

Abstract
Nerve growth factor (NGF) promotes the survival of embryonic sensory neurons and maintains the phenotypic characteristics of primary nociceptive neurons postnatally. NGF also contributes to nociceptor activation and hyperalgesia during inflammatory pain states. The purpose of this study was to determine whether NGF might have an additional pronociceptive action by interfering with opioid-mediated analgesia in primary nociceptive neurons. Sensory neurons were isolated from the dorsal root ganglia of weanling rats and kept in standard culture conditions either with or without exogenous NGF (50 ng/ml). Currents through voltage-gated calcium channels were recorded from individual neurons using the whole cell patch clamp technique with Ba2+ as the charge carrier (IBa). The μ-opioid agonist fentanyl (1 μM) and the GABAB agonist baclofen (50 μM) were used to test G protein-dependent inhibition of IBa. Fentanyl inhibited IBa by an average of 38±4% in untreated cells vs. 25±2% in NGF-treated cells (P<0.01). NGF had no effect on IBa current magnitude or kinetics. The NGF-induced attenuation of opioid action was observed as early as 4 h after exposure, but was not seen when NGF was applied by bath perfusion for up to 40 min, suggesting that the effect was not mediated by a rapid phosphorylation event. The effect of NGF was prevented by K-252a (100 nM), an inhibitor of TrkA autophosphorylation. Baclofen-induced inhibition of IBa, on the other hand, was not affected by NGF treatment, suggesting that NGF modulation of opioid-mediated inhibition occurred upstream from the G protein. This was supported by the finding that GTP-γ-S, an agonist independent G protein activator, inhibited IBa similarly in both untreated and NGF treated cells. The results show that NGF selectively attenuated opioid-mediated inhibition of IBa via TrkA receptor activation, possibly by altering opioid receptor function.
Keywords: neurotrophins, DRG neurons, G proteins, patch clamp
Nerve growth factor (NGF) is a neurotrophin required for the survival of most primary nociceptive neurons during embryonic development. A large number of nociceptive neurons remain sensitive to NGF postnatally, and in adult animals NGF plays a role in both physiological and pathological pain conditions. In the resting state, low levels of NGF are needed for the expression of nociceptor markers such as the neuropeptides substance P and calcitonin-gene-related peptide (CGRP; Molliver and Snider, 1997; Patel et al., 2000), and for nociceptive afferents to retain their ability to respond to noxious stimuli (Ritter et al., 1991; Lewin and Mendell, 1994; McMahon et al., 1995; Bennett et al., 1998). During tissue inflammation NGF levels increase and contribute to inflammatory pain by inducing nociceptor sensitization and hyperalgesia (Donnerer et al., 1992; Woolf et al., 1994; McMahon et al., 1995; Koltzenburg et al., 1999).
NGF may sensitize nociceptors in several ways. NGF has been shown to upregulate the TRPV1 channel, one of the molecular transducers of noxious heat which is also activated by capsaicin, and to increase capsaicin-induced currents in nociceptors (Nicholas et al., 1999; Shu and Mendell, 2001; Ji et al., 2002), thereby contributing to thermal hyperalgesia. NGF also increases currents through voltage-gated calcium channels (VGCC) in a wide variety of cells (Lesser and Lo, 1995; Levine et al., 1995; Wildering et al., 1995; Lei et al., 1997; Jia et al., 1999) including sensory neurons (Fitzgerald and Dolphin, 1997). Upregulation of VGCC may enhance nociceptor excitability, and promote the release of excitatory substances from nociceptor terminals. It is not entirely clear which intracellular mediators may be involved in these effects, but some evidence points to several well known NGF-activated signaling cascades, the phosphoinositide 3 (PI3) kinase/Akt pathway, the Ras/mitogen activated protein (MAP) kinase pathway, and phospholipase C (PLC)-γ (Fitzgerald, 2000; Kaplan and Miller, 2000; Ji et al., 2002).
Opioid receptors couple to G proteins in many cell types and activate a variety of signaling pathways to exert their analgesic effects. One of these effects, mediated directly by G protein subunit interactions, is a modulation of VGCC in sensory neurons which leads to marked decreases in currents through VGCC. Opioid-mediated inhibition of VGCC is regulated by several kinases, including protein kinase C and PLC (Swartz, 1993; Zhu and Ikeda, 1994; Zamponi et al., 1997; Zhu and Yakel, 1997; King et al., 1999; Xie et al., 1999; Barrett and Rittenhouse, 2000). In addition, PI3 kinase and MAP kinases may be involved in opioid receptor desensitization (Schmidt et al., 2000; Tan et al., 2003). Because NGF activates these signaling cascades, it is conceivable that increased levels of NGF might attenuate opioid receptor function, thus antagonizing opioid-mediated analgesia.
This study was designed to test the hypothesis that NGF interferes with opioid-mediated inhibition of VGCC in sensory neurons. Experiments were designed to evaluate the effects of NGF on different aspects of G protein signaling. In addition, to determine whether NGF altered the function, distribution and opioid responsiveness of the heat sensitive nociceptor population (McDowell, 2003), capsaicin responses were also measured and correlated with opioid sensitivity.
EXPERIMENTAL PROCEDURES
Isolation and culture of dorsal root ganglion neurons
Sprague–Dawley weanling rats of either sex were killed with pentobarbital (0.2 mg/g, i.p.) according to a protocol approved by the Animal Care Use Committee at the University of Wisconsin and in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were used to minimize the number of animals used and their suffering. Dorsal root ganglia were removed from the spinal column using a dissecting microscope and placed in 0.25% trypsin solution (Sigma, St. Louis, MO, USA) containing 2 mg/ml collagenase (Sigma) at 35 °C for 45 min. The ganglia were washed and resuspended in the basic culture medium consisting of Dulbecco’s Modified Eagle medium (Gibco, Gaithersburg, MD, USA) containing 10% fetal bovine serum, 50 U/ml penicillin, and 50 μg/ml streptomycin (Sigma). After triturating the ganglia using flame polished Pasteur pipettes, the cells were centrifuged and resuspended in either basic culture medium or the basic culture medium supplemented with NGF (50 ng/ml). In some experiments, either NGF antiserum, or K-252a or its vehicle, were also added to the cells at this time. Cells were plated on glass coverslips coated with poly-L-lysine (Sigma) and maintained in an incubator at 35 °C and an atmosphere of 5% CO2 for between 3 and 36 h after isolation.
Whole-cell patch clamp recording
Patch pipettes were made from borosilicate glass using a two-stage pipette puller (Narishige USA, East Meadow, NY, USA) and heat-polished on a microforge (ALA Scientific Instruments, Westbury, NY, USA) to a resistance of approximately 1–3 MΩ. Pipettes were coated with Sylgard to within 100 μm of the tip. Coverslips with cells attached were transferred to the recording chamber and perfused with external solutions. Whole cell currents were recorded in voltage clamp mode using a patch clamp amplifier (Axopatch 200B; Axon Instruments, Foster City, CA, USA). Smaller sensory neurons were chosen preferentially as nociceptive neurons are generally thought to have smaller cell somata. Data acquisition and analysis were performed using a Digidata 1200 A/D converter and pClamp versions 6.0 and 9.0 (Axon) running on a microcomputer. Immediately after obtaining a tight seal and rupturing the cell membrane to achieve the whole-cell recording configuration, series resistance and membrane capacitance were estimated from the settings on the amplifier obtained after eliminating the capacitative current transient recorded during a small voltage step, and the series resistance was typically compensated by 70–80%. Currents were digitized at 5–10 kHz and filtered at 1–2 kHz. Leak and capacitative currents were subtracted from current records online by adding the currents obtained from four hyperpolarizing pulses each equal to one-fourth the magnitude of the depolarizing test pulse (−P/4 protocol).
Both barium current (IBa) and capsaicin-induced currents were recorded from individual cells at room temperature from a membrane holding potential of −80 mV. To examine the inhibition of IBa by fentanyl and baclofen, currents were elicited every 10 s by depolarizing square wave voltage steps of either 20 or 90 ms duration to a test potential of −10 mV, which is at or near the peak of the current voltage relation under these conditions without correction of junction potentials. Current voltage relations were determined by recording currents during voltage steps of 90 ms duration from a holding potential of −80 mV to a range of test potentials from −60 to +30 mV. In some experiments, reversal of G protein-mediated inhibition was examined using a voltage protocol featuring two steps of 20 ms duration to a test potential of −10 mV, separated by a depolarizing pulse of 40 ms duration to a potential of +70 mV. Voltage protocols similar to this have previously been used to demonstrate relief of G protein-mediated inhibition (Swartz, 1993; Zhu and Ikeda, 1994; Herlitze et al., 1996; Zhu and Yakel, 1997).
Solutions
The pipette solution contained (in mM): CsCl 80, CsOH 40, EGTA 10, HEPES 10, MgATP 2, LiGTP 0.3, and di-Tris-phosphocreatine 11. The pH was adjusted to 7.2 with HCl and osmolality was adjusted to 290 with sucrose. In some experiments, GTP in the pipette solution was replaced with an equal concentration of the nonhydrolyzable analog GTP-γ-S. The external solution used to perfuse the cells between experiments and to record capsaicin-induced currents contained (in mM): NaCl 130, KCl 5, MgCl2 1, CaCl2 2, glucose 10, and HEPES 10. The pH was adjusted to 7.4 with NaOH and osmolality was adjusted to 310 with sucrose. For recording IBa, the solution was switched to one containing (in mM): tetraethylammonium chloride 135, BaCl2 1, MgCl2 1, glucose 10, and HEPES 10. The pH was adjusted to 7.4 with CsOH and osmolality adjusted to 310 with sucrose. Cells were gravity perfused with solutions flowing through one of three large capillary tubes glued together in a row. The tubes were rapidly moved with a motor (Warner, Hamden, CT, USA) allowing solution exchange rates of less than 1 s as estimated by changes in pipette tip potentials.
A capsaicin stock solution was made in ethanol and refrigerated until use. For 10 μM capsaicin, the concentration of ethanol in the working solution was 0.1% (v/v). Stock solutions of fentanyl citrate and baclofen were made in distilled water, frozen until use, and diluted in the external recording solution prior to use. Stock solutions of NGF and anti-NGF antiserum (Sigma) were made in standard culture medium, frozen until use, and diluted in culture medium. K-252a (Calbiochem, San Diego, CA, USA) was dissolved in DMSO and frozen, and dissolved in the standard culture medium before use.
Data analysis
The magnitude of IBa was calculated by integrating the total current recorded during the first 20 ms of the test pulse. Linear regression analysis of current magnitudes recorded before and after application of test drug was used to estimate changes in IBa over time. The effect of the test drug was then calculated as the percent change from the expected current magnitude at that time as calculated from the regression equation. During the course of a typical experiment, current rundown led to an average decrease in IBa of less than 10% of the initial current magnitude. Post-pulse facilitation was used to quantify the recovery of IBa from G protein-mediated inhibition after a large depolarizing voltage pulse, and was calculated by dividing the difference in current magnitudes measured before and after the depolarizing pulse by the current magnitude measured before the depolarizing pulse. The magnitude of the capsaicin-induced current was taken as the peak inward current measured during a 5 s application of capsaicin (10 μM, a concentration that produces a near maximal response; McDowell, 2003) at a membrane potential of −80 mV at the end of the experiment. Agonist-induced decreases in IBa of >10% and capsaicin-induced inward currents of >25 pA were considered positive responses and were averaged.
Data are expressed as means±S.E.M. Differences between groups were compared using one-way analysis of variance followed by Dunnett’s post hoc test or by unpaired t-tests where appropriate (GraphPad Software, San Diego, CA, USA). The significance level was 0.05.
RESULTS
The μ-opioid agonist fentanyl decreased IBa in most of the sensory neurons studied. In cells maintained in normal culture medium, fentanyl inhibited IBa in 21 of 25 cells, by an average (±S.E.M.) of 37.7±4.1% in responding cells (Fig. 1). The effect of fentanyl was observed within 10 s of application, was maximal by 20–30 s, and recovered within approximately 60 s after washout. In the presence of fentanyl, activation of IBa was slowed as has been described with agonists of the μ-opioid receptor, likely reflecting voltage-dependent relief of the G protein-mediated inhibition of the VGCC during the test pulse (Ikeda, 1996). When cells maintained in NGF-supplemented culture medium were tested, fentanyl decreased IBa by an average of just 24.6±1.5% in the 18 of 23 cells that responded to fentanyl (P<0.01 vs. untreated cells). Notably, in none of the NGF treated cells was the magnitude of inhibition >35%, while 10 of 21 untreated cells demonstrated >35% inhibition (Fig. 1). NGF treatment had no obvious effect on the kinetics of activation or inactivation of IBa. The magnitude of IBa recorded at −10 mV was not different in NGF-treated cells (−61.6±5.7 nA×ms, n=29) compared with untreated cells (−56.0±5.1 nA×ms, n=33). In separate experiments, the IBa current-voltage relation was not altered by NGF treatment (Fig. 2).
Fig. 1.
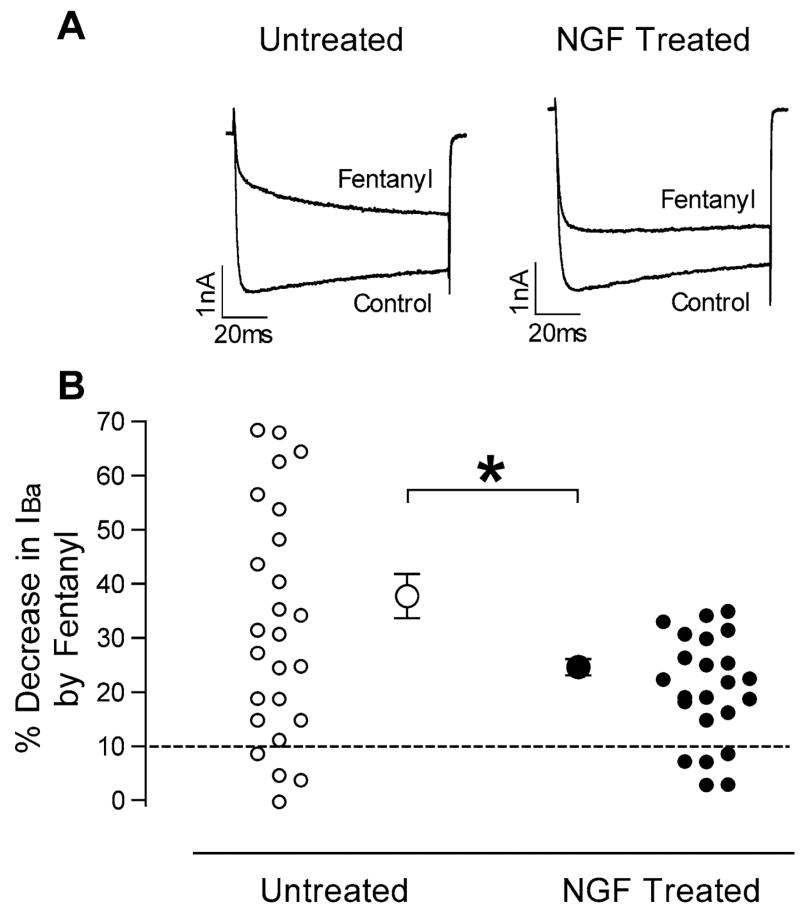
Fentanyl inhibition of IBa is greater in untreated than in NGF treated sensory neurons. (A) IBa current traces from two cells, one untreated and one NGF treated, in the absence and presence of fentanyl. (B) Magnitudes of IBa inhibition by fentanyl, expressed as percentage decreases in IBa, for untreated cells (left, open circles) and NGF-treated cells (right, closed circles). Responses of individual cells to fentanyl are shown by the smaller circles. The dashed line denotes 10% inhibition, below which cells were considered to be insensitive to fentanyl. Average values (±S.E.M.) for the fentanyl sensitive cells are shown by the larger circles with error bars. * P<0.01.
Fig. 2.

IBa current-voltage relations were not different in NGF-treated cells. (A) IBa current traces from two cells, one untreated and one NGF treated, at test potentials ranging from −60 mV to −10 mV. (B) Averaged current voltage relations in untreated (open circles, n=6) and NGF-treated (closed circles, n=6) cells.
The fentanyl-induced inhibition of IBa was attenuated after incubating the cells with NGF-supplemented culture medium for as little as 4 h. NGF has been reported by others to have effects on sensory neurons after just a few minutes (Shu and Mendell, 2001). Separate experiments were therefore performed to ascertain whether NGF might also have more acute effects on opioid-induced inhibition of IBa. An example of the experimental protocol and the responses of one cell are shown in Fig. 3. IBa was recorded from untreated cells and the effect of fentanyl was determined. The cell was then perfused with a solution containing NGF (50 ng/ml), and the effect of fentanyl was determined again at regular intervals in the continuous presence of NGF. Only cells with large responses to fentanyl (>30% inhibition of IBa) were selected for these experiments. In four cells, the inhibition did not change after 20 min of NGF exposure (49.8±4.8% vs. 49.5±8.7% before NGF). In three of these cells, it was possible to continue recording for another 20 min of NGF exposure, but still no change in the fentanyl-induced inhibition could be seen (53.3±9.0% inhibition after 40 min of NGF vs. 54.7±9.9% before NGF).
Fig. 3.

An example from one cell showing that acute treatment with NGF did not alter inhibition of IBa by fentanyl. (A) IBa current traces recorded, from left to right, before NGF; after approximately 30 min of NGF exposure; after approximately 60 min of NGF exposure; and following NGF exposure. Currents were recorded before (control) and during exposure to fentanyl at each time point. The current traces have been scaled so that control IBa magnitudes at each time point are similar. (B) Each point represents the relative magnitude of IBa recorded during a test pulse to −10 mV, expressed as a percentage of the control IBa magnitude recorded 20 s before each application of fentanyl. Fentanyl was applied for 20–30 s approximately every 5 min as indicated by the lines at the bottom of the graph. The time of NGF application is shown by the bar at the top of the graph. Fentanyl inhibited IBa by approximately 75% in this cell, and this inhibition was not altered by NGF, even after 60 min of exposure.
Untreated cells, while not deliberately cultured with NGF, may have been exposed to NGF after isolation, either from serum added to the culture medium or from glia or other cells present in the acute culture that may have secreted NGF. Thus sensory neurons that were not treated with exogenous NGF may still have been exposed to NGF. To test this possibility, the effect of fentanyl was determined in cells maintained in the standard culture medium to which anti-NGF antiserum (1:1000 dilution) was added. As shown in Fig. 4A, the magnitude of fentanyl-induced inhibition when anti-NGF antiserum was added to the culture medium was not different from that observed in cells grown in the standard culture medium alone, indicating that there is a negligible amount of endogenous NGF present in these culture conditions. The antiserum was effective, since it prevented the previously observed effects of exogenous NGF on the fentanyl-induced inhibition (Fig. 4A).
Fig. 4.

(A) Anti-NGF antiserum does not alter the inhibition of IBa by fentanyl in untreated cells. The magnitude of inhibition is expressed as a percentage decrease in IBa by fentanyl for, from left to right, untreated cells (n=21), NGF-treated cells (n=18), cells treated with anti-NGF antiserum (n=15), and cells treated with both NGF and anti-NGF antiserum (n=6). * P<0.01 NGF-treated vs. untreated. (B) The kinase inhibitor K-252a prevents the attenuation of fentanyl-induced inhibition of IBa by NGF. The magnitude of inhibition is expressed as a percentage decrease in IBa by fentanyl for, from left to right, untreated cells (n=21), NGF-treated cells (n=18), cells treated with NGF and DMSO (vehicle for K-252a; 0 nM; n=9), cells treated with NGF and K-252a (10 nM; n=13), and cells treated with NGF and K-252a (100 nM; n=5). * P<0.01 NGF-treated vs. untreated. † P<0.05, NGF+K-252a (100 nM) was significantly different from NGF+K-252a (10 nM), NGF+K-252a (0 nM), and NGF alone.
NGF signals via two receptors, the p75 receptor which binds most of the neurotrophins, and the NGF specific receptor TrkA (Chao, 2003). Activation of the TrkA receptor initiates a phosphorylation cascade initiated by TrkA receptor autophosphorylation. The kinase inhibitor K-252a blocks TrkA receptor autophosphorylation and thus prevents NGF signaling through the TrkA receptor (Berg et al., 1992). To test whether the effect of NGF on fentanyl-induced inhibition of IBa is dependent on activation of TrkA, cells were incubated with NGF and K-252a, or with NGF and DMSO (vehicle for K-252a, 0.01–0.1%). At a concentration of 100 nM, but not 10 nM, K-252a prevented NGF from attenuating the fentanyl-induced inhibition of IBa (Fig. 4B), indicating that the effect of NGF was mediated by the TrkA receptor.
To determine whether the observed effects of NGF were specific for signaling through the μ-opioid receptor, the effect of the GABAB agonist baclofen on IBa was measured with and without prior exposure of sensory neurons to NGF. As shown in Fig. 5, baclofen produced moderate inhibition of IBa in most cells examined, with slowing of IBa activation similar to that observed with fentanyl. Inhibition of IBa by baclofen, however, was not altered by NGF exposure.
Fig. 5.

Baclofen inhibition of IBa is similar in untreated and NGF-treated sensory neurons. (A) IBa current traces from two cells, one untreated and one NGF-treated, in the absence and presence of baclofen. (B) Magnitudes of IBa inhibition by baclofen, expressed as percentage decreases in IBa, for untreated cells (left, open circles) and NGF-treated cells (right, closed circles). Responses of individual cells to baclofen are shown by the smaller circles. The dashed line denotes 10% inhibition, below which cells were considered to be insensitive to baclofen. Average values (±S.E.M.) for the baclofen sensitive cells are shown by the larger circles with error bars.
The lack of effect of NGF on baclofen-induced inhibition of IBa suggests that G protein coupling to the VGCC is not affected by NGF treatment. To test this more directly, the G protein activator GTP-γ-S (0.3 mM) was included in the pipette solution during whole-cell voltage clamp. IBa recording was initiated as quickly as possible after patch rupture after changing the external solution and compensating for cell capacitance and series resistance. About 1 min after patch rupture, the effect of GTP-γ-S on IBa was apparent in all cells, and by about 3 min the inhibition of IBa was maximal. The decrease in IBa was typical of G protein mediated inhibition, as both kinetic slowing and relief of inhibition by a large depolarizing prepulse were prominent (see Fig. 6A). After GTP-γ-S, responses to fentanyl and baclofen were occluded. There were no significant differences in either the magnitude or the time course of the G protein mediated inhibition between untreated and NGF-treated groups (Fig. 6B). Relief from inhibition (facilitation) with a depolarizing prepulse in the presence of GTP-γ-S was also similar in untreated and NGF-treated neurons (Fig. 6C), suggesting normal G protein-VGCC coupling in the presence of NGF.
Fig. 6.

GTP-γ-S inhibits IBa similarly in untreated and NGF-treated cells. (A) IBa current traces from two cells, one untreated and one NGF treated, at 80 and 200s after patch rupture when GTP was replaced with GTP-γ-S in the pipette solution. The voltage protocol is shown at the top, with two test pulses to −10 mV separated by a depolarizing step to +70 mV as described in the Experimental Procedures. (B) IBa magnitudes (mean±S.E.M.) measured during the test pulse to −10 mV before the depolarizing pulse to +70 mV every 10 s beginning 80 s after patch rupture. Untreated (open squares, n=6) and NGF-treated (closed squares, n=8) cells, when the pipette solution contained GTP, exhibited a slow, steady rundown. When the pipette solution contained GTP-γ-S, both untreated (open circles, n=13) and NGF-treated (closed circles, n=10) cells showed marked inhibition of IBa that was maximal at about 3 min after patch rupture. (C) Post-pulse facilitation is used as an index of recovery from G protein-mediated inhibition. In untreated (open squares, n=6) and NGF-treated (closed squares, n=8) cells, when the pipette solution contained GTP, facilitation was minimal, indicating little G protein-mediated inhibition of IBa. When the pipette solution contained GTP-γ-S, facilitation was evident in both untreated (open circles, n=13) and NGF-treated (closed circles, n=10) cells, and increased as the G protein-mediated inhibition increased over time. There were no significant differences in either IBa magnitude or post-pulse facilitation between untreated and NGF-treated cells.
NGF and capsaicin sensitivity
NGF has been shown to increase the proportion of sensory neurons that respond to capsaicin in vitro (Nicholas et al., 1999; Stucky and Lewin, 1999), as well as the magnitude of capsaicin- or heat-induced responses (Nicholas et al., 1999; Stucky and Lewin, 1999; Shu and Mendell, 2001). In the present study, the proportion of cells tested that responded to capsaicin with an inward current was high and not different between groups (24/29 in untreated cells vs. 21/25 in NGF treated cells). The magnitude of the currents elicited by capsaicin was not significantly different in the NGF-treated cells compared with the untreated cells (Fig. 7).
Fig. 7.

Capsaicin-induced currents were similar in untreated and NGF-treated sensory neurons. (A) Representative membrane current traces from two cells, one untreated and one NGF treated, during a 5 s application of capsaicin (CAP, 10 μM), indicated by the bar. (B) Magnitudes (mean±S.E.M.) of the capsaicin-induced current in untreated (open bar, n=19) and NGF-treated (closed bar, n=20). The difference was not statistically significant.
Because capsaicin responsive neurons may be more likely to express opioid sensitive VGCC (Borgland et al., 2001; McDowell, 2003), the proportions of untreated and NGF-treated cells that responded to both capsaicin and fentanyl was recorded. The proportion of cells responding to both capsaicin and fentanyl was similar between the untreated and NGF-treated groups (Table 1), with capsaicin sensitive cells being much more likely to respond to fentanyl than capsaicin insensitive cells in both groups.
Table 1.
Classification of individual sensory neurons according to capsaicin response and fentanyl sensitivity in untreated and NGF treated cellsa
| Untreated
|
NGF treated
|
|||
|---|---|---|---|---|
| Capsaicin sensitive | Capsaicin insensitive | Capsaicin sensitive | Capsaicin insensitive | |
| Fentanyl sensitive | 13 | 3 | 14 | 0 |
| Fentanyl insensitive | 0 | 3 | 1 | 4 |
Individual neurons were classified according to capsaicin sensitivity (capsaicin-induced inward current >25 pA) and fentanyl sensitivity (>10% inhibition of IBa) in both untreated and NGF-treated groups.
The number of neurons in each group is shown.
DISCUSSION
The results of the present study show that NGF can interfere with opioid-mediated inhibition of VGCC in sensory neurons via its high affinity receptor TrkA. NGF did not interfere with VGCC inhibition by another G protein-coupled receptor, the GABAB receptor, or by the direct G protein activator GTP-γ-S, indicating an effect upstream of the receptor-G protein interaction. Because the effect of NGF was only observed after 4 h or more of exposure, a change in transcription may be involved.
NGF is the prototypical neurotrophin, originally described over 50 years ago as a soluble factor required for the survival and growth of both sensory and sympathetic neurons. In recent years, NGF has also been shown to be an important mediator and/or promoter of nociception through actions on primary nociceptive neurons (Mendell et al., 1999). Nociceptors depend on NGF for survival as well as for innervation of peripheral cutaneous targets and for the expression of phenotypic markers characteristic of nociceptors such as TRPV1 receptors and neuropeptides such as substance P and CGRP (Molliver and Snider, 1997; Patel et al., 2000). Shortly after birth, NGF is not required for sensory neuron survival in rodents, although a large population of nociceptors continues to express the NGF receptor TrkA (Molliver and Snider, 1997; Mendell et al., 1999; Patel et al., 2000). Postnatally NGF continues to be expressed by target tissues such as the skin, and this endogenous NGF maintains the nociceptor phenotype of afferents innervating the tissue. In postnatal animals, for example, sequestration of endogenous NGF using anti-NGF antiserum or a TrkA–IgG fusion protein causes loss of nociceptor function (Ritter et al., 1991; Lewin and Mendell, 1994; McMahon et al., 1995; Bennett et al., 1998). Experimentally increasing levels of NGF in the skin, either by administration of exogenous NGF or in transgenic mice selectively overexpressing NGF in skin, enhances the sensitivity of nociceptors to noxious stimuli (Lewin et al., 1993; Lewin and Mendell, 1994; Rueff and Mendell, 1996; Stucky et al., 1999). Furthermore, NGF has been shown to be an important mediator of hyperalgesia in inflammatory pain states. NGF levels in the skin increase after an inflammatory insult (Weskamp and Otten, 1987; Donnerer et al., 1992; Woolf et al., 1994), and administration of either an anti-NGF antiserum or a TrkA-IgG fusion protein prevents the development of inflammatory hyperalgesia and nociceptor activation without changing the amount of tissue inflammation (Donnerer et al., 1992; Woolf et al., 1994; McMahon et al., 1995; Koltzenburg et al., 1999).
The present results support a pronociceptive role for NGF in primary sensory neurons by demonstrating its ability to attenuate opioid-mediated inhibition of VGCC in sensory neurons. Sites where this regulation of opioid action may occur include both the peripheral and central terminals of the nociceptive neuron. In the peripheral terminals, opioid inhibition of VGCC may decrease neuronal excitability and the release of algogenic substances such as substance P from nociceptor nerve endings, particularly during inflammation when opioid receptors are upregulated in the terminals (Sawynok, 2003). At central nociceptor terminals in the spinal dorsal horn, opioids inhibit nociceptive transmission by reducing calcium influx into the presynaptic terminal through VGCC, thereby decreasing excitatory neurotransmitter release (Jessell and Iverson, 1977; Macdonald and Nelson, 1978; Aimone and Yaksh, 1989; Ueda et al., 1995). By preventing opioid actions at these sites, NGF could promote nociceptor activation, sensitization, and central neurotransmission. Although NGF is primarily derived from peripheral sources and may have local actions, the NGF signal is also transported from the periphery centrally to the nucleus (Miller and Kaplan, 2001), where transcriptional events presumably take place that would have effects throughout the cell.
NGF signaling is accomplished by a multitude of pathways. Although NGF binds to both the nonspecific p75 neurotrophin receptor and its specific, high affinity TrkA receptor, most of the cell survival and trophic effects of NGF are mediated by the TrkA receptor (Kaplan and Miller, 2000). The TrkA receptor is a receptor tyrosine kinase that autophosphorylates when activated by NGF binding. This initiates several phosphorylation cascades, including the PI3 kinase/Akt pathway and the Ras/MAP kinase pathway, as well as activation of PLC-γ (Kaplan and Miller, 2000). The final mediator of NGF effects is often a change in transcription, but phosphorylation of cellular proteins may also lead to rapid functional changes after NGF exposure. For example, NGF has been shown to rapidly increase capsaicin-induced currents in primary afferent nociceptors (Shu and Mendell, 2001) as well as increase currents through VGCC in some cells (Wildering et al., 1995; Jia et al., 1999) within a few minutes. On the other hand, TRPV1 receptors can also be upregulated by NGF with a time course of hours to days (Nicholas et al., 1999; Ji et al., 2002), as can VGCC (Lesser and Lo, 1995; Levine et al., 1995; Fitzgerald and Dolphin, 1997; Lei et al., 1997). The lack of a rapid effect on opioid-mediated inhibition of IBa when NGF was applied acutely in the present study suggests that this effect was not directly mediated by protein phosphorylation or dephosphorylation, but involved a longer term, possibly transcriptional event.
NGF has been reported to increase the magnitude of currents through VGCC in embryonic and neonatal neurons (Levine et al., 1995; Fitzgerald and Dolphin, 1997) as well as undifferentiated sympathetic cell lines such as PC12 cells and neuroblastoma cells (Lesser and Lo, 1995; Jia et al., 1999). In these types of cells, NGF promotes survival, differentiation, and cell growth. NGF also increases currents through voltage-gated Na+ and K+ channels in PC12 cells (Lesser and Lo, 1995), suggesting a generalized trophic effect of NGF in these immature cells. One reason for the lack of effect of NGF on VGCC current magnitudes in the present study may be that the cells used were acutely cultured from adult animals, and thus fully differentiated and no longer dependent on NGF for survival. Developmental changes in the signaling pathways for NGF may direct the consequences of NGF binding to its receptor from general trophic effects to more specific modulatory effects in these neurons.
NGF treatment may have disrupted opioid-mediated inhibition in several ways. G protein-mediated inhibition of VGCC involves interactions among three major proteins, namely the GPCR, the G protein, and the VGCC. In the case of the μ-opioid receptor, agonist binding causes activation of G proteins, primarily of the Go subtype (Moises et al., 1994; Jiang et al., 1998). The βγ subunits of the G protein heterotrimer dissociate from the α subunit and mediate inhibition of VGCC by direct binding to the α subunit of the VGCC (Herlitze et al., 1996; Ikeda, 1996; Zamponi et al., 1997; Delmas et al., 1998; Zamponi and Snutch, 1998). Because the GABAB receptor has also been shown to inhibit VGCC by activating Go (Menon-Johansson et al., 1993; Kajikawa et al., 2001), the lack of effect of NGF on baclofen-mediated inhibition of IBa in the present study indicates that NGF did not alter the function of G protein subunits or their coupling to VGCC. Further evidence for normal function downstream of the opioid receptor is the lack of effect of NGF on direct G protein activation with GTP-γ-S.
The results of the present study indicate that the μ-opioid receptor is the target of NGF signaling. The nature of the effect and the molecular pathways involved are not known, but there are several possibilities. NGF may have induced desensitization and internalization of the opioid receptor, similar to that produced by prolonged opioid agonist exposure, which has been reported to lead to activation of either the PI3 kinase pathway or the MAP kinase pathway (Schmidt et al., 2000; Tan et al., 2003). However, activation of these pathways during agonist exposure also alters the G protein-VGCC coupling (Tan et al., 2003), which was not seen during NGF exposure. The cellular protein arrestin, which has been suggested to couple the β-adrenergic receptor to G proteins and is required for receptor internalization (Law et al., 2000), may also be involved in opioid receptor internalization and may be up-regulated by NGF. More experiments will be needed to help clarify the roles these signaling pathways play in opioid receptor regulation and to ascertain how NGF exerts its effects in primary nociceptive neurons.
Acknowledgments
Supported by an NIH K08 Award (GM64672-01), a New Investigator Award from the Foundation for Anesthesia Education and Research and the American Society of Regional Anesthesia, and the Department of Anesthesiology, the University of Wisconsin, Madison, WI. Thanks to Craig Levenick for expert technical assistance.
Abbreviations
- CGRP
calcitonin-gene-related peptide
- EGTA
ethylene glycol-bis (2-amino ethylether)-N,N,′,N′-tetraacetic acid
- HEPES
N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid)
- GTP-γ-S
Guanosine-5′-[γ-thio] triphosphate
- IBa
barium current
- MAP
mitogen activated protein kinase
- NGF
nerve growth factor
- PI3
phosphoinositide 3 kinase
- PLC
phospholipase C
- TrkA
tropomysin-related kinase A
- TRPV1
transient receptor potential vanilloid receptor type 1
- VGCC
voltage-gated calcium channels
References
- Aimone LD, Yaksh TL. Opioid modulation of capsaicin-evoked release of substance P from rat spinal cord in vivo. Peptides. 1989;10:1127–1131. doi: 10.1016/0196-9781(89)90003-x. [DOI] [PubMed] [Google Scholar]
- Barrett CF, Rittenhouse AR. Modulation of N-type calcium channel activity by G-proteins and protein kinase C. J Gen Physiol. 2000;115:277–286. doi: 10.1085/jgp.115.3.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DL, Koltzenburg M, Priestley JV, Shelton DL, McMahon SB. Endogenous nerve growth factor regulates the sensitivity of nociceptors in the adult rat. Eur J Neurosci. 1998;10:1282–1291. doi: 10.1046/j.1460-9568.1998.00139.x. [DOI] [PubMed] [Google Scholar]
- Berg MM, Sternberg DW, Parada LF, Chao MV. K-252a inhibits nerve growth factor-induced trk proto-oncogene tyrosine phosphorylation and kinase activity. J Biol Chem. 1992;267:13–16. [PubMed] [Google Scholar]
- Borgland SL, Connor M, Christie MJ. Nociceptin inhibits calcium channel currents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse. J Physiol. 2001;536:35–47. doi: 10.1111/j.1469-7793.2001.t01-1-00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein bg subunits in N-type Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. J Physiol. 1998;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnerer J, Schuligoi R, Stein C. Increased content and transport of substance P and calcitonin gene-related peptide in sensory nerves innervating inflamed tissue: evidence for a regulatory function of nerve growth factor in vivo. Neuroscience. 1992;49:693–698. doi: 10.1016/0306-4522(92)90237-v. [DOI] [PubMed] [Google Scholar]
- Fitzgerald EM. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras-mitogen-activated protein kinase pathway. J Physiol. 2000;527:433–444. doi: 10.1111/j.1469-7793.2000.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald EM, Dolphin AC. Regulation of rat neuronal voltage-dependent calcium channels by endogenous p21-ras. Eur J Neurosci. 1997;9:1252–1261. doi: 10.1111/j.1460-9568.1997.tb01480.x. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein beta gamma subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Ikeda S. Voltage-dependent modulation of N-type calcium channels by G-protein bg subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jessell TM, Iverson LL. Opiate analgesics inhibit substance P release from rat trigeminal nucleus. Nature. 1977;268:549–551. doi: 10.1038/268549a0. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Jia M, Li M, Liu XW, Jiang H, Nelson PG, Guroff G. Voltage-sensitive calcium currents are acutely increased by nerve growth factor in PC12 cells. J Neurophysiol. 1999;82:2847–2852. doi: 10.1152/jn.1999.82.6.2847. [DOI] [PubMed] [Google Scholar]
- Jiang M, Gold MS, Boulay G, Spicher K, Peyton M, Brabet P, Srinivasan Y, Rudolph U, Ellison G, Birnbaumer L. Multiple neurological abnormalities in mice deficient in the G protein Go. Proc Natl Acad Sci USA. 1998;95:3269–3274. doi: 10.1073/pnas.95.6.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajikawa Y, Saitoh N, Takahashi T. GTP-binding protein bg subunits mediate presynaptic calcium current inhibition by GABAB receptor. Proc Natl Acad Sci USA. 2001;98:8054–8058. doi: 10.1073/pnas.141031298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- King AP, Hall KE, Macdonald RL. k- And m-opioid inhibition of N-type calcium currents is attenuated by 4b-phorbol 12-myristate 13-acetate and protein kinase C in rat dorsal root ganglion neurons. J Pharmacol Exp Ther. 1999;289:312–320. [PubMed] [Google Scholar]
- Koltzenburg M, Bennett DL, Shelton DL, McMahon SB. Neutralization of endogenous NGF prevents the sensitization of nociceptors supplying inflamed skin. Eur J Neurosci. 1999;11:1698–1704. doi: 10.1046/j.1460-9568.1999.00590.x. [DOI] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- Lei S, Dryden WF, Smith PA. Regulation of N- and L-type Ca2+ channels in adult frog sympathetic ganglion B cells by nerve growth factor in vitro and in vivo. J Neurophysiol. 1997;78:3359–3370. doi: 10.1152/jn.1997.78.6.3359. [DOI] [PubMed] [Google Scholar]
- Lesser SS, Lo DC. Regulation of voltage-gated ion channels by NGF and ciliary neurotrophic factor in SK-N-SH neuroblastoma cells. J Neurosci. 1995;15:253–261. doi: 10.1523/JNEUROSCI.15-01-00253.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Differential effects of NGF and BDNF on voltage-gated calcium currents in embryonic basal forebrain neurons. J Neurosci. 1995;15:3084–3091. doi: 10.1523/JNEUROSCI.15-04-03084.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Regulation of cutaneous C-fiber heat nociceptors by nerve growth factor in the developing rat. J Neurophysiol. 1994;71:941–949. doi: 10.1152/jn.1994.71.3.941. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Ritter AM, Mendell LM. Nerve growth factor-induced hyperalgesia in the neonatal and adult rat. J Neurosci. 1993;13:2136–2148. doi: 10.1523/JNEUROSCI.13-05-02136.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald RL, Nelson PG. Specific-opiate-induced depression of transmitter release from dorsal root ganglion cells in culture. Science. 1978;199:1449–1451. doi: 10.1126/science.204015. [DOI] [PubMed] [Google Scholar]
- McDowell TS. Fentanyl decreases Ca2+ currents in a population of capsaicin-responsive sensory neurons. Anesthesiology. 2003;98:223–231. doi: 10.1097/00000542-200301000-00034. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Bennett DL, Priestley JV, Shelton DL. The biological effects of endogenous nerve growth factor on adult sensory neurons revealed by a trkA-IgG fusion molecule. Nat Med. 1995;1:774–780. doi: 10.1038/nm0895-774. [DOI] [PubMed] [Google Scholar]
- Mendell LM, Albers KM, Davis BM. Neurotrophins, nociceptors, and pain. Microsc Res Tech. 1999;45:252–261. doi: 10.1002/(SICI)1097-0029(19990515/01)45:4/5<252::AID-JEMT9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Menon-Johansson AS, Berrow N, Dolphin AC. Go transduces GABAB-receptor modulation of N-type calcium channels in cultured dorsal root ganglion neurons. Pflugers Arch. 1993;425:335–343. doi: 10.1007/BF00374184. [DOI] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. On Trk for retrograde signaling. Neuron. 2001;32:767–770. doi: 10.1016/s0896-6273(01)00529-3. [DOI] [PubMed] [Google Scholar]
- Moises HC, Rusin KI, Macdonald RL. m-Opioid receptor-mediated reduction of neuronal calcium current occurs via a Go-type GTP-binding protein. J Neurosci. 1994;14:3842–3851. doi: 10.1523/JNEUROSCI.14-06-03842.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliver DC, Snider WD. Nerve growth factor receptor TrkA is down-regulated during postnatal development by a subset of dorsal root ganglion neurons. J Comp Neurol. 1997;381:428–438. doi: 10.1002/(sici)1096-9861(19970519)381:4<428::aid-cne3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Nicholas RS, Winter J, Wren P, Bergmann R, Woolf CJ. Peripheral inflammation increases the capsaicin sensitivity of dorsal root ganglion neurons in a nerve growth factor-dependent manner. Neuroscience. 1999;91:1425–1433. doi: 10.1016/s0306-4522(98)00706-4. [DOI] [PubMed] [Google Scholar]
- Patel TD, Jackman A, Rice FL, Kucera J, Snider WD. Development of sensory neurons in the absence of NGF/TrkA signaling in vivo. Neuron. 2000;25:345–357. doi: 10.1016/s0896-6273(00)80899-5. [DOI] [PubMed] [Google Scholar]
- Ritter AM, Lewin GR, Kremer NE, Mendell LM. Requirement for nerve growth factor in the development of myelinated nociceptors in vivo. Nature. 1991;350:500–502. doi: 10.1038/350500a0. [DOI] [PubMed] [Google Scholar]
- Rueff A, Mendell LM. Nerve growth factor and NT-5 induce increased thermal sensitivity of cutaneous nociceptors in vitro. J Neurophysiol. 1996;76:3593–3596. doi: 10.1152/jn.1996.76.5.3593. [DOI] [PubMed] [Google Scholar]
- Sawynok J. Topical and peripherally acting analgesics. Pharmacol Rev. 2003;55:1–20. doi: 10.1124/pr.55.1.1. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Schulz S, Klutzny M, Koch T, Handel M, Hollt V. Involvement of mitogen-activated protein kinase in agonist-induced phosphorylation of the m-opioid receptor in HEK 293 cells. J Neurochem. 2000;74:414–422. doi: 10.1046/j.1471-4159.2000.0740414.x. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol. 2001;86:2931–2938. doi: 10.1152/jn.2001.86.6.2931. [DOI] [PubMed] [Google Scholar]
- Stucky CL, Koltzenburg M, Schneider M, Engle MG, Albers KM, Davis BM. Overexpression of nerve growth factor in skin selectively affects the survival and functional properties of nociceptors. J Neurosci. 1999;19:8509–8516. doi: 10.1523/JNEUROSCI.19-19-08509.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucky CL, Lewin GR. Isolectin B4-positive and -negative nociceptors are functionally distinct. J Neurosci. 1999;19:6497–6505. doi: 10.1523/JNEUROSCI.19-15-06497.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swartz KJ. Modulation of Ca2+ channels by protein kinase C in rat central and peripheral neurons: disruption of G protein-mediated inhibition. Neuron. 1993;11:305–320. doi: 10.1016/0896-6273(93)90186-u. [DOI] [PubMed] [Google Scholar]
- Tan M, Groszer M, Tan AM, Pandya A, Liu X, Xie CW. Phosphoinositide 3-kinase cascade facilitates m-opioid desensitization in sensory neurons by altering G-protein-effector interactions. J Neurosci. 2003;23:10292–10301. doi: 10.1523/JNEUROSCI.23-32-10292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda M, Sugimoto K, Oyama T, Kuraishi Y, Satoh M. Opioidergic inhibition of capsaicin-evoked release of glutamate from rat spinal dorsal horn slices. Neuropharmacology. 1995;34:303–308. doi: 10.1016/0028-3908(94)00160-t. [DOI] [PubMed] [Google Scholar]
- Weskamp G, Otten U. An enzyme-linked immunoassay for nerve growth factor (NGF): a tool for studying regulatory mechanisms involved in NGF production in brain and in peripheral tissues. J Neurochem. 1987;48:1779–1786. doi: 10.1111/j.1471-4159.1987.tb05736.x. [DOI] [PubMed] [Google Scholar]
- Wildering WC, Lodder JC, Kits KS, Bulloch AG. Nerve growth factor (NGF) acutely enhances high-voltage-activated calcium currents in molluscan neurons. J Neurophysiol. 1995;74:2778–2781. doi: 10.1152/jn.1995.74.6.2778. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D. Genetic alteration of phospholipase C b3 expression modulates behavioral and cellular responses to m opioids. Proc Natl Acad Sci USA. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel a1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gbg subunit. Proc Natl Acad Sci USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Modulation of Ca2+-channel currents by protein kinase C in adult rat sympathetic neurons. J Neurophysiol. 1994;72:1549–1560. doi: 10.1152/jn.1994.72.4.1549. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Yakel JL. Modulation of Ca2+ currents by various G protein-coupled receptors in sympathetic neurons of male rat pelvic ganglia. J Neurophysiol. 1997;78:780–789. doi: 10.1152/jn.1997.78.2.780. [DOI] [PubMed] [Google Scholar]