

Abstract
Chemokines and their receptors play important roles in normal physiological functions and the pathogeneses of a wide range of human diseases, including the entry of human immunodeficiency virus type 1 (HIV-1). However, the use of natural chemokines to probe receptor biology or to develop therapeutic drugs is limited by their lack of selectivity and the poor understanding of mechanisms in ligand-receptor recognition. We addressed these issues by combining chemical and structural biology in research into molecular recognition and inhibitor design. Specifically, the concepts of chemical biology were used to develop synthetically and modularly modified (SMM) chemokines that are unnatural and yet have properties improved over those of natural chemokines in terms of receptor selectivity, affinity, and the ability to explore receptor functions. This was followed by using structural biology to determine the structural basis for synthetically perturbed ligand-receptor selectivity. As a proof-of-principle for this combined chemical and structural-biology approach, we report a novel d-amino acid-containing SMM-chemokine designed based on the natural chemokine called viral macrophage inflammatory protein II (vMIP-II). The incorporation of unnatural d-amino acids enhanced the affinity of this molecule for CXCR4 but significantly diminished that for CCR5 or CCR2, thus yielding much more selective recognition of CXCR4 than wild-type vMIP-II. This d-amino acid-containing chemokine also showed more potent and specific inhibitory activity against HIV-1 entry via CXCR4 than natural chemokines. Furthermore, the high-resolution crystal structure of this d-amino acid-containing chemokine and a molecular-modeling study of its complex with CXCR4 provided the structure-based mechanism for the selective interaction between the ligand and chemokine receptors and the potent anti-HIV activity of d-amino acid-containing chemokines.
Protein-protein interactions play important roles in a wide variety of physiological and pathological processes. The inhibition or promotion of these interactions, by either small or relatively large synthetic molecules, is of great interest for understanding the mechanism of biological recognition and developing novel therapeutic agents. In this regard, much progress has been made in recent years (3, 8, 29). This type of chemical research in protein-protein interactions is becoming increasingly important, especially in the postgenomic era, as chemically synthesized regulators of protein-protein interactions can be used to study the functions of new proteins uncovered by genomic-research efforts.
One of the most important and challenging questions in the field of protein-protein interactions and development of intervening agents is selectivity in protein-protein interactions. Specifically, what are the mechanisms that dictate how one protein recognizes another out of a myriad of biological molecules, especially when the interacting partners in the same protein family share structural or functional homology? Alternatively, the question can be asked in terms of how some proteins can interact with multiple protein partners from the same protein family, leading to the functional cross-activity and redundancy that, like monospecificity, is also commonly observed in protein-protein interaction networks involved in biological recognition or signal transduction. Understanding the mechanism for such selectivity or nonselectivity in protein-protein interactions at the structural and chemical levels is crucial if one seeks to engineer de novo selectivity into natural, nonselective protein-protein interfaces to develop protein functional probes or therapeutic inhibitors for which high selectivity is of the utmost importance. Here, the interactions among chemokines and their receptors were used as model systems to address the issue of selectivity in protein-protein interactions. An integrated approach combining chemical and structural biology was utilized to probe the chemical and structural bases of selectivity versus nonselectivity of chemokine ligands for their receptors and to design de novo ligand molecules with higher receptor selectivity.
Chemokines and their receptors play important roles in normal physiology and the pathogeneses of a wide range of human diseases, including multiple neurological disorders, cancer, and most notably AIDS (3, 4, 8, 29, 35). Chemokine receptors belong to the superfamily of G-protein-coupled receptors (GPCRs). As the natural ligands of chemokine receptors, chemokines act as chemoattractants of various types of leukocytes to sites of inflammation and to secondary lymphoid organs, and they can be divided into two main subfamilies, CXC and CC proteins, based on the positions of two conserved cysteine residues in the amino (N) terminus (3, 4, 29, 35). Two chemokine receptors, CXCR4 and CCR5, act as the principal coreceptors for human immunodeficiency virus type 1 (HIV-1) entry (16, 20, 44, 46). While M-tropic strains of HIV-1 primarily use CCR5 as an entry coreceptor during the asymptomatic stage of disease (1, 12, 14), in 40 to 50% of HIV-infected individuals, T-tropic strains that use CXCR4 eventually replace M-tropic strains, which is associated with rapid disease progression (7, 38, 41). Natural chemokine ligands of CXCR4 or CCR5 can inhibit HIV-1 infection (5, 32) by blocking virus-binding sites on the receptor and/or inducing receptor internalization (2, 16).
Due to the importance of chemokines in numerous physiological and pathological processes, the use of these ligands as research probes to analyze the functions of their receptors and as potential therapeutic agents to prevent relevant disease processes has been the subject of intense research. However, such efforts are greatly hindered by a number of intrinsic limitations of natural chemokines. Most notably, there is a lack of selectivity in chemokine-receptor interactions. With the exception of some natural chemokines, such as stromal cell-derived factor 1α (SDF-1α), which is specific for its receptor, CXCR4, many of the 50 identified natural chemokines recognize more than one receptor among the known chemokine receptors (8, 35). The lack of selectivity is best exemplified by viral macrophage inflammatory protein II (vMIP-II), which recognizes a variety of CC and CXC chemokine receptors, including CXCR4, CCR5, and CCR2 (28). Thus, the cross-activity of natural chemokines makes it difficult to use them to analyze and dissect the roles of a particular ligand-receptor pair among the complicated chemokine and receptor networks. Due to the lack of receptor selectivity, natural chemokines may have unwanted side effects in clinical applications, as they may react with multiple receptors. Also, there is cause for concern regarding undesired side effects of blocking the normal CXCR4 function, since knockout mice lacking either CXCR4 (40, 50) or its only natural ligand, SDF-1α (30), die during embryogenesis, with evidence of hematopoietic, cardiac, vascular, and cerebellar defects. Consequently, the development of new inhibitors engineered with higher selectivity for specific regions of CXCR4 that are selective for HIV-1 coreceptor function only, but not the normal function of SDF-1α, is clearly desirable. In fact, we have recently reported potentially different determinants for CXCR4 interactions with HIV-1 gp120 and SDF-1α, which provided a basis for the development of new inhibitory agents that modulate the functional sites or conformations of CXCR4 for the purpose of reducing or avoiding the limitations and side effects caused by nonselective inhibitors of this important coreceptor (9, 42). Although a number of natural chemokines have been shown to inhibit human diseases, such as HIV-1 infection (5, 22, 32), these natural chemokines may not be suitable for clinical applications unless they are modified to increase the target receptor selectivity and to reduce side effects.
To address this issue of selectivity in chemokine ligand-receptor interactions, we have developed a chemical and protein structure-based strategy that employs synthetically and modularly modified (SMM) chemokines (8, 9, 13, 23). In this approach, synthetic chemistry is applied to introduce unnatural amino acids or novel chemical modifications into the important functional-sequence modules of native chemokines, such as vMIP-II and SDF-1α, to yield new molecules with high receptor selectivity and other improved biological and pharmacological properties. To demonstrate a proof of the principle, we applied this SMM chemokine approach to convert the nonspecific vMIP-II into a highly selective ligand of CXCR4 by replacing the first N-terminal (amino acids 1 to 10) sequence module of vMIP-II with unnatural d-amino acids. This new molecule, termed RCP168, displayed enhanced selectivity and potency for binding CXCR4 and inhibiting HIV-1 infection via this coreceptor. The high-resolution crystal structure of RCP168, compared with that of vMIP-II, revealed that the enhanced selectivity of RCP168 was associated with structural changes, not only at the N terminus due to the d-amino acid modification, but also, surprisingly, in the 30s loop through a mechanism involving conformational changes in the 30s loop propagated from the N terminus by a disulfide bridge linking these two sequentially distal regions. This provided a structural mechanism for the enhanced selectivity of the ligand for CXCR4 and suggested new approaches in designing receptor-selective chemokine analogs. Finally, molecular-modeling studies were performed on possible models of RCP168-CXCR4 and SDF-1α-CXCR4 complexes to determine the origin of differential binding requirements for RCP168 and SDF-1α. These studies may explain the enhanced anti-HIV-1 potency of RCP168 over SDF-1α.
MATERIALS AND METHODS
Total chemical synthesis of SMM chemokines.
The automated stepwise incorporation of protected amino acids was performed using an Applied Biosystems 433A peptide synthesizer (Foster City, CA) with a CLEAR amide resin (Peptides International, Louisville, KY) as the solid support, as described previously (9, 13, 23). 9-Fluorenylmethoxy carbonyl chemistry was employed for the synthesis (13, 23). 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate and N-hydroxybenzotriazole were used as coupling reagents in the presence of diisopropylethylamine. In certain coupling steps with potentially low reaction rates, double coupling, followed by capping of the unreacted amino functional groups, was performed. After incorporation of the 50th residue, 2% (vol/vol) dimethyl sulfoxide was introduced into the solution to enhance the coupling reaction. After N-terminal 9-fluorenylmethoxy carbonyl protection was removed, the protein was cleaved from the resin support by adding a cleavage cocktail comprised of phenol (4% [wt/vol]), thioanisole (5% [vol/vol]), water (5% [vol/vol]), ethanedithiol (2.5% [vol/vol]), triisopropylsilane (1.5% [vol/vol]), and trifluoroacetic acid (TFA) (82% [vol/vol]). The protein was precipitated by adding ice-cold tert-butyl methyl ether and washed repeatedly in cold ether. The crude protein was dissolved in 25% CH3CN in water containing 0.1% TFA before being lyophilized, and it was dissolved in water and purified using semipreparative reverse-phase high-performance liquid chromatography. Folding of the purified protein was performed in 1 M guanidinium hydrochloride and 0.1 M trisma base at pH 8.5 (1 mg protein/ml folding buffer) and was monitored by analytical reverse-phase high-performance liquid chromatography using a Vydac C18 column (0.46 by 15 cm; 5 μm) with a flow rate of 1 ml/min (solvent A, water with 0.1% TFA; solvent B, 20% water in CH3CN with 0.1% TFA) and a linear gradient of 30 to 70% B over 30 min. Protein desaltation and purification were then performed. The purified protein was characterized by matrix-assisted laser desorption ionization-time of flight mass spectrometry.
Single-round virus inhibition assay.
293T human embryonic kidney and Cf2Th canine thymocytes (ATCC, Manassas, VA) were grown at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium (CAMBREX, Walkersville, MD) containing 10% fetal bovine serum (CAMBREX) and 100 μg/ml penicillin-streptomycin (CAMBREX). Cf2Th cells stably expressing human CD4 and CXCR4 (15) were grown in the medium supplemented with 0.4 mg/ml G418 (CAMBREX) and 0.15 mg/ml hygromycin B (Roche Diagnostics, Basel, Switzerland). The 293T cells were cotransfected with vectors expressing the pCMVΔP1Δenv HIV Gag-Pol packaging construct (34), the envelope glycoproteins of HIV-1 isolates (HXBc2 or JR-FL), and a firefly luciferase reporter gene at a DNA ratio of 1:1:3 μg using Effectene transfection reagent (QIAGEN, Valencia, CA). Cotransfection produced single-round, replication-defective viruses. Virus-containing supernatants were harvested 24 to 30 h after transfection, filtered (0.45 μm), aliquoted, and frozen at −80°C until further use. The reverse transcriptase activities of all the viruses were measured as described previously (37). To determine infection by single-round luciferase viruses, Cf2Th-CD4-CXCR4 target cells were seeded at a density of 6 × 103 cells/well in 96-well luminometer-compatible tissue culture plates (Dynex, Chantilly, VA) 24 h before infection. On the day of infection, synthetic chemokines were added to the target cells and incubated for 1 h at 37°C. Following incubation, recombinant viruses (10,000 RT units), to a final volume of 50 μl, were added to the chemokine-cell mixtures and incubated for 48 h at 37°C. The medium was removed from each well, and the cells were lysed with 30 μl passive lysis buffer (Promega, Madison, WI) and by three freeze-thaw cycles. An EG&G Berthold Microplate Luminometer LB 96V was used to measure the luciferase activity of each well after the addition of 100 μl luciferin buffer (15 mM MgSO4, 15 mM KPO4, pH 7.8, 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM d-luciferin potassium salt (BD Pharmingen, San Diego, CA).
Virus infection assay.
To assess the inhibition of replication-competent HIV-1 infection by RCP168, virus infection assays were performed using MAGI X4 cells (Division of AIDS, National Institute of Allergy and Infectious Disease, National Institutes of Health). MAGI X4 cells express high levels of CXCR4 and contain one copy of the HIV-1 long terminal repeat promoter that drives the expression of the β-galactosidase gene upon Tat transactivation. A day before the assay, MAGI X4 cells were plated on 96-flat-well plates. On the day of the assay, the media were removed while RCP168 and a known titer of virus (HIV-1IIIB) were added to the cells. The cells were incubated for 48 h, after which they were washed with phosphate-buffered saline. β-Galactosidase enzyme expression was determined by chemiluminescence (Perkin-Elmer Applied Biosystems) according to the manufacturer's instructions. Also, to determine cell viability and the toxicity of RCP168, the cells were stained with MTS (Promega, Madison, WI) at the termination of the assay. The conversion of MTS into soluble formazan is accomplished by dehydrogenase enzymes found only in metabolically active cells. Thus, the quantity of the formazan product (solubilized MTS) is directly proportional to the number of living cells in culture. The plates were incubated at 37°C for 4 to 6 h. The plates, sealed with adhesive plate sealers, were inverted several times to mix the soluble formazan before being read spectrophotometrically at 490/650 nm with a Molecular Devices Vmax or SpectraMax Plus plate reader. Antiviral compounds, AMD-3100 and Chicago Sky Blue, were used as positive controls.
Crystallization, data collection, and structure determination.
RCP168 was crystallized by the hanging-drop vapor diffusion method, and its structure was solved using the molecular-replacement method. The reservoir solution was 0.2 M (NH4)2HPO4 (pH = 7.9), 20% (wt/vol) polyethylene glycol 4000. RCP168 (15 mg/ml) was mixed with an equal volume of reservoir solution and 10% (vol/vol) polyethylene glycol 400. The plate was incubated at 15°C. Crystals began to form after 2 to 3 weeks. To collect the diffraction data, the crystals were first briefly soaked in cryoprotectant (35% sucrose) and frozen in liquid nitrogen. The data were collected at the SBC 19BM beamline of the Advanced Photon Source at Argonne National Laboratory (Argonne, IL) and processed with HKL2000 software (33). The data were 96.9% complete in the resolution range of 30.0 to 2.0 Å. The coordinates of residues 11 to 71 from the A chain of vMIP-II dimer (Protein Data Bank code, 1CM9.pdb) (18) were used as the search model. Rigid-body refinement was applied to the model using AmoRe (31), which was followed by structural refinement with Shelxl-97 (39). After the refinement, the coverage of residues by electron density was checked on a 2Fo-Fc map. The backbone atoms of most residues were well covered by the electron density, except the residues in the 30s loop region (such as Gln33 and Leu34), which were partially or not covered. The positions of these residues were manually adjusted in program O (21). The structure was then refined in Shelxl-97. To determine the electron density for the d-amino acid residues, the omit map was generated using the coordinates of residues 11 to 71. The backbone atoms of residues Trp5 to Arg7 and all the atoms of Pro8 to Lys10 were added according to the electron density on the omit map. The structure was further refined by simulated annealing with XPLOR (6) in the resolution range of 10.0 to 2.0 Å. Solvent molecules were added and verified by the electron density map. After the temperature factor refinement, solvent molecules with B factors greater than 70 were removed. The final structure of RCP168 gave an R factor of 0.235 and an Rfree of 0.290.
Molecular modeling of ligand-CXCR4 interactions.
Molecular-modeling studies of ligand-CXCR4 complexes were carried out using a set of procedures previously developed by our group (19, 47, 49). The same protocol was used to develop the homology model. The molecular-dynamics (MD) simulation was performed using Sybyl 7.2 and a Tripos force field (43). During the MD simulations, only residues in the extracellular loops of CXCR4 and all residues in ligands were allowed to move, whereas the rest of the CXCR4 residues were frozen to their positions in the homology model. A distance constraint of ∼1.35 Å was applied to the amide bonds at the interface between the atoms that were allowed to move and atoms that were frozen. In each MD simulation, the temperature of the complex was initially increased from 0 K to 300 K in 600 fs. The system was equilibrated at 300 K for an additional 200 fs. Finally, the MD simulation was performed for 200 ps while the temperature of the system was maintained at 300 K. The X-ray structures of RCP168 as reported here and wild-type SDF-1α (11) were used to generate complexes with the CXCR4 model.
RESULTS
RCP168 selectively binds CXCR4 as an antagonist and potently inhibits HIV-1 entry via CXCR4.
To demonstrate the concept of SMM chemokines and develop nonselective natural chemokines into both receptor-selective inhibitors and mechanistic probes of ligand-receptor interactions, we used vMIP-II as a template, as the engineering of selectivity into the nonselective vMIP-II would provide a proof of principle. We previously found that d-amino acid-containing peptides derived from the N terminus of vMIP-II displayed selective binding to CXCR4 and not to other chemokine receptors (48), suggesting that d-amino acids in this region might increase selectivity for CXCR4. Therefore, we synthesized RCP168, a new analog of vMIP-II with the first 10 N-terminal residues changed from l- to d-amino acids (vMIP-II, LGASWHRPDKCCLGYQKRPLPQVLLSSWYPTSQLCSKPGVIFLTKRGRQVCADKSKDWVKKLMQQLPVTAR; RCP168, LGASWHRPDKCCLGYQKRPLPQVLLSSWYPTSQLCSKPGVIFLTKRGRQVCADKSKDWVKKLMQQLPVTAR [the d-amino acids are shown in italics]). Interestingly, despite the incorporation of d-amino acids and the expected conformational changes, RCP168 displayed very high binding affinity for CXCR4 (50% inhibitory concentration [IC50] = 5 nM in competition with radioisotope-labeled SDF-1α), about four times stronger than vMIP-II (IC50 = 22 nM), as reported previously (23). Similar results were obtained from antibody competition binding assays using a CXCR4 monoclonal antibody, 12G5, where RCP168 displayed higher binding affinity for CXCR4 (IC50 = 5 nM) than vMIP-II (IC50 = 10 nM) (data not shown). More importantly, in sharp contrast to vMIP-II, which binds multiple chemokine receptors, including CXCR4, CCR5, and CCR2, RCP168 showed drastically improved CXCR4 selectivity, as RCP168 had much lower binding activities toward CCR5 (IC50 = 43 nM) and CCR2 (IC50 = 513 nM), as we previously reported (23). As for the signaling activity, like the parent molecule, vMIP-II, which is a known antagonist of CXCR4, RCP168 induced neither calcium (Ca2+) mobilization nor CXCR4 internalization, as we previously reported (23). This is in contrast to the natural CXCR4 ligand, SDF-1α, which is capable of inducing both, suggesting that RCP168 acts as an antagonist of CXCR4.
The ability of RCP168 to inhibit HIV-1 entry via CXCR4 was previously examined in our laboratory (23) using a panel of HIV-1 strains resistant to commonly used reverse transcriptase inhibitors, such as zidovudine. Here, we further tested the activity of RCP168 in inhibiting additional HIV-1 isolates and compared the potency of RCP168 with that of the natural chemokine SDF-1α or an approved HIV-1 drug, T20, which inhibits HIV-1 entry by targeting the viral gp41 glycoprotein. In single-round virus inhibition assays, RCP168 was very active in blocking HIV-1 entry and more potent than SDF-1α (Fig. 1A). The anti-HIV activity of RCP168 was also demonstrated in different antiviral assays, namely, virus infection assays, which gave the IC50 of RCP168 as 50 nM (Fig. 1B). Consistent with its receptor selectivity, RCP168 did not block a CCR5-preferring, M-tropic HIV-1 strain from infecting target cells (Fig. 1A). Note that the inhibitory activity of RCP168 was not due to its toxicity, as it did not have any effect on cell viability at concentrations up to 100 μM (23). To further assess the potential of RCP168 for clinical applications, we compared its antiviral activity with that of the HIV-1 entry inhibitor drug T20. As shown in Fig. 1C, RCP168 showed antiviral activity comparable to that of T20.
FIG. 1.

Antiviral activity of RCP168. (A) RCP168 was more potent in blocking HIV-1 entry than SDF-1α and was highly selective in blocking T-tropic HIV-1 strains by single-round virus inhibition assays. (B) The strong antiviral activity of RCP168 was also demonstrated by virus infection assays. (C) RCP168 showed antiviral effects comparable to those of the T20 peptide, a recently approved drug targeting viral gp41 protein-mediated HIV-1 entry. The error bars indicate standard deviations.
Structural basis for the CXCR4 selectivity of RCP168.
To understand the structure-based mechanism of action of RCP168, we determined the crystal structure of RCP168. The structure includes residues 5 to 71 and 23 water molecules. Residues 1 to 4 were missing in the structure, as the electron density for these four residues was not visible on the density map. The tertiary structure of RCP168 displayed the typical chemokine fold, consisting of a flexible N terminus followed by three antiparallel β-strands (residues 25 to 30, 39 to 44, and 49 to 53) arranged in a Greek key motif with one C-terminal α-helix (residues 57 to 65) laid on the top (Fig. 2A). The overall structure is stabilized by two disulfide bonds (Cys11-Cys35 and Cys12-Cys51) and a conserved hydrophobic core formed around the side chain of Phe42 (18). Most l-amino acid residues (80.4%) are distributed in the most favored regions on the Ramachandran space, while the rest of the l-amino acids are in the additional allowed regions. As for the d-amino acids, Pro8 and Lys10 are located in the “invert beta” area on the Ramachandran space, while His6 and Arg7 are located in the vicinity of the invert beta area. None of the d-amino acids occupied the “inverted α-helix (L)” region around −60° and −45°, which is consistent with the alpha helix-destabilizing properties of d-amino acids and the propensity of d-amino acids to promote β-turn formation (27, 45). All of the amino acid residues, including both d and l forms, adopt the trans conformation for the amide bond. The flexibility of the polypeptide chain is reflected in the average B factors of the backbone atoms in the crystal structure. Comparing the B factor of RCP168 with that of the previously reported 74-residue vMIP-II, the average B factor for the main chain of RCP168 is significantly lower than that of vMIP-II.
FIG. 2.

(A) Superposition of the crystal structures of 2.1-Å vMIP-II (green) and RCP168 (blue). (B) Structural comparison of the N termini of vMIP-II (green) and RCP168 (blue). (C) Structural comparison of the 30s loops of vMIP-II and RCP168. Only the backbone atoms of residues Lys10 to Tyr15 and Pro30 to Gly39 are shown for clarity.
To understand why RCP168 containing d-amino acids at its N terminus displays higher affinity and selectivity for CXCR4 than wild-type vMIP-II, we compared the crystal structure of RCP168 as described above to that of vMIP-II, previously reported by others and our laboratory (18, 24, 25), to find the structural difference between the two ligands that could account for their different selectivities for CXCR4. Although RCP168 and vMIP-II adopt similar folds in their core structures, as displayed by the well-superimposed C-terminal helix and three antiparallel β-strands, significant structural differences were found in the N-terminal and the 30s loop regions (Fig. 2A). As shown in Fig. 2B, the incorporation of d-amino acids in RCP168 induced significant changes in both main and side chain torsional angles of the N terminus compared with vMIP-II. In RCP168, Lys10 to Cys11 is the transition point from d- to l-amino acids. The structural changes in d-amino acid residues 5 to 10 also affected the conformation of the adjacent residues, Cys11, Cys12, and Leu13 to Tyr15, within the N-terminal region.
Besides the structural changes for the modified N-terminal region, it was interesting to find the conformational changes in the 30s loop of RCP168 (residues Thr31 to Lys37), which is distal from the d-amino acid region of residues 1 to 10 (Fig. 2C). These changes in the 30s loop can be related to those in the d-amino acid region through the disulfide bridge between Cys11 at the N terminus and Cys35 in the 30s loop. As discussed above, a structural change was seen in the main chain and side chain of Cys11 in RCP168. As a result, the orientation of the disulfide bond connecting Cys11 to Cys35 was also changed, which could lead to the movement of the 30s loop in RCP168. Note that Thr31 and Lys37 are flanked on each end by two proline residues, Pro30 and Pro38. Proline is known to have more restricted conformational freedom than other amino acids. This may explain why the structural impact relayed from the N terminus to the 30s loop through the Cys11-Cys35 disulfide bond is localized within the Thr31-to-Lys37 region.
Molecular modeling of RCP168-CXCR4 and SDF-1α-CXCR4 complexes.
The experimental result that RCP168 is very active in blocking HIV-1 entry with higher potency than SDF-1α prompted us to investigate the mechanism of the recognition of RCP168 by CXCR4. Our previous mutagenesis study showed that RCP168 and wild-type SDF-1α interact with CXCR4 very differently (9) (Fig. 3A). The study revealed that mutations at CXCR4 residues Tyr45, Phe87, Asp97, Tyr121, Asp171, Trp252, Tyr255, Glu288, and Phe292 diminish the affinity of RCP168 by 30 to 100%. While most of these mutations have no effect on SDF-1α binding, mutations at Phe87, Asp171, and Phe292 drastically reduce the binding activity of SDF-1α. Interestingly, visual inspection of the CXCR4 model shows that four of the residues (Tyr121, Trp252, Tyr255, and Glu288) that affect RCP168 binding are located in a region defined as site A inside a groove in the transmembrane (TM) domain (Fig. 3B). Furthermore, Phe87 and Phe292 in CXCR4, which affect SDF-1α binding, are located in the same groove in the TM domain but in a different region defined as site B (Fig. 3B). Thus, the mutational data (9), together with analysis of the CXCR4 model, suggest that the N termini of both ligands possibly bind CXCR4 differently, as the N terminus of RCP168 binds to site A whereas that of SDF-1α binds to site B. The conformational change introduced by the incorporation of unnatural d-amino acids in the N terminus of wild-type vMIP-II probably resulted in a binding mode of the N terminus of RCP168 different from that of SDF-1α. In order to probe the conformational properties of the N termini of RCP168 and SDF-1α, we conducted MD simulations of RCP168 and SDF-1α in complex with CXCR4.
FIG. 3.

(A) Schematic illustration of the locations of residues found to be important for RCP168 or SDF-1α in CXCR4 TM and ECL2 domains (9). The residues selectively involved in SDF-1α binding are shown as white spots, while those that are selectively involved in RCP168 binding are shown as black spots. Reprinted from reference 9. (B) Cartoon view of CXCR4 from the extracellular domain displaying two regions, site A (red circle) and site B (yellow circle), inside the TM domain of CXCR4. The residues in site A (Tyr121, Trp252, Tyr255, and Glu288) that are important for CXCR4 binding of RCP168 are shown in cyan, whereas residues in site B (Phe87 and Phe292) that are important for CXCR4 binding of SDF-1α are shown in green.
While X-ray crystallography is of limited use in exploring the interactions of ligands with GPCRs like CXCR4 due to the difficulties in crystallizing membrane proteins, the X-ray structure of RCP168 combined with the homology model structure of CXCR4 provided an opportunity to predict the mode of interaction between RCP168 and CXCR4, particularly in the context of the N terminus of the ligand. CXCR4 belongs to family A of GPCRs, which represents proteins homologous to rhodopsin. The X-ray structure for bacteriorhodopsin has been reported (19, 26), and it has been the basis for the development of homology models for GPCRs belonging to family A. In the case of chemokine receptors, the structure of bacteriorhodopsin was used as a template by our group to develop possible CXCR4 and CCR5 homology models (47, 49). Here, we have used homology modeling and MD simulation to predict the interactions between RCP168 and CXCR4.
Initially, the N terminus of RCP168 was manually docked into the groove between site A and site B. The complex was minimized and subjected to a 200-ps MD simulation. During the entire MD simulation, RCP168 displayed some flexibility, with an average root mean square deviation of 4.14 Å from the starting structure. As the simulation progressed, the N terminus moved toward site A. This movement seemed to be the result of the hydrophobic interactions between the side chain of Leu1 in RCP168 and those of His113, Val114, and Ile259 in site A of CXCR4 (Fig. 4A). This orientation of the N terminus is also stabilized by different interactions by some of the residues in the N terminus of RCP168. For instance, Ser4 in RCP168 forms a hydrogen bond with the main chain oxygen of Tyr28 in RCP168, while His6 in RCP168 forms van der Waals interactions with Ile269 in site A of CXCR4. According to the mutational data (9), at least four TM domain residues (Tyr121, Trp252, Tyr255, and Glu288) in site A affect the binding affinity of RCP168. Thus, the tendency of the N terminus of RCP168 to populate site A, as suggested by the modeling study, is in agreement with the previously reported mutational data.
FIG. 4.

Snapshots from the MD simulations of RCP168 (A) and SDF-1α (B) in complex with CXCR4. These snapshots are representative of other states predicted during the MD simulations of the ligand-CXCR4 complexes, and they reveal that the N terminus of RCP168 (green) assumes different orientations from that of SDF-1α (red). In the case of RCP168, the N terminus populates site A (blue), whereas in the case of SDF-1α, the N terminus populates site B (cyan) in CXCR4.
In addition to the N terminus of RCP168, some of the interactions by residues in the 30s loop of RCP168 with residues in the second extracellular loop 2 (ECL2) of CXCR4 were present during the MD simulation. The interactions involved are primarily hydrophobic interactions: Lys37, Pro38, and Gly39 in RCP168 interacting with Tyr190 and Pro191 in CXCR4. As stated above, the 30s loop of RCP168 exhibited a different conformation than the 30s loop of vMIP-II. This conformational difference may bring the 30s loop of RCP168 closer to the ECL2 of CXCR4. Interestingly, the mutations at Asp187, Phe189, and Pro191 in the ECL2 drastically reduce HIV-1 entry (30 to 100%) (9). Thus, the interactions between the 30s loop of RCP168 and the ECL2 of CXCR4, as predicted from the MD simulation, may be important for blocking HIV-1 entry into the target cell.
To probe the structural basis for the different binding behaviors of RCP168 and SDF-1α as revealed by our previous mutational study (9), we also modeled SDF-1α using a similar procedure. Initially, we superimposed the N terminus of SDF-1α onto that of RCP168 bound to CXCR4 to ensure very similar starting geometries of the N termini of both ligands. Subsequently, the complex was minimized and subjected to a 200-ps MD simulation. As the MD simulation progressed, the N terminus of SDF-1α attained a different orientation than that in the RCP168-CXCR4 complex, even though we started from a similar geometry. In the case of SDF-1α, the N terminus populates site B (Fig. 4B). This orientation of the N terminus is stabilized by several interactions between the N-terminal residues and the residues in site B of CXCR4. The interactions include the hydrogen bond between the NH of Lys1 in SDF-1α and the hydroxyl oxygen of Ser285 in CXCR4 and the hydrogen bond between the carbonyl oxygen of Lys1 in SDF-1α and the hydroxyl oxygen of Thr117. Furthermore, the side chain of Lys1 in SDF-1α forms van der Waals interactions with the side chain of Leu120 in site B in CXCR4. It must be noted that Phe87 and Phe292, which are located in site B, affect the binding of SDF-1α. These residues form a hydrophobic binding pocket with Leu120. During the entire MD simulation, the side chain of Lys1 in SDF-1α lay in close proximity to this binding pocket, suggesting that the N terminus of SDF-1α probably makes favorable interactions with residues in site B of CXCR4. The mutations at Phe87 and Phe292 potentially disturb the three-dimensional structure of this binding pocket, thereby hampering the affinity of SDF-1α for CXCR4 (9). In this respect, the preferential movement of the N terminus of SDF-1α to interact with residues in site B, as suggested by the modeling studies, is consistent with the experimental results.
To have a quantitative measure of the movements of the N termini of both ligands during the MD simulations, as described above, we measured the distance of the CG atom of Leu1 in RCP168 and the NZ atom of Lys1 in SDF-1α from the centers of sites A and B. As shown in Fig. 5, this analysis clearly shows that the N terminus of RCP168 primarily populates site A, with an average distance of 7.7 Å for CG atoms in Leu1 from the center of the site. On the other hand, the N terminus of SDF-1α mainly populates site B, as the average distance of NZ in Lys1 from the center of the site is 4.7 Å. These differential binding profiles of the N termini of the two ligands are also evident from the superimposition of average structures derived from the MD simulations of the ligand-CXCR4 complexes (Fig. 6).
FIG. 5.

Analysis of the movement of the N termini of RCP168 and SDF-1α in complex with CXCR4. (A) In the case of RCP168, the distance of the CG atom of Leu1 from the center of site A (blue) is shorter than that from the center of site B (magenta), suggesting that the N terminus of RCP168 primarily populates site A. (B) A similar analysis of the trajectory of the SDF-1α-CXCR4 complex suggests that the N terminus of SDF-1α mainly populates site B, as the distance of the NZ atom of Lys1 from the center of site B is shorter than that from site A.
FIG. 6.
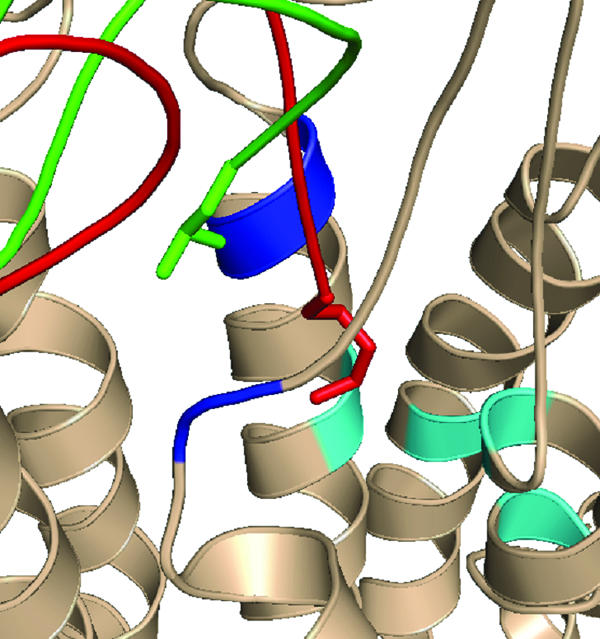
Superimposition of the average structures obtained from the 200-ps MD simulations of RCP168-CXCR4 and SDF-1α-CXCR4 complexes. The superimposition displays the difference in the binding of the N termini of ligands. In the case of RCP168 (green), the N terminus populates site A (blue), whereas in the case of SDF-1α (red), the N terminus populates a hydrophobic region in site B (cyan) in CXCR4. The first N-terminal residue for either ligand is displayed in a stick model.
DISCUSSION
Understanding the structural and chemical bases of selectivity in protein-protein interactions and developing methods to engineer selectivity in these interactions are of great importance in studying protein functions and intervening in disease processes mediated by these protein-protein interactions. Here, we investigated these issues using chemokines as the model systems. All the chemokines with known structures adopt similar three-dimensional folding patterns, which include three antiparallel β-strands packed against a C-terminal α-helix (17). However, the interaction of chemokines with their corresponding receptors has not yet been fully revealed due to the difficulty in crystallizing membrane receptors. Mutagenesis studies of chemokines in combination with structure determinations through nuclear magnetic resonance, crystallization, and molecular modeling revealed that the flexible N-terminal residues, including the N loop, a short fragment immediately after the first two conserved cysteines, are thought to be the primary determinants of receptor binding affinity and specificity (10). Another important functional determinant was hypothesized to be the 30s loop, the region between the first and second β-strands of the chemokine (36). However, it has yet to be determined whether and how the N terminus (including the N loop) and the 30s loop might be structurally connected and participate in receptor recognition in a coordinated manner. Here, we investigated this question by combining chemistry and structural biology to generate RCP168, which has the N terminus of vMIP-II replaced with d-amino acids.
The crystal structure of RCP168 revealed that significant structural changes at the N terminus due to the d-amino acid replacement can exert a long-range effect on the distal 30s loop through the disulfide bridge connecting the N terminus and the 30s loop. While this disulfide bridge, which is highly conserved among all chemokines, was previously thought to stabilize the structures of chemokines, our new finding suggests another important role of the disulfide bridge in structurally coordinating the N terminus and the 30s loop, two key modules for the recognition of chemokines by their receptors. In light of this finding, one may rationalize a structural basis for the conformational-change cascade in chemokine-receptor interactions, which might include (i) the initial binding of the N terminus of a chemokine to the receptor; (ii) the resulting conformational changes in the N terminus (including the N loop) and subsequently the 30s loop, as facilitated by the disulfide bridge; and finally, (iii) the triggered recognition between the 30s loop and the receptor, leading to the multipoint (at least including the N terminus and the 30s loop) contact between the chemokine and its receptor. This mechanism is consistent with the experimental observations made in this study.
The crystal structure of RCP168, compared with that of wild-type vMIP-II, also revealed a structure-based mechanism for ligand-receptor selectivity. As vMIP-II is highly nonselective in binding many chemokine receptors (28), it was most interesting to find that RCP168, by virtue of incorporating only 10 d-amino acid residues at its N terminus, selectively lost its binding to other receptors while gaining higher affinity for CXCR4. There were only two regions, the N terminus and the 30s loop, that showed concerted conformational changes due to the d-amino acid incorporation. While the N terminus has been shown to be the major binding site for CXCR4, the N terminus seems to have a less prominent role in binding to other receptors, such as CCR5 and CCR2, since the N-terminally truncated vMIP-II analog still retains reasonable binding affinity for CCR5 and CCR2, which is in sharp contrast to its complete loss of binding to CXCR4 (23). This suggests that another domain(s) of the ligand may be important for recognizing CCR5 or CCR2. The 30s loop, as revealed in this study, may be such a critical domain for ligand binding to CCR5 and CCR2, since the conformational changes in the 30s loop of RCP168, either alone or together with those of the N terminus of RCP168, seem to be a major cause of the loss of CCR5 or CCR2 binding. To further investigate the receptor binding mechanism of RCP168 compared with SDF-1α, we conducted MD simulation studies for RCP168-CXCR4 and SDF-1α-CXCR4 complexes. These studies suggested that the N terminus of RCP168 occupies a binding site different from that of SDF-1α. This is consistent with our previous findings of the differential binding profiles of RCP168 and SDF-1α by mutational studies (9).
In addition to suggesting a potential mechanism for ligand-receptor selectivity based on the conformational changes of the ligand at the N terminus and the 30s loop, the present study unveiled an interesting property of CXCR4, i.e., the flexibility of the CXCR4 surface in recognizing ligands of different chiralities or, more precisely, different conformations. We previously found that all d-amino acid peptides derived from the N terminus of vMIP-II selectively bind CXCR4 (48). The results with RCP168 provide further support for the notion that CXCR4 is capable of interacting with diverse conformations of a ligand. The fact that CXCR4 can recognize both d- and l-amino acid-containing ligands is unprecedented in the GPCR superfamily, as we are not aware of similar cases reported in the literature. Our previous studies to map the receptor binding sites using site-directed mutants of CXCR4 have shown distinctive regions involved in d- versus l-ligand binding (9). Interestingly, d-amino acid-containing ligands, such as RCP168, recognize sites on CXCR4 shared by HIV-1 gp120, but not by SDF-1α. In light of these results, the unusual flexibility of the CXCR4 ligand binding surface raises an intriguing question about whether HIV-1 gp120 might exploit this feature of CXCR4-ligand interaction for its entry into the target cell. It is well known that gp120 can frequently mutate itself, especially in the V3 loop region that is thought to recognize the coreceptor. The sequence mutations presumably bring about changes in the conformation of gp120, which could serve as a strategy for the virus to evade recognition by HIV-1-neutralizing antibodies. However, it is puzzling that despite these changes, gp120 retains the ability to recognize host cell receptors, such as CXCR4. Our findings may shed some new light on a possible mechanism through which the varied structures of gp120 could be accommodated by the flexible CXCR4 ligand binding surface.
RCP168 is a prototype molecule for a novel family of SMM chemokines that are highly selective and potent CXCR4 receptor inhibitors compared with the natural chemokines. An important goal of the SMM chemokine approach is to engineer de novo receptor selectivity into nonselective natural chemokines. This was clearly demonstrated by the generation of RCP168. In contrast to the broad activity of vMIP-II, RCP168 has higher selectivity and affinity for CXCR4 binding. As a result, RCP168 is more potent in inhibiting HIV-1 infection than SDF-1α. In addition, the anti-HIV activity of RCP168 is also comparable to that of T20, a currently marketed anti-HIV drug targeting a gp41-mediated HIV entry mechanism. Furthermore, we previously reported that RCP168 has much weaker activity in interfering with SDF-1α signaling, in contrast to its potent anti-HIV activity (23). These disparate inhibitory-activity profiles of RCP168 in differentiating HIV-1 coreceptor function versus the normal function of CXCR4 suggest that this chemically engineered molecule may be used to selectively disrupt the coreceptor activity of CXCR4 without inducing unwanted Ca2+ signaling or interfering with SDF-1α signaling important for normal physiological functions at the concentrations used for inhibiting HIV-1 infection. In fact, the mechanistic basis for the disparate activities of RCP168 was recently investigated and shown by our mutational-mapping analysis of binding sites of RCP168 and other d-amino acid-containing SMM-chemokines on CXCR4, revealing that RCP168 binding sites on CXCR4 overlap significantly with HIV-1 but differ from SDF-1α (9, 42). These results strongly suggest that RCP168 may serve as a prototype molecule for the development of highly selective and effective anti-HIV agents to be used in combination with other currently available drugs, such as T20 and/or drugs targeted to HIV-1 protease or reverse transcriptase.
Acknowledgments
We thank Douglas Richman of the UCSD Center for AIDS Research for helpful discussion of this study and for performing further antiviral studies for our compounds. We also thank Rongguang Zhang, Randy Alkire, Norma E. C. Duke, and Andrzej Joachimiak at the SBC 19BM beamline, Argonne National Laboratory, for their help with data collection.
The use of the Argonne National Laboratory Structural Biology Center beamlines at the Advanced Photon Source is supported by the Office of Energy Research, U.S. Department of Energy, under contract no. W-31-109-ENG-38. This work is supported by grants from the National Institutes of Health. All SMM chemokines, including RCP168, used in this study were provided by Raylight Corporation and Chemokine Pharmaceutical Inc., La Jolla, CA.
Footnotes
Published ahead of print on 8 August 2007.
REFERENCES
- 1.Alkhatib, G., C. Combadiere, C. C. Broder, Y. Feng, P. E. Kennedy, and P. M. Murphy. 1996. CC CKR5: a RANTES, MIP-1α, MIP-1β receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 272:1955-1958. [DOI] [PubMed] [Google Scholar]
- 2.Amara, A., S. L. Gall, O. Schwartz, J. Salamero, M. Montes, P. Loetscher, M. Baggiolini, J.-L. Virelizier, and F. Arenzana-Seisdedos. 1997. HIV coreceptor downregulation as antiviral principle: SDF-1α-dependent internalization of the chemokine receptor CXCR4 contributes to inhibition of HIV replication. J. Exp. Med. 186:139-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baggiolini, M., B. Dewald, and B. Moser. 1997. Human chemokines: an update. Annu. Rev. Immunol. 15:675-705. [DOI] [PubMed] [Google Scholar]
- 4.Berger, E. A., P. M. Murphy, and J. M. Farber. 1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev. Immunol. 17:657-700. [DOI] [PubMed] [Google Scholar]
- 5.Bleul, C. C., M. Farzan, H. Choe, C. Parolin, I. Clark-Lewis, and J. Sodroski. 1996. The lymphocyte chemoattractant SDF-1 is a ligand for LESTR/fusin and blocks HIV-1 entry. Nature 382:829-833. [DOI] [PubMed] [Google Scholar]
- 6.Brunger, A. T. 1992. XPLOR manual. Yale University, New Haven, CT.
- 7.Cheng-Mayer, C., D. Seto, M. Tateno, and J. A. Levy. 1988. Biologic features of HIV-1 that correlate with virulence in the host. Science 240:80-82. [DOI] [PubMed] [Google Scholar]
- 8.Choi, W. T., Y. Kumaki, J. An, D. D. Richman, J. Sodroski, and Z. W. Huang. 2007. Basic and translational research of chemokine ligands and receptors and development of novel therapeutics, p. 300-335. In Z. W. Huang (ed.), Drug discovery research: new frontiers in the post-genomic era. John Wiley & Sons, Hoboken, NJ.
- 9.Choi, W. T., S. Tian, C. Z. Dong, S. Kumar, D. Liu, N. Madani, J. An, J. G. Sodroski, and Z. Huang. 2005. Unique ligand binding sites on CXCR4 probed by a chemical biology approach: implications for the design of selective human immunodeficiency virus type 1 inhibitors. J. Virol. 79:15398-15404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crump, M. P., J. H. Gong, P. Loetscher, K. Rajarathnam, A. Amara, F. Arenzana-Seisdedos, J. L. Virelizier, M. Baggiolini, B. D. Sykes, and I. Clark-Lewis. 1997. Solution structure and basis for functional activity of stromal cell-derived factor-1; dissociation of CXCR4 activation from binding and inhibition of HIV-1. EMBO J. 16:6996-7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dealwis, C., E. J. Fernandez, D. A. Thompson, R. J. Simon, M. A. Siani, and E. Lolis. 1998. Crystal structure of chemically synthesized [N33A] stromal cell-derived factor 1α, a potent ligand for the HIV-1 “fusin” coreceptor. Proc. Natl. Acad. Sci. USA 95:6941-6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng, H., R. Liu, W. Ellmeier, S. Choe, D. Unutmaz, M. Burkhart, P. D. Marzio, S. Marmon, R. E. Sutton, C. M. Hill, C. B. Davis, S. C. Peiper, T. J. Schall, D. R. Littman, and N. R. Landau. 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature 381:661-666. [DOI] [PubMed] [Google Scholar]
- 13.Dong, C. Z., S. Kumar, W. T. Choi, N. Madani, S. Tian, J. An, J. G. Sodroski, and Z. Huang. 2005. Different stereochemical requirements for CXCR4 binding and signaling functions as revealed by an anti-HIV, d-amino acid-containing SMM-chemokine ligand. J. Med. Chem. 48:7923-7924. [DOI] [PubMed] [Google Scholar]
- 14.Dragic, T., V. Litwin, G. P. Allaway, S. R. Martin, Y. Huang, K. A. Nagashima, C. Cayanan, P. J. Maddon, R. A. Koup, J. P. Moore, and W. A. Paxton. 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 381:667-673. [DOI] [PubMed] [Google Scholar]
- 15.Farzan, M., T. Mirzabekov, P. Kolchinsky, R. Wyatt, M. Cayabyab, N. P. Gerard, C. Gerard, J. Sodroski, and H. Choe. 1999. Tyrosine sulfation of the amino terminus of CCR5 facilitates HIV-1 entry. Cell 96:667-676. [DOI] [PubMed] [Google Scholar]
- 16.Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872-877. [DOI] [PubMed] [Google Scholar]
- 17.Fernandez, E. J., and E. Lolis. 2002. Structure, function, and inhibition of chemokines. Annu. Rev. Pharmacol. Toxicol. 42:469-499. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez, E. J., J. Wilken, D. A. Thompson, S. C. Peiper, and E. Lolis. 2000. Comparison of the structure of vMIP-II with eotaxin-1, RANTES, and MCP-3 suggests a unique mechanism for CCR3 activation. Biochemistry 39:12837-12844. [DOI] [PubMed] [Google Scholar]
- 19.Henderson, R., J. M. Baldwin, T. A. Ceska, F. Zemlin, E. Beckmann, and K. H. Downing. 1990. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 213:899-929. [DOI] [PubMed] [Google Scholar]
- 20.Hill, C. M., and D. R. Littman. 1996. Natural resistance to HIV? Nature 382:668-669. [DOI] [PubMed] [Google Scholar]
- 21.Jones, T. A., J. Y. Zou, S. W. Cowan, and M. Kjeldgaard. 1991. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47:110-119. [DOI] [PubMed] [Google Scholar]
- 22.Kledal, T. N., M. M. Rosenkilde, F. Coulin, G. Simmons, A. H. Johnsen, S. Alouani, C. A. Power, H. R. Luttichau, J. Gerstoft, P. R. Clapham, I. Clark-Lewis, T. N. C. Wells, and T. W. Schwartz. 1997. A broad-spectrum chemokine antagonist encoded by Kaposi's sarcoma-associated herpesvirus. Science 277:1656-1659. [DOI] [PubMed] [Google Scholar]
- 23.Kumar, S., W. T. Choi, C. Z. Dong, N. Madani, S. Tian, D. Liu, Y. Wang, J. Pesavento, J. Wang, X. Fan, J. Yuan, W. R. Fritzsche, J. An, J. G. Sodroski, D. D. Richman, and Z. Huang. 2006. SMM-chemokines: a class of unnatural synthetic molecules as chemical probes of chemical receptor biology and leads for therapeutic development. Chem. Biol. 13:69-79. [DOI] [PubMed] [Google Scholar]
- 24.Li, Y., D. X. Liu, R. Cao, S. Kumar, C. Z. Dong, J. An, S. R. Wilson, Y. G. Gao, and Z. W. Huang. 2007. Crystal structure of chemically synthesized vMIP-II. Proteins 67:243-246. [DOI] [PubMed] [Google Scholar]
- 25.Liwang, A. C., Z. X. Wang, Y. Sun, S. C. Peiper, and P. J. Liwang. 1999. The solution structure of the anti-HIV chemokine vMIP-II. Protein Sci. 8:2270-2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luecke, H., B. Schobert, H. T. Richter, J. P. Cartailler, and J. K. Lanyi. 1999. Structure of bacteriorhodopsin at 1.55 Å resolution. J. Mol. Biol. 291:899-911. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell, J. B., and J. Smith. 2003. d-Amino acid residues in peptides and proteins. Proteins 50:563-571. [DOI] [PubMed] [Google Scholar]
- 28.Moore, P. S., C. Boshoff, R. A. Weiss, and Y. Chang. 1996. Molecular mimicry of human cytokine and cytokine response pathway genes by KSHV. Science 274:1739-1744. [DOI] [PubMed] [Google Scholar]
- 29.Murphy, P. M. 1994. The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol. 12:593-633. [DOI] [PubMed] [Google Scholar]
- 30.Nagasawa, T., S. Hirota, K. Tachibana, N. Takakura, S. Nishikawa, Y. Kitamura, N. Yoshida, H. Kikutani, and T. Kishimoto. 1996. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature 382:635-638. [DOI] [PubMed] [Google Scholar]
- 31.Navaza, J. 1994. Amore—an automated package for molecular replacement. Acta Crystallogr. A 50:157-163. [Google Scholar]
- 32.Oberlin, E., A. Amara, F. Bachelerie, C. Bessia, J. L. Virelizier, F. Arenzana-Seisdedos, O. Schwartz, J. M. Heard, I. Clark-Lewis, D. F. Legler, M. Loetscher, M. Baggiolini, and B. Moser. 1996. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature 382:833-835. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski, Z., and W. Minor. 1997. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. A 276:307-326. [DOI] [PubMed] [Google Scholar]
- 34.Parolin, C., B. Taddeo, G. Palu, and J. Sodroski. 1996. Use of cis- and trans-acting viral regulatory sequences to improve expression of human immunodeficiency virus vectors in human lymphocytes. Virology 222:415-422. [DOI] [PubMed] [Google Scholar]
- 35.Proudfoot, A. 2002. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2:106-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qian, Y. Q., K. O. Johanson, and P. McDevitt. 1999. Nuclear magnetic resonance solution structure of truncated human GROβ [5-73] and its structural comparison with CXC chemokine family members GROα and IL-8. J. Mol. Biol. 294:1065-1072. [DOI] [PubMed] [Google Scholar]
- 37.Rho, H. M., B. Poiesz, F. W. Ruscetti, and R. C. Gallo. 1981. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology 112:355-360. [DOI] [PubMed] [Google Scholar]
- 38.Schellekens, P. T., M. Tersmette, M. T. Roos, R. P. Keet, F. de Wolf, R. A. Coutinho, and F. Miedema. 1992. Biphasic rate of CD4+ cell count decline during progression to AIDS correlates with HIV-1 phenotype. AIDS 6:665-669. [DOI] [PubMed] [Google Scholar]
- 39.Sheldrick, G. M., and T. R. Schneider. 1997. SHELXL: high-resolution refinement. Macromol. Crystallogr. B 277:319-343. [PubMed] [Google Scholar]
- 40.Tachibana, K., S. Hirota, H. Iizasa, H. Yoshida, K. Kawabata, Y. Kataoka, Y. Kitamura, K. Matsushima, N. Yoshida, S. Nishikawa, T. Kishimoto, and T. Nagasawa. 1998. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393:591-594. [DOI] [PubMed] [Google Scholar]
- 41.Tersmette, M., J. M. Lange, R. E. de Goede, F. de Wolf, J. K. Eeftink-Schattenkerk, P. T. Schellekens, R. A. Coutinho, J. G. Huisman, J. Goudsmit, and F. Miedema. 1989. Association between biological properties of human immunodeficiency virus variants and risk for AIDS and AIDS mortality. Lancet i:983-985. [DOI] [PubMed] [Google Scholar]
- 42.Tian, S., W. T. Choi, D. Liu, J. Pesavento, Y. Wang, J. An, J. G. Sodroski, and Z. Huang. 2005. Distinct functional sites for human immunodeficiency virus type 1 and stromal cell-derived factor 1a on CXCR4 transmembrane helical domains. J. Virol. 79:12667-12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tripos, I. 2007. SYBYL, 7.2 ed. Tripos, Inc., St. Louis, MO.
- 44.Trkola, A., T. Dragic, J. Arthos, J. M. Binley, W. C. Olson, G. P. Allaway, C. Cheng-Mayer, J. Robinson, P. J. Maddon, and J. P. Moore. 1996. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature 384:184-187. [DOI] [PubMed] [Google Scholar]
- 45.Uray, K., J. Kajtar, E. Vass, M. R. Price, M. Hollosi, and F. Hudecz. 2000. Effect of d-amino acid substitution in a mucin 2 epitope on mucin-specific monoclonal antibody recognition. Arch. Biochem. Biophys. 378:25-32. [DOI] [PubMed] [Google Scholar]
- 46.Wu, L., N. P. Gerard, R. Wyatt, H. Choe, C. Parolin, N. Ruffing, A. Borsetti, A. A. Cardoso, E. Desjardin, W. Newman, C. Gerard, and J. Sodroski. 1996. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 6605:179-183. [DOI] [PubMed] [Google Scholar]
- 47.Zhou, N., Z. Luo, J. W. Hall, J. Luo, X. Han, and Z. Huang. 2000. Molecular modeling and site-directed mutagenesis of CCR5 reveal residues critical for chemokine binding and signal transduction. Eur. J. Immunol. 30:164-173. [DOI] [PubMed] [Google Scholar]
- 48.Zhou, N., Z. Luo, J. Luo, X. Fan, M. Cayabyab, M. Hiraoka, D. Liu, X. Han, J. Pesavento, C. Z. Dong, Y. Wang, J. An, H. Kaji, J. G. Sodroski, and Z. Huang. 2002. Exploring the stereochemistry of CXCR4-peptide recognition and inhibiting HIV-1 entry with d-peptides derived from chemokines. J. Biol. Chem. 277:17476-17485. [DOI] [PubMed] [Google Scholar]
- 49.Zhou, N., Z. Luo, J. Luo, D. Liu, J. W. Hall, R. J. Pomerantz, and Z. Huang. 2001. Structural and functional characterization of human CXCR4 as a chemokine receptor and HIV-1 co-receptor by mutagenesis and molecular modeling studies. J. Biol. Chem. 276:42826-42833. [DOI] [PubMed] [Google Scholar]
- 50.Zou, Y., A. Kottmann, M. Kuroda, I. Taniuchi, and D. Littman. 1998. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393:595-599. [DOI] [PubMed] [Google Scholar]