

Abstract
Cyanide produces degeneration of the nervous system in which different modes of cell death are activated in the vulnerable brain areas. In brain, the mechanism underlying the cell death is not clear. In this study, an immortalized dopaminergic cell line was used to characterize the cell death signaling cascade activated by cyanide. Cyanide-treated cells exhibited a time- and concentration-dependent apoptosis that was caspase-independent. Cyanide induced a rapid surge of intracellular reactive oxygen species (ROS) generation, followed by p38 mitogen-activated protein kinase (MAPK) activation and nuclear accumulation of hypoxia-inducible factor-1α (HIF-1α). Activation of p38 MAPK and HIF-1α accumulation were attenuated by N-acetyl-L-cysteine (antioxidant), catalase (hydrogen peroxide scavenger) or a selective p38 MAPK inhibitor (SB203580). Cyanide activated the hypoxia response element (HRE) promoter, which was also blocked by the antioxidants and SB203580. HRE activation was followed by increased BNIP3 gene transcription, as reflected by elevated BNIP3 mRNA and protein levels. BNIP3 upregulation was reduced by selective RNAi knockdown of HIF-1α. Overexpression of BNIP3 produced mitochondrial dysfunction (reduced membrane potential), caspase-independent apoptosis, and sensitization of the cells to cyanide-induced toxicity. Expression of a dominant negative mutant or RNAi knockdown of BNIP3 protected the cells from cyanide. It was concluded that cyanide activated the HIF-1α-mediated pathway of BNIP3 induction through a redox-sensitive process. Increased BNIP3 expression then served as an initiator of mitochondrial-mediated death.
Keywords: cyanide, HIF-1α, BNIP3, cell death, reactive oxygen species, RNA interference
INTRODUCTION
Cyanide is a potent neurotoxin for which the central nervous system is the primary target organ. At the cellular level, cyanide produces chemical hypoxia (histotoxic anoxia) by inhibiting cytochrome c oxidase in complex IV of the mitochondrial oxidative phosphorylation chain to markedly reduce ATP [1,2]. In neurons, cyanide increases cytosolic-free Ca2+ by stimulating Ca2+ influx, accompanied by rapid surge of ROS generation at complex I and III [3,4]. The mitochondrial dysfunction and ionic imbalance activates the cell death cascades in which apoptotic death occurs in cortical cells and necrosis in mesencephalic cells [5–7].
Hypoxia-mediated apoptosis is closely linked to induction of pro-death Bcl-2 proteins by stimulating hypoxia inducible factor-1 (HIF-1). HIF-1 is a heterodimeric basic helix-loop-helix transcription factor composed of HIF-1α and HIF-1β subunits (also termed ARNT for arylhydrocarbon receptor nuclear translocator). Under normoxic conditions, HIF-1β is constitutively expressed, whereas HIF-1α undergoes rapid ubiquitination and degradation by proteasomes [8]. In hypoxia, proteasomal degradation of HIF-1α is reduced, leading to HIF-1α accumulation and translocation to the nucleus. In the nucleus, HIF-1α heterodimerizes with partner proteins from the ARNT family, followed by binding to the hypoxia response element (HRE) to activate transcription of pro-death target genes [9].
HIF-1 can also respond to a variety of non-hypoxic stimuli, including vasoactive peptides, cytokine and hormones [10–12]. These stimuli can initiate ROS production which then can activate the HIF-1 cascade. Thus, non-hypoxic stimuli can also activate HIF-1α through redox signaling by activation of specific kinases or inactivation of phosphatases [13]. For instance, overexpression of the p38 MAPK kinases can enhance HIF-1α protein levels under either normoxic or hypoxic conditions [14]. In the case of cyanide, we have shown in rat primary cortical cells that p38 MAPK is phosphorylated through a ROS-mediated pathway [15]. Thus, it is possible that the cyanide-induced stimulation of p38 MAPK could lead to activation of the HIF-1 pathway to initiate a cell death cascade.
In hypoxia, BNIP3 (Bcl-2/adenovirus E1B 19kDa interacting protein 3) is one of the primary pro-death genes activated by the HRE promoter [9,16]. BNIP3 shares limited homology with the Bcl-2 family in the Bcl-2 homology 3 (BH3) domain and C-terminal transmembrane (TM) domain. However, unlike other BH3 containing proteins, deletion of the BH-3 domain in BNIP3 does not abrogate its pro-death activity [17]. The TM domain is critical for BNIP3 translocation to the mitochondria where it heterodimerizes with pro-survival proteins Bcl-2 and Bcl-XL to neutralize their anti-apoptotic function. In ischemia, BNIP3 can undergo rapid upregulation associated with a delayed neuronal cell death [16]. Recently, we have shown that cyanide can induce BNIP3 and stimulate mitochondrial translocation in primary cortical cells [18].
Since cyanide produces a histotoxic anoxia in which cellular oxygen concentrations are normal but oxygen metabolism is inhibited, it was of interest to determine if cyanide can act through redox-mediated HIF-1α activation and upregulation of the pro-death gene product BNIP3. In the current study, Mes 23.5, an immortalized dopaminergic cell line, was used to investigate cyanide neurotoxicity. This cell line displays properties of substantia nigra zona compacta neurons and is considered a dopaminergic cell that expresses tyrosine hydroxylase and synthesizes dopamine [19]. Mes 23.5 cells exhibit a unique susceptibility to oxidative stress-induced toxicity and have been use as a cell model to study free-radical-mediated cell damage [20].
MATERIALS AND METHODS
Cell culture
The Mes 23.5 cell line, obtained from Dr. C. Rochet, Purdue University, was derived from somatic cell fusion of rat embryonic mesencephalic cells and the murine neuroblastoma-glioma cell line N18TG2 [19].
Cells were seeded on poly-l-lysine-precoated plates and maintained in DMEM supplemented with 5% fetal bovine serum (FBS), 2% new born calf serum (NBS), 15 mM HEPES and SATO components (insulin 5 μg/ml, transferrin 5 μg/ml, pyruvic acid 48.6 μg/ml, putrescine 4 μg/ml, sodium selenite 5 ng/ml, progesterone 6.3 ng/ml) at 37°C in an atmosphere of 5% CO2 and 95% air.
Cell death assay
Cell viability was determined by trypan blue exclusion assay [21]. Cells were rinsed twice with PSB and suspended in complete medium with 0.4% trypan. The ability of the cells to exclude trypan blue was determined after 5 min, using a hemocytometer under a light microscope. The cell viability was defined as percentage of cells that excluded the trypan blue.
Apoptosis was detected by nuclear staining with Hoechst 33258 as described previously [22]. Cells were washed twice with phosphate-buffered saline (PBS), and fixed in 4% paraformaldehyde in PBS, then treated with 2 μM Hoechst 33258 dye and examined under fluorescence microscopy. Cells with condensed and fragmented DNA were considered apoptotic. The apoptosis was quantified by averaging cell counts in four random 400 × fields.
The in situ terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling (TUNEL) assay was used to confirm apoptosis. The TUNEL assay was performed on paraformaldehyde (4% in PBS) fixed cells using the Apoptag™ in situ apoptosis detection kit (Oncor, Gaithersburg, MD) as described previously [23].
Measurement of ROS generation
The 2′,7′-dichlorofluorescin-diacetate (DCF-DA) assay was used to measure cellular generation of ROS [23]. Cells were loaded with 30 μM DCF-DA (Molecular Probes, Eugene, OR) for 30 min at 37°C in the dark and then washed with PBS to remove free DCF-DA. After cyanide treatment, the culture medium was removed and cells washed twice with PBS. Fluorescence intensity was monitored with a microtiter plate reader at excitation wavelength of 485 nm and emission wavelength of 535 nm. Values were expressed as percent of control groups (without cyanide treatment).
Western blotting
Following treatments, whole cell lysates were prepared using a lysis buffer containing 220 mM mannitol, 68 mM sucrose, 20 mM HEPES, pH 7.4, 50 mM KCl, 5 mM EGTA, 1 mM EDTA, 2 mM MgCl2, 1 mM dithiothreitol, 0.1% Triton X-100 and protease inhibitors. Western blotting was carried out using the ECF Western blot kit (Amersham Biosciences, Piscataway, NJ) as described by the manufacture. The primary antibodies were: anti-BNIP3 antibody, anti-β-actin antibody (Sigma Chemical Co, St. Louis, Mo), anti-phospho-p38 MAPK antibody, anti-p-38 MAPK antibody, anti-Bax and anti-Bcl-2 antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
For detection of nuclear HIF-1α, nuclear extracts of the cells were prepared. Briefly, cells were harvested and lysed in buffer containing 10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 1 mM DTT, 1 mM PMSF, supplemented with a protease inhibitor cocktail and centrifuged at 14,000 rpm for 5 min at 4 °C. Supernatants were harvested as cytosolic extracts. The pellets were further lysed with buffer containing 20 mM HEPES, 1.5 mM MgCl2, 0.42 mM NaCl, 0.2 mM EDTA, 0.5 mM DTT, 25% glycerol, supplemented with a protease inhibitor cocktail. Thirty minutes later, the supernatants (nuclear extracts) were harvested after centrifugation at 14,000 rpm at 4°C for 10 min. HIF-1α expression in nuclear extracts was detected by Western blotting as described above. The primary antibodies were anti-HIF-1α (Novus, Littleton, CO) and anti-histone H3 antibodies (Cell Signaling, Danvers, MA).
Real-time reverse transcription-PCR
Total RNA was isolated from cells using the RNeasy Mini Kit (QIAGEN, Valencia, CA) and was reverse transcribed into cDNA. The forward and reverse primers for target genes were obtained from Integrated DNA Technologies (Coralville, IA). The primers are: BNIP3, forward, 5′-GGA CGA AGC AGC TCC AAG AG-3′ and reverse, 5′-TCA CCA AAG CTG TGG GTG TCT-3′; Bax, forward, 5′-CTC GTG GTT GCC CTC TT-3′ and reverse 5′-TGA TCA GCT CGG GCA CTT TA-3′; Bcl-2, forward, 5′-TCG TCA CCC CAC CCT TAC AT-3′ and reverse 5′-AGT GCC TGT CTG GGA TGC A-3′; β-actin, forward, 5′-TCC TCC TGA GCG CAA GTA CTC T-3′ and reverse, 5′-GCT CAG TAA CAG TCC GCC TAG AA-3′. The Absolute QPCR SYBR Green Mix kit (ABgene, Rochester, NY) was used for real-time RCR analyses. The amplification was performed in the Mx3000P Real-Time PCR System (Atratagene, La Jolla, CA) in which the conditions were 15 min at 95°C, followed by 40 cycles of 30 s at 95°C, 1 min at 60°C and 30 s at 72°C.
Plasmid constructs and transient transfection
Nucleotide sequences encoding human BNIP3 and BNIP3ΔTM (transmembrane domain deleted form of BNIP3) [24] were subcloned into the pCMV-HA mammalian expression vector (Clontech Laboratories, Inc) with a N-terminal hemagglutinin (HA) epitope tag, via utilizing EcoRI/KpnI restriction enzyme sites. To visualize the exogenously expressed protein directly in transiently transfected cells, enhanced yellow fluorescent protein (EYFP) fusion proteins of BNIP3 and BNIP3ΔTM were constructed by inserting EYFP encoding sequence to the C-terminus of HA-BNIP3 and HA-BNIP3ΔTM through KpnI/NotI restriction enzyme sites with a flexible Gly-Gly-Gly-Gly-Ser-Gly-Gly-Gly-Gly-Ser-Gly-Gly-Gly-Gly-Ser linker sequence in between. Transient transfections were performed using Lipofectamine 2000 from Invitrogen (Carlsbad, CA) following the manufacturer’s recommendations and the transfection efficiency was approximately 80% when assessed using EYFP (data not shown).
HRE reporter gene assay
Cells were transiently co-transfected with 1 μg of a 60:1 mixture of the constructs containing HRE-luciferase reporter gene (a kind gift from Dr. Navdeep S. Chandel, Northwestern University) and the pRL-CMV reporter vector containing Renilla luciferase [25]. Twenty four hours after transfection, cells were subjected to different treatments as described, luciferase activities were then measured in a luminometer using the Dual-Luciferase Reporter Assay System (Progmega, Madison, WI) according to the manufacturer’s instructions.
RNA interference
Small interfering RNA (siRNA) corresponding to BNIP3 was designed and synthesized by Ambion, Inc. (Austin, TX). The gene-specific sequences were: sense, 5′-GCU ACU CUC AGC AUG AGA Att-3′ and antisense, 5′-UUC UCA UGC UGA GAG UAG Ctg. Pre-designed siRNA for HIF-1α (Santa Cruz Biotechnology, Santa Cruz, CA) was used to knock down gene expression. The silencer negative control siRNA, which does not target rat, mouse or human genes, was used as a negative control (Ambion, Austin, TX). Transient transfection of siRNA was performed with Lipofectamine 2000™ (Invitrogen, Carlasbad, CA).
Mitochondrial membrane potential (ΔΨm)
Changes in ΔΨm were estimated using the cationic fluorescent dye (JC-1) as described previously [23]. JC-1 is a dual-emission potential-sensitive probe that measures ΔΨm since the ratio of green to red fluorescence of JC-1 is dependent on the mitochondrial membrane potential. After treatment, cells were incubated with JC-1 (3.0 μM) for 30 min at 37°C in the dark and then washed twice with PBS. Fluorescence was measured with a plate reader (TECAN Spectra Fluor plus, Zurich, Switzerland) at excitation 485 nm, emission 525 nm and 595 nm, respectively. The fluorescence signal represents the average signal of the total cell population and the magnitude of ΔΨm changes in cells exhibiting marked mitochondrial dysfunction may not be captured by the this approach. JC-1 may also respond to plasma membrane depolarization, thus the intensity of JC-1 florescence ratio (red/green) was measured after dissipation of ΔΨm with FCCP (p-trifluoromethoxy carbonyl cyanide phenyl hydrazone), a potent uncoupler of oxidative phosphorylation. JC-1 intensity was significantly abolished by FCCP, confirming the probe is specific marker for mitochondrial potential at the concentration used in current study. Parallel results were obtained with rhodamine 123, thus confirming the specificity of the JC-1 method for ΔΨm (data not shown).
Caspase-3 protease activity
The cleavage of the substrate Ac-DEVD-pNA was used to determine caspase-3 protease activity by following the manufacture’s protocol (BioVision Inc., Mountain View, CA). After the treatments, cells were harvested in PBS and centrifuged at 500 × g for 5 min. The cell pellet was further lysed on ice with lysis buffer for 10 min and then centrifuged at 13,000 rpm for 1 min at 4° C. The supernatant was harvested and 80 μg total protein incubated with buffer containing 10 mM dithiothreitol and 5 μl of Ac-DEVD-pNA (final concentration 200 μM) at 37°C. The chromophore P-nitroanilide was determined at 405 nm with a microtiter plate reader.
Statistics
Data were expressed as mean ± SEM. One-way analysis of variance (ANOVA) followed by Tukey-Kramer procedure for multiple comparisons were used to determine statistical differences between treatments. Differences were considered significant at P<0.05.
RESULTS
Cyanide-induced oxidative stress and cell death
In Mes 23.5 cells, KCN induced a concentration-dependent cell death over the range of 200–600 μM, as determined by the trypan blue exclusion assay (Fig. 1A). The mode of death was identified by Hoechst 33258 staining; a concentration-dependent apoptosis was induced within 12 h, and progressively increased over the following 12 h (Fig. 1B). ROS generation was detected within 30 min of cyanide and was reduced by the free radical scavenger N-acetyl-L-cysteine (NAC) or the hydrogen peroxide (H2O2) scavenger, catalase (CAT) (Fig. 1C). To identify the sources of intracellular ROS generation, FCCP, an uncoupler of oxidative phosphorylation that causes loss of ΔΨm and abolishes ROS generation from mitochondria was used. Incubation of the cultures with FCCP reduced the basal level of ROS production, suggesting that the majority or ROS generated in the control cells was mitochondrial in origin (Fig. 1D). In addition, KCN-induced ROS was also decreased significantly by FCCP, indicating the contribution of mitochondria to total ROS production. NAC and CAT also reduced the apoptotic cell death, indicating involvement of ROS (Fig. 1E). It was concluded that oxidative stress was an initiator of the apoptosis.
Fig. 1.

Cyanide-induced cell death and ROS generation. (A) Cells were treated with varying concentrations of KCN (200–600 μM) for 24 h, followed by analysis of cell viability by trypan blue exclusion test. (B) Cells were treated with KCN (200–600 μM) for varying times (0–24 h), followed by analysis of apoptosis by Hoechst 33258 staining. (C) Cells were pretreated with NAC (0.5 mM), CAT (0.2 mM), SB203580 (20 μM) or SB202474 (20 μM) for 30 min prior to incubation with KCN (400 μM) for 30 min, then ROS generation determined. (D) KCN-induced ROS generation was determined in the presence or absence of FCCP (1 μM). (E) Apoptotic death was determined in the presence or absence of different concentrations of NAC or CAT for 30 min and then KCN (400 μM) treatment for 24 h. Data represent mean ± SEM of 3–8 separate determinations. *Significantly different from control (no treatment) group; #significantly different from KCN (control) group. P<0.05
HIF-1α activation is mediated by p38 MAPK pathway
We have shown that the p38 MAPK activation is an upstream initiator of cyanide-induced toxicity in cortical cells [15]. In Mes 23.5 cells, cyanide activated p38 MAPK within 30 min (Fig. 2A). The activation, as measured by protein phosphorylation, was blocked by SB203580, a p38 MAPK antagonist, whereas the control peptide SB202474 did not alter phospho-p38 expression (Fig. 2B). Pretreatment with NAC or CAT also reduced p38 MAPK activation, showing that ROS is an activation signal of the kinase. On the other hand, SB203580 had no effect on cellular ROS levels, indicating p38 MAPK activation was downstream of oxidative stress (Fig. 1C).
Fig. 2.
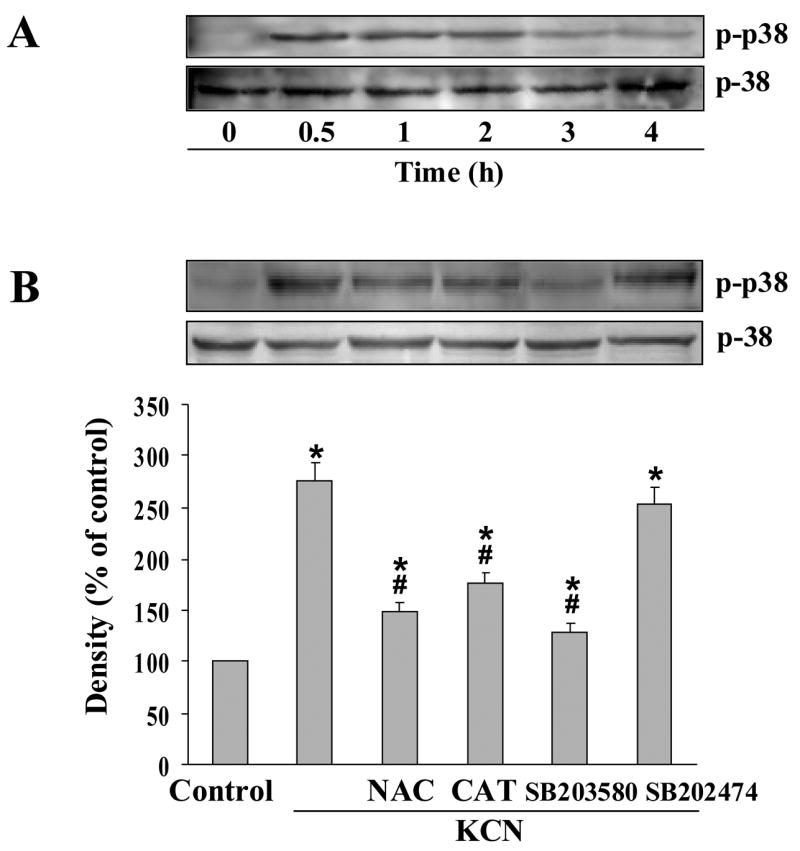
Cyanide-induced activation of p38 MAP kinase. (A) Cells were treated with 400 μM cyanide (0.5–4 h), and whole cell lysates subjected to Western blot analysis with anti-phospho-p38 MAP kinase and anti-p38 MAP kinase antibodies. (B) Blockade of cyanide-induced activation of p38 MAP kinase. Cells were pretreated for 30 min with NAC (0.5 mM), CAT (0.2 mM), SB203580 (20 μM) or SB202474 (20 μM), then with 400 μM KCN for 30 min. p38 and phospho-p38 MAP kinase proteins were determined by Western blotting. Densitometric analysis is the mean ± SEM of three separate determinations. *Significantly different from control group; #significantly different from KCN group. P<0.05.
Since p38 MAPK can regulate HIF-1α accumulation [25], it was determined whether HIF-1α was activated by cyanide. At varying times of cyanide treatment, HIF-1α protein level was determined in the nuclear fraction. Increased HIF-1α expression was observed within 1 h of cyanide exposure, and maximal levels detected at 3 h (Fig. 3A). Also, total cellular HIF-1α levels were increased by KCN (data not shown). The increased HIF-1α expression was markedly suppressed by NAC, CAT and SB203580 (Fig. 3B). HIF-1α transcriptional activity was then examined by a luciferase reporter assay under control of a promoter containing three hypoxia response element sites (HRE-luciferase). Cyanide increased HRE-dependent luciferase induction, whereas NAC, CAT and SB203580 blocked HIF-1α-mediated promoter activation (Fig. 3C). Thus KCN-induced HIF-1α accumulation and HRE-promoter activation are dependent on oxidative stress and p38 MAPK activation. It should be noted that at 20 μM, SB203580 was specific for p38 MAPK and did not affect the activity of other kinases including ERK1/2 or JNK (data not shown).
Fig. 3.

Cyanide-induced HIF-1α activation. (A) Cells were treated with 400 μM KCN (0.5–12 h), and cell lysates subjected to nuclear fractionation followed by Western blotting to determine HIF-1α levels. (B) Effects of antioxidants and a p38 inhibitor on HIF-1α protein expression. Cells were pretreated for 30 min with NAC (0.5 mM), CAT (0.2 mM), SB203580 (20 μM) or SB202474 (20 μM) prior to 400 μM KCN for 3 h. HIF-1α was determined by Western blotting. Densitometric data represent the mean±SEM of three separate determinations. (C) Cells expressing the HRE-luciferase reporter gene construct were exposed to KCN (400 μM) in the presence or absence of the compounds as indicated above. The reporter assay was conducted 3 h after cyanide addition. *Significantly different from control group; #significantly different from KCN alone group, P<0.05.
Upregulation of BNIP3 is transcriptionally regulated by the HIF-1α pathway
To determine involvement of the Bcl-2 family in cyanide-induced cell death in Mes 23.5 cells, gene expression of the pro-cell death regulators, BNIP3 and BAX, and the anti-apoptotic regulator, Bcl-2, was estimated after cyanide treatment. Quantitative real-time PCR showed no difference in BAX expression between control and cyanide-treated cells (Fig. 4A). In contrast, BNIP3 mRNA levels increased within 3 h, peaked at 9 h and remained elevated over the 24 h observation period. Western blotting showed that BNIP3 expression was time- (3–24 h) and concentration-dependent (200–600 μM KCN) (Fig. 4B, C). Notably, cyanide slightly increased (1.5 fold as compared to control) Bcl-2 gene expression (mRNA), which was elevated within 3 h and returned to basal levels with 24 h (Fig. 4A). Bcl-2 and BAX protein levels were not influenced by cyanide (Fig. 4B). BAX mRNA and protein levels were not changed by cyanide. Based on these observations, it was concluded that induction of BNIP3 expression by cyanide was transcriptionally regulated.
Fig. 4.

Effect of cyanide on BNIP3, BAX and Bcl-2 gene expression. (A) The mRNA levels of BNIP3, BAX and Bcl-2 in cells treated with 400 μM KCN as determined by real-time PCR. Levels of mRNA are presented relative to β-actin. (B) BNIP3, BAX and Bcl-2 expression was determined by Western blotting at different durations of 400 μM KCN exposure. (C) Cells were treated with different concentrations of KCN or transfected with BNIP3 cDNA for 24 h, followed by Western blotting of BNIP3. Data represent mean ± SEM of 3 separate determinations. *Significantly different from control (without KCN treatment) group, P<0.05.
To determine if BNIP3 upregulation was mediated by the HIF-1α pathway, HIF-1α gene silencing was produced by transient transfection with RNAi specific for HIF-1α. As shown in Fig. 5A, cyanide-induced nuclear localization of HIF-1α was significantly reduced by RNAi. Importantly, when HIF-1α expression was knocked down, cyanide upregulation of both BNIP3 mRNA and protein expression was reduced (Fig 5B, C). These observations confirm involvement of HIF-1α in cyanide-induced upregulation of BNIP3 expression.
Fig. 5.

HIF-1α silencing reduced BNIP3 expression in cyanide treated cells. (A) Cells were transfected with either control siRNA or siRNA specific for HIF-1α for 36 h, then treated with 400 μM KCN for 3 h. The HIF-1α level was determined by Western blotting. (B) HIF-1α siRNA transfected cells were treated with 400 μM KCN for 9 h, then BNIP3 mRNA levels were determined by real-time RCR. (C) Cells were transfected as in (B), then exposed to 400 μM KCN for 24 h. The level of BNIP3 expression was determined by Western blotting. Data represent mean ± SEM of 3 separate determinations. *Significantly different from control RNAi treatment group; #signficantly different from the KCN + RNAi control group. P<0.05.
BNIP3 and cell death
To determine the association of cyanide-induced death with BNIP3 upregulation, cells were transiently transfected with wild-type BNIP3 (BNIP3+) or a transmembrane domain deleted mutant of BNIP3 (BNIP3ΔTM). Both BNIP3+ cells and cyanide-treated cells exhibited TUNEL positive staining and characteristic apoptotic morphology with Hoechst 33258 staining, including condensed and segmented nuclei (Fig. 6A). BNIP3+ cells treated with cyanide for 24 h exhibited a significantly higher level of death as compared to empty vector (EV) transfected cells treated with cyanide, indicating BNIP3 overexpression sensitized the cells to KCN. In contrast, transfection with BNIP3ΔTM markedly reduced cyanide-induced cell death (Fig. 6A, B). Since BNIP3ΔTM functions as a dominant negative mutant of BNIP3, these observations show that BNIP3 is a pro-death factor in cyanide-induced cell death.
Fig. 6.

BNIP3-mediated mitochondrial dysfunction and cell death. (A) Cells were transfected with empty vector control (EV), BNIP3 or exposed to 400 μM KCN for 24 h. Photomicrographs (400 x) of apoptotic cells stained by TUNEL (dark cells) or Hoechst 33258 stain (bright cells). (B, C) Cells were transiently transfected with empty vector control (EV), BNIP3 or BNIP3ΔTM mutant and then exposed to 400 μM KCN for 24 h in the presence or absence of CsA (1 μM). Then apoptosis was determined (B) and the relative ΔΨm monitored (C). Data represent mean ± SEM of 3–6 separate determinations. *Significantly different from EV-control group; #significantly different from BNIP3-control group; +Signficantly different from BNIP3+KCN group. P<0.05.
Since mitochondria are the primary site of action of Bcl-2 proteins [26], the effect of BNIP3 on ΔΨm was determined. BNIP3+ cells exhibited a reduced ΔΨm, which was further reduced by cyanide (Fig. 6C). In contrast, BNIP3ΔTM-transfected cells maintained ΔΨm, even following exposure to cyanide. Since collapse of ΔΨm is associated with opening of the mitochondrial permeability transition (MPT) pore, a blocker of the MPT pore was used to preserve ΔΨm. Cyclosporin A (CsA) blocked both cyanide and BNIP3-induced collapse of ΔΨm and reduced the apoptosis (Fig. 6B, C), thus indicating involvement of MPT in the mitochondrial dysfunction.
To confirm that BNIP3 is an upstream regulator of the cell death, cyanide up-regulation of BNIP3 expression was knocked down by RNAi. The RNAi treatment markedly reduced the induction of BNIP3 expression and rescued the cells from cyanide-induced cell death (Fig. 7A, B). Under control conditions, RNAi did not alter the low, constitutive BNIP3 expression which apparently undergoes a slow turnover, rather RNAi blocked the induction by cyanide, reflecting the transcriptional up-regulation of BNIP3 expression.
Fig. 7.

BNIP3 expression and caspase-independent cell death. (A, B) Cells were transfected with siRNA control or siRNA specific for BNIP3 and 24 h post-transfection cells were treated with 400 μM KCN for 24 h. BNIP3 expression as determined by Western blotting and apoptotic cell death by Hoechst 33258 staining. (C) Cells were transfected with EV control or BNIP3 cDNA for 24 h, followed by 400 μM KCN treatment for 24 h and then caspase-3 activity was determined. H2O2 (100 μM) treatment group was used as positive control. (D) Cells were transfected as in (C), followed by exposure to 400 μM KCN for 24 h in the presence or absence of zVAD-fmk (25 μM). Apoptosis was then determined. Data represent mean ± SEM of three to six separate determinations. *Significantly different from control group; #Signficantly different from KCN+RNAi control group. P<0.05.
To further characterize the mode of cell death induced by cyanide, caspase activity was determined following transient transfection with plasmids encoding EV, BNIP3, or BNIP3ΔTM in the presence or absence of cyanide. Although H2O2 (positive control) induced a 3.5 fold increase in caspase substrate cleavage, lysates from cells transfected with the plasmids showed no difference in caspase activity (Fig. 7C). To further determine if BNIP3-induced cell death was caspase-mediated, the effect of the broad-spectrum caspase inhibitor zVAD-fmk on the apoptosis was evaluated (Fig. 7D). In cells transfected with BNIP3 and treated with KCN, the apoptosis was not affected by zVAD-fmk. It was concluded that in Mes 23.5 cells, BNIP3-mediated cell death was caspase-independent apoptosis that involves MPT pore opening and collapse of the ΔΨm.
DISCUSSION
Cyanide induces selective degeneration of the nervous system in which two distinct modes of cell death have been characterized; apoptosis in the cortex and necrosis in substantia nigra [5]. The apoptotic cell death is initiated by mitochondrial dysfunction in which cytochrome c oxidase is inhibited and an increased ROS generation is produced [22]. The present study clarifies the underlying signaling cascade in a dopaminergic cell model and shows that cyanide activates the HIF pathway to upregulate BNIP3. Cyanide induced a rapid surge of ROS generation, followed by activation of p38 MAPK and HIF-1 accumulation. As a target gene transcriptionally regulated by HIF-1, BNIP3 participated in the cell death by disrupting mitochondrial function to produce a caspase-independent apoptosis.
HIF-1 can be upregulated under both hypoxia and normoxic conditions. Oxidative stress is a non-hypoxic stimulus that induces HIF-1 activation. For instance, growth factor, thrombin or insulin are able to promote the HIF-1 response following stimulation of cellular ROS generation [10,27,28]. These responses were inhibited by antioxidants or overexpression of redox-modifying enzymes, showing that an antioxidant state reduces levels of free radicals to limit activation of the HIF pathway, whereas elevated cellular levels of oxidative radicals promote the pathway. In this study the initiation signal for HIF-1 activation by cyanide was ROS, whereas treatment with a free radical scavenger reduced ROS accumulation and inhibited HIF-1 pathway activation.
Cyanide-induced cellular oxidative stress appears to arise through multiple pathways. In current study, DCF-DA was used to detect the intracellular ROS production. Although DCF-DA has been widely applied to determine the free radicals generation, it should be noted that this probe is non-specific and detects oxidative radicals generated from multiple intracellular organelles, including mitochondria, endoplasmic reticulum and peroxisomes. To differentiate the possible sources of ROS, cells were treated with mitochondrial uncoupler FCCP. It was observed in KCN treated cells that ROS generated from sources other than mitochondria represented a small portion of the total cellular ROS. This is in agreement with previous work indicating mitochondria may be the primary source of ROS following cyanide [4]. It is well characterized that cyanide inhibits cytochrome oxidase, which in turn stimulates ROS formation at complex I and III. On the other hand, an alternative origin of ROS may exist since FCCP did not completely abolish ROS generation compared to control cells. An additional source of the cyanide-induced intracellular oxidative stress may be the activation of phospholipase A2 and subsequent metabolism of arachidonic acid [29]. In primary mesencephalic cells, cyanide depletes mitochondrial reduced glutathione, a vital component of the cellular antioxidant defense, to produce an oxidative stress-mediated cell death [30]. Excess production of ROS may damage cells by either directly oxidizing cellular macromolecules and/or initiating death signaling cascades, such as the HIF-1α pathway. It is likely that the level of oxidative stress produced by cyanide varies with brain area and perhaps accounts for the selective vulnerability characteristic of cyanide by activating different cell death pathways [5].
Present results indicate that p38MAPK signaling is necessary for induction of HIF-1α by cyanide. The p38 MAPK, a member of the MAP kinase family involved in apoptotic cell death, can be activated by dual phosphorylation on Thr-180 and Tyr-182 [31,32]. Numerous studies have shown that stimulation of cellular ROS generation can lead to MAP kinase activation whereas antioxidants can inhibit activation. In the case of Mes 23.5 cells, cyanide-induced activation of p38 MAP kinase preceded nuclear HIF-1α accumulation since pharmacologic inhibition of p38 MAP kinase blocked both induction of HIF-1α and the increased transcriptional activity. Although the mechanism underlying HIF-1α regulation by p38 MAPK is unclear, it appears that p38 MAPK acts as a redox-sensitive factor to induce HIF-1α stabilization, followed by nuclear translocation to promote transcription of target genes. The functional result is a transfer of the redox signal to the nucleus to initiate transcription of pro-death gene products.
As a transcription factor, HIF-1α regulates a series of target genes encoding proteins that promote cell survival [33]. On the other hand, HIF-1α can also participate in cell death by activating pro-death genes, including BNIP3, and Nix [34]. BNIP3 expression can be upregulated under both hypoxic and non-hypoxic conditions in cell lines derived from carcinomas, fibroblasts and macrophages [34,35]. In the nervous system, BNIP3 functions as a pro-death factor in select brain regions following subarachnoid hemorrhage [36] or focal cerebral ischemia [16]. Recently, BNIP3 was linked to brain developmental apoptosis in which BNIP3 mRNA increased in parallel with developmental cell death in neonatal rat brain [37].
The data show that HIF-1α activation is essential for BNIP3 transcription after cyanide treatment. Rodent BNIP3 promoter is activated by HIF-1α [38] and HIF-1α can bind to a HRE site on the BNIP3 promoter to activate BNIP3 transcription [39]. It was concluded that this occurred following cyanide since HIF-1α protein accumulation (1 h) preceded BNIP3 mRNA upregulation. HIF-1α transcriptional activity was increased within 3 h after cyanide treatment. Moreover, HIF-1α knockdown by RNAi markedly reduced cyanide-mediated upregulation of BNIP3 mRNA and protein expression, further supporting the role of HIF-1α in regulation of BNIP3 in this cell death model.
In figure 4, BNIP3 was transcriptionally induced within 3 h after cyanide treatment. Accumulation of the BNIP3 protein correlated with onset of cell death which was initially observed at 6 h and reached a maximum at 24 h. Similar induction of BNIP3 by cyanide in primary cortical cells was also reported [18]. Knockdown of BNIP3 by siRNA transfection protected cells from cytotoxicity, providing strong support for the role of BNIP3 in the cell death. It was interesting to note that RNAi did not influence the low, constitutive BNIP3 expression in wildtype cells, apparently due to low BNIP3 turnover under control conditions. However, RNAi produced a marked reduction of BNIP3 induction by cyanide which could be explained by inhibiting the induction of BNIP3 transcription by cyanide.
Unlike the classic Bcl-2 protein-mediated apoptosis, cell death was not caspase-dependent, although the cells displayed morphological characteristics of apoptosis. BNIP3-mediated atypical cell death has also been reported in other cell systems, where cells exhibited nuclear apoptotic change in the absence of Apaf-1, cytochrome c release and caspase activation [40,41]. We have also observed that cyanide can produce apoptosis in rat primary cortical cells in which BNIP3 released cytochrome c from mitochondria, followed by caspase-dependent apoptosis [18]. It is possible that the cyanide-induced increase in BNIP3 may activate different execution cascades in cortical and mesencephalic cells.
In mediating cell death, the target of BNIP3 is mitochondria [42]. Forced overexpression of BNIP3 in Mes 23.5 cells decreased ΔΨm, which was prevented by CsA, an MPT inhibitor. Previous studies have shown that upon overexpression, BNIP3 integrates into the outer mitochondrial membrane with a special orientation of the N-terminus in the cytoplasm and C-terminus in the membrane [41]. After integration into the mitochondrial membrane, BNIP3 may interact with components of the MPT pore to induce rapid opening of the MPT pore to dissipate ΔΨm. Expression of the transmembrane domain deleted form of BNIP3 (BNIP3ΔTM) reduced the cyanide-induced cell death. BNIP3ΔTM functions as a dominant negative mutant to block integration of wildtype BNIP3 into the mitochondrial membrane [43].
In summary, it was shown that in cyanide-induced cell death, oxidative stress initiates upregulation of BNIP3 expression via a HIF-1α signaling regulated transcription. Subsequently, increased BNIP3 expression produced ΔΨm dissipation, followed by execution of a caspase-independent apoptotic cell death.
Acknowledgments
This work was supported by National Institutes of Health grant ES 04140.
Abbreviations
- ARNT
arylhydrocarbon receptor nuclear translocator
- BNIP3
Bcl-2/adenovirus E1B 19kDa interacting protein 3
- BNIP3ΔTM
transmembrane domain deleted form of BNIP3
- BH3
Bcl-2 homology 3
- CAT
catalase
- CsA
cyclosporin A
- DCF-DA
2′,7′-dichlorofluorescin-diacetate
- EV
empty vector
- EYFP
enhanced yellow fluorescence protein
- FBS
fetal bovine serum
- HIF-1α
hypoxia-inducible factor-1α
- H2O2
hydrogen peroxide
- HRE
hypoxia response element
- KCN
potassium cyanide
- p38 MAPK
p38 mitogen-activated protein kinase
- MKP
MAPK phophatases
- MPT
mitochondrial permeability transition
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NAC
N-acetyl-L-cysteine
- NBS
new born calf serum
- NMDA
N-methyl-D-aspartate
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
- siRNA
small interfering RNA
- TM
C-terminal transmembrane
- TUNEL
in situ terminal deoxynucleotidyl transferase-mediated DNA nick-end labeling
- ΔΨm
mitochondrial membrane potential
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Way JL. Cyanide intoxication and its mechanism of antagonism. Annu Rev Pharmacol Toxicol. 1984;24:451–481. doi: 10.1146/annurev.pa.24.040184.002315. [DOI] [PubMed] [Google Scholar]
- 2.Pearce LL, Bominaar EL, Hill BC, Peterson J. Reversal of cyanide inhibition of cytochrome c oxidase by the auxiliary substrate nitric oxide: an endogenous antidote to cyanide poisoning. J Biol Chem. 2003;278:52135–52145. doi: 10.1074/jbc.M310359200. [DOI] [PubMed] [Google Scholar]
- 3.Maduh EU, Borowitz JL, Isom GE. Cyanide-induced alteration of the adenylate energy pool in rat neuroscretory cell line. J Appl Toxicol. 1991;11:97–101. doi: 10.1002/jat.2550110205. [DOI] [PubMed] [Google Scholar]
- 4.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 5.Mills EM, Gunasekar PG, Li L, Borowitz JL, Isom GE. Differential susceptibility of brain areas to cyanide involves different modes of cell death. Toxicol Appl Pharmacol. 1999;156:6–16. doi: 10.1006/taap.1999.8630. [DOI] [PubMed] [Google Scholar]
- 6.Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE. Calcineurin-mediated Bad translocation regulates cyanide-induced neuronal apoptosis. Biochem J. 2004;379:805–813. doi: 10.1042/BJ20031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prabhakaran K, Li L, Borowitz JL, Isom GE. Cyanide induces different modes of death in cortical and mesencephalon cells. J Pharmacol Expl Therap. 2002;303:510–519. doi: 10.1124/jpet.102.039453. [DOI] [PubMed] [Google Scholar]
- 8.Hirota K, Semenza GL. Regulation of hypoxia-inducible factor 1 by prolyl and asparaginyl hydroxylases. Biochem Biophys Res Commun. 2005;338:610–616. doi: 10.1016/j.bbrc.2005.08.193. [DOI] [PubMed] [Google Scholar]
- 9.Greijer AE, van der Groep P, Kemming D, Shvarts A, Semenza GL, Meijer GA, van de Wiel MA, Belien JA, van Diest PJ, van der Wall E. Up-regulation of gene expression by hypoxia is mediated predominantly by hypoxia-inducible factor 1 (HIF-1) J Pathol. 2005;206:291–304. doi: 10.1002/path.1778. [DOI] [PubMed] [Google Scholar]
- 10.BelAiba RS, Djordjevic T, Bonello S, Flugel D, Hess J, Kietzmann T, Gorlach A. Redox-sensitive regulation of the HIF pathway under non-hypoxic conditions in pulmonary artery smooth muscle cells. Biol Chem. 2004;385:249–257. doi: 10.1515/BC.2004.019. [DOI] [PubMed] [Google Scholar]
- 11.Haddad JJ, Land SC. A non-hypoxic, ROS-sensitive pathway mediates TNF-alpha-dependent regulation of HIF-1alpha. FEBS Lett. 2001;505:269–274. doi: 10.1016/s0014-5793(01)02833-2. [DOI] [PubMed] [Google Scholar]
- 12.Biswas S, Gupta MK, Chattopadhyay D, Mukhopadhyay CK. Insulin induce activation of Hypoxia inducible factor-1 requires generation of reactive oxygen species by NADPH oxidase. Am J Physiol Heart Circ Physiol. 2006 doi: 10.1152/ajpheart.00718.2006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 13.Ueda S, Masutani H, Nakamura H, Tanaka T, Ueno M, Yodoi J. Redox control of cell death. Antioxid Redox Signal. 2002;4:405–414. doi: 10.1089/15230860260196209. [DOI] [PubMed] [Google Scholar]
- 14.Kietzmann T, Jungermann K, Gorlach A. Regulation of the hypoxia-dependent plasminogen activator inhibitor 1 expression by MAP kinases. Thromb Haemost. 2003;89:666–673. [PubMed] [Google Scholar]
- 15.Shou Y, Li L, Prabhakaran K, Borowitz JL, Isom GE. p38 Mitogen-activated protein kinase regulates Bax translocation in cyanide-induced apoptosis. Toxicol Sci. 2003;75:99–107. doi: 10.1093/toxsci/kfg157. [DOI] [PubMed] [Google Scholar]
- 16.Althaus J, Bernaudin M, Petit E, Toutain J, Touzani O, Rami A. Expression of the gene encoding the pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible factor-1alpha (HIF-1alpha) protein following focal cerebral ischemia in rats. Neurochem Int. 2006;48:687–695. doi: 10.1016/j.neuint.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Ray R, Chen G, Vande Velde C, Cizeau J, Park JH, Reed JC, Gietz RD, Greenberg AH. BNIP3 heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2 homology 3 (BH3) domain at both mitochondrial and nonmitochondrial sites. J Biol Chem. 2000;275:1439–1448. doi: 10.1074/jbc.275.2.1439. [DOI] [PubMed] [Google Scholar]
- 18.Prabhakaran K, Li L, Zhang L, Borowitz JL, Isom GE. Upregulation of BNIP3 and translocation to mitochondria mediates cyanide-induced apoptosis in cortical cells. J Neurochem. 2007 doi: 10.1016/j.neuroscience.2007.07.033. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le WD, Colom L, Xie WJ, Smith RG, Alexianu M, Appel SH. Cell death induced by β-amyloid 1–40 in MES 23.5 hybrid clone: the role of nitric oxide and NMDA-gated channel activation leading to apoptosis. Brain Res. 1995;686:49–60. doi: 10.1016/0006-8993(95)00450-5. [DOI] [PubMed] [Google Scholar]
- 20.Le WD, Rowe D, Xie W, Ortiz I, He Y, Appel SH. Microglial activation and dopaminergic cell injury: an in vitro model relevant to Parkinson’s disease. J Neurosci. 2001;21:8447–8455. doi: 10.1523/JNEUROSCI.21-21-08447.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khan N, Wilmot CM, Rosen GM, Demidenko E, Sun J, Joseph J, O’Hara J, Kalyanaraman B, Swartz HM. Spin traps: in vitro toxicity and stability of radical adducts. Free Radic Biol Med. 2003;34:1473–1481. doi: 10.1016/s0891-5849(03)00182-5. [DOI] [PubMed] [Google Scholar]
- 22.Jones DC, Prabhakaran K, Li L, Gunasekar PG, Shou Y, Borowitz JL, Isom GE. Cyanide enhancement of dopamine-induced apoptosis in mesencephalic cells involves mitochondrial dysfunction and oxidative stress. Neurotoxicol. 2003;24:333–342. doi: 10.1016/S0161-813X(03)00042-1. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Li L, Prabhakaran K, Borowitz JL, Isom GE. Trimethyltin-induced apoptosis is associated with upregulation of inducible nitric oxide synthase and Bax in a hippocampal cell line. Toxicol Appl Pharmacol. 2006;216:34–43. doi: 10.1016/j.taap.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Chen G, Ray R, Dubik D, Shi L, Cizeau J, Bleackley RC, Saxena S, Gietz RD, Greenberg AH. The E1B 19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein that activates apoptosis. J Exp Med. 1997;186:1975–1983. doi: 10.1084/jem.186.12.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Mol Cell Biol. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gustafsson AB, Gottlieb RA. Bcl-2 Family Members and Apoptosis, Taken to Heart. Am J Physiol Cell Physiol. 2006 Aug 30; doi: 10.1152/ajpcell.00229.2006. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 27.Calvani M, Rapisarda A, Uranchimeg B, Shoemaker RH, Melillo G. Hypoxic induction of an HIF-1alpha-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood. 2006;107:2705–2712. doi: 10.1182/blood-2005-09-3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leek RD, Stratford I, Harris AL. The role of hypoxia-inducible factor-1 in three-dimensional tumor growth, apoptosis, and regulation by the insulin-signaling pathway. Cancer Res. 2005;65:4147–4152. doi: 10.1158/0008-5472.CAN-04-2184. [DOI] [PubMed] [Google Scholar]
- 29.Gunasekar PG, Borowitz JL, Isom GE. Cyanide-induced generation of oxidative species: involvement of nitric oxide synthase and cyclooxygenase-2. J Pharmacol Expl Therap. 1998;285:236–241. [PubMed] [Google Scholar]
- 30.Prabhakaran K, Li L, Borowitz JL, Isom GE. Inducible nitric oxide synthase up-regulation and mitochondrial glutathione depletion mediate cyanide-induced necrosis in mesencephalic cells. J Neurosci Res. 2006;84:1003–1011. doi: 10.1002/jnr.20998. [DOI] [PubMed] [Google Scholar]
- 31.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 32.Ho TC, Yang YC, Cheng HC, Wu AC, Chen SL, Chen HK, Tsao YP. Activation of mitogen-activated protein kinases is essential for hydrogen peroxide -induced apoptosis in retinal pigment epithelial cells. Apoptosis. 2006;11:1899–1908. doi: 10.1007/s10495-006-9403-6. [DOI] [PubMed] [Google Scholar]
- 33.Yeo EJ, Chun YS, Park JW. New anticancer strategies targeting HIF-1. Biochem Pharmacol. 2004;68:1061–1069. doi: 10.1016/j.bcp.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 34.Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001;61:6669–6673. [PubMed] [Google Scholar]
- 35.Vengellur A, LaPres JJ. The role of hypoxia inducible factor 1alpha in cobalt chloride induced cell death in mouse embryonic fibroblasts. Toxicol Sci. 2004;82:638–646. doi: 10.1093/toxsci/kfh278. [DOI] [PubMed] [Google Scholar]
- 36.Ostrowski RP, Colohan AR, Zhang JH. Mechanisms of hyperbaric oxygen-induced neuroprotection in a rat model of subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2005;25:554–571. doi: 10.1038/sj.jcbfm.9600048. [DOI] [PubMed] [Google Scholar]
- 37.Sandau US, Handa RJ. Localization and developmental ontogeny of the pro-apoptotic Bnip3 mRNA in the postnatal rat cortex and hippocampus. Brain Res. 2006;1100:55–63. doi: 10.1016/j.brainres.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 38.Bruick RK. Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad Sci USA. 2000;97:9082–9087. doi: 10.1073/pnas.97.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kothari S, Cizeau J, McMillan-Ward E, Israels SJ, Bailes M, Ens K, Kirshenbaum LA, Gibson SB. BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene. 2003;22:4734–4744. doi: 10.1038/sj.onc.1206666. [DOI] [PubMed] [Google Scholar]
- 40.Wan J, Martinvalet D, Ji X, Lois C, Kaech SM, Von Andrian UH, Lieberman J, Ahmed R, Manjunath N. The Bcl-2 family pro-apoptotic molecule, BNIP3 regulates activation-induced cell death of effector cytotoxic T lymphocytes. Immunology. 2003;110:10–17. doi: 10.1046/j.1365-2567.2003.01710.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee H, Paik SG. Regulation of BNIP3 in normal and cancer cells. Mol Cells. 2006;21:1–6. [PubMed] [Google Scholar]
- 43.Burton TR, Henson ES, Baijal P, Eisenstat DD, Gibson SB. The pro-cell death Bcl-2 family member, BNIP3, is localized to the nucleus of human glial cells: Implications for glioblastoma multiforme tumor cell survival under hypoxia. Intl J Cancer. 2006;118:1660–1669. doi: 10.1002/ijc.21547. [DOI] [PMC free article] [PubMed] [Google Scholar]