

Summary
The L11 binding site is one of the most important functional sites in the ribosome. The N-terminal domain of L11 has been implicated as a “reversible switch” in facilitating the coordinated movements associated with EF-G–driven GTP hydrolysis. The “reversible switch” mechanism has been hypothesized to require conformational flexibility involving re-orientation and re-positioning of the two L11 domains, and warrants a close examination of the structure and dynamics of L11. Here we report the solution structure of free L11, and relaxation studies of free L11, L11complexed to its 58 nt RNA recognition site, and L11 in a ternary complex with the RNA and thiostrepton antibiotic. The binding site of thiostrepton on L11 was also defined by analysis of structural and dynamics data and chemical shift mapping. The conclusions of this work are as follows: First, the binding of L11 to RNA leads to sizable conformation changes in the regions flanking the linker and in the hinge area that links a β-sheets and a 310-helix-turn-helix element in the N-terminus. Concurrently, the change in the relative orientation may lead to re-positioning of the N-terminus, as implied by a decrease of radius of gyration from 18.5 Å to 16.2 Å. Second, the regions, which undergo large conformation changes, exhibit motions on ms-μs or ns-ps time scales. Third, binding of thiostrepton results in more rigid conformations near the linker (Thr71) and near its putative binding site (Leu12). Lastly, conformational changes in the putative thiostrepton binding site are implicated by the re-emergence of cross-correlation peaks in the spectrum of the ternary complex, which were missing in that of the binary complex. Our combined analysis of both the chemical shift perturbation and dynamics data clearly indicates that thiostrepton binds to a pocket involving residues in the 310-helix in L11.
Keywords: ribosome protein, thiostrepton inhibition, dynamics, NMR, L11
Introduction
One of the remaining important questions in the post-ribosome structure era is the mechanism of translocation in protein synthesis.1-3 During the elongation cycle of protein synthesis, both elongation factors, EF-G and EF-Tu, play essential roles. EF-Tu forms a complex with aminoacyl-tRNA and GTP, and delivers the aminoacyl-tRNA to the A site,4,5 whereas EF-G in complex with GTP drives the translocation reaction. Both factors interact with a site that consists of the highly conserved ribosomal protein L11 and a stretch of 58 nucleotides (nt) in 23S rRNA. The functional importance of this site on the ribosome is also suggested by the fact that it is a target for the thiazole class of antibiotics, e.g. thiostrepton and micrococin, which inhibit EF-Tu– and EF-G–dependent reactions.5-8 L11-deficient mutants of both Escherichia coli (E. coli) and Bacillus megaterium are viable but sick5 due to dramatically slowed protein synthesis.9 The putative interaction site on L11 for thiostrepton is believed to be centered around Pro21 and Pro22, which are conserved among bacterial and archaeal proteins.10,11 The lack of these two prolines in eukaryotic L11 provides a plausible explanation for the insensitivity of eukaryotic ribosomes to thiostrepton.
The C-terminal domain (L11-CTD) is responsible for tight binding in the distorted minor groove of the highly conserved L11 binding domain RNA (L11 BD RNA),12,13 whereas the N-terminal domain (L11-NTD) has limited interaction with the rRNA.12,14 In L11 structures derived from the crystal structure of either intact ribosomes or a binary L11 – rRNA fragment complex,12 the relative position of the L11-NTD to the L11-CTD is not well-defined. A poor electron density of the L11-NTD may be caused by the rigid body movement of the L11-NTD.12 If so, this movement of the L11-NTD implies flexibility in the linker between the two domains, even though the linker is short and inherently rigid because of the presence of two proline residues.12Coordinated movements among the C-terminal domain of L7/L12, the G’ domain of EF-G and the L11-NTD have been proposed to play an important role in the translocation reaction.15,16
Initial fits of L11 to lower-resolution cryo-EM electron density maps of the ribosome suggested that a substantial rotation (~40°) and a shift (~7 Å) of the L11-NTD had occurred relative to a conformation seen in a crystal structure of the binary L11–58mer rRNA complex,12,16 and that further changes in the relative position of the L11-NTD took place in response to GTP hydrolysis.16 A recent fitting of the L11 binary crystal structure into a density map from a higher resolution cryo-EM study required smaller adjustments in the orientation of the L11-NTD, although inhibition of L11-NTD movements by thiostrepton remains the leading potential mechanism for thiostrepton action.17 Given the importance of alternative L11-NTD orientations and relative positioning of the two domains to the ribosome function, it is essential to accurately define the free L11 structure in terms of both the relative orientation and positioning, and to carefully examine the dynamics profiles of L11 in free state, in the binary and ternary complexes in the context of the structure and its functions. Moreover, this study would provide insight into the behavior of a functionally relevant section of the ribosome, at a level of detail not possible in experiments with intact ribosomes.
In this paper, we present the high-resolution structure of free L11 of Thermus thermophilis (T. thermophilis), together with comparative studies of the dynamics of L11 in the free-state, and in the binary and ternary complexes. The solution structure and dynamics of L11 presented here complement and extend previous structural work, which includes a crystal structure of the L11-RNA complex,12 and a recent NMR study of L11 of Thermotoga maritima (T. maritime).18
Results
Structure determination of free L11
The structure determination of L11 using conventional NOE distance and torsion angle restraints was straightforward (Figure S1, Supporting Materials). The backbone-heavy atom root-mean-square deviations (rmsds) relative to the average structures were 0.75 and 0.55 Å for the L11-NTD (residues 1–66) and the L11-CTD (residues 73–138, excluding a known flexible RNA binding loop (RBL, residues 83–96)), respectively. However, the linker that connects the two domains (residues 67–72) was poorly defined, possibly due to flexibility or lack of restraints to define the structure of the region, or both. The relaxation parameters T1, T1ρ and 15N-[1H] NOEs of L11 backbone amides do not suggest that the linker is any more flexible than the rest of the protein (Figure S2, Supporting Material). Model-free analysis of 15N relaxation data yields the generalized order parameter, S2, and the effective internal correlation time, τe; the profiles of these parameters are plotted in Figure S3 in Supporting Materials. The generalized order parameter of the linker residues is ~0.75 on average (Figure S3), and confirmed that the linker residues are not particularly dynamic. Therefore, the apparent structure disorder in this linker is likely due to the lack of restraints, resulting in the under-defined relative orientation and position of the two domains. We therefore applied both alignment and diffusion tensor analysis to characterize the relative orientation.
We first analyzed residual dipolar couplings (RDC) data of L11 using the singular value decomposition method19. An identical alignment tensor for both protein domains was obtained, suggesting that the two domains act as a single rigid body. We then refined the structure by including the RDCs of NH, CαHα and CαC’ in the structure calculation (Table 1). Moreover, since the number of non-dynamic RDCs that we could measured accurately was less that the twice of the number of the residues in the protein, the error in applying the RDC for the structure calculation may still exist.20 For this reason, we resorted to the orientation diffusion tensor analysis, which is independent from alignment tensor analysis, to characterize the relative orientation.
Table 1.
Restraints and Structural Statistics
| Restraints | |
| Total experimental restraints | |
| Total distance restraints | 1306 |
| Intraresidue (i = j) | 492 |
| Sequential (∣i – j∣ =1) | 361 |
| Short range (1 < ∣i – j∣ ≤ 4) | 144 |
| Long range (∣i – j∣ > 4) | 259 |
| Hydrogen bond | 50 |
| Total dihedral restraintsa | 373 |
| Phi | 217 |
| Psi | 162 |
| Chi1 | 180 |
| Total dipolar couplings | |
| HNN | 58 |
| CαHα | 51 |
| CαC’ | 41 |
| NOE violation > 0.5 Å | 0 |
| Dihedral angle violation > 5 deg. | 1 |
| SANS (D2O)b Range | 0.03-0.2577 Å-1 |
|
| |
| Rmsds | |
|
| |
| Deviation from idealized geometry | |
| Bonds (Å) | 0.0015 ± 0.0002 |
| Angles (deg.) | 0.35 ± 0.03 |
| Impropers (deg.) | 0.31 ± 0.1 |
| Backbone rmsds in regular secondary structurec (Å) | 0.3 3 ±0.05 |
| Nonhydrogen atoms in regular secondary structure (Å) | 0.85 ± 0.2 |
| Rmsds from Residual dipolar coupling | |
| NH | 0.49 ±0.03 |
| CαHα | 0.79 ±0.04 |
| C’Cα | 0.63 ±0.03 |
| χ2 (Exp. SANS vs. bkCalc SANS)d | 1.23 ± 0.14 |
|
| |
| Energies | |
| E(NOE) (kcal/mol) | 56.2 ± 3.0 |
| E(dihed.) (kcal/mol) | 9.2 ± 1.0 |
| E(repel) (kcal/mol) | 52.5 ± 7.0 |
| E(RDC) (kcal/mol) | 106.33 ± 10 |
| E(SANS) (kcal/mol) | 20.21 ± 0.5 |
Note: Torsion angles are restrained ambiguously due uncertainty in stereo-specific assignments and in NOE assignment.61
The SANS in D2O was used for the refinement.
Regular secondary structure elements are the α-helices 1 (residues 35–47), 2 (75–82), 3 (101–109), 4 (120–134); 310-helix (24–26); β-strands 1 (6–13), 2 (52–60), 3 (64–66), 4 (98–99) and 5 (137–139).
The χ2 between experimental and back-calculated SANS. The χ2 is defined in the text.
If the relative orientation between the domains is rigid and correctly defined, the RDC refined structure should agree with the diffusion tensor analysis of the relaxation data. The calculated rotational diffusion tensors of the individual domains and the full length protein, using the standard linear least-squares optimization, T1/T1ρ ratio21-25 and the solution structure coordinates, are listed in Table 2. Based on the calculation using the axial symmetric model, the overall rotational correlation times for the L11-NTD, the L11-CTD and the overall structure were 11.1, 11.2 and 11.1 ns, respectively. These values are expected for the L11 molecular weight of ~16 kDa. In contrast, an overall rotational correlation time of ~6.0 ns would have been expected if the individual domains were to rotate independently.26 The average angles between the principle inertia tensor axes of the individual domains in their rotational diffusion tensor frames, from the longest to the shortest axes among the ensemble of the structures, were approximately 74°, 81° and 66°. These angles are similar to those calculated based on the structures refined with RDCs (88°, 81° and 65° Figure S4). Therefore, in terms of the relative oritentation between the two domains, the diffusion tensor analysis of the relaxation data agrees with the structure that was refined with RDCs, and both the diffusion tensor analysis and RDC data suggest that the L11 acts as a single body.
Table 2.
Diffusion parameters for L11 from 15N relaxationa
| Tensor | Diso (10-7 s-1) | 2Dzz/(Dxx+Dyy) | Dxx/Dyy | |
|---|---|---|---|---|
| Overall | Isotropicb | 1.41±0.01 | - | - |
| Axialc | 1.50±0.01 | 1.53±0.01 | - | |
| Anisotropic | 1.49±0.01 | 1.52±0.01 | 0.88±0.03 | |
|
| ||||
| NTD | Isotropicb | 1.40±0.01 | - | - |
| Axialc | 1.50±0.01 | 1.60±0.01 | - | |
| Anisotropic | 1.50±0.01 | 1.61±0.01 | 0.91±0.01 | |
|
| ||||
| CTD | Isotropicb | 1.42±0.01 | - | - |
| Axialc | 1.49±0.01 | 1.53±0.02 | - | |
| Anisotropic | 1.49±0.01 | 1.52±0.02 | 0.92±0.05 | |
Values of D for 107, 55 and 52 residues were fitted using the local diffusion approximation for overall, L11-NTD and L11-CTD, respectively.
Diso=Dxx=Dyy=Dzz.
D:=Dzz, Df =Dxx=Dyy, Diso=(D:+2 Df )/3, D:/ Df =2Dzz/(Dxx+Dyy).
The ensemble of the 20 lowest-energy RDC-refined structures is shown in Figure 1(a). The global rmsd values relative to the mean coordinates were 0.75 Å and 1.22 Å, respectively, for the backbone and all heavy atoms of residues 5–140, excluding the RBL. The L11-NTD consists of two integral parts: a 310-helix-turn-helix (310HTH; residues 14–50), depicted in magenta in Figure 1(c), and β-sheets, depicted in cyan in Figure 1(c). “Hinges” link these two parts, allowing possible movements within the L11-NTD itself (see later sections of this paper).
Figure 1. Solution structure of L11 from Thermus thermophilus.

(a) The polypeptide backbone represented by a bundle of the 20 lowest-energy conformers calculated using NOE, dihedral angle and RDC constraints. The N-, C- termini and the RBL are indicated. (b) A bundle of the 20 lowest-energy conformers using a radius of gyration (Rg) from the small-angle neutron scattering (SANS) data, together with NOE, dihedral angle and RDC constraints. (c) Ribbon drawing of one of the 20 lowest-energy conformers from (b). The regular secondary structure elements are labeled. The L11-NTD consists of β-sheets (cyan) and a 310HTH motif (magenta) that are connected by two hinges centered around residues Leu12 and Asp50.
Refining the L11 structure using small-angle neutron scattering (SANS) data
As discussed in the Introduction, another important question pertinent to the “reversible switch” is the relative positioning of the two domains, which can normally be well defined with enough NOE distance restraints. However, only a very few NOE distance restraints between the two domains of the L11 from T. thermophilis were extracted in our hands, and the same problem has also been recognized by Ilin et al. in their L11 from T. maritime.18 This lack of sufficient inter-domain distance restraints may lead to uncertainty in defining the relative positions of the two domains.
Small angle scattering curves contain information about dimensionality in the form of radius of gyration (Rg) and global shape of molecules27, and have been used to generate low-resolution structural models,28-31 or used to complement NMR data to derive the solution structures of multi-domain proteins.32,33. Very recently, a protocol has been implemented to directly refine NMR structures using small-angle X-ray scattering (SAXS) data.20 We resorted to small-angle neutron scattering (SANS) measurements 34 to resolve the global shape, specifically the relative position of the domains of L11.
The Rg values for free L11 in D2O and H2O buffers extracted from the SANS data, were 18.2±0.3 and 18.5±0.3 Å, respectively. These values are significantly different from those calculated based on the RDC-refined NMR structures (16.7 Å), suggesting a deviation in the overall dimension of the RDC-defined structure from that of the true solution structure. This deviation is likely caused by possible mis-placement of the relative position of the two domains. We then adopted two approaches to refine the structure that make use of the SANS data from the SANS data. Using a reported protocol,35 we first refined the RDC-refined NMR structure using Rg. Figure 2 shows the average SANS data for the samples measured in the D2O buffer, along with the SANS curves back-calculated from the RDC-refined NMR structure (χ2=4.2 ). With the simple refinement of using experimental Rg as one of restraints, the χ2 improves to ~2.0 (Figure 2).
Figure 2.

Normalized SANS intensity, I(Q) vs. Q, of free L11 in D2O buffer (red with error bars), together with back-calculated SANS curves based on the RDC (blue)-, Rg(purple)- and SANS (black)-refined minimized average structures. We also plot the GNOM67 preprocessed curve (yellow) of the experimental data in the figure for comparison. The nearly identical SANS curves of the Rg- and SANS refined structures suggest that the relative position of the two domains can be sufficiently restrained using Rg in the presence of the distance, torsion angle and RDC restraints in the case of L11 where the two domains are linked by a short linker.
Since small-angle scattering scattering data contains information about not only Rg but also the overall shape of a molecule,27 a SAXS data was recently utilized in combination with NMR data to directly refine the solution structure of γ-crystallin to better define relative position of the two domains and the overall packing of the protein.20 The advantage of using SANS over SAXS is that the former measures “dry volume” and the interpretation of the data is relatively straightforward, whereas the latter measures “wet volume”36 and the interpretation requires maing an assumption about the hydration water.37 This difference in the two approaches is important because the size of the hydration layer is often comparable to the subtle change in Rg due to a switch in conformation, and uncertainty in estimating the hydration layer may obscure the change in dimension due to conformation switch and may complicate the interpretation of SAXS data. We have implemented the protocol of direct refinement of structures using NMR and SANS data in Xplor-NIH. The χ2 value was improved to be about 1.23 between the experimental and back-calculated SANS. While the backbone RMSDs of domain-wise comparison between the RDC- and Rg-refined, and between the RDC-refined and SANS-refined minimized average structures were rather small, about 0.35 Å for the both domains, the overall backbone RMSD, ~1.3 Å, between the two structures was significantly large. Because the relative orientation between the two domains was the same in all three structures because of RDC restraints, the difference in the overall backbone RMSDs between the RDC-refined and either Rg-refined or SANS-refined structures was attributed to the difference in the relative positioning gin the structures.
L11 changes conformations near the ends of the linker and in the hinges upon binding to RNA
To determine whether free and RNA-bound L11 are structurally different, we compared the structure of free L11 described above with that of the L11 in the binary complex crystal structure.12 These findings are important because they may reveal residues or regions that may be susceptible to conformation changes and may be critical to the L11-NTD movements necessary for its function. Comparisons of the individual domains in the solution structure with those of the crystal structure show nearly identical folds, with backbone RMSD values of 1.45±0.11 and 1.16±0.04 Å for the regular secondary structures in the L11-NTD and -CTD, respectively (Figure S5). However, when the regular secondary structures of both the domains are simultaneously superimposed, the backbone RMSD value increased to 7.20±0.12 Å. The large RMSD difference between the two structures is attributed roughly to a rotation of the L11-NTD by ~70 ° around the longest principal component of the inertia tensor, and to a tilt of the domain by ~40° along one of the horizontal axes relative to the L11-CTD (Figure 3). In detail, the conformation change upon binding to RNA was accomplished via a series of change in both φ and ψ angles. We calculated the average difference of both φ/ψ angles between the two structures as defined using the following formula:
Figure 3.
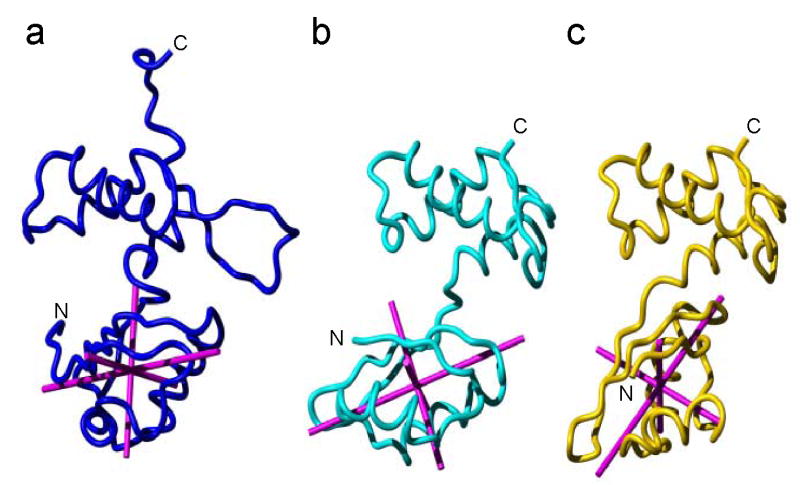
Comparison of the relative orientation between L11-NTD and L11-CTD among the solution structure (a), the structure of L11 in the complex with 58 nt RNA fitted into cryo-EM density of 70S ribosome (1JQS)16 (b) and the crystal structure of L11 in the complex with 58 nt RNA (1MMS) 12 (c). The backbone atoms for L11-CTD (residues 75–140, excluding the flexible RBL) have been superimposed for the best fit. The principal components of inertia tensors of L11-NTD of the solution structure, the cryo-EM structure and the crystal structure were calculated using the MolMol program and are shown in magenta. The N- and C- termini are indicated.
Δang clearly revealed the residues where the φ/ψ torsion angles underwent substantial changes upon L11 binding to the RNA (Figure 4). Those residues include not only those located in the L11-CTD regions in direct contact with the RNA (Ala82-Gly97, Met112, Leu115 and Thr118) as expected, but also residues in or around the linker (Thr71 and Ala74) and in the L11-NTD. The latter residues include those in the two hinges (Ala14-Lys16 in one, and Met48 and Asp50 in another, Figure 4). The large changes in the torsion angles of the hinge residues Ala14-Lys16, Met48 and Asp50 allowed the 310 HTH motif to reorient relative to the β-sheets in the L11-NTD when bound to rRNA. These large changes were also accompanied by changes in the torsion angles in the post–310-helix residues in a loop (Gly28 and Ala32). The regions that exhibited large changes in φ/ψ angles were also those that showed motions on various time scales (see the following sections).
Figure 4.

Comparison of torsion angles between the solution structure and the crystal and the structure of L11. Residue regions showing Δang>~50° are indicated.
Dynamics of L11 in the free state and in the binary and the ternary complexes
We have optimized solution conditions for NMR investigations of L11 in binary (L11-RNA) and ternary (L11-RNA-thiostrepton) complexes. Almost complete assignments of the backbone 1H/13C/15N chemical shifts of the L11 in both complexes have been obtained by using a combination of 3D TROSY experiments. For the binary complex, the ratio of RNA:L11 had to be at least 2:1 due to a fast exchange between bound and free states in order to obtain a high-quality spectrum. The weaker binding between the RNA and L11, which is estimated to be on a μm-scale, may complicate the interpretation of the T1 and T1ρ of the binary complex due to contributions from chemical exchanges. The ratio of RNA:L11 in the ternary complex was kept to 1:1, with an excess of thiostrepton. The TROSY spectra of the L11 in all three states are illustrated in Figure 5.
Figure 5.

2D [15N,1H]-TROSY spectra of (a) L11; (b) L11-RNA complex; and (c) L11-RNA-thiostrepton complex. The concentration of 15N,2H-labeled L11 in all samples was 0.7 mM. For both complexes, non-labeled RNA was used. For the L11-RNA-thiostrepton, non-labeled thiostrepton was used. All spectra were recorded at pH 6.5 and 40 °C on a Bruker Avance600 spectrometer. In order to achieve the maximum sensitivity, all three spectra were recorded using different delays for polarization transfer, as well as different t1max and t2max.
Due to in part of the large molecular weights, ~40 kDa, and in part chemical exchanges between the bound and free L11 in complex samples, it was impossible to apply the conventional array of experiments to record relaxation data for L11 in all three states. The estimated low end of the overall rotational correlation time was approximately 17 ns for both the binary and ternary complexes using the 1D [15N,1H]-TRACT experiment.38 We then applied TROSY-type T1, T1ρ and 15N-[1H] NOE measurements39,40 in order to compare the dynamic properties of all states (Figure 6). However, due to a poor signal-to-noise ratio in the T1ρ measurements of the binary and ternary complexes, we then resorted to measuring T1ρ of the TROSY-component of L11 in all three states using Palmer’s scheme.41 These relaxation parameters are sensitive to motions on the ps and ns time scale. However, the quantitative interpretation of the relaxation parameters of these TROSY components has not been developed. Therefore, we present in this paper only the qualitative interpretation of the data. Nevertheless, we have measured the relaxation of free L11 using conventional experiments (Figure S2) and the results showed basically the same trend as that for free L11 using the TROSY-based experiments (Figure 6). In addition, we have also performed quantitative analysis of the relaxation parameters of free L11, and the results are presented in supplementary materials (Figure S3).
Figure 6.
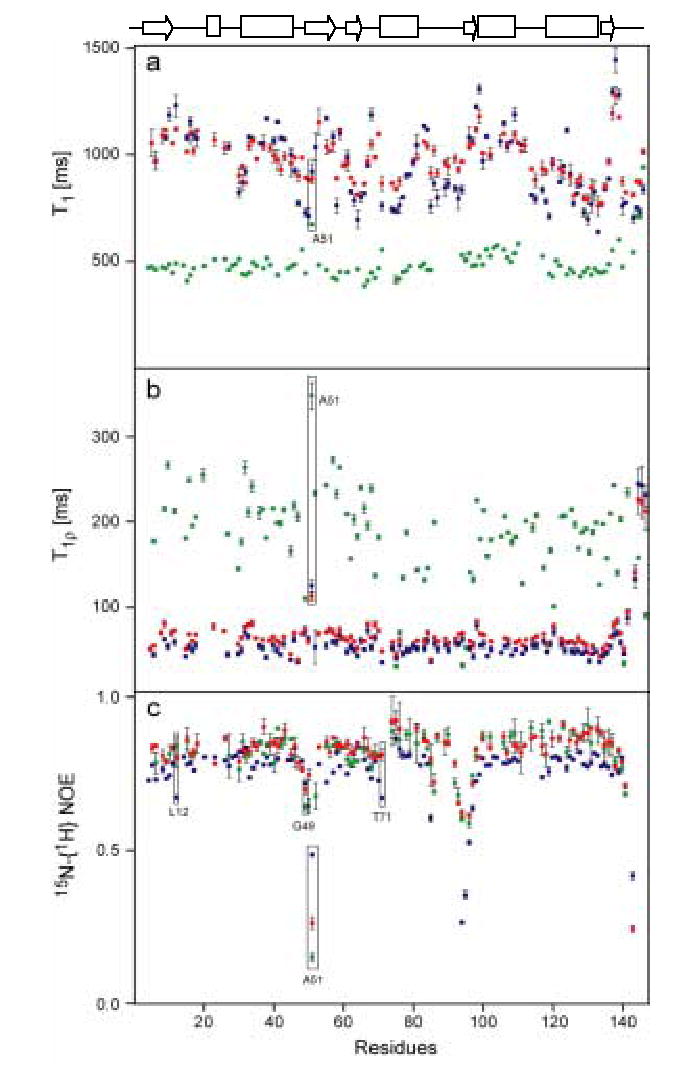
Comparison of the polypeptide backbone amide 15N (a) T1; (b) T1ρ; and (c) 15N-[1H] NOE of free L11 (green); L11 in the binary (blue); and L11 in the ternary complexes (red). All spectra were acquired with the selection of a slowly relaxing component of the 15N doublet, using a pulse scheme.39-41 The errors were estimated based on duplicating the measurements that were recorded several days apart. The concentrations of 15N,2H-labeled L11 in all samples were about 0.7 mM. Non-labeled RNA was used to prepare the L11-RNA complex and L11-RNA-thiostrepton complexes. The secondary structure of L11 is drawn on the top of the figure. Excess non-labeled RNA and thiostrepton were used to prepare the binary and ternary complexes, respectively. All spectra were recorded at 40 °C on a Bruker Avance600 spectrometer.
The TROSY-type T1, T1ρ and NOE data, referred as T1, T1ρ and NOE in the rest of the text, for free L11 were plotted against residue numbers as shown in green in Figure 6. The average NOE value, excluding outliers, was about 0.83. The NOE values for the L11-NTD were relatively uniform, except in the region around Ala51. In contrast, the L11-CTD contains the large unstructured RBL, part of which shows significantly depressed NOE values (residues 90–95), agreeing with previous measurements of the L11-C76 fragment (from Bacillus stearothermophilus, comprising residues 64–139).42,43 The C-terminal tail showed strongly depressed NOEs from residues 143–147. The depression of the NOEs, which reflects internal motion on the sub-ns timescale, was also reflected in depressed T1 and elevated T1ρ values for 90–95 in the RBL. Additionally, the C-terminal tail and residue Ala51 in the L11-NTD showed substantially increased T1ρ, which, together with substantially depressed NOE at these positions, indicate possible motions on the sub-ns timescale. It is interesting to note that in contrast to those in the C-terminus, the residues in the N-terminus, which make β1 strand, are not particularly dynamic (Figure 6).
For comparison, the T1, T1ρ, and NOE data for the binary and ternary complexes were plotted next to those for free L11 (Figure 6). The average T1ρ of the TROSY components of L11 in the binary complex was approximately 60 ms, about 5 ms shorter than that of L11 in the ternary complex (Figure 6(c)), and may be attributed to the slow chemical exchange due to transient inter-complex interaction. Compared to the T1ρ of free L11, the T1ρ of L11 in the complexes showed relatively small fluctuations around their average mean values, with the exception of Ala51. The T1 of L11 in both the binary and ternary complexes varied in concert, although the T1 of L11 in the binary complex exhibits a larger variation (Figure 6). The NOE values of the binary are consistently lower than those of the ternary complexes. The NOE values for the binary complex are virtually identical in two sets of measurements recorded with 3 and 6 second recycling time (data not shown). Furthermore, the T1ρ, T1 and 15N-[1H] NOE values indicate the RBL in complexes is flexible and still exhibits motion faster than their overall tumbling time in the complexes (Figure 6c).
The L11-NTD in free L11 was more dynamic than the CTD. The average T1ρ of the L11-NTD in free L11 was ~210 ms, whereas that of the L11-CTD was ~180 ms, suggesting that, on average, the residues in the L11-NTD undergo internal motions on a time scale faster than those in the L11-CTD. This result appears to be in agreement with a recent molecular dynamics study, in which the L11-NTD was seen to fluctuate significantly more than the L11-CTD.44 This difference between the domains disappeared in L11 bound to the RNA in both the binary and ternary complexes, possibly due to the changes in dynamics upon the binding in the regions flanking the linker (see the following discussion).
We also examined the relaxation data in the linker between the two domains and the regions flanking the linker in the three states. The linker may serve as a hinge for the L11-NTD movement, and the relaxation data may provide a clue to the dynamics of the linker. First, the residues in the linker appeared to have an average value of NOE in all three states (Figure 6c). Second, Ala51 in all three states showed a deeply depressed NOE and a greatly elevated T1ρ, indicating internal motion on the sub-ns time scale. Ala51 in free L11 also showed a greatly elevated T1, comparable only to residues at the end of the L11-CTD, suggesting a very fast motion of Ala51. This is in contrast to the complexes in which the Ala51 T1 value is about same as the mean value of T1. Last, the NOE value of the L11 Thr71 was depressed in the binary complex but was about the average value in the ternary complex (Figure 6(c)), suggesting that thiostrepton binding resulted in an altered dynamics, most likely more rigid conformation. It is noteworthy that Leu12 showed a depressed value in the binary complex but had an average NOE value in the ternary complex (Figure 6(c)), indicating a more rigid conformation, which might be attributed directly to thiostrepton binding as Leu12 was located right across from the 310-helix, the putative binding site for the antibiotic. We also attempted to investigate the slow motion dynamics of L11 in the three states. The slow motion dynamics of free L11, using the Car-Puercell-Meiboom-Gill technique (CPMG) dispersion experiments41,45 indicate residues Ser75 at the end of the linker along Gly49 in the hinge region in free L11 showed internal motions on a μs-ms time scale. Ser75 is located at the beginning of α-helix 2 in the L11-CTD. We have performed the same experiments for L11 in the binary and ternary complexes but were unable to obtain consistent sets of results due to a poor signal-to-noise ratio of the spectra.
It is clear that residues that exhibit dynamic motion in the free state also have large changes the φ/ψ torsion angles when compared with those in the binary complex (Figures 4 and 6).12 In particular, Ser75, located at the beginning of α2, shows ms-μs time scale motions; Thr71, located at the end of the linker, shows an ns-ps time scale motion (Figures 4 and 6). Both of the regions undergo large conformation changes (Figure 4). Furthermore, binding of L11 to the RNA causes sizable changes in the torsion angles of the hinge residues that hold the 310HTH and β-sheets (Figures 1(c) and 4). As a result, Pro21 and Pro22 in the motif begin to face toward A1067 and A1095 of the RNA, as shown in the binary complex.12 The ability of torsion angles to change in the region around G49 also appears to be associated with motions of μs-ms (G49) and ns-ps (residues 48–52) time scales (Figures 4 and 5).
The mode of action of thiostrepton
The chemical shift differences of the L11 amide groups between the free protein and binary complex indicate, as expected from RNA binding studies with truncated L1114,46 and from crystal structures of L11-RNA complexes,12,13 that L11 binds to the RNA via the L11-CTD (Figure 8(a) and (b)). The largest chemical shift perturbations were seen in the RBL between residues Gly83 and Gly97. Besides the large perturbations of chemical shifts in the L11-CTD, smaller perturbations of chemical shifts also occurred in the L11-NTD residues.
Figure 8.

1H and 15N chemical shift differences in the backbone amide groups of L11 in the free state and the binary complex (a) and (b), respectively, and L11 in the binary and ternary complexes (c) and (d), respectively. The secondary structure of L11 is drawn on the top of the figure.
In contrast to the difference between chemical shifts in free and RNA-bound L11, a comparison of the binary and ternary complexes suggests that binding of thiostrepton mainly perturbs residues within the L11-NTD, while those in the L11-CTD remain largely unchanged (Figures 8(c) and (d)). Some of the largest perturbations of chemical shifts in the L11-NTD occurred in amides of residues Val5, Val7, Leu10, Ala32, Gly49-Ala51, Val53 and Ser75, among which residues Gly49-Ala51 are located in one of the hinges that bridge the β-sheets and the 310HTH motif. The same region also showed one of largest changes in the torsion angles when bound to the RNA (Figure 4), and ms-ps motions (Figure 6(c)). The amide chemical shifts of Ile52 were assigned in the free L11 but the cross peak was missing in both the spectra of the binary and ternary complexes. We could not obtain chemical shift perturbation data in the putative binding site for thiostrepton because it contains two prolines (Pro21 and Pro22); the cross peaks of residues next to these two prolines were also missing in the binary complex spectrum, possibly due to exchange line broadening. However, many of these missing cross peaks in the spectrum of L11 in the binary complex, including two residues immediately following these prolines, re-emerged in the spectrum of the ternary complex. These residues were Lys3, Val4, Gln11, Val23, Gly24, Ala26, Gly28 and Gln29. The re-emergence of these missing cross peaks was the direct result of altered chemical exchange/dynamics of the region caused by thiostrepton binding. These affected residues are located in β1 and the 310-helix (Figure 9).
Figure 9. The molecular surface and ribbon diagram of L11.
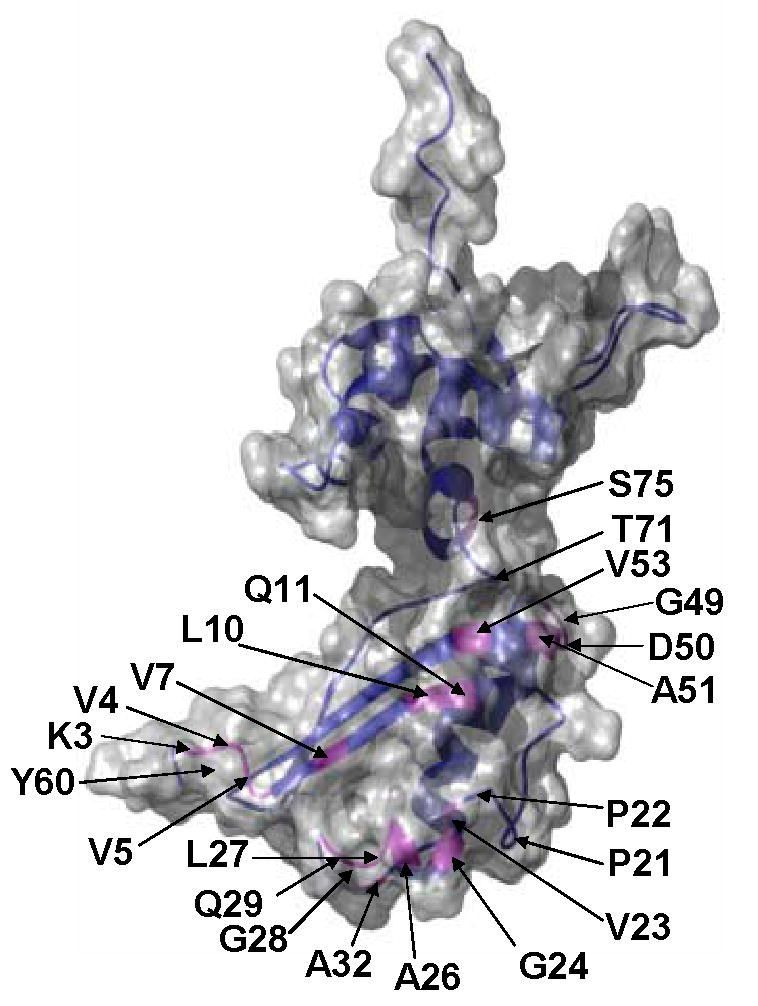
Residues whose chemical shifts of backbone amide groups either re-emerged or were significantly perturbed upon thiostrepton binding are indicated by their residue numbers and magenta color in the ribbon diagram. The Pro21 and Pro22 positions in the structure are also indicated. The position of Tyr60 is indicated at the back of the N-terminus.
Discussion
In the bacterial ribosome, the complex between protein L11 and the associated 58mer nt domain of rRNA is involved in several important, factor-dependent processes.47 During the elongation cycle of protein synthesis, EF-Tu–aminoacyl-tRNA complexes and EF-G, which share structural similarities,48 alternately interact with the same region of the ribosome.49 It has been hypothesized that the L11-NTD, which only weakly interacts with rRNA,12 may be able intrinsically to change its conformation or position, and function as a molecular switch in synchronization with the binding and release of EF-Tu and EF-G during the elongation cycle.12,16,50 The antibiotic thiostrepton may affect the ribosome function by altering the conformations accessible to the L11-NTD.10,11 In this article, we present experimental evidence that may reveal the structural and dynamic basis for the mobility of the L11-NTD.
The question of the relative orientation and position of the two domains is the central to the “reversible switch” hypothesis. The L11-NTD in the free-state and in the binary complex adopts different conformations and orientations relative to its CTD. The different orientations are due to alternative conformations in the regions flanking the linker and in one of the hinges in the L11-NTD, both of which exhibit ns-ps motions. In the crystal structure of the binary complex,12 interface residues forming hydrogen bonds (the equivalent of Thr71 is bonded to Lys111 and Asp114) are highly conserved. However, these hydrogen bonds are not present in our solution structure of the free L11, and our relaxation data indicate that the amide 15N of Thr71 of L11 in the binary complex exhibits ns-ps motions (Figure 6). The major structural differences in terms of the torsion angles can be seen in the residues flanking the linker and the residues in the hinges between the β-sheets and the 310HTH motif in the L11-NTD, in addition to those expected in the L11-CTD (Figure 4). These structural differences correlate well with the chemical shift perturbation data (Figure 8 (a) and (b)): wherever a large change in torsion angles occures between the two structures, a large change in chemical shifts occurs in the same region, suggesting that these structural differences are not artifacts of crystal packing. Furthermore, the change in the Rg value, from 18.5 Å for free L11 to 16.2 Å for the L11 in the binary complex (1MMS12), with the χ2 = 9.4 between the experimental and the back-calculated SANS, indicates a decrease in overall dimension, likely due to a shift of the relative position between the two domains, upon binding to the RNA. We suggest that the inherent dynamics in the regions flanking the linker and in the hinges allows them to adopt different conformations and the relative position with little penalties in free energy when L11 binds to rRNA. While this work was in progress, an NMR study of L11 from T. maritima18 also concluded that the L11 domain interface in solution is rigid and differs from that in the crystal structure. Unfortunately, the coordinates of that structure are not availabel for a detailed comparison with the T. thermophilis structure reported here.
Thiostrepton is believed to be the only antibiotic drug to bind to both rRNA and a protein in ribosome. Its binding site on 23S rRNA, which involves A1067 and A10957,51, is relatively well defined. In contrast, the interface between L11 and thiostrepton is less clear. A mutagenesis study suggests that both Pro21 and Pro22 may be involved in the binding.10 A recent proposal suggests that the antibiotic primarily binds to rRNA, inhibiting protein synthesis by blocking a conformational change in the L11-NTD.11,47 Indirect experimental evidence came from the altered protease susceptibility of Tyr62.11,47 Tyr62 (Tyr60 in T. thermophilis) is located at the end of β2, which is adjacent to the N-terminus of the protein. The chemical shift perturbation data indicate that the residues in the N-terminus were among the most affected by thiostrepton binding (Figure 8 (c) and (d), and Figure 9), suggesting that significant conformation change took place in the region. It is possible that the thiostrepton-induced conformational change in the N-terminus renders Tyr62 less accessible to the protease.
Moreover, the residues that were affected by thiostrepton binding can be divided into two groups. The first group is located either in the vicinity of the putative binding site, and the second group in the remote region at the hinge and the end of the linker. The perturbed residues near the putative binding site include those in the 310-helix, in the opposing β1 and the rest of the N-terminus (Figure 9). The 310-helix and the opposing β1 strand form a shallow pocket. Interestingly, almost all residues following, but none of those preceding the Pro21 and Pro22, were affected by the binding. One plausible explanation is that thiostrepton may be in close contact to the residues in the 310-helix, which directly faces RNA residues A1067 and A1095 in the binary complex.12 This explanation is consistent with the proposed “thiostrepton pocket” model, in which the antibiotic is sandwiched between A1067 and A1095, and the 310-helix.47 The second group of the perturbed residues include those in the hinge and those at the end of the link (Figure 9). Both Ala51 in the hinge and Thr71 at the end of the linker exhibit ns-ps dynamics in the binary complex (Figure 6(c)), and Thr71 appears to become rigid upon thiostrepton binding in the ternary complex (Figure 6(c)). The chemical shift perturbation of these amide groups in the hinge and at the end of the linker is likely due to the conformation change taking place in these two dynamic regions to accommodate the antibiotic insertion into the pocket formed between the A1067 and A1095, and the 310-helix. Furthermore, restricted motions of Thr71 and elsewhere throughout the L11 backbone, as suggested by an increase in average values of NOE upon thiostrepton binding, may limit the conformation space that is required for a concerted movement involving L11 and several other factors during the elongation cycle in protein synthesis
Materials and Methods
Sample preparation
The 58mer rRNA fragment was prepared using a protocol similar to one previously described by other researchers.52 RNA was transcribed in vitro using purified His6-tagged T7 RNA polymerase and plasmid DNA, one end of which was generated by digestion using Rsa I to give the exact 5′ ending sequence of the RNA fragment.52 The RNA was purified by denaturing 20% polyacrylamide gel electrophoresis and excised from the gel. RNA was eluted from the gel using an Elutrap (Schleicher & Schuell, Keene, NH), ethanol-precipitated, and desalted using 3K molecular cutoff spin filters (Millipore, Billerica, MA). The purified RNA was lyophilized and suspended in a buffer containing 50 mM NaCl, 20 mM MES (pH 6.5) and 0.5 mM MgCl2 to make a 1.2 mM stock RNA solution. The protein L11 sample was prepared in a way similar to that previously reported,53 with the following exception: a his-tag followed by a thrombin digestion site was cloned into the N-terminus of the protein; therefore, a cobalt column was used in the first step of purification. The his-tag was cleaved by thrombin digestion (0.01 unit/μg) at ambient room temperature overnight. The digestion mixture was passed through a C-18 reverse-phase column, and the L11 fractions were lyophilized and suspended in the same buffer as the RNA sample to make a 1.5 mM stock solution. Thiostrepton was purchased from Sigma (St. Louis, MO) and was dissolved in DMSO to make a concentrated stock of ~100 mM. To make the ternary complex of L11-RNA-thiostrepton, we first titrated the L11 solution with the RNA using amide 2D [15N, 1H]-TROSY spectra54 until the peaks of L11 in the free form were not observed. We then added thiostrepton stock solution to the L11-RNA complex to confirm the ternary complex by monitoring with 2D [15N,1H]-TROSY. The final ternary complex solution contained 5% DMSO. As a control, up to 10% DMSO was added to the L11-RNA complex sample, but the signal from the protein in 2D [15N,1H]-TROSY spectra was not changed. The ternary complex solution was then spin-filtered in a 10K MWCO filter to concentrate it to 0.7 mM in about 330 μl volume. Using a previously reported procedure,55 a preparation of 85% perdeuterate, 13C/15N-labeled L11 was used for assigning the backbone of L11 in the ternary complex. The RDCs of L11 were recorded using a 6% polyacrylamide gel as described in a published procedure.56 The ratio of stretching was 6:4.8.
Solution structure determination of free L11
The 1H/13C/15N chemical shift assignments of the free L11 were made in 10 mM KPi (pH 6.5), 70 mM KCl,53 which is different from our current buffer. We verified the assignments using the normal array of through-bond assignment experiments.57 For the structure determination of the free L11, we obtained distance constraints from three NOESY experiments with a mixing time of 90 ms, i.e. 3D 15N-resolved [1H,1H]-NOESY, and 3D 13C-resolved [1H,1H]-resolved NOESY, with the 13C carrier frequency in the aliphatic and aromatic region, respectively. All NMR data were processed with the program nmrPipe,58 and the program CARA was used for the spectral analysis.59
The three NOESY spectra were picked and assigned by automated NOESY peak picking and the NOE assignment method ATNOS/CANDID.60 The input for the ATNOS/CANDID approach consisted of the chemical shift list obtained from a sequence-specific assignment and the three NOESY spectra. A total of 1,256 meaningful NOE upper limits obtained from ATNOS/CANDID calculation, together with 565 torsion angle constraints, was used for structure calculation with the program CYANA.61 Based on the 20 lowest target function conformers from the aforementioned structure calculation, 51 backbone hydrogen bonds were identified by CYANA and used for the final structure calculation.
This NOE-based solution structure of L11 was further refined with RDC, using Xplor-NIH version 2.6. About 150 RDCs, for residues with order parameters greater than 0.7 and well resolved cross peaks, were used in the refinement. The initial Da and R values were estimated from a histogram,62 and the final values, 14.0 and 0.55, and 14.3 and 0.5, were determined using the SA and SVD methods, respectively.19,62 Because L11 consists of two domains joined by a short linker, we also evaluated the Da and R of the two domains individually, using both approaches. Since the results were very similar for each domain using both approaches, one alignment was sufficient to characterize L11. The structure validation was performed using standard procedures 63,64 and the average Peterson correlation coefficients, Rp, were approximately 0.95. The calculated structures have good covalent geometries, and the residues in the most favoured, additionally allowed, generously allowed and disallowed regions are 64.8%, 26.2%, 6.6% and 2.5%, respectively. It is noteworthy that more than 50% of the L11 residues are in non-regular, secondary structure regions. The residues that are in generously allowed and disallowed regions are mostly those in the flexible loops where there is little restraint.
Chemical shift assignment of L11 in binary and ternary complexes
Assigning the backbone heteronuclear chemical shifts of the L11 in the binary and ternary complexes was challenging due to unfavorable solution behavior of the complexes. This was especially true for the binary complex and was likely due to “rigid body” movements of the L11-NTD relative to the rest of the complex. The overall rotational correlation times of L11 in the complexes were estimated to be ~17 ns using the 1D [15N,1H]-TRACT.38 We applied a combination of 3D TROSY-type triple resonance experiments,65 namely HNCA, HNCACB and 3D15N-resolved [1H,1H]-NOESY spectra to assign more than 98% of the backbone amide group, 13Cα and 13Cβ of L11 in the binary and the ternary complexes using 15N,13C,2H-triplely labeled L11.
Relaxation experiments
For detailed analysis of the dynamic properties of L11 in the free form, we recorded conventional T1, T1ρ and 15N-[1H] NOE at 30 °C on a Bruker Avance600 spectrometer operating at a 1H frequency of 600.133 MHz. The T1 data was obtained using 15N relaxation delays of 8, 64, 136, 232, 336, 472, 664 and 800 ms. The T1ρ data was obtained using 15N relaxation delays of 8, 24, 48, 72, 96, 120, 160 and 192 ms with the 625 Hz field strength CPMG. For the 15N-[1H] NOE measurement, two 2D spectra were acquired in an interleaved manner with and without the saturation of protons.
To compare the dynamic properties among L11, the L11-RNA and L11-RNA-thiostrepton complexes, we recorded T1, T1ρ, and 15N-[1H] NOE of the TROSY component39,41 at 40 °C on the Bruker Avance600 spectrometer. For L11, the T1 data was obtained using relaxation delays of 10.7, 85.3, 192, 320, 480, 687.3 and 981.3 ms, and T1ρ was obtained using relaxation delays of 32, 64, 96, 128, 192, 288, 320, 384 and 448 ms with the 625 Hz field strength CPMG. For the L11-RNA complex and L11-RNA-thiostrepton complex, the 15N T1 was obtained using relaxation delays of 10.7, 85.3, 192, 320, 480, 687.3, 981.3 and 1440 ms; T1ρ was obtained from experiments with relaxation delays of 3.2, 6.4, 9.6, 12.8, 19.2, 28.8, 32.0, 38.4, 44.8, 57.6, 76.8 and 96.0 ms, with a 625 Hz field strength CPMG. The values of 15N-[1H] NOE for all samples were from an intensity ratio derived from two data sets taken with and without saturation of the protons.
For the relaxation dispersion measurement of L11, we recorded two sets of T1ρ41 with τCP of 1 and 10 ms refocusing CPMG at 30 °C. For T1ρ with the 1 ms refocusing CPMG, the relaxation delays of 4, 8, 16, 32, 64, 128, 256 and 512 ms were measured, while the relaxation delays of 40, 80, 160, 240, 320 and 440 ms were measured with the 10 ms refocusing CPMG. All relaxation measurements were duplicated several days apart.
All data were processed with the NMRPipe software package,58 and peak integrations were performed with the CARA software package.59 The peak intensities versus relaxation delays for T1 and T1ρ were fitted to a single exponential decay to obtain the relaxation times.
Determination of components of the rotational diffusion tensor of the free L11
The rotational diffusion tensors were calculated by standard linear least-squares optimization using the structures of the individual domains (55 and 52 residues were used for the L11-NTD and the L11-CTD, respectively) and the full-length L11 (107 residues) solution structure.24,25 Even though the relative lengths of the principal axes of the inertia tensor for L11 (1.00:0.69:0.41) imply that the rotational diffusion tensor may be anisotropic, the rotational diffusion tensors calculated using the axial symmetric model and the anisotropic model are very similar (Table 2). Thus, we used the axial symmetric model for the detailed analysis of 15N relaxation22 (Figure S3, Supplementary Materials). The rotational diffusion tensor D was determined by a linear least-squares fit to the T1/T1ρ ratio of a set of 15N-1H vectors24,25 in the individual domains and overall L11, using the 20 lowest-energy conformers representing the solution structure. Residues with 15N-[1H] NOE values less than 0.65 were excluded from the calculation, due to fast internal motions. In addition, residues that are subject to conformational exchange were excluded with the following criteria:22
| (1) |
where σ is the standard deviation of , and the brackets indicate average overall residues.
SANS measurements and refinement of the structures with SANS data
SANS measurements were performed on the 30-meter SANS instruments at the National Institute of Standards and Technology (NIST) Center for Neutron Research in Gaithersburg, MD.53 The neutron wavelength, λ, was 5Å, with a wavelength spread, Δλ/λ, of 0.15. Scattered neutrons were detected with a 64×64 cm 2-dimensional position-sensitive detector with 128×128 pixels at a resolution of 0.5 cm/pixel. Raw counts were normalized to a common monitor count and corrected for empty cell counts, ambient room background counts and non-uniform detector response. Data were placed on an absolute scale by normalizing the scattered intensity to the incident beam flux. Finally, the data were radially averaged to produce scattered intensity, I(Q), vs. Q curves, where Q=4πsin(θ)/λ and 2θ is the scattering angle. A sample-to-detector distance of 1.5 m was used to cover the range 0.03Å-1≤Q≤0.3Å-1. The scattered intensities from the samples were then further corrected for buffer scattering and incoherent scattering from hydrogen in the samples.
Initial data analysis was performed using the Guinier approximation, on the low-Q
| (2) |
portions of the data to obtain values for the radius of gyration, Rg, and the forward scattering intensity, I(0), of the samples. This analysis is valid only in the region where QRg ~1. SANS scattered intensity curves were compared to model curves calculated from high-resolution X-ray crystal or NMR structures using the program XTAL2SAS.54,55 The Rg values from the model SANS intensity curves were obtained by XTAL2SAS using the following relation:
| (3) |
where P(r) is the distance distribution function. The Rg values calculated from the model structures were compared to the average Rg calculated from the combined H2O and D2O data. In addition, the model scattered intensity curves were compared to the average scattered intensity curve for the data obtained in D2O buffer. The I(Q) vs. Q SANS data were recorded for L11 at concentrations ranging from 2 mg/ml to 5 mg/ml in H2O buffer and from 1 mg/ml to 5 mg/ml in D2O buffer, along with the Rg and I(0) values calculated using the Guinier analysis. We used the same buffer for both NMR and SANS experiments.
Refinement of the NMR-structures against the SANS data was performed using the newly implemented protocol in the Xplor-NIH package. While the details of the protocol will be described elsewhere to avoid diluting the main focus of this article, a brief outline follows: A harmonic energy potential, along with other standard multiple energy terms,66 was used for the refinement against the scattering intensity,
| (4) |
where ωscat is an overall scale factor on the energy term, ωj is a per q weighting, Iobs(qj) is the observed scattering intensity, and the sum is over the data points of qj . The protocol consists of 20 ps of dynamics at 3000 K, followed by annealing from 3000 K to 25 K at 12.5K increments, with 0.2ps of dynamics run at each temperature. Final gradient minimization was performed in torsion-angle space, followed by all-degree of freedom minimization. The 20 lowest-energy (omitting TADB and non-bonded terms) structures were used for analysis. Atomic masses were set to 300 amu. With the regard to the fitness of the structures to the SANS, χ2 values were calculated for the top 20 structures using the equation where NI is the total number of data points, and are the observed and calculated scattering intensities at point j, and and are the scattering intensities normalized to their q = 0 values.
| (5) |
The RDC-refined structure was used as the starting coordinates for the refinement with the SANS data. The force constants on the various energy terms, Table 3, were either scaled geometrically during refinement, or held constant, while the atomic radius used in the nonbonded interaction was scaled down. Xplor-NIH versions 2.16 and later contain support for SANS refinement. The input scripts used in these calculations can be obtained from the authors.
Table 3.
Scaling force constants used in the refinement
| Term | high temperature | initial | final | units |
|---|---|---|---|---|
| RDC | 0.001 | 0.001 | 1 | kcal/mol/Hz2 |
| NOE | 30 | 2 | 30 | kcal/mol/Å2 |
| Dihedral | 10 | 200 | 200 | kcal/mol/rad2 |
| TADB | 0.002 | 0.002 | 1 | kcal/mol |
| SANS | 100 | 100 | 100 | kcal/mol |
| Bond | 1 | 1 | 1 | kcal/mol/ Å2 |
| Angle | 0.4 | 0.4 | 1 | kcal/mol/rad2 |
| Improper | 0.1 | 0.1 | 1 | kcal/mol/ rad2 |
| Nbond | 0.004 | 0.004 | 4 | kcal/mol/ Å4 |
| Radius | 1.2 | 0.9 | 0.8 | Å |
The L11 structures have been deposited with PDB accession codes 2H8W (NOE-dihedral-refined), 2E34 and 2E35 (Rg-refined) and 2E36 (SANS-refined). The assignments of the backbone amide 1H and 15N, 13Cα and 13Cβ have been deposited with accession codes 7314 for the binary, 7315 for the ternary complexes.
Supplementary Material
Figure 7.
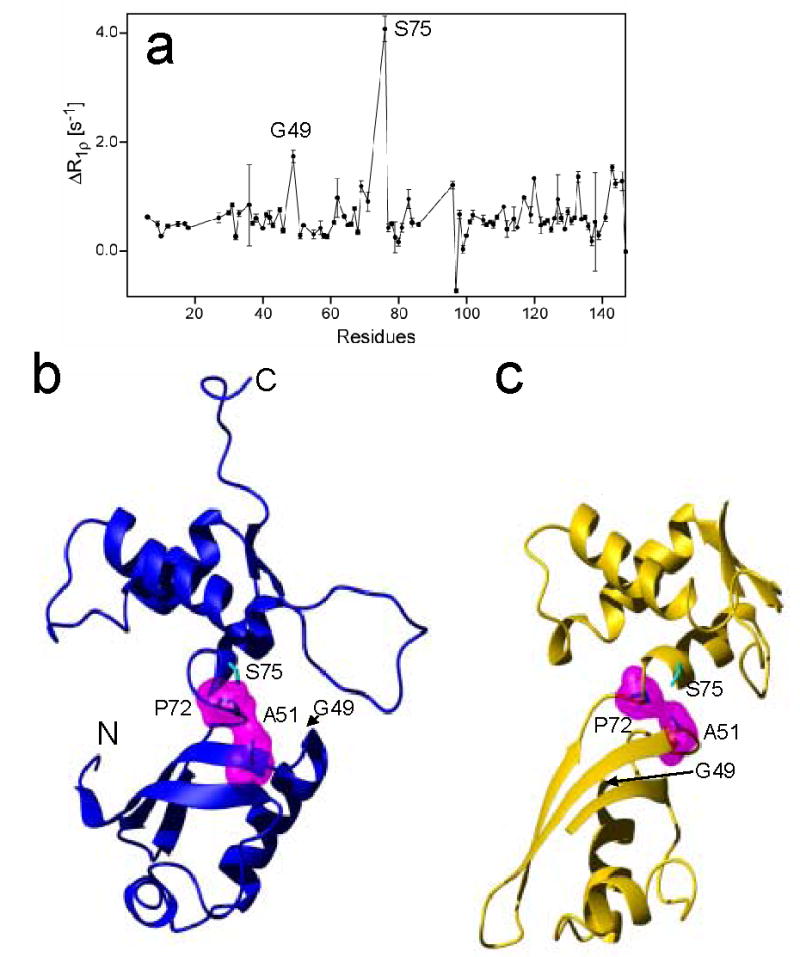
(a) Plot of the difference ΔR1ρ=R1ρ(τCP=10 ms) R1ρ(τCP=1 ms) of L11 versus the residue number, and the ribbon drawings of (b) the solution structure and (c) the crystal structure of L11. The surface areas of Ala51 and Pro72 in magenta, which were calculated using program MolMol, are in close hydrophobic contact in both the free state and binary complex.. The residues showing conformational exchange are indicated (Gly49 and Ser75).
Acknowledgments
We thank Dr. Bruce Shapiro for providing valuable discussion. This research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials, instruments, or equipment identified are necessarily the best availabel for the purpose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moore PB, Steitz TA. The ribosome revealed. Trends Biochem Sci. 2005;30:281–283. doi: 10.1016/j.tibs.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000;289:920–930. doi: 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- 3.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science. 2000;289:905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 4.Agrawal RK, Spahn CM, Penczek P, Grassucci RA, Nierhaus KH, Frank J. Visualization of tRNA movements on the Escherichia coli 70S ribosome during the elongation cycle. J Cell Biol. 2000;150:447–460. doi: 10.1083/jcb.150.3.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cundliffe E, Thompson J. Ribose methylation and resistance to thiostrepton. Nature. 1979;278:859–861. doi: 10.1038/278859a0. [DOI] [PubMed] [Google Scholar]
- 6.Egebjerg J, Douthwaite S, Garrett RA. Antibiotic interactions at the GTPase-associated centre within Escherichia coli 23S rRNA. Embo J. 1989;8:607–611. doi: 10.1002/j.1460-2075.1989.tb03415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson J, Cundliffe E, Stark M. Binding of thiostrepton to a complex of 23-S rRNA with ribosomal protein L11. Eur J Biochem. 1979;98:261–265. doi: 10.1111/j.1432-1033.1979.tb13184.x. [DOI] [PubMed] [Google Scholar]
- 8.Modolell J, Cabrer B, Parmeggiani A, Vazquez D. Inhibition by siomycin and thiostrepton of both aminoacyl-tRNA and factor G binding to ribosomes. Proc Natl Acad Sci USA. 1971;68:1796–1800. doi: 10.1073/pnas.68.8.1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stoffler G, Cundliffe E, Stoffler-Meilicke M, Dabbs ER. Mutants of Escherichia coli lacking ribosomal protein L11. J Biol Chem. 1980;255:10517–10522. [PubMed] [Google Scholar]
- 10.Cameron DM, Thompson J, Gregory ST, March PE, Dahlberg AE. Thiostrepton-resistant mutants of Thermus thermophilus. Nucleic Acids Res. 2004;32:3220–3227. doi: 10.1093/nar/gkh644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Porse BT, Leviev I, Mankin AS, Garrett RA. The antibiotic thiostrepton inhibits a functional transition within protein L11 at the ribosomal GTPase centre. J Mol Biol. 1998 ;276:391–404. doi: 10.1006/jmbi.1997.1541. [DOI] [PubMed] [Google Scholar]
- 12.Wimberly BT, Guymon R, McCutcheon JP, White SW, Ramakrishnan V. A detailed view of a ribosomal active site: the structure of the L11-RNA complex. Cell. 1999;97:491–502. doi: 10.1016/s0092-8674(00)80759-x. [DOI] [PubMed] [Google Scholar]
- 13.Conn GL, Draper DE, Lattman EE, Gittis AG. Crystal structure of a conserved ribosomal protein-RNA complex. Science. 1999;284:1171–1174. doi: 10.1126/science.284.5417.1171. [DOI] [PubMed] [Google Scholar]
- 14.Bausch SL, Poliakova E, Draper DE. Interactions of the N-terminal domain of ribosomal protein L11 with thiostrepton and rRNA. J Biol Chem. 2005 doi: 10.1074/jbc.M504182200. [DOI] [PubMed] [Google Scholar]
- 15.Datta PP, Sharma MR, Qi L, Frank J, Agrawal RK. Interaction of the G’ domain of elongation factor G and the C-terminal domain of ribosomal protein L7/L12 during translocation as revealed by cryo-EM. Mol Cell. 2005;20:723–731. doi: 10.1016/j.molcel.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 16.Agrawal RK, Linde J, Sengupta J, Nierhaus KH, Frank J. Localization of L11 protein on the ribosome and elucidation of its involvement in EF-G-dependent translocation. J Mol Biol. 2001;311:777–787. doi: 10.1006/jmbi.2001.4907. [DOI] [PubMed] [Google Scholar]
- 17.Seo HS, Abedin S, Kamp D, Wilson DN, Nierhaus KH, Cooperman BS. EF-G-dependent GTPase on the ribosome. conformational change and fusidic acid inhibition. Biochemistry. 2006;45:2504–2514. doi: 10.1021/bi0516677. [DOI] [PubMed] [Google Scholar]
- 18.Ilin S, Hoskins A, Ohlenschlager O, Jonker HR, Schwalbe H, Wohnert J. Domain reorientation and induced fit upon RNA binding: solution structure and dynamics of ribosomal protein L11 from Thermotoga maritima. Chembiochem. 2005;6:1611–8. doi: 10.1002/cbic.200500091. [DOI] [PubMed] [Google Scholar]
- 19.Losonczi JA, Andrec M, Fischer MWF, Prestegard JH. Order matrix analysis of residual dipolar couplings using singular value decomposition. J Magn Reson. 1999;138:334–342. doi: 10.1006/jmre.1999.1754. [DOI] [PubMed] [Google Scholar]
- 20.Bax A, Grishaev A. Weak alignment NMR: a hawk-eyed view of biomolecular structure. Curr Opinion Struct Biol. 2005;15:563–570. doi: 10.1016/j.sbi.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 21.Copie V, Tomita Y, Akiyama SK, Aota S, Yamada KM, Venable RM, Pastor RW, Krueger S, Torchia DA. Solution structure and dynamics of linked cell attachment modules of mouse fibronectin containing the RGD and synergy regions: comparison with the human fibronectin crystal structure. J Mol Biol. 1998;277:663–682. doi: 10.1006/jmbi.1998.1616. [DOI] [PubMed] [Google Scholar]
- 22.Tjandra N, Feller SE, Pastor RW, Bax A. Rotational diffusion anisotropy of human ubiquitin from N-15 NMR relaxation. 1995;117:12562–6. [Google Scholar]
- 23.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by N-15 inverse-detected heteronuclear Nmr spectroscopy - Application to staphylococcal nuclease. Biochem. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 24.Lee LK, Rance M, Chazin WJ, Palmer AG. Rotational diffusion anisotropy of proteins from simultaneous analysis of N-15 and C-13(alpha) nuclear spin relaxation. 1997;9:287–298. doi: 10.1023/a:1018631009583. [DOI] [PubMed] [Google Scholar]
- 25.Bruschweiler R, Liao XB, Wright PE. Long-range motional restrictions in a multidomain zinc-finger protein from anisotropic tumbling. 1995;268:886–889. doi: 10.1126/science.7754375. [DOI] [PubMed] [Google Scholar]
- 26.Wüthrich K. NMR of Proteins and Nucleic Acids. John Wiley; New York: 1986. [Google Scholar]
- 27.Koch MH, Vachette P, Svergun DI. Small-angle scattering: a view on the properties, structures and structural changes of biological macromolecules in solution. Q Rev Biophys. 2003;36:147–227. doi: 10.1017/s0033583503003871. [DOI] [PubMed] [Google Scholar]
- 28.Grossmann JG, Ali SA, Abbasi A, Zaidi ZH, Stoeva S, Voelter W, Hasnain SS. Low-resolution molecular structures of isolated functional units from arthropodan and molluscan hemocyanin. Biophys J. 2000;78:977–981. doi: 10.1016/s0006-3495(00)76655-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krueger JK, Gallagher SC, Wang CA, Trewhella J. Calmodulin remains extended upon binding to smooth muscle caldesmon: a combined small-angle scattering and fourier transform infrared spectroscopy study. Biochemistry. 2000;39:3979–3987. doi: 10.1021/bi992638x. [DOI] [PubMed] [Google Scholar]
- 30.Bernocco S, Finet S, Ebel C, Eichenberger D, Mazzorana M, Farjanel J, Hulmes DJ. Biophysical characterization of the C-propeptide trimer from human procollagen III reveals a tri-lobed structure. J Biol Chem. 2001;276:48930–48936. doi: 10.1074/jbc.M108611200. [DOI] [PubMed] [Google Scholar]
- 31.Aparicio R, Fischer H, Scott DJ, Verschueren KH, Kulminskaya AA, Eneiskaya EV, Neustroev KN, Craievich AF, Golubev AM, Polikarpov I. Structural insights into the beta-mannosidase from T. reesei obtained by synchrotron small-angle X-ray solution scattering enhanced by X-ray crystallography. Biochemistry. 2002;41:9370–9375. doi: 10.1021/bi025811p. [DOI] [PubMed] [Google Scholar]
- 32.Sunnerhagen M, Olah GA, Stenflo J, Forsen S, Drakenberg T, Trewhella J. The relative orientation of Gla and EGF domains in coagulation factor X is altered by Ca2+ binding to the first EGF domain. A combined NMR-small angle X-ray scattering study. Biochemistry. 1996;35:11547–11559. doi: 10.1021/bi960633j. [DOI] [PubMed] [Google Scholar]
- 33.Mattinen ML, Paakkonen K, Ikonen T, Craven J, Drakenberg T, Serimaa R, Waltho J, Annila A. Quaternary structure built from subunits combining NMR and small-angle x-ray scattering data. Biophys J. 2002;83:1177–1183. doi: 10.1016/S0006-3495(02)75241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Glinka CJ, Barker JG, Hammouda B, Krueger S, Moyer JJ, Orts WJ. The 30 m small-angle neutron scattering instruments at the National Institute of Standards and Technology. J Appl Cryst. 1998;31:430–445. [Google Scholar]
- 35.Kuszewski J, Gronenborn AM, Clore GM. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J Am Chem Soc. 1999;121:2337–2338. [Google Scholar]
- 36.Jacrot B. The study of biological structures by neutron scattering from solution. Rep Prog Phys. 1976;39:911–953. [Google Scholar]
- 37.Grishaev A, Wu J, Trewhella J, Bax A. Refinement of multidomain protein structures by combination of solution small-angle X-ray scattering and NMR data. J Am Chem Soc. 2005;127:16621–16628. doi: 10.1021/ja054342m. [DOI] [PubMed] [Google Scholar]
- 38.Lee D, Hilty C, Wider G, Wuthrich K. Effective rotational correlation times of proteins from NMR relaxation interference. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 39.Zhu G, Xia Y, Nicholson LK, Sze KH. Protein dynamics measurements by TROSY-based NMR experiments. J Magn Reson. 2000;143:423–6. doi: 10.1006/jmre.2000.2022. [DOI] [PubMed] [Google Scholar]
- 40.Zhu G, Xia Y, Lin D, Gao X. TROSY-based NMR experiments for the study of macromolecular dynamics and hydrogen bonding. Methods Mol Biol. 2004;278:161–184. doi: 10.1385/1-59259-809-9:161. [DOI] [PubMed] [Google Scholar]
- 41.Loria JP, Rance M, Palmer AG. A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999;15:151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- 42.Markus MA, Hinck AP, Huang S, Draper DE, Torchia DA. High-resolution solution structure of ribosomal protein L11-C76, a helical protein with a flexible loop that becomes structured upon binding to RNA. Nat Struct Biol. 1997;4:70–77. doi: 10.1038/nsb0197-70. [DOI] [PubMed] [Google Scholar]
- 43.Hinck AP, Markus MA, Huang S, Grzesiek S, Kustonovich I, Draper DE, Torchia DA. The RNA binding domain of ribosomal protein L11: three-dimensional structure of the RNA-bound form of the protein and its interaction with 23 S rRNA. J Mol Biol. 1997;274:101–113. doi: 10.1006/jmbi.1997.1379. [DOI] [PubMed] [Google Scholar]
- 44.Li W, Sengupta J, Rath BK, Frank J. Functional conformations of the L11-ribosomal RNA complex revealed by correlative analysis of cryo-EM and molecular dynamics simulations. Rna. 2006 doi: 10.1261/rna.2294806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang C, Rance M, Palmer AG., 3rd Mapping chemical exchange in proteins with MW > 50 kD. J Am Chem Soc. 2003;125:8968–8969. doi: 10.1021/ja035139z. [DOI] [PubMed] [Google Scholar]
- 46.Xing Y, Draper DE. Stabilization of a ribosomal RNA tertiary structure by ribosomal protein L11. J Mol Biol. 1995;249:319–331. doi: 10.1006/jmbi.1995.0299. [DOI] [PubMed] [Google Scholar]
- 47.Porse BT, Garrett RA. Ribosomal mechanics, antibiotics, and GTP hydrolysis. Cell. 1999;97:423–426. doi: 10.1016/s0092-8674(00)80751-5. [DOI] [PubMed] [Google Scholar]
- 48.Nissen P, Kjeldgaard M, Thirup S, Polekhina G, Reshetnikova L, Clark BF, Nyborg J. Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science. 1995;270:1464–1472. doi: 10.1126/science.270.5241.1464. [DOI] [PubMed] [Google Scholar]
- 49.Cundliffe E. On the nature of antibiotic binding sites in ribosomes. Biochimie. 1987;69:863–869. doi: 10.1016/0300-9084(87)90213-6. [DOI] [PubMed] [Google Scholar]
- 50.Rawat UB, Zavialov AV, Sengupta J, Valle M, Grassucci RA, Linde J, Vestergaard B, Ehrenberg M, Frank J. A cryo-electron microscopic study of ribosome-bound termination factor RF2. Nature. 2003;421:87–90. doi: 10.1038/nature01224. [DOI] [PubMed] [Google Scholar]
- 51.Rosendahl G, Douthwaite S. The antibiotics micrococcin and thiostrepton interact directly with 23S rRNA nucleotides 1067A and 1095A. Nucleic Acids Res. 1994;22:357–363. doi: 10.1093/nar/22.3.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Draper DE, Xing Y, Laing LG. Thermodynamics of RNA unfolding: stabilization of a ribosomal RNA tertiary structure by thiostrepton and ammonium ion. J Mol Biol. 1995;249:231–238. doi: 10.1006/jmbi.1995.0291. [DOI] [PubMed] [Google Scholar]
- 53.Markus MA, Triantafillidou D, Choli-Papadopoulou T, Torchia DA. Letter to the Editor: H-1, N-15, and C-13 assignments and secondary structure identification for full-length ribosomal protein L11 from Thermus thermophilus. 2001;20:293–294. doi: 10.1023/a:1011275602138. [DOI] [PubMed] [Google Scholar]
- 54.Pervushin K, Riek R, Wider G, Wuthrich K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garrett DS, Seok YJ, Liao DI, Peterkofsky A, Gronenborn AM, Clore GM. Solution structure of the 30 kDa N-terminal domain of enzyme I of the Escherichia coli phosphoenolpyruvate:sugar phosphotransferase system by multidimensional NMR. Biochemistry. 1997;36:2517–2530. doi: 10.1021/bi962924y. [DOI] [PubMed] [Google Scholar]
- 56.Chou JJ, Gaemers S, Howder B, Louis JM, Bax A. A simple apparatus for generating stretched polyacrylamide gels, yielding uniform alignment of proteins and detergent micelles. J Biomol NMR. 2001;21:377–382. doi: 10.1023/a:1013336502594. [DOI] [PubMed] [Google Scholar]
- 57.Grzesiek S, Bax A. An efficient experiment for sequential backbone assignment of medium-sized isotopically enriched proteins. J Magn Reson. 1992;99:201–207. [Google Scholar]
- 58.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe - A multidimensional spectral processing system based on Unix pipes. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 59.Keller R. The computer aided resonance tutorial. CANTINA Verlag; Goldau, Switzerland: 2004. [Google Scholar]
- 60.Herrmann T, Guntert P, Wuthrich K. Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR. 2002;24:171–189. doi: 10.1023/a:1021614115432. [DOI] [PubMed] [Google Scholar]
- 61.Guntert P, Mumenthaler C, Wuthrich K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J Mol Biol. 1997;273:283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 62.Clore GM, Gronenborn AM, Bax A. A robust method for determining the magnitude of the fully asymmetric alignment tensor of oriented macromolecules in the absence of structural information. J Magn Reson. 1998;133:216–221. doi: 10.1006/jmre.1998.1419. [DOI] [PubMed] [Google Scholar]
- 63.Bax A, Kontaxis G, Tjandra N. Nuclear Magnetic Resonance of Biological Macromolecules, Pt B. Vol. 339. 2001. Dipolar couplings in macromolecular structure determination; pp. 127–174. [DOI] [PubMed] [Google Scholar]
- 64.Wu Y, Migliorini M, Walsh J, Yu P, Strickland DK, Wang YX. NMR structural studies of domain 1 of receptor-associated protein. J Biomol NMR. 2004;29:271–279. doi: 10.1023/B:JNMR.0000032507.96325.6d. [DOI] [PubMed] [Google Scholar]
- 65.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. TROSY in triple-resonance experiments: New perspectives for sequential NMR assignment of large proteins. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schwieters CD, Kuszewski J, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:66–74. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 67.Semenyuk AV, I SD. GNOM - a program package for small-angle scattering data processing. J Appl Cryst. 1991;24:537–540. [Google Scholar]
- 68.Abragam A. The Principles of Nuclear Magnetism. Clarendon Press; Oxford: 1961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.