC lipids is based mainly on studies with murine macrophage-like P3888D1 tumor cells (24, 25). Although Figs. 9 and 10 illustrate that GPC lipids of native peritoneal macrophages recently harvested from wild-type or iPLA2β-null mice do not differ in content of arachidonate-containing species, it could be argued that a defect in arachidonate incorporation into GPC lipids might be demonstrable with isolated cells incubated with exogenous arachidonic acid even though compensatory mechanisms recruited in vivo could overcome any such defect in intact mice.
We therefore compared ex vivo incorporation of exogenous arachidonic acid added to culture medium into GPC lipids of wild-type of iPLA2β-null peritoneal macrophages (Fig. 11). With wild-type macrophages, the abundance of 16:0/20:4-GPC (m/z 788) increased markedly after 2 h of incubation with supplemental arachidonic acid, and this was followed by an increase in 18:0/20:4-GPC (Fig. 11, A–C) that reflects sequential sn-2 and then sn-1 remodeling (60, 84, 85). Arachidonate incorporation into GPC lipids of iPLA2β-null macrophages was indistinguishable from that of wild-type macrophages in rate, extent, or pattern (Fig. 11D). We thus find no evidence that iPLA2β is required for arachidonate incorporation into GPC lipids of native mouse peritoneal macrophages.
FIGURE 11. Electrospray ionization mass spectrometric analyses of glycerophosphocholine lipid species in wild-type and iPLA2β-null mouse peritoneal macrophages incubated with supplemental arachidonic acid for various intervals.

Wild-type (WT) (A–C) or iPLA2β-null (D) macrophages were incubated with arachidonic acid for 0 h (A), 2 h (B), or 6 h (B and D). Lipids were then extracted, and GPC lipids were analyzed by positive ion ESI/MS/MS as Li+ adducts for constant neutral loss of 183.
Composition of Sphingolipids and Anionic Phospholipids in Peritoneal Macrophages from Wild-type and iPLA2β-Null Mice
Accumulation of sphingomyelin and PC are reciprocally regulated by sphingomyelin synthase, which converts PC and ceramide to sphingomyelin and diacylglycerol (86–89). It is thus possible that a cell in which PC metabolism was perturbed might compensate by altering sphingolipid metabolism to preserve an optimal PC content and composition. Sphingomyelin avidly binds cholesterol (90), and the sphingomyelin content of mouse peritoneal macrophages affects their ability to survive cholesterol loading (91). We therefore compared sphingolipid content and composition of wild-type and iPLA2β-null macrophages and observed no major differences between the two genotypes, as illustrated in supplemental Fig. S1 and Table S2.
Arachidonic acid or other polyunsaturated fatty acids initially incorporated into PC can be transferred subsequently to other phospholipid classes in part via a CoA-independent transacylase (60, 92). It is thus possible that a cell in which PC metabolism was perturbed might compensate by altering metabolism of other phospholipid head group classes to preserve an optimal PC content and composition. We therefore compared the content and composition of glycerophosphoethanolamine, glycerophosphoglycerol, glycerophosphoserine, and glycerophosphoinositol lipids in wild-type and iPLA2β-null macrophages and again observed no major differences between the two genotypes, as illustrated in supplemental Figs. S2 and S3 and Table S1.
Mitochondrial Phospholipid Composition of Wild-type and iPLA2β-null Mouse Macrophages
Although the global phospholipid compositions of wild-type and iPLA2β-null mouse macrophages are virtually identical, differences in their mitochondrial membrane composition might confer differential susceptibility to apoptosis, but positive ion ESI/MS analyses of Li+ adducts indicate that the GPC lipid and sphingomyelin composition of isolated mitochondria from wild-type and iPLA2β-null macrophages are virtually identical (supplemental Fig. S5 and Table S3).
Induction of ER stress by incubating wild-type macrophages with thapsigargin resulted in a decline in mitochondrial GPC lipids (Fig. 12 and supplemental Table S4) that was not observed with iPLA2β-null cells (supplemental Table S4). Negative ion ESI/MS analyses indicated that the mitochondrial content of glycerophosphoethanolamine and glycerophosphoglycerol lipids also fell in thapsigargin-treated wild-type but not iPLA2β-null cells (supplemental Table S4).
FIGURE 12. Electrospray ionization mass spectrometric analyses of glycerophosphocholine lipids in wild-type mouse peritoneal macrophages incubated under control conditions or subjected to ER stress with thapsigargin.

Macrophages were incubated (24 h) without (A) or with (B) thapsigargin (0.5 μM), and their mitochondria were then isolated. Mitochondrial lipids were extracted, mixed with 14:0/14:0-GPC internal standard, and analyzed by positive ion ESI/MS/MS as Li+ adducts for constant neutral loss of 189.
DISCUSSION
Fig. 13 is a model for participation of iPLA2β in free cholesterol loading-induced apoptosis of mouse peritoneal macrophages that incorporates findings reported here into a scheme developed by Tabas and co-workers (52, 53). Free cholesterol loading induced by incubating macrophages with Ac-LDL and ACAT inhibitor 58035 activates multiple signaling pathways (52), all of which are necessary and none of which is sufficient alone to cause apoptosis. One pathway is ER stress that results from free cholesterol accumulation in ER membranes, which increases membrane order and results in SERCA inhibition and ER Ca2+ store depletion (51).
FIGURE 13. Model for involvement of iPLA2β in apoptosis in macrophages induced by free cholesterol loading.
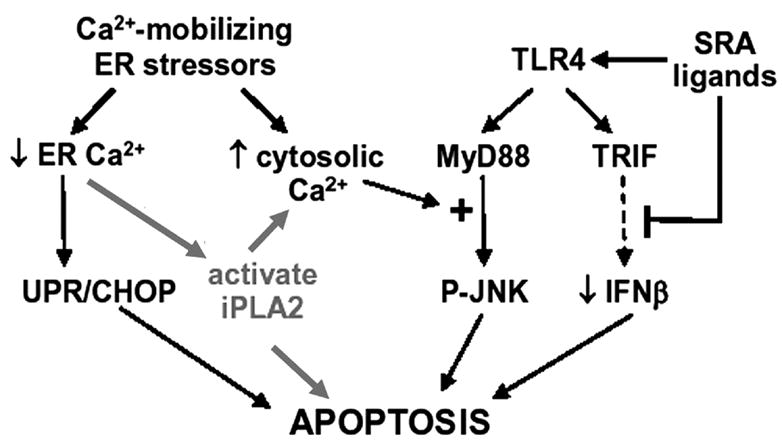
The model of Tabas and coworkers (52, 53) is revised to incorporate data in this report. Free cholesterol accumulates in macrophage ER membranes, alters their physical properties, inhibits SERCA, and causes ER Ca2+ loss. This activates UPR-CHOP pathway, causes a rise in cytosolic [Ca2+], which amplifies the TLR4-MyD88-JNK pathway, and activates iPLA2β, which associates with mitochondria to facilitate cytochrome c release. In coordination with signals from SRA and TL4 receptor engagement, the combinatorial convergence of multiple events induces apoptosis (53).
Several other events are also required to induce apoptosis, probably because the main goal of ER stress is to activate cell repair mechanisms, and death occurs only when damaging insults are multiple and overwhelming. With ER-stressed macrophages, other events required for apoptosis include CHOP induction (50), TLR4-MyD88 activation, proapoptotic JNK induction (52), engagement of SRA and the pattern recognition receptor TLR4, silencing of the TLR4-TRIF-IRF3-IFNβ cell-survival pathway (77), and ER stress-induced rises in cytosolic [Ca2+] (50, 52, 53).
Our findings suggest that another consequence of ER stress is activation of iPLA2β and its association with mitochondria. This may facilitate cytochrome c release, which along with the permeability transition (81) does occur when mouse macrophages are loaded with cholesterol (82). Cytochrome c released into cytosol activates the caspase cascade and execution of apoptosis (93–95). This sequence of events could rationalize several previous observations by us (45, 46, 66) and others (39, 44, 78–81, 96–99).
Our findings here indicate that iPLA2β-null mouse macrophages are more resistant than wild-type cells to apoptosis induced either by free cholesterol loading or by combined SERCA inhibition with thapsigargin and SRA engagement with fucoidan, as visualized by phosphatidylserine externalization, caspase-3 activation, and DNA laddering. Free cholesterol loading of wild-type macrophages also induces arachidonic acid hydrolysis from membrane phospholipids, which is consistent with PLA2 activation, and this is attenuated with iPLA2β-null macrophages.
These findings contribute additional evidence that iPLA2β is activated by ER Ca2+ loss (34, 45, 97–99) and participates in ER stress-induced apoptosis (29, 42, 44, 46). A role for iPLA2β in apoptosis was first suggested by the finding that inducing human U937 promonocyte apoptosis is associated with arachidonic acid hydrolysis from membrane phospholipids by a mechanism that is blunted by pharmacologic inhibition of iPLA2β (42) and may involve proteolytic activation of iPLA2β by caspase-3 (29). Overexpressing this iPLA2β cleavage product amplifies both thapsigargin-induced arachidonate release and cell death (29).
The SERCA inhibitor thapsigargin induces ER Ca2+ loss and apoptosis in many cells by ER stress pathways. In pancreatic islet β-cells, thapsigargin induces apoptosis in a manner that does not require a rise in cytosolic [Ca2+] but does require arachidonic acid hydrolysis from phospholipids and its metabolism (44). Thapsigargin-induced arachidonic acid hydrolysis from islet phospholipids occurs by a Ca2+-independent mechanism that is suppressed by pharmacologic inhibition of iPLA2β (45). Overexpressing iPLA2β amplifies thapsigargin-induced apoptosis of INS-1 insulinoma cells, and this is also suppressed by iPLA2β inhibition (46). The magnitude of thapsigargin-induced INS-1 cell apoptosis correlates with iPLA2β expression level in various INS-1 cell lines, and apoptosis is associated with iPLA2β stimulation and subcellular redistribution (46). Pharmacologic and biochemical evidence indicates that ER Ca2+ store depletion also results in iPLA2β activation in vascular smooth muscle cells (30, 34, 97).
Accumulating evidence suggests that a mechanism for iPLA2β to participate in apoptosis involves its association with mitochondria and facilitation of cytochrome c release (78–81, 96). Isolated mitochondria from cardiomyocytes, brain, and rat liver all contain immunoreactive, catalytically competent iPLA2β (78–81). Ischemia reperfusion-induced cardiac myocyte death is associated with iPLA2β subcellular redistribution, mitochondrial phospholipid hydrolysis, and mitochondrial dysfunction (81), and iPLA2β inhibition reduces mitochondrial phospholipid loss and cell death (81, 100).
Association of BAX and truncated BID with brain mitochondria also induces cytochrome c release that is associated with mitochondrial iPLA2β activation and attenuated by iPLA2β inhibition (80). Rat liver mitochondria release cytochrome c and undergo permeability transition in response to Ca2+ loading and depolarization, and this is also associated with iPLA2β activation and reduced by iPLA2β inhibition (79, 81). All of these findings are consistent with our observations that cytochrome c release into cytosol in response to apoptogenic stimuli is reduced in iPLA2β-null macrophages compared with wild-type cells, that immunoblotting studies indicate that iPLA2β associates with mitochondria in macrophages subjected to ER stress, and that the ER stress-induced mitochondrial phospholipid loss observed with wild-type macrophages is blunted or absent with iPLA2β-null macrophages.
Widely cited hypotheses are that iPLA2β plays the house-keeping roles of providing LPC acceptors for incorporating arachidonic acid into PC (24, 25) and maintaining membrane phospholipid homeostasis by degrading excess PC (26, 27. This could have important implications for macrophage function because arachidonate-containing phospholipids are more abundant in macrophages than in many other cells (101), and the physical properties of such phospholipids influence the ability of macrophages to survive cholesterol loading (51).
Nonetheless, we observe a PC composition of iPLA2β-null mouse macrophages that is similar or identical to that of wild-type macrophages, and this is also the case for the PC composition of testes, brain, and pancreatic islets from iPLA2β-null and wild-type mice (54, 70). In particular, there is no deficiency in arachidonate-containing PC species in any of these tissues or in peritoneal macrophages from iPLA2β-null mice.
Our ESI/MS findings that the PC content and composition of macrophages from iPLA2β-null and wild-type mice are virtually identical are consistent with the lack of effects of pharmacologic inhibition of iPLA2β activity (60, 102) or of molecular biologic manipulation of iPLA2β expression (70, 84) on these parameters in insulinoma cells and islets. Neither stable overexpression (84) nor suppression (70) of iPLA2β expression in insulinoma cells affects their PC content or composition or rates of arachidonic acid incorporation into PC. Moreover, we observe no differences between wild-type and iPLA2β-null macrophages in the content or composition of sphingolipids or of anionic glycerophospholipids with -ethanolamine, -serine, -inositol, or -glycerol head groups.
Although we find no evidence that iPLA2β plays housekeeping roles in PC homeostasis (26, 27) and remodeling (24, 25) in mouse peritoneal macrophages, iPLA2β is widely expressed and might have multiple functions that vary among tissues and cell types. The function of iPLA2β in a given setting might depend in part on which splice variants (40, 41) and proteolytic processing products (29, 102, 103) of iPLA2β are expressed in a given cell and on what interacting proteins (98) are present in the cell compartment (84, 101) in which the iPLA2β isoform resides.
It should be noted that evidence for iPLA2β participation in arachidonate incorporation into membrane phospholipids is based primarily on studies with P388D1 mouse macrophage-like tumor cells (24, 25) or with other transformed cells of monocyte/macrophage lineage (32). The fact that iPLA2β does not appear to participate in this process in native mouse macrophages raises the possibility that any such role for iPLA2β might be confined to transformed, cultured cells.
The findings reported here thus contribute to a growing body of evidence that iPLA2β plays a role in ER stress-induced apoptosis and indicate that homozygous iPLA2β gene disruption produces phenotypic abnormalities in several tissues and cells that include mouse peritoneal macrophages in addition to testes (54) and pancreatic islets (57).
Supplementary Material
Acknowledgments
We thank Sheng Zhang and Min Tan for their excellent technical assistance and Dr. Richard Gross for supplying iPLA2β antibody 509.
Footnotes
This work was supported by United States Public Health Service Grants R37-DK34388, P01-HL57278, P41-RR00954, P60-DK20579, RO1– 69455, and P30-DK56341.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Materials, Experimental Procedures, Results, Figs. 1–5, Tables 1– 4, and additional references.
The abbreviations used are: PLA2, phospholipase A2; BEL, bromoenol lactone suicide substrate; CAD, collisionally activated dissociation; ESI, electrospray ionization; GPC, glycerophosphocholine; iPLA2β, group VIA phospholipase A2; LPC, lysophosphatidylcholine; MS, mass spectrometry; MS/MS, tandem MS; PAF, platelet-activating factor; PC, phosphatidylcholine; RT, reverse transcriptase; WT, wild type; ER, endoplasmic reticulum; SERCA, sarcoplasmic reticulum Ca2+-ATPase; PBS, phosphate-buffered saline; BSA, bovine serum albumin; AA, arachidonic acid; Ac-LDL, acetyl-low density lipoprotein; ACAT, acyl-CoA:cholesterol O-acyltransferase; JNK, c-Jun NH2-terminal kinase; SRA, scavenger receptor A; sPLA2, secretory PLA2; cPLA2, cytosolic PLA2; KO, knock out.
References
- 1.Six DA, Dennis EA. Biochim Biophys Acta. 2000;1488:1–19. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 2.Ma Z, Turk J. Prog Nucleic Acids Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 3.Brash AR. J Clin Investig. 2001;107:1339–1345. doi: 10.1172/JCI13210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Radu CG, Yang LV, Riedinger M, Au M, Witte ON. Proc Natl Acad Sci U S A. 2004;101:245–250. doi: 10.1073/pnas.2536801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murphy RC, Sala A. Methods Enzymol. 1990;187:90–98. doi: 10.1016/0076-6879(90)87013-s. [DOI] [PubMed] [Google Scholar]
- 6.Gijon MA, Spencer DM, Kaiser AL, Leslie CC. J Cell Biol. 1999;145:1219–1232. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. J Biol Chem. 1998;273:21926–21932. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 8.Pickard RT, Strifler BA, Kramer RM, Sharp JD. J Biol Chem. 1999;274:8823–8831. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]
- 9.Song C, Chang XJ, Bean KM, Proia MS, Knopf JL, Kriz RW. J Biol Chem. 1999;274:17063–17067. doi: 10.1074/jbc.274.24.17063. [DOI] [PubMed] [Google Scholar]
- 10.Ohto T, Uozumi N, Hirabayashi T, Shimizu T. J Biol Chem. 2005;280:24576–24583. doi: 10.1074/jbc.M413711200. [DOI] [PubMed] [Google Scholar]
- 11.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 12.Balboa MA, Balsinde J, Jones SS, Dennis EA. J Biol Chem. 1997;272:8576–8580. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 13.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson J, Turk J. J Biol Chem. 1997;272:11118–11127. [PubMed] [Google Scholar]
- 14.Hazen SL, Zupan LA, Weiss RH, Getman DP, Gross RW. J Biol Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- 15.Balsinde J, Dennis EA. J Biol Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- 16.Ma Z, Ramanadham S, Hu Z, Turk J. Biochim Biophys Acta. 1998;1391:384–400. doi: 10.1016/s0005-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 17.Balsinde J, Dennis EA. J Biol Chem. 1996;271:6758–6765. doi: 10.1074/jbc.271.12.6758. [DOI] [PubMed] [Google Scholar]
- 18.Mancuso DJ, Jenkins CM, Gross RW. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka H, Takeya R, Sumimoto H. Biochem Biophys Res Commun. 2000;272:320–326. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 20.van Tienhoven M, Atkins J, Li Y, Glynn P. J Biol Chem. 2002;277:20942–20948. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 21.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. J Biol Chem. 2004;279:48968–48975. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 22.Balsinde J. Biochem J. 2002;364:695–702. doi: 10.1042/BJ20020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Balsinde J, Balboa MA. Cell Signal. 2005;17:1052–1062. doi: 10.1016/j.cellsig.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 24.Balsinde J, Bianco ID, Ackermann EJ, Conde-Frieboes K, Dennis EA. Proc Natl Acad Sci U S A. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Balsinde J, Balboa MA, Dennis EA. J Biol Chem. 1997;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 26.Baburina I, Jackowski S. J Biol Chem. 1999;274:9400–9408. doi: 10.1074/jbc.274.14.9400. [DOI] [PubMed] [Google Scholar]
- 27.Barbour SE, Kapur A, Deal CL. Biochim Biophys Acta. 1999;1439:77–88. doi: 10.1016/s1388-1981(99)00078-5. [DOI] [PubMed] [Google Scholar]
- 28.Akiba S, Mizunaga S, Kume K, Hayama M, Sato T. J Biol Chem. 1999;274:19906–19912. doi: 10.1074/jbc.274.28.19906. [DOI] [PubMed] [Google Scholar]
- 29.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 30.Jenkins CM, Han X, Mancuso DJ, Gross RW. J Biol Chem. 2002;277:32807–32814. doi: 10.1074/jbc.M202568200. [DOI] [PubMed] [Google Scholar]
- 31.Seegers HC, Gross RW, Boyle WA. J Pharmacol Exp Ther. 2002;302:918–923. doi: 10.1124/jpet.302.3.918. [DOI] [PubMed] [Google Scholar]
- 32.Perez R, Melero R, Balboa MA, Balsinde J. J Biol Chem. 2004;279:40385–40391. doi: 10.1074/jbc.M402562200. [DOI] [PubMed] [Google Scholar]
- 33.Yellaturu CR, Rao GN. J Biol Chem. 2003;278:43831–43837. doi: 10.1074/jbc.M301472200. [DOI] [PubMed] [Google Scholar]
- 34.Smani T, Zakharov SI, Csutora P, Leno E, Trepakova ES, Bolotina VM. Nat Cell Biol. 2004;6:113–120. doi: 10.1038/ncb1089. [DOI] [PubMed] [Google Scholar]
- 35.Martinson BD, Albert CJ, Corbett JA, Wysolmerski RB, Ford DA. J Lipid Res. 2003;44:1686–1691. doi: 10.1194/jlr.M300018-JLR200. [DOI] [PubMed] [Google Scholar]
- 36.Moran JM, Buller RM, McHowat J, Turk J, Wohltmann M, Gross RW, Corbett JA. J Biol Chem. 2005;280:28162–28168. doi: 10.1074/jbc.M500013200. [DOI] [PubMed] [Google Scholar]
- 37.Guo Z, Su W, Ma Z, Smith GM, Gong MC. J Biol Chem. 2003;278:1856–1863. doi: 10.1074/jbc.M211075200. [DOI] [PubMed] [Google Scholar]
- 38.Balboa MA, Saez Y, Balsinde J. J Immunol. 2003;170:5276–5280. doi: 10.4049/jimmunol.170.10.5276. [DOI] [PubMed] [Google Scholar]
- 39.Song K, Zhang X, Zhao C, Ang NT, Ma ZA. Mol Endocrinol. 2005;19:504–515. doi: 10.1210/me.2004-0169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Larsson Forsell PK, Kennedy BP, Claesson HE. Eur J Biochem. 1999;262:575–585. doi: 10.1046/j.1432-1327.1999.00418.x. [DOI] [PubMed] [Google Scholar]
- 41.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 1999;274:9607–9616. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. J Biol Chem. 1998;273:13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 43.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou YP, Teng D, Dralyuk F, Ostrega D, Roe MW, Philipson L, Polonsky K. J Clin Investig. 1998;101:1623–1632. doi: 10.1172/JCI1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nowatzke W, Ramanadham S, Ma Z, Hsu FF, Bohrer A, Turk J. Endocrinology. 1998;139:4073–4085. doi: 10.1210/endo.139.10.6225. [DOI] [PubMed] [Google Scholar]
- 46.Ramanadham S, Hsu FF, Zhang S, Jin C, Bohrer A, Ma Z, Turk J. Biochemistry. 2004;43:918–930. doi: 10.1021/bi035536m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ross R. Annu Rev Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- 48.Ball RY, Stowers ED, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Atherosclerosis. 1995;114:45–54. doi: 10.1016/0021-9150(94)05463-s. [DOI] [PubMed] [Google Scholar]
- 49.Yao PM, Tabas I. J Biol Chem. 2000;275:23807–23813. doi: 10.1074/jbc.M002087200. [DOI] [PubMed] [Google Scholar]
- 50.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Rong D, Tabas I. Nat Cell Biol. 2003;5:781–792. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 51.Li Y, Ge M, Ciani L, Kuriakose G, Westover EJ, Dura M, Covey DF, Freed JH, Mayfield FR, Lytton J, Tabas I. J Biol Chem. 2004;279:37030–37039. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- 52.De Vries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. J Cell Biol. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Proc Natl Acad Sci U S A. 2006;103:19794–19799. doi: 10.1073/pnas.0609671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194–38200. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ross AC, Go KJ, Heider JG, Rothblat GH. J Biol Chem. 2004;259:815–819. [PubMed] [Google Scholar]
- 56.Basu SK, Goldstein JL, Anderson RGW, Brown MS. Proc Natl Acad Sci U S A. 1976;73:3178–3182. doi: 10.1073/pnas.73.9.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bao S, Song H, Wohltmann M, Bohrer A, Turk J. J Biol Chem. 2006;281:20958–20973. doi: 10.1074/jbc.M600075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 59.Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 60.Ramanadham S, Hsu FF, Bohrer A, Ma Z, Turk J. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 61.Qiu ZH, de Carvalho MS, Leslie CC. J Biol Chem. 1993;32:24506–24513. [PubMed] [Google Scholar]
- 62.Gijon MA, Spener DM, Siddiqi AR, Bonventre JV, Leslie CC. J Biol Chem. 2000;275:20146–20156. doi: 10.1074/jbc.M908941199. [DOI] [PubMed] [Google Scholar]
- 63.Krippner A, Matsuno-Yagi A, Gottlieb RA, Babior BM. J Biol Chem. 1996;271:21629–21636. doi: 10.1074/jbc.271.35.21629. [DOI] [PubMed] [Google Scholar]
- 64.Sayan BS, Sayan AE, Knight RA, Melino G, Cohen GM. J Biol Chem. 2006;281:13566–13573. doi: 10.1074/jbc.M512467200. [DOI] [PubMed] [Google Scholar]
- 65.Barrett LE, Van Vockstaele EJ, Sul JY, Takano H, Haydan PG, Eberwine JH. Proc Natl Acad Sci U S A. 2006;103:5155–5160. doi: 10.1073/pnas.0510477103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hallberg A. Biochim Biophys Acta. 1984;796:328–335. doi: 10.1016/0005-2760(84)90134-6. [DOI] [PubMed] [Google Scholar]
- 67.Hsu FF, Bohrer A, Turk J. J Am Soc Mass Spectrom. 1998;9:516–526. doi: 10.1016/S1044-0305(98)00012-9. [DOI] [PubMed] [Google Scholar]
- 68.Hsu FF, Turk J, Thukkani AK, Messner MC, Wildsmith KR, Ford DA. J Mass Spectrom. 2003;38:752–763. doi: 10.1002/jms.491. [DOI] [PubMed] [Google Scholar]
- 69.Hsu FF, Turk J. J Am Soc Mass Spectrom. 2003;14:352–363. doi: 10.1016/S1044-0305(03)00064-3. [DOI] [PubMed] [Google Scholar]
- 70.Bao S, Bohrer A, Ramanadham S, Jin W, Zhang S, Turk J. J Biol Chem. 2006;281:187–198. doi: 10.1074/jbc.M509105200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramanadham S, Hsu FF, Zhang S, Bohrer A, Ma Z, Turk J. Biochim Biophys Acta. 2000;1484:251–266. doi: 10.1016/s1388-1981(00)00022-6. [DOI] [PubMed] [Google Scholar]
- 72.Yang HC, Mosior M, Ni B, Dennis EA. J Neurochem. 1999;73:1278–1287. doi: 10.1046/j.1471-4159.1999.0731278.x. [DOI] [PubMed] [Google Scholar]
- 73.Zinszner H, Kuroda M, Wang Z, Batchvarova N, Lightfoot R, Remotti H, Stevens JL, Ron D. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. Biochem Biophys Res Commun. 2006;340:286–290. doi: 10.1016/j.bbrc.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 75.Li J, Lee B, Lee AS. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- 76.Hsu FF, Ma Z, Wohltmann M, Bohrer M, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 2000;275:16579–16589. doi: 10.1074/jbc.M908342199. [DOI] [PubMed] [Google Scholar]
- 77.Han X, Yang K, Yang J, Fikes KN, Cheng H, Gross RW. J Am Soc Mass Spectrom. 2005;17:264–274. doi: 10.1016/j.jasms.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 78.Williams SD, Gottlieb RA. Biochem J. 2002;362:23–32. doi: 10.1042/0264-6021:3620023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Broekemeier KM, Iben JR, LeVan EG, Crouser ED, Pfeiffer DR. Biochemistry. 2002;41:7771–7780. doi: 10.1021/bi020157z. [DOI] [PubMed] [Google Scholar]
- 80.Brustovetsky T, Antonsson B, Jemmerson R, Dybinsky JM, Brustovetsky N. J Neurochem. 2005;94:980–984. doi: 10.1111/j.1471-4159.2005.03248.x. [DOI] [PubMed] [Google Scholar]
- 81.Gadd ME, Broekemeier KM, Crouser ED, Kumar J, Graff G, Pfeiffer DR. J Biol Chem. 2006;282:6931–6939. doi: 10.1074/jbc.M510845200. [DOI] [PubMed] [Google Scholar]
- 82.Yao PM, Tabas I. J Biol Chem. 2001;45:42468–44247. doi: 10.1074/jbc.M101419200. [DOI] [PubMed] [Google Scholar]
- 83.Zhang D, Tang W, Yao PM, Yang C, Xie B, Jackowski S, Tabas I. J Biol Chem. 2000;275:35368–35376. doi: 10.1074/jbc.M007099200. [DOI] [PubMed] [Google Scholar]
- 84.Ma Z, Ramanadham S, Wohltmann M, Bohrer A, Hsu FF, Turk J. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 85.Kuwae T, Schmid PC, Schmid HHO. Biochim Biophys Acta. 1997;1344:74–86. doi: 10.1016/s0005-2760(96)00135-x. [DOI] [PubMed] [Google Scholar]
- 86.Luberto C, Hannun YA. J Biol Chem. 1998;272:14550–14559. doi: 10.1074/jbc.273.23.14550. [DOI] [PubMed] [Google Scholar]
- 87.Vivekananda J, Smith D, King RJ. Am J Physiol. 2001;281:L98–L107. doi: 10.1152/ajplung.2001.281.1.L98. [DOI] [PubMed] [Google Scholar]
- 88.Yamaoka S, Miyaji M, Kitano T, Umehara H, Okasaki T. J Biol Chem. 2004;279:18688–18693. doi: 10.1074/jbc.M401205200. [DOI] [PubMed] [Google Scholar]
- 89.Miyaji M, Jin ZX, Yamaoka S, Amakawa R, Fukuhara S, Sato SB, Kobayashi T, Domae N, Mimori T, Bloom ET, Oakzaki T, Umehara H. J Exp Med. 2005;202:249–259. doi: 10.1084/jem.20041685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Slotte JP. Chem Phys Lipids. 1999;102:13–17. doi: 10.1016/s0009-3084(99)00071-7. [DOI] [PubMed] [Google Scholar]
- 91.Leventhal A, Chen W, Tall AR, Tabas I. J Biol Chem. 2001;276:44976–44983. doi: 10.1074/jbc.M106455200. [DOI] [PubMed] [Google Scholar]
- 92.Surette ME, Winkler JD, Fonteh AN, Chilton FH. Biochemistry. 1996;35:9187–9196. doi: 10.1021/bi9530245. [DOI] [PubMed] [Google Scholar]
- 93.Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR, Poirier GG, Salvesen GS, Dixit VM. Cell. 1995;81:801–809. doi: 10.1016/0092-8674(95)90541-3. [DOI] [PubMed] [Google Scholar]
- 94.Ivana Scovassi A, Diederich M. Biochem Pharmacol. 2004;68:1041–1047. doi: 10.1016/j.bcp.2004.04.023. [DOI] [PubMed] [Google Scholar]
- 95.Wang ZB, Liu YQ, Cui YF. Cell Biol Int. 2005;29:489–496. doi: 10.1016/j.cellbi.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 96.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. J Biol Chem. 2006;281:22275–22288. doi: 10.1074/jbc.M604330200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wolf MJ, Wang J, Turk J, Gross RW. J Biol Chem. 1997;272:1522–1526. doi: 10.1074/jbc.272.3.1522. [DOI] [PubMed] [Google Scholar]
- 98.Wang Z, Ramanadham S, Ma Z, Bao S, Mancuso DJ, Gross RW, Turk J. J Biol Chem. 2005;280:6840–6849. doi: 10.1074/jbc.M405287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Roe MW, Worley JF, Qian F, Tamarina N, Mittal AA, Dralyuk F, Blair NT, Mertz RJ, Philipson LH, Dukes ID. J Biol Chem. 1998;273:10402–10410. doi: 10.1074/jbc.273.17.10402. [DOI] [PubMed] [Google Scholar]
- 100.Williams SD, Hsu FF, Ford DA. J Lipid Res. 2000;41:1585–1595. [PubMed] [Google Scholar]
- 101.Scott WA, Zrike JM, Hamill AL, Kempe J, Cohn ZA. J Exp Med. 1980;152:324–335. doi: 10.1084/jem.152.2.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ramanadham S, Song H, Hsu FF, Zhang S, Crankshaw M, Grant GA, Newgard CB, Bao S, Ma Z, Turk J. Biochemistry. 2003;42:13929–13940. doi: 10.1021/bi034843p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song H, Hecimovic S, Goate A, Hsu FF, Bao S, Vidavsky I, Ramanadham S, Turk J. J Am Soc Mass Spectrom. 2004;15:1780–1793. doi: 10.1016/j.jasms.2004.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.