

Abstract
Ionizable planar interfaces and linear polyelectrolytes show markedly different proton-binding behavior. Planar interfaces protonate in a single broad step, whereas polyelectrolytes mostly undergo a two-step protonation. Such contrasting behavior is explained using a discrete-charge Ising model. This model is based on an approximation of the ionizable groups by point charges that are treated within a linearized Poisson–Boltzmann approximation. The underlying reason as to why planar interfaces exhibit mean-field-like behavior, whereas linear polyelectrolytes usually do not, is related to the range of the site–site interaction potential. For a planar interface, this interaction potential is much more long ranged if compared with that of the cylindrical geometry as appropriate to a linear polyelectrolyte. The model results are in semi-quantitative agreement with experimental data for fatty-acid monolayers, water-oxide interfaces, and various linear polyelectrolytes.
Ionization of proteins, weak polyelectrolytes, or water–solid interfaces is a central theme across many disciplines. The understanding of such polyprotic systems is crucial for the assessment of various important processes, such as buffering of protons and metal ions, protein-folding mechanisms, antigen–antibody interactions, formation of surfactant aggregates, particle coagulation dynamics, and crystal growth. Based on the seminal work of Tanford and Kirkwood (1, 2), much progress has been made in the development of quantitative models for the dissociation of ionizable residues in proteins. The electrostatic interactions between charged residues are treated using a Poisson–Boltzmann (or other) approximation, which provides the basis for the evaluation of the thermal statistics of protonation equilibria (3–6). Because the results of such models sensitively depend on the primary, secondary, and tertiary protein structure, the derived protonation patterns appear to be rather protein-specific. The main reasons for this behavior are the heterogeneities introduced by the different proton affinities of different amino acid residues and the specific effects due to geometrical arrangement of the ionizable groups. Any generic features in the ionization process of polyprotic protein systems are difficult to recognize.
Generic features in the ionization patterns can be recognized, however, for simpler polyprotic systems such as planar ionizable interfaces and weak linear polyelectrolytes, which will be the focus of this paper. Such systems may involve a high degree of homogeneity due to the presence of identical ionizable groups and simpler geometrical structure. Moreover, such systems often contain a large number of ionizable sites, so that it is sufficient to consider the limit of an infinite number of sites. The detailed description of the ionization process for planar interfaces and linear polyelectrolytes represents an essential step in the understanding of ionization of complex polyprotic systems; a development that is not only of relevance to biochemistry but to other disciplines as well. The fact that the ionization patterns of planar interfaces and linear polyelectrolytes are markedly different has been documented repeatedly. A single, broad protonation step is observed for planar interfaces, whereas linear polyelectrolytes show two distinct protonation steps. This behavior is generic; it is observed for systems that are entirely different chemically. For planar interfaces such behavior has been documented for cationic and anionic monolayers at the air–water interface, and surfaces of latex or metal oxide particles (7–13). The corresponding data for linear polyelectrolytes include polymaleic acid, polyfumaric acid, poly(vinyl)amine, or poly(ethylene)imine (14–16).
The titration behavior of planar interfaces is commonly approached with electrostatic double-layer models, such as the constant capacitance model, diffuse layer model, or variants thereof (11–13). Such models invoke a mean-field approximation for the interaction potential between the ionizable residues. The magnitude of this mean field increases monotonically with the degree of charging, and therefore this model leads to a broad, featureless titration curve. Furthermore, the overall strength of this mean field is rationalized within a smearing-out approximation for the charge distribution at the surface.
Such an approach usually fails for a linear polyelectrolyte; a mean-field model cannot explain the two-step titration curve. Titration behavior of weak polyelectrolytes is therefore interpreted on the basis of an Ising model on a linear chain. In this model, one assigns to each site a two-valued state variable, which indicates whether the site is protonated or not. Thermal averages over all states are weighted by an interaction energy, which is assumed to be dominated by nearest-neighbor repulsive (antiferromagnetic) pair interactions (15–20). Indeed, this model implies the stabilization of an intermediate state of alternating protonated and deprotonated groups, which leads to an intermediate plateau in the titration curve. The two-step protonation behavior of linear polyelectrolytes is thus explained.
What remains unexplained, however, is why the mean-field approximation does work for the planar interface, while it is inappropriate for linear polyelectrolytes. From the statistical mechanical literature it is well known that the mean-field approximation becomes more accurate as one increases the system dimensionality or the range of the site–site interaction potential (21). For a successful description of weak polyelectrolytes, on the other hand, the site–site interaction potential (between the ionizable residues) must be very short ranged and dominated by nearest-neighbor pair interactions. However, the pronounced difference in the protonation behavior between a two-dimensional planar interface, where mean-field approximation applies, and the one-dimensional linear polyelectrolyte, where the same approximation fails, is startling. Indeed, Monte Carlo simulations of antiferromagnetic Ising models show that for a two-dimensional lattice with nearest-neighbor pair interactions a mean-field description is poor (18). This observation strongly indicates that in the ionization problem the site–site interaction potential must be distinctly different for the two geometries.
The major purpose of this paper is to show that this difference between the site–site interaction potentials indeed provides the fundamental reason for the contrasting ionization behavior of the planar interface and linear polyelectrolyte. The discrete-charge Ising model discussed here is entirely analogous to the model of the ionization process of proteins proposed by Kirkwood and Tanford (1, 2). The site–site interaction potentials are deduced by treating the ionizable groups as point charges. These charges are arranged within a dielectric medium of low dielectric constant, which is in contact with an electrolyte solution treated within the linearized Poisson–Boltzmann equation. Whereas Kirkwood and Tanford (1, 2) have considered a spherical dielectric cavity, we shall discuss the analogous situations for planar and cylindrical geometries to model the planar interface and the linear polyelectrolyte (Fig. 1). Thus, the discrete-charge Ising model treats both types of systems equally but distinguishes them through their geometries. Clearly, such a model is too crude to be quantitative. Due to its simplicity, however, it provides much physical insight and captures the essence of the process in a semi-quantitative fashion.
Figure 1.

Pictorial representation of the discrete-charge Ising model. Solid circles represent the ionizable sites, which are assumed to be arranged within dielectric medium with a dielectric constant ɛd. This dielectric is in contact with an aqueous electrolyte solution with a dielectric constant ɛw and a Debye screening length κ−1. (a) The water–solid interface is modeled by a planar geometry with ionizable sites arranged on a regular, two-dimensional lattice. (b) The linear polyelectrolyte is modeled by arranging the ionizable sites equidistantly along the cylinder axis.
Qualitatively, the difference in the interaction potentials between the planar and cylindrical geometries is readily understood (Fig. 2). The interaction potential between two point charges in a dielectric medium is Coulombic, whereas in an electrolyte it is both weaker (due to the high dielectric constant of water) and short ranged (due to the screening by the electrolyte ions). The interaction potential for the planar and cylindrical geometry must lie between these two extremes. Considering the relative volumes occupied by the dielectric and the electrolyte, respectively, it follows that the interaction potential for the planar interface lies closer to the potential in a uniform dielectric, whereas the potential for the linear polyelectrolyte lies closer to the potential in an electrolyte. As we shall demonstrate, it is this difference between the interaction potentials for the planar interface and the linear polyelectrolyte that is responsible for the contrasting proton titration behavior of these two classes of system (Fig. 3).
Figure 2.

Accurate numerical results for the site–site interaction potentials W(r) between two elementary charges as a function of their separation distance r in units of kT. The charges are located in a dielectric with ɛd = 3, which is in contact with aqueous electrolyte of concentration cs = 0.5 M (κ−1 ≃ 0.43 nm) and with ɛw = 80. The interface is modeled as a semi-infinite planar dielectric with charges at a depth of a = 0.25 nm from the dielectric–electrolyte interface. The polyelectrolyte is approximated by a dielectric cylinder of radius a = 0.25 nm, with charges arranged along its longitudinal axis. The limiting law for small distances is the Coulomb potential in the dielectric; for large distances the interaction potential decays like r−3. For the cylinder the large distance behavior is given by a screened Coulomb law in water.
Figure 3.

Titration curves derived from discrete-charge Ising models. Comparison of Monte Carlo simulations (symbols) with the mean-field approximation (solid line, Eq. 5). Parameters are the same as in Fig. 2. The sites have pK = 10 and are uncharged in the deprotonated state. (a) Ionizable sites are arranged within a planar interface on a square lattice with lattice spacing of 1.1 nm, and (b) along the axis arranged along the axis of the cylinder with a spacing of 0.35 nm. The broken line represents the result without interactions. In b, the thin line is the analytical solution of the Ising model on a linear chain with nearest-neighbor pair interactions.
Electrostatic Energy of Discrete-Charge Arrays
We now derive the electrostatic energy of an array of discrete charges, in both the planar and linear geometries, treating the electrolyte within the linearized Poisson–Boltzmann (Debye–Hückel) approximation. As indicated in Fig. 1, we considered an array of discrete charges of magnitude ξi (measured in units of elementary charge e) that are located at positions ri within a dielectric medium of dielectric constant ɛd. This dielectric medium is in contact with an aqueous electrolyte solution with dielectric constant ɛw and Debye screening length κ−1 (κ2 = 2βe2cs/ɛ0ɛw, β−1 = kT is the thermal energy, ɛ0 the permittivity of vacuum, cs the concentration of monovalent electrolyte). To model the planar interface, we assume the charges to be located within the semi-infinite dielectric at a distance a from the planar interface, while the linear polyelectrolyte is modeled by discrete charges, which are aligned along the central axis of an infinitely long cylinder of the same radius a.
The electrostatic energy of such an array of charges can be obtained by solving for the electrostatic potential ψ(r). Within the electrolyte the electrostatic potential is assumed to satisfy the linearized Poisson–Boltzmann equation
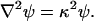 |
1 |
In the dielectric, the right-hand side of Eq. 1 vanishes for r ≠ ri. The boundary conditions at the dielectric–electrolyte interface are stipulated by the continuity of the electrostatic potential and the normal component of the electric displacement.
Site–Site Interaction Potential
The total interaction energy can be obtained, through superposition, by a summation of all pair interaction energies. The electrostatic energy of two charges i and j is given by ξiξjW(r), where W(r) is the electrostatic energy of two elementary charges separated by a distance r = |ri − rj|. In the following, we shall refer to W(r) as the site–site interaction potential.
Let us first discuss the case of a planar interface. This problem can be solved in a straightforward fashion by introducing a two-dimensional Fourier transform (23–25) leading to the site–site interaction potential
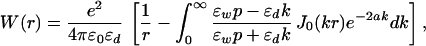 |
2 |
where p = (k2 + κ2)1/2 introduces the dependence on the Debye length and J0(z) is the zeroth-order Bessel function of the first kind (26). The first term corresponds to a bare Coulomb interaction in the dielectric medium, while the second term represents the polarization term.
For the cylinder, which is used to model the linear polyelectrolyte, the analogous problem can be solved by introducing a one-dimensional Fourier transform of the electrostatic potential along the longitudinal axis of the cylinder and expanding the angular contributions into a Fourier series (27). The spirit of the calculation represents a combination of the techniques applied in the analogous problem for a sphere (1, 2) and for a plane (23–25). The result can be simplified considerably if the charges are placed at the center of a cylinder of radius a, in which case the site–site interaction potential becomes
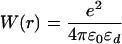 |
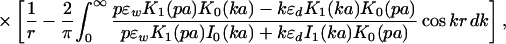 |
3 |
where In(z) and Kn(z) are modified Bessel functions (26). Note again the decoupling into bare Coulomb and polarization terms.
The site–site interaction potential between two charges for a planar interface and a linear polyelectrolyte (compare Eqs. 2 and 3) were evaluated numerically and are compared in Fig. 2. Indeed, the interaction potential is stronger and more long ranged for a planar interface than for a linear polyelectrolyte. At small separation distances, the polarization term gives a constant contribution and thus both interaction potentials are dominated by the bare Coulomb potential. Substantial differences arise at large distances, however. In this limit the Coulomb contribution is almost canceled by the polarization term, which causes the interaction potential to decay much faster than the Coulomb potential. As evident from Fig. 2, this decay at larger separations is significantly slower in the planar than in the cylindrical geometry. For the plane, it can be shown that this decay always scales as r−3 by expanding the integrand in Eq. 2. For the cylinder, the decay at larger separations is dominated by the classical screened Coulomb potential, which is proportional to e−κr/r. The latter result derives from a numerical analysis of Eq. 3. Qualitatively, this different behavior can be understood as follows. In the planar case, the point charge inside the dielectric generates its image charge of opposite sign in the electrolyte. The electrostatic potential induced by these charges decays dipole-like at large distances. For the model of a linear polyelectrolyte, on the other hand, the very presence of the cylinder can be ignored at distances that are much larger than cylinder radius, and the screened Coulomb potential is obtained. For the linear polyelectrolyte one thus obtains full screening at large distances, whereas for the planar interface full screening is never obtained.
Ising Model
In the statistical mechanics literature the Ising model is commonly used for the description of magnets or gas adsorption phenomena (21). However, the very same model represents the proper framework for the description of proton-binding equilibria in polyprotic systems (1–6, 15–22). The protonation state of an ionizable site i can be characterized by its charge ξi. Assuming all sites are equivalent, this variable can attain only two values, namely ξi = z if the site is deprotonated and ξi = z + 1 when the site is protonated. The ionization properties can now be derived from a free energy, which has the form of a classical Ising Hamiltonian (21)
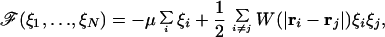 |
4 |
where the repulsive (antiferromagnetic) pair interaction energies are given either by Eq. 2 or Eq. 3. The chemical potential μ can be expressed as βμ/ln 10 = pK − pH, where pK and pH are the negative common logarithms of the intrinsic dissociation constant of the ionizable group and of the proton activity, respectively.
Thermal averages are obtained by summing over all possible ionization states and weighting each configuration according to the Ising Hamiltonian, Eq. 4. The titration (magnetization) curve is given by the average charge ξ plotted as a function of the chemical potential μ (magnetic field), or more commonly, the pH. The average degree of protonation θ = ξ − z is often reported instead.
Mean-Field Approximation
In the mean-field approximation, one replaces all of the state variables ξi by their average ξ. In the present context of ionization equilibria, the mean-field approximation leads to the classical constant capacitance model (11, 12)
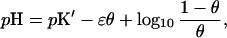 |
5 |
where pK′ = pK + zɛ and ɛ = βE/ln 10. Whereas for E = 0 the classical titration curve of noninteracting sites is recovered, in the general case, this parameter characterizes the overall strength of the mean field and is given by
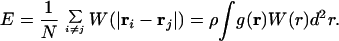 |
6 |
The right-hand side of the equation applies to a planar interface only. Thereby, ρ is the interfacial site density (per unit area) and g(r) is the pair distribution function of the sites in the interface. An analogous expression also applies for the linear arrangement. One simply replaces ρ by the line site density (per unit length) and the integral by a one-dimensional integral along the cylinder axis.
Monte Carlo Simulations
Without introducing any approximations, thermal averages for the Ising model with a general form of site–site interaction potential can be obtained by the Monte Carlo simulation technique (21). We have used this technique to evaluate titration curves of ionizable sites on planar interfaces and linear polyelectrolyte with the interaction potentials discussed earlier (see Fig. 2). In both cases, we have assumed all groups to be neutral in the deprotonated state (z = 0) and characterized by pK = 10. For both geometries, the resulting titration curves are compared with the mean-field approximation in Fig. 3. Note that within our linearized model, the titration curves retain their shape for other values of z and pK; changes of these parameters will only cause a horizontal displacement of the curves.
For the planar interface, which is here modeled by a square lattice with a lattice constant of 1.1 nm (ρ ≃ 0.8 nm−2), the agreement between Monte Carlo results and the mean-field approximation is very good (see Fig. 3a). In spite of the strong effect of interactions, the mean-field model remains a good approximation if we change the lattice spacings or use different, physically realistic interaction potential parameters. The same has been verified for other types of lattices (e.g., honeycomb, triangular) and is expected to hold for a disordered (glass-like) arrangement of sites. For the planar interface, we were unable to find a realistic situation where the mean-field approximation breaks down.
On the other hand, the mean-field approximation fails entirely for the linear polyelectrolyte (see Fig. 3b). Before a full discussion of this situation is given, we note first that if we arrange ionizable sites along the polyelectrolyte with the same spacing as we have used for the planar interface (1.1 nm), there is a minimal effect due to interactions. This feature is immediately understandable from the differences between the cylindrical and planar scenarios—the site–site interaction potential along the polyelectrolyte decays considerably faster than the planar analog and, moreover, there are fewer nearest neighbors in the polyelectrolyte. Therefore, the overall strength of the interactions will be much weaker in the polyelectrolyte than in the planar interface for a given intergroup distance. To observe appreciable effects of interactions in the polyelectrolyte case, we must thus reduce the intersite separation relative to that in the set-up for the planar interface.
For a polyelectrolyte with sites at smaller separation distances—we have used 0.35 nm in this case—the Monte Carlo data are poorly described by the mean-field model (see Fig. 3b). Variation of parameters shows that this behavior is again rather typical for most situations. Only for larger intergroup spacing and, particularly, at lower ionic strengths, can mean-field behavior also be observed for the linear geometry. In such situations, however, the overall effect of interactions is only moderate.
As further shown in Fig. 3b, we have also compared the resulting titration curve with the linear nearest-neighbor Ising model (15–21). This simple model reproduces the Monte Carlo results reasonably well. Neglecting all but nearest-neighbor interactions is a reasonable approximation for the linear polyelectrolyte; for the planar interface the same approximation would fail entirely.
Smearing-Out Approximation
For mathematical convenience one often invokes the approximation that the charges are not discrete but uniformly smeared out. The mean-field model discussed here reduces precisely to this situation if spatial correlations between the ionizable sites are neglected.
Consider the planar geometry first. Setting g(r) = 1 in Eq. 6 and using Eq. 2 we recover the usual smeared-charge result for the plane (11–13)
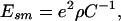 |
7 |
where C is the capacitance per unit area. This capacitance can be interpreted as arising from two parallel plate capacitors in series
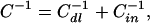 |
8 |
where Cdl = κɛ0ɛw originates from the diffuse layer, while Cin = ɛ0ɛd/a arises from the dielectric medium between the smeared-out charge and the planar interface. In the double-layer model (12), the second contribution in Eq. 8 is neglected and the capacitance is given by the double-layer capacitance Cdl only.
As known from the electrochemical literature (28), the smearing-out approximation does fail for a lattice, where substantial (excluded volume type) correlations exist. The strength of the mean field E depends on the geometrical arrangement through the pair correlation function g(r) in a nontrivial fashion and thus this parameter will depend on the lattice geometry. For the square lattice (the situation shown in Fig. 3) from Eq. 6 we obtain ɛ ≃ 7.37. For other lattices with the same site density, the corresponding lattice sums give ɛ ≃ 7.23 for a triangular lattice and ɛ ≃ 7.71 for a honeycomb lattice. The strength of the mean field is poorly estimated by the smeared-out charge model, which predicts ɛsm ≃ 22.4 (overestimate), as well as the classical double-layer model, which yields ɛdl ≃ 1.37 (underestimate).
For the linear polyelectrolyte, as we have shown, the mean-field approximation is poor. However, one could still employ the smearing-out approximation to estimate the overall effect of the interaction (i.e., strength of the mean field). In this case, the capacitance entering Eq. 7 must be interpreted per unit length and ρ replaced by the line density. Moreover, smearing out the charges along the longitudinal axis of a cylinder leads to a divergence of the electrostatic energy. Thus, we can only meaningfully discuss the contribution of the diffuse layer (29). From our treatment we recover the double-layer capacitance per unit length Cdl = 2πɛ0ɛwaκK1(κa)/K0(κa). For the case shown in Fig. 3b, the mean-field interaction energy is characterized by a pair interaction parameter ɛ ≃ 8.27. Smearing the charges along the cylinder axis we obtain ɛsm → ∞, whereas smearing the charges on the cylinder surface gives ɛdl ≃ 1.76. As in the planar case, both approximations bound the correct value but do not provide useful estimates of the effect of the interactions.
Comparison with Experiment
In a semi-quantitative fashion, numerous experimental data sets support the picture put forward here. Experimental titration curves reveal the same generic trends as predicted by the discrete-charge Ising model. In the case of planar interfaces, the effect of site–site interactions is pronounced even at site separations of approximately 1 nm (see Fig. 3a). The resulting titration curves are of the mean-field type and in semi-quantitative agreement with the ionization behavior of ionizable monolayers or latex particles (7–10). At smaller separations, the effect of site–site interactions is so strong that the linearized Poisson–Boltzmann breaks down and it becomes impossible to titrate a given site fully within the experimentally accessible pH window; water–oxide interfaces with intergroup distances below 0.5 nm are the primary examples of this behavior (13).
For polyelectrolytes, on the other hand, interaction effects set in at smaller distances. These interactions become only dominant for rather small separations between ionizable groups (below 0.5 nm), and one observes marked deviations from the mean-field behavior and a characteristic plateau in the titration curve around θ = 1/2 (see Fig. 3b). Various linear polyelectrolytes with closely spaced ionizable groups behave in this fashion, for example, poly(maleic acid), poly(fumaric acid) poly(vinylamine) or poly(ethyleneimine) (14–16). For linear polyelectrolytes, at intergroup distances somewhere below 1 nm, effects of interactions become observable. This situation is exemplified by polyacrylic acid and hyaluronic acid, where the effects of interactions are weak and titration curves are of the mean-field type, as also revealed by the discrete-charge Ising model (14, 30, 31).
Even though all of these generic features are reproduced by our model semi-quantitatively, no quantitative comparisons were attempted. For obvious reasons, the model is too crude to explain the behavior of individual systems in detail. Within the Poisson–Boltzmann approximation, a certain level of molecular detail can be introduced by solving for the electrostatic energies of realistic geometries. This approach has reached a substantial degree of sophistication for proteins (3–6). However, a quantitative treatment calls for the consideration of additional effects: (i) accurate estimation of intrinsic microscopic pK values of ionizable groups in the appropriate molecular environment (3, 4,); (ii) possible breakdown of the linear or nonlinear Poisson–Boltzmann approximation, Stern-layer, and dielectric saturation effects (4, 6, 11–13); (iii) contributions from conformational degrees of freedom (31, 32).
Conclusion
Typical differences between the ionization properties of planar interfaces and linear polyelectrolytes (as displayed in Fig. 3) have their origin in the marked contrast between the site–site interaction potentials of two point charges near a planar interface and close to a polymer backbone. The interaction potential between two point charges is stronger and more long ranged for the planar interface than for the cylinder (see Fig. 2). This difference originates from the contrasting behavior of the interaction potentials at large separations: for the plane, the potential decays as r−3, while for the linear polyelectrolyte as e−κr/r. If we now consider the protonation of such systems, the range and strength of the site–site interaction potential plays a key role. In this case, the range of the interaction potential primarily determines whether the system will behave in a mean-field like fashion or not. For the planar interface the interaction potential is long ranged and, therefore, the system behaves mean-field-like. For the polyelectrolyte, the interactions are short ranged and most important for nearest neighbors; for this one-dimensional system the mean-field approximation fails. Similar reasoning applies to the overall strength of the interactions. For a given distance between ionizable sites, the effect of interactions is much stronger for the planar interface than for the linear polyelectrolyte.
References
- 1.Tanford C, Kirkwood J C. Am J Chem Soc. 1957;79:5333–5339. [Google Scholar]
- 2.Kirkwood J G. J Chem Phys. 1934;7:351–361. [Google Scholar]
- 3.Bashford D, Karplus M. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- 4.Honig B, Nicholls A. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 5.Beroza P, Fredkin D R, Okamura M Y, Feher G. Proc Natl Acad Sci USA. 1991;88:5804–5808. doi: 10.1073/pnas.88.13.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ullner M, Woodward C E, Jönsson B. J Chem Phys. 1996;105:2056–2065. [Google Scholar]
- 7.Zhao X, Ong S, Wang H, Eisenthal K B. Chem Phys Lett. 1993;214:203–207. [Google Scholar]
- 8.Lovelock B, Grieser F, Healy T W. Langmuir. 1986;2:443–448. [Google Scholar]
- 9.Drummond C J, Grieser F, Healy T W. J Phys Chem. 1988;92:2604–2613. [Google Scholar]
- 10.Schubin V E, Isakova I V, Sidorova P M, Men’shikova P M, Evseeva T G. Kolloid Zh. 1990;52:935–941. [Google Scholar]
- 11.Healy T W, White L R. Adv Colloid Interface Sci. 1978;9:303–345. [Google Scholar]
- 12.Westall J, Hohl H. Adv Colloid Interface Sci. 1980;12:265–294. [Google Scholar]
- 13.Hiemstra T, van Riemsdijk W H, Bolt G H. J Colloid Interface Sci. 1989;133:91–104. [Google Scholar]
- 14.Kitano T, Kawaguchi S, Ito K, Minakata A. Macromolecules. 1987;20:1598–1606. [Google Scholar]
- 15.Katchalsky A, Mazur J, Spitnik P. J Polym Sci. 1957;23:513–532. [Google Scholar]
- 16.Smits R G, Koper G J M, Mandel M. J Phys Chem. 1993;97:5745–5751. [Google Scholar]
- 17.Marcus R A. J Phys Chem. 1954;58:621–623. [Google Scholar]
- 18.Reiss H. J Phys Chem. 1988;92:3657–3662. [Google Scholar]
- 19.Borkovec M, Koper G J M. J Phys Chem. 1994;98:6038–6045. [Google Scholar]
- 20.Borkovec M, Koper G J M. Ber Bunsenges Phys Chem. 1996;100:764–769. [Google Scholar]
- 21.Chandler D. Introduction to Modern Statistical Mechanics. New York: Oxford Univ. Press; 1987. [Google Scholar]
- 22.Jäger I. Surf Sci. 1991;254:300–308. [Google Scholar]
- 23.Stillinger F H. J Chem Phys. 1961;35:1584–1589. [Google Scholar]
- 24.Richmond P. J Chem Soc Faraday Trans 2. 1974;70:1067–1073. [Google Scholar]
- 25.Medina-Noyolam M, Ivlev B I. Phys Rev E. 1995;52:6281–6288. doi: 10.1103/physreve.52.6281. [DOI] [PubMed] [Google Scholar]
- 26.Gradsteyn I S, Ryzhik I M. Table of Integrals, Series, and Products. New York: Academic; 1980. [Google Scholar]
- 27.Soumpasis D. J Chem Phys. 1978;69:3190–3196. [Google Scholar]
- 28.Barlow C A, Macdonald J R. Adv Electrochem Electrochem Eng. 1967;6:1–199. [Google Scholar]
- 29.Hill T L. Arch Biochem Biophys. 1955;57:229–239. doi: 10.1016/0003-9861(55)90195-8. [DOI] [PubMed] [Google Scholar]
- 30.Cleland R L. Macromolecules. 1982;15:386–395. [Google Scholar]
- 31.Ullner M, Jönsson B, Peterson C. J Chem Phys. 1996;104:3048–3057. [Google Scholar]
- 32.Figueirido F, Del Buono G S, Levy R M. J Phys Chem. 1996;100:6389–6392. [Google Scholar]