

Abstract
The complex cellular functions of an organism frequently rely on physical interactions between proteins. A map of all protein-protein interactions, an interactome, is thus an invaluable tool. We present an interactome for Arabidopsis (Arabidopsis thaliana) predicted from interacting orthologs in yeast (Saccharomyces cerevisiae), nematode worm (Caenorhabditis elegans), fruitfly (Drosophila melanogaster), and human (Homo sapiens). As an internal quality control, a confidence value was generated based on the amount of supporting evidence for each interaction. A total of 1,159 high confidence, 5,913 medium confidence, and 12,907 low confidence interactions were identified for 3,617 conserved Arabidopsis proteins. There was significant coexpression of genes whose proteins were predicted to interact, even among low confidence interactions. Interacting proteins were also significantly more likely to be found within the same subcellular location, and significantly less likely to be found in conflicting localizations than randomly paired proteins. A notable exception was that proteins located in the Golgi were more likely to interact with Golgi, vacuolar, or endoplasmic reticulum sorted proteins, indicating possible docking or trafficking interactions. These predictions can aid researchers by extending known complexes and pathways with candidate proteins. In addition we have predicted interactions for many previously unknown proteins in known pathways and complexes. We present this interactome, and an online Web interface the Arabidopsis Interactions Viewer, as a first step toward understanding global signaling in Arabidopsis, and to whet the appetite for those who are awaiting results from high-throughput experimental approaches.
High-throughput experiments have resolved genome scale networks of protein-protein interactions (PPIs; interactomes) in yeast (Saccharomyces cerevisiae), fruitfly (Drosophila melanogaster), nematode worm (Caenorhabditis elegans), and human (Homo sapiens; Uetz et al., 2000; Giot et al., 2003; Li et al., 2004; Miller et al., 2005; Rual et al., 2005; Gandhi et al., 2006). These interactomes have revealed protein transactions in biological processes and relatedness of interacting partners. Interactomics is quickly becoming a valuable new area of systems biology by comprehensively deducing the networks of PPIs that form the basis for much of signaling and regulatory control as well as the machinery of cellular function.
Where the cost of a high-throughput experimental approach is prohibitive, a computational alternative is often a useful preliminary step, especially when combined with literature extraction of all published protein interactions. The Online Predicted Human Interaction Database (Brown and Jurisica, 2005) combines extensive literature search from the Human Protein Resource Database (http://www.hprd.org) and predictions of interacting orthologs (interologs) derived from yeast and fruitfly (Krogan et al., 2006). Predicted interactomes are deduced from experimental interactomes of other species. A pair of interologs in the reference species predicts an interaction in the test species. This method relies on accurately predicting orthologous genes using similarity cutoffs and prediction algorithms such as INPARANOID (http://inparanoid.cgb.ki.se), and not simply best blast score (O'Brien et al., 2005). Because of this limitation, an interactome predicted from interologs will show interactions among the most conserved proteins. Fortunately many pathways such as endomembrane trafficking and small GTPase signaling actively being studied show significant conservation among eukaryotes (Carter et al., 2004; Chang and Philips, 2006).
In a similar approach, the gene-coexpression network can be built by examining coexpression of genes across a wide number of tissues and experiments (Hanisch et al., 2002). This provides useful information about genes likely to be involved in the same biological processes in humans, mouse, Escherichia coli, and yeast (Bhardwaj and Lu, 2005).
Although plant protein interaction networks based on literature mining and coexpression of neighboring Arabidopsis (Arabidopsis thaliana) genes have been recently reviewed, there is currently no publicly available large-scale plant interactome (Uhrig, 2006; Williams and Bowles, 2006). In this article, we present predicted Arabidopsis protein interactome based on the interolog method. We have shown that these predicted interacting proteins are significantly colocalized and coexpressed by analyzing existing experimental data from Arabidopsis. We have recapitulated many known signaling pathways and protein complexes in Arabidopsis and have extended by adding new and often unknown proteins into existing networks. In this way we provide an avenue to expand the current understanding of signaling and cellular function by enabling hypothesis generation based on our predicted Arabidopsis interactome.
RESULTS
Building a Predicted Interactome
PPIs, which are the basis of intracellular signaling and regulation, were predicted on the assumption that evolutionarily conserved proteins would tend to have conserved interactions. The process began by using the ortholog predicting algorithm INPARANOID (Remm et al., 2001; O'Brien et al., 2005) and using automatic annotation from ENSEMBL (Birney et al., 2004; Curwen et al., 2004) to identify Arabidopsis proteins orthologous to proteins in yeast, nematode worm, fruitfly, and human. Partial interactomes from these four species have been experimentally determined (Giot et al., 2003; Li et al., 2004; Rual et al., 2005; Gandhi et al., 2006). A predicted interaction was established for Arabidopsis where orthologs existed for both interactive proteins in one of these four established interactomes. This process, outlined in Figure 1, is known as interaction-ortholog (or interolog) mapping and is an established method of predicting interactomes (Lehner and Fraser, 2004; Yu et al., 2004). Interologs are thus a prediction without direct experimental verification, but none the less a good place to begin. Using this method we identified 19,979 predicted interactions for 3,617 Arabidopsis proteins (Supplemental Table S1). Of these 611 are predicted self interactions (homodimers) and 19,368 are interactions between different proteins (heterodimers).
Figure 1.

Flowchart for the predicted Arabidopsis interactome. A list of Arabidopsis orthologs were identified using INPARANOID and ENSEMBL algorithms (see “Materials and Methods”) from genome databases of yeast, nematode, fruitfly, and human. Where orthologs were found for both partners of a known protein interaction in the reference species, that interaction was mapped to (i.e. replaced with) corresponding Arabidopsis genes. This generated the Arabidopsis predicted interactome and a CV based on the amount of supporting evidence. Subsequent verification and analysis examined each interaction protein pair using Pearson correlation of gene expression profiles in an Arabidopsis transcriptome database (AtGenExpress) and checked for colocalization using SUBA. [See online article for color version of this figure.]
As an internal quality control we established a confidence value (CV; Supplemental Table S1). Our CV is established individually for each pair of interacting proteins and is based on the product of: (1) In how many different datasets was the interaction predicted; (2) How many different kinds of experiments supported this interaction; and (3) In how many (out of four) species was this interaction found. With this assessment, we have identified 1,159 high confidence interactions (CV > 10), 5,913 medium confidence (CV between 2 and 10), and 12,907 low confidence interactions (CV = 1).
Predicted Arabidopsis interacting protein pairs (from Supplemental Table S1) were loaded into the network building programs OSPREY and CYTOSCAPE (see “Materials and Methods”) to visualize interaction pathways. Surprisingly, 3,482 of the set of 3,617 conserved proteins were connected into a single interconnected network (Fig. 2A). Many proteins have a high number of interacting partners, including perhaps predictably ubiquitin-related proteins and members of the 26S proteosome, but also a Ras-related GTPase (At2g2290) and CDC2A, members of known signaling pathways (Table I). To analyze the topology of the network, proteins were divided into free ends (with only one interaction), pipes (two interactions), and hubs of different size (demonstrated in Fig. 2C). The distribution of hub sizes was logarithmic (Supplemental Table S1), however, when broken down by class, the largest class of proteins were medium hubs (Fig. 2D). Interacting proteins had an average of 11 interacting partners, which is smaller than is found in yeast (average 22 interacting partners), but comparable to Drosophila (average nine interacting partners). As we are only looking at evolutionarily conserved interactions and not any plant-specific interactions, pipes (two interacting partners) and free ends (single interacting partner) could easily be underrepresented (see “Materials and Methods”). When super and major hubs (>50 interactions; 116 proteins total) were removed from the interactome and the network was reconstructed, 3,230 (92%) of the remaining proteins still held together in a single network, with dozens of disconnected subnets of two to five proteins (data not shown). While this is similar to the observation in yeast that the network integrity is held together by smaller hubs called the stratus structure (Batada et al., 2006), this should perhaps not be surprising as many of these interologs are based on yeast. Only 292 interactions have been found in two interactome datasets, usually yeast and Drosophila. The nematode worm and human interactome data experimentally derived is relatively incomplete, thus accounting for poor overlap with other datasets. At a minimum the proteins that generated the stratus structure in yeast have orthologs in Arabidopsis, and thus our prediction is for a stratus structure.
Figure 2.

Visualizing the Arabidopsis predicted interactome. A, Giant hairy ball of all 19,979 interactions visualized by Cytoscape. B, Enlargement showing example of some detail captured by visualization. C, Different types of protein nodes classified as major hubs when interacting with 50 to 100 other proteins, medium hubs 11 to 50, minor hub three to five, pipes two, free end one, and unconnected zero interacting proteins. D, Frequency distribution of different node classes based on number of interacting partners.
Table I.
Twenty most highly connected protein interaction hubs
Each edge represents a unique predicted PPI.
| Loci | Edges | Protein Description |
|---|---|---|
| At4g26840 | 172 | Ubiquitin-like protein (SMT3) |
| At1g14400 | 119 | UBIQUITIN-CONJUGATING ENZYME1 |
| At1g80410 | 115 | Acetyltransferase related |
| At5g02530 | 112 | RNA and export factor-binding protein |
| At5g13780 | 112 | GCN5-related N-acetyltransferase |
| At1g02690 | 108 | Importin α-2 subunit |
| At4g38630 | 108 | 26S proteasome regulatory subunit S5A (RPN10) identical to multiubiquitin chain binding protein (MBP1) |
| At5g26680 | 108 | Endonuclease |
| At4g25630 | 107 | FIBRILLARIN2 |
| At3g48750 | 102 | CELL DIVISION CONTROL PROTEIN2 HOMOLOG A |
| At3g58560 | 101 | Endonuclease/exonuclease/phosphatase family protein similar to Glc-repressible alcohol dehydrogenase transcriptional effector |
| At5g20850 | 100 | DNA repair protein RAD51 |
| At1g04730 | 97 | AAA-type ATPase family protein |
| At3g22590 | 97 | RNA pol II accessory factor Cdc73 family protein |
| At2g31970 | 94 | DNA repair-recombination protein (RAD50) |
| At1g29990 | 93 | Prefoldin |
| At3g42660 | 92 | Transducin family protein/WD-40 repeat family protein |
| At2g34210 | 91 | KOW domain-containing transcription factor family protein |
| At3g06720 | 90 | Importin α-1 subunit |
| At2g22290 | 89 | Ras-related GTP-binding protein |
Extending Known Pathways and Complexes
Many of the Arabidopsis interologs were predicted from multiple species and interacting proteins fall into known complexes such as DNA repair and RNA splicing (Table II). Notably, some of the most evolutionarily conserved interactions included proteins with no previously known function, such as At5g27740, whose orthologs in human, yeast, and fruitfly interact with an AAA-type (ATPase associated with a variety of cellular activities) ATPase, also found in Arabidopsis (Table II, line 9). Thus it may be possible to extend known pathways or identify unknown members of protein complexes in Arabidopsis and assign putative function on the basis of its interacting partner's function. This type of annotation (i.e. predicted to interact with X) would extend functional annotation of the Arabidopsis genome.
Table II.
Twenty most conserved interactions
| Locus A | Locus B | Protein A | Protein B | Species | CV |
|---|---|---|---|---|---|
| At2g47640 | At4g30220 | Small nuclear ribonucleoprotein D2 | Small nuclear ribonucleoprotein F | 4 | 192 |
| At5g17310 | At5g17310 | UTP—Glc-1-P uridylyltransferase | UTP—Glc-1-P uridylyltransferase | 4 | 40 |
| At3g18524 | At4g02070 | DNA mismatch repair protein MSH2 | DNA mismatch repair protein MSH6-1 | 3 | 1,155 |
| At1g21690 | At5g27740 | Replication factor C 37 kD | Unknown expressed protein | 3 | 540 |
| At2g03870 | At5g48870 | Small nuclear ribonucleoprotein | Small nuclear ribonucleoprotein | 3 | 192 |
| At1g21690 | At1g77470 | Replication factor C 37 kD | Replication factor C 36 kD | 3 | 315 |
| At2g18510 | At4g21660 | Pre-mRNA splicing factor | Proline-rich spliceosome-associated (PSP) family protein | 3 | 210 |
| At4g20330 | At1g03280 | Transcription initiation factor-related contains weak similarity to transcription initiation factor IIE | Transcription initiation factor IIE (TFIIE) α-subunit family protein/general transcription factor TFIIE | 3 | 273 |
| At5g27740 | At5g22010 | Unknown expressed protein | AAA-type ATPase family protein/BRCT domain-containing protein | 3 | 198 |
| At5g67100 | At1g67630 | DNA-directed DNA polymerase α-catalytic subunit | DNA polymerase α-subunit B family | 3 | 150 |
| At1g63160 | At1g77470 | Replication factor C 40 kD | Replication factor C 36 kD | 3 | 135 |
| At1g63780 | At5g66540 | Brix domain-containing protein | Unknown expressed protein | 3 | 108 |
| At2g20140 | At2g20580 | 26S protease regulatory complex subunit 4 | 26S proteasome regulatory subunit S2 (RPN1) | 3 | 108 |
| At1g24180 | At5g50850 | Pyruvate dehydrogenase E1 component α-subunit | Pyruvate dehydrogenase E1 component β-subunit | 3 | 45 |
| At2g27020 | At1g47250 | 20S proteasome α-subunit G (PAG1; PRC8) | 20S proteasome α-subunit F2 (PAF2; PRC2B; PRS1) | 3 | 30 |
| At5g22330 | At5g67630 | TATA box-binding protein-interacting protein related | DNA helicase | 3 | 24 |
| At2g27020 | At5g35590 | 20S proteasome α-subunit G (PAG1; PRC8) | 20S proteasome α-subunit A1 (PAA1; PRC1) | 3 | 27 |
| At2g33370 | At2g44510 | 60S ribosomal protein L23 (RPL23B) | p21Cip1-binding protein related | 3 | 18 |
| At5g22370 | At4g21800 | ATP-binding family protein | ATP-binding family protein | 3 | 9 |
| At4g02460 | At4g09140 | DNA mismatch repair protein | DNA mismatch repair protein MLH1 | 2 | 720 |
A small interactome for Arabidopsis built by extensively mining the literature is available in the BIND database (Bader et al., 2001, 2003) and contains some 356 proteins and 711 interactions. Only 95 of the BIND proteins are found among the orthologs used in our predicted interactome. These 95 proteins had 85 interactions in BIND, of which 30 are also predicted by our method, which is significant overlap compared to an expected value of 1.3 (see “Materials and Methods”). Known interactions between OSMOTIC SENSITIVE1, syntaxin, and v-SNARE proteins formed a small network of vacuolar and Golgi localized proteins (Fig. 3, blue edges). When this network was extended based on interolog prediction, 20 new proteins were putatively added to this network, including many other Golgi and vacuolar proteins, more syntaxins, SNAREs, and SNAP (soluble NSF attachment protein) proteins, but also an ATPase, heat shock protein-83, protein phosphatases, and the RAS-related protein ARA5 (Fig. 3, red edges). When extended with interologs, known pathways for RAS and RHO-like GTPases (Supplemental Fig. S1) gained several not so surprising members (i.e. ROP and other RHO-GAPs). A few interesting members were also identified. For example an unknown NCK1-like SH3 domain protein, a key protein interaction and signaling domain in humans (Wu et al., 2007), and associated with vesicle trafficking in Arabidopsis (Lam et al., 2001). Also a PH (pleckstrin homology) domain protein was identified that may also have a role in vesicle trafficking or lipid signaling (Lee et al., 2002; Tang et al., 2005). Similarly, interactions for the KNAT/STM/BELL homeotic transcription factors and the RNA splicing machinery were extended using interolog prediction (Supplemental Figs. S2 and S3). These new protein interactions are only predictions and demonstrate that interologs can be of some immediate use in generating a list of candidate genes when trying to reassemble protein complexes and signaling pathways for experimental verification.
Figure 3.

SNARE-syntaxin network expanded by predicted interactions. Proteins with known, experimentally determined interactions (blue lines) from the BIND dataset formed an initial set. This was expanded one layer outwards by identifying all proteins that are predicted to interact with proteins from the initial set. All predicted interactions are rated by CV (line thickness) and coexpression (line color). Nodes are color coded with predicted subcellular localizations and sized according to the number of predicted interacting protein partners throughout the entire predicted interactome. Note that the interaction between OSM1 and VTI12 is both predicted and experimentally determined (both red and blue lines connect these nodes).
Subcellular Localization of Interologs
To interact, interacting proteins should in general reside in the same subcellular location, although some proteins will interact across adjacent subcellular locations (i.e. cytosol-membrane associated) and some will migrate between compartments and could have interaction partners in both locations (i.e. nucleus-cytosol). Proteins in the predicted Arabidopsis interactome were assigned to a subcellular location using data from The Arabidopsis Subcellular Database (SUBA; Heazlewood et al., 2005, 2007). We then found those interologs for which both interacting proteins possessed data from SUBA on subcellular localization and were not self-interacting proteins. Subcellular localization data was available for 2,623 interologs, corresponding to 918 unique proteins. Figure 4 shows the numbers of interologs as a function of the subcellular localization of their interacting proteins. P values associated with the deviation of these counts from a random interactome network with the same properties (see “Materials and Methods”) are illustrated in Figure 4. There is a statistically significant enrichment of interologs for which both proteins pairs are in the same compartment for all compartments except the extracellular space, for which there is very little data. These results indicate that as expected, pairs of proteins predicted to interact tend to reside in the same location. The only other pairs of locations in Figure 4 with an enrichment of interologs are the Golgi apparatus/endoplasmic reticulum (ER) and Golgi apparatus/vacuole. In both of these cases the enrichment can be partially explained by the experimental difficulties in distinguishing proteins in these compartments (Dunkley et al., 2006), which will be reflected in the data in SUBA used for the analysis.
Figure 4.

Subcellular localization of protein interactions. A, A network subset of medium confidence interacting proteins where proteins were assigned to a subcellular compartment in the SUBA database. B, Analysis of all interacting protein pairs in which both partners were assigned to a subcellular compartment. The numbers of individual protein numbers is in italics beside compartment names. Compartment pairs that showed enriched or depleted numbers of interactions (compared to chance) are color coded. For example, there is a significant (P < 0.01) enrichment of interactions in which both partners are nuclear localized, while there is a significant depletion of interactions between nuclear and vacuolar localized proteins. Chloro, Chloroplast; Cyskel, cytoskeleton; Excell, extracellular; Mito, mitochondria; Perox, peroxisome.
Some of the Golgi/ER and Golgi/vacuole protein interaction enrichment can also be attributed to real interactions between members of complexes involved in the endomembrane trafficking pathway (i.e. in Fig. 3). Gandhi et al. (2006) also note a strong enrichment of interacting proteins between these organelles in their recent study of the human interactome. In contrast to the enrichment found, there is a significant depletion of interologs for which one protein is nuclear and the other from the cytoplasm, ER, Golgi apparatus, mitochondria, peroxisome, or vacuole. As interactions between proteins in these compartments are unlikely, these results also confirm expectations.
Coexpression of Interologs
Proteins that interact could be expected to possess similar or complementary gene expression profiles (for example, see Ge et al., 2001 or Fraser et al., 2004). Consequently, if an interolog pair is positively coexpressed, this strengthens the confidence in the prediction that the pair interacts. The lack of correlation does not necessarily imply that the interologs do not interact. Specifically, it could be the case that one member is constitutively expressed while the other interacting partner is only expressed under certain conditions. It is also possible that an existing protein might relocalize to a new compartment, undergo allosteric regulation, or even move extracellularly or translocate to other tissues, thus propagating a signal or interaction without a change in mRNA level. Coexpression of genes was computed by applying the Pearson correlation coefficient (r) to expression data for a gene pair. The gene expression analysis exhibited a strong and statistically significant trend (P < 10−21, using a two-sample Kolmogorov-Smirnov test, see “Materials and Methods”) of coexpression for the interolog pairs when compared to random gene pairs drawn from all Arabidopsis coding sequences (Fig. 5A). As well, we were able to visualize a positive correlation between interolog pair coexpression and the interolog CV (Fig. 5B). We have incorporated known interactions (Bader et al., 2003) and our predicted interolog data into the Arabidopsis Interaction Viewer at http://bbc.botany.utoronto.ca/interactions/ and into the outputs from the Expression Browser and Expression Angler tools of the Botany Array Resource (Toufighi et al., 2005), see Figure 6. It is thus possible to easily see if two or more genes that are coexpressed are interologs or interactors. Additionally, genes with unknown function that are flagged as interologs and coexpress with known genes are very likely to be involved in the gene of known function's biological process, thus aiding hypothesis generation.
Figure 5.

Coexpression of interologs. A, The PCC for 19,979 predicted interaction pairs was calculated and plotted as the number of pairs in each Pearson correlation coefficient range, with an r unit bin size of 0.1 (blue points). The correlation coefficient calculation was also performed for 20,000 randomly selected pairs of Arabidopsis genes from within our interactome (green points), from all AGI IDs on the ATH1 GeneChip (red points), or from all AGI IDs on the ATH1 GeneChip such that the topology of the random network was the same as that of our predicted interactome (magenta points). Note that not all gene pairs mapped to probe sets on the Affymetrix ATH1 Gene Chip. The gene expression set used is an compendium of the four smaller AtGenExpress compendia displayed in the Expression Browser tool at http://bbc.botany.utoronto.ca. These include data sets generated by Schmid et al. (2005), Kilian et al. (2007), and other members of the AtGenExpress consortium. Genes with a high PCC are considered to be coexpressed. The interolog distribution is shown to contain many coexpressed pairs. B, The interolog CV was plotted against the correlation coefficient for each pair, demonstrating that a high confidence score (score ≥ 11) may suggest that the interolog pair is coexpressed. Significant values (P < 0.05) lie above and below the dotted lines. [See online article for color version of this figure.]
Figure 6.

Interolog database and integration with BAR Expression Browser output. A, The top 32 interologs were displayed in the output of a query on the Schmid et al. (2005) data set as present in the BAR (Toufighi et al., 2005). The left arrow highlights the expression clustering results, indicating high degrees of coexpression, while the loops joining two AGI identifiers highlighted by the right arrow denote interolog pairs. The color of the loop indicates the interolog CV. The AGI identifiers are colored according to their biological functions: light green, transcription initiation; dark green, DNA mismatch repair; light blue, pyruvate dehydrogenase E1a and E1b subunits; dark blue, proteosomal complex components; magenta, spliceosomal components; orange, DNA replication; white, unknown. B, Clicking on the interolog loops in the above output will open an output window for an Arabidopsis Interaction Viewer query, providing more detailed information on the predicted and experimentally identified interactions present in the database.
DISCUSSION
How to Use the Predicted Interactome
A predicted interactome has been made for Arabidopsis, based on evolutionary conservation of protein interactions across species. Each interaction has been assigned a CV based on the number of organisms and experiments it is supported by. Assignments for subcellular localization and coexpression can be used as further indicators of confidence in a predicted interaction. Interacting proteins tend to be colocalized to the same compartment, or to adjacent compartments such as Golgi vacuole and Golgi ER. As many of these proteins are part of the endomembrane trafficking complexes such as SNARE-SNAP-syntaxin (Fig. 3), these intercompartment interactions could represent docking or trafficking interactions. Interacting proteins also tend to be highly coexpressed across tissues and organs and in response to hormone and stress treatments.
To make use of this resource, an interactome network file is built using a network assembly and visualization tool. Alternately, users may query the Arabidopsis Interaction Viewer at http://bbc.botany.utoronto.ca/interactions/. The raw data to build the network has been provided (Supplemental Table S1). Both Osprey 1.2 (http://biodata.mshri.on.ca; Breitkreutz et al., 2003) and Cytoscape 2.4.1 (http://www.cytoscape.org; Shannon et al., 2003) are publicly available tools well suited to browse the Arabidopsis predicted interactome. Network files have been prebuilt for both of these tools and have been included as Supplemental Data (api.osp and api.cys), and are also available upon request. These can simply be loaded into the appropriate tool and one can begin browsing or searching the interactome right away. Cytoscape, used in this work, offers more visualization options although Osprey is a little easier to navigate for the uninitiated. In addition to the Cytoscape network file, other files containing layout, node, and edge attributes are available. In our visualization, nodes are given both The Arabidopsis Information Resource (TAIR) annotation (AtXgNNNNN) and common gene names as interchangeable attributes, the size of the node is related to the number of interactions, while the color of the node is its SUBA assignment for subcellular localization. Edges are colored according to coexpression correlation and the line thickness correlates to our internal CV. It is also possible to map gene ontology categories and other attributes by making a simple text file out of Supplemental Table S1 and including a column of whatever attribute is to be assigned to each interaction or each protein. While the interactome in its entirety initially appears as a giant hairy ball, it is possible to zoom into each section, or more profitably to use filters to look at specific genes and all surrounding interactions (i.e. by using depth filter in Osprey or filter dialog in Cytoscape). This allows researchers the ability to build and extend their own pathway or protein complex using this Arabidopsis predicted interactome. For custom queries using the Arabidopsis Interactions Viewer, the user may also download a file for use in Cytoscape, or explore the network within a SVG-plugin-enabled Web browser.
Universality and Ancestry of Some Protein Interaction Pathways
Predicting the Arabidopsis interactome relies on some universality of protein function among eukaryotes, especially as these predictions are based on interactions of nonplant species. Using the INPARANOID ortholog prediction algorithm, we identified 3,206 genes for which orthologs were found in all five eukaryotic species (Arabidopsis, yeast, nematode worm, fruitfly, and humans), which is a significant fraction (approximately 10%) of the genome, and another 7,570 Arabidopsis genes had an ortholog in at least one other species. We also identified 292 interactions that were present in at least two different species and orthologous genes found in Arabidopsis (Supplemental Table S1), which is large considering the incomplete nature of interactome datasets and small pool of overlapping orthologs. We have estimated that conservatively there are 100,000 to 200,000 interactions if we assume that the number of noninteracting proteins is proportionately similar to yeast, and that conserved interologs discovered here are significantly more interactive than nonconserved genes.
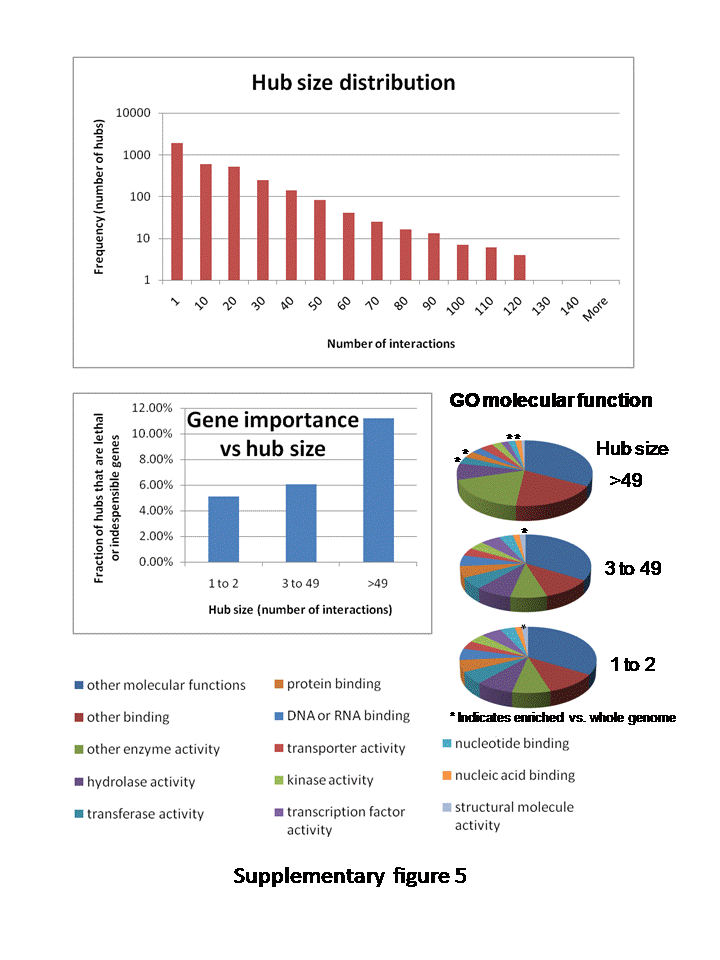
Interestingly, the most highly conserved interactions tend to be those between two highly connected hubs. Is there an increased likelihood that the duplication or loss of that hub is lethal or deleterious when one protein interacts with many others compared to loss or duplication of a single protein interaction? If so, highly connected hubs may thus be under more evolutionary pressure to remain conserved, while smaller hubs are free to duplicate and diverge. Hubs with 50 or more interacting partners tended to be enriched in nuclear localization, DNA, and RNA metabolism, although not transcription factors themselves, and 11% of all large hubs are lethal genes or otherwise indispensable (Tzafrir et al., 2004), while this applies to only 6% of intermediate hubs and 5% of pipes and loose ends (Supplemental Fig. S5). Core members of a protein complex are highly coexpressed and often lethal if mutated (Dezso et al., 2003). These core proteins are surrounded by a cloud of transiently docking peripheral proteins that are less likely to be lethal if individually deleted. Highly connected hubs may thus represent these conserved cores of signaling complexes.
Another surprise was the chloroplast localization of many of the interologs. These are all nuclear encoded, but chloroplast localized proteins. While photosynthesis is absent from the species used to build the orthologs, the complex phylogenetic origin of proteins found in the chloroplasts of higher plants (Leister, 2003) means that many proteins found in the chloroplast today have close orthologs in species from other evolutionary lineages. Not only were orthologs to chloroplast genes found, but they were enriched (P < 0.1; Fig. 4) for interactions with orthologs to genes that were also chloroplast localized in Arabidopsis. This suggests that interacting pathways have been moved to the chloroplast from other cellular compartments at some time postendosymbiosis. Closer inspection of this list of 55 chloroplast located interolog pairs reveals it includes components of a number of well characterized metabolic pathways that are known from the literature to have chloroplast located versions in plants, but to be mitochondrial or cytosolic in yeast and animals (Supplemental Table S1). This includes enzymes of biosynthetic pathways for purines, pyrimidines, heme, and riboflavin, but also enzymes in or associated with glycolysis and a range of proteins involved in posttranscriptional and translational machinery. These proteins have not simply been cannibalized to make new plant-specific pathways, but the original function of these pathways and complexes is likely to be preserved, and thus the predicted interactions of chloroplast protein orthologs are probably still functionally related.
MATERIALS AND METHODS
Interolog Construction
Ortholog data for generating interologs were obtained from INPARANOID (http://inparanoid.cgb.ki.se/) and ENSEMBL (http://www.ensembl.org/index.html) through BIOMART (http://www.biomart.org/), and loaded into a MySQL database. To obtain as many functional orthologs as possible in the dataset, especially for large gene families, we chose to include only individual ortholog pairs from each family. This reduced the size of the potential interactions, many of which are probably real, but many more, especially those involving divergent in paralogs, are likely false positives. More recent methods for predicting functional orthologs using evolutionary conservation of partners (Bandyopadhyay et al., 2006) or coevolution were not used due to the high computational demands in applying this method for entire genomes. Interactome databases were obtained from BIND (08-11-2005 release), MIPS (November 2005 release), BIOGRID (version 20), and DIP (November 2005 release). These interactome datasets can be found at http://www.unleashedinformatics.com, http://mips.gsf.de; http://www.thebiogrid.org, and http://dip.doe-mbi.ucla.edu, respectively. Interactome and ortholog data included many different types of gene identifiers, so cross-identification tables were constructed from BIOMART, TAIR (www.arabidopsis.org), and from a kindly donation of data from Tanya Berardini, these tables are available upon request. Orthologs were mapped onto interactome data, and where both interacting proteins in a reference species had orthologs in Arabidopsis (Arabidopsis thaliana), an interolog prediction was recorded. The raw table of interologs is found in Supplemental Table S1 as an Excel spreadsheet. This includes the Arabidopsis proteins, the reference species, and the reference interactome for 37,235 predicted interactions, and has many duplicates where the same interaction was predicted from different species or interactome datasets. A separate sheet was generated containing 19,979 unique interacting protein pairs (entered in both orientations), along with the CV, the Pearson correlation coefficient of coexpression (PCC), and the predicted subcellular localization. A third sheet was added that includes the identification of each Arabidopsis protein in the dataset and the number of unique interacting partners that protein is predicted to have. The average number of interactions per interacting protein we predict by interologs in Arabidopsis is 10.9, while in the yeast (Saccharomyces cerevisiae) interactome the average number of interactions per interacting protein is 22.3, and in Drosophila 9.02. Due to the incomplete nature of interolog mapping, we expect some bias toward intermediate and large hub detection. For example, if we suppose interologs pick up 10% to 50% of all interactions among conserved proteins, a large hub in humans has 100 interactions and in Arabidopsis has 100 interactions, through mapping interologs we might detect 10 or 50 of those interactions and declare this Arabidopsis protein to be an intermediate or major hub. With the same detection rate, a human protein with just one interaction has a 10% to 50% chance of appearing in Arabidopsis and a 50% to 90% chance of not being included, thus proteins with only one or two interacting partners will likely be undercounted by the interolog method.
Calculation of the CV for Experimental Support
To estimate the strength of experimental support for each predicted interaction, we have calculated a CV. It is more convincing if different experimental methods predict the same interaction and the interaction is likely more conserved if it appears in multiple species. Our CV began with the total number of datasets the interactome appears in (N), and it was given a bonus multiplier if different experimental methods predicted the same interaction (E) and another multiplier if it was found in multiple eukaryote species (S). The formulation CV = N × E × S was thus our best attempt at determining the level of experimental support. The distribution of interactions by confidence was calculated for the CV and each component variable (presented in Supplemental Table S1; Fig. S4). Interactions can be thus divided low confidence (CV = 1) that will contain some false positives, especially as some high-throughput techniques such as yeast two hybrid have likely generated many artifactual interactions especially in early yeast interactomes (Cornell et al., 2004). False-positive interacting pairs are unlikely to be repeated, discovered using different experimental techniques, or found in other species, and so are generally limited this low confidence dataset. Some experimental techniques such as phenotypic enhancement or suppression offer only indirect evidence for a physical interaction between proteins, and are as likely to represent genetic interactions. A total of 3,967 interactions are based on these indirect kinds of evidence, while 16,012 interactions have direct evidence for a physical interaction (e.g. affinity capture, yeast two hybrid, etc). A total of 1,268 interactions have both direct and indirect evidence. Experimental evidence type is included in Supplemental Table S1, on the sources of interactions worksheet, and each predicted interaction is flagged with direct/indirect/both on the Arabidopsis interactome worksheet to allow researchers to exclude or include each subgroup.
Comparison of Interologs to Experimentally Derived Arabidopsis Interactions
A gold standard of experimentally generated interactions was established from 711 interactions of 356 Arabidopsis proteins mined from the literature by BIND (Bader et al., 2003). Of these proteins, 95 were found in the predicted interactome. Of the common set of 95 proteins, 85 interactions were identified experimentally (BIND) and 70 were predicted by our method. There were 30 interactions common to both sets, whereas 1.30 interactions expected to match by chance given the total possible number of 4,560 interactions between 95 proteins (=95 × 94/2 for unique heterointeractions + 95 self interactions) and extracting a random subset of 85 and 70 interactions (85/4,560 × 70 = 1.30). Using a χ2 test with 3 degrees of freedom, we calculated the observed number of interactions (30 overlap, 55 BIND only, 40 our method only, and 4,435 noninteractors) and compared to the expected distributions (1.3 overlap, 83.7 BIND only, 68.7 our method only, 4,406.3 noninteractors). This gave a P value of 10−142, meaning that the observed distribution is very unlikely to occur by chance.
Subcellular Localization
Protein localization data was taken from SUBA (http://www.suba.bcs.uwa.edu.au; Heazlewood et al., 2005). SUBA contains direct or indirect experimental data on the localization of 6,743 Arabidopsis proteins from five sources of information: GFP fusion experiments, mass spectrometry studies, AmiGO annotation, Swiss-Prot annotation, and localization based on TAIR gene descriptions. From these data proteins are localized to the following 11 distinct cellular compartments: cell plate, chloroplast, cytoskeleton, cytosol, ER, extracellular space, Golgi, mitochondria, nucleus, peroxisome, plasma membrane, and vacuole.
Occasionally, two or more of the information sources in SUBA annotate a protein as located in different compartments. In this study, a winner-takes-all approach was adopted in which a protein was designated in a compartment if a plurality of the five information sources above annotated it as belonging to that compartment. For example, if both the GFP and AmiGO fields in SUBA contain a localization of mitchondria but the Swiss-Prot field contains that of cytosol, the protein is designated as mitochondrial. Proteins for which no subcellular compartment was designated by a plurality of information sources were ignored. Note that for a large majority of proteins, SUBA contained data from only one source or that multiple sources of data agreed on localization. The winner-takes-all approach was used to resolve conflicting subcellular localizations for 358 proteins. The processing of the SUBA localization data assigned a single subcellular localization to 5,832 Arabidopsis proteins.
Enrichment Analysis
Statistical tests for the observed numbers of interologs with respect to the subcellular localization of interacting proteins were computed following the methods of Gandhi et al. (2006). The P value for the observed number of interologs nab, where one protein is in subcellular location a and the other in b, is calculated using a Poisson distribution:
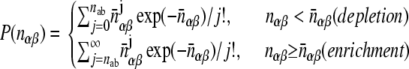 |
Here 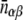 is the expected number of interologs with one protein in location α and the other in location β for the ensemble of random protein networks that maintain the following properties as the observed network: the annotation of proteins in their subcellular compartments, the degree (k) of each protein (the number of proteins that interact with it), and the total number of interacting pairs (E).
is the expected number of interologs with one protein in location α and the other in location β for the ensemble of random protein networks that maintain the following properties as the observed network: the annotation of proteins in their subcellular compartments, the degree (k) of each protein (the number of proteins that interact with it), and the total number of interacting pairs (E). 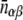 is given by
is given by
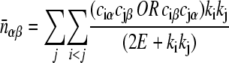 |
where the indices i and j run over all interacting proteins and the indices ciα equal 1 or 0 if protein i is in compartment α or not, respectively. The term OR indicates that if both proteins are in the same compartment the term within the parentheses is 1. Self-interacting proteins in the Arabidopsis interactome were ignored in this analysis, to avoid spurious enrichment.
The P values are finally subject to a multiple-testing correction P (multi) = 1 − (1 − P)m where for enrichment m equals the number of αβ pairs with at least one observed interolog and for depletion m equals the number of αβ pairs possible in the ensemble of random networks. The reader is referred to Supplemental Materials and Methods S1 or Gandhi et al. (2006) for more details on the analysis.
Coexpression Analysis
We examined Arabidopsis microarray data on the Affymetrix ATH1 chip from 945 AtGenExpress data sets (Schmid et al., 2005). Coexpression between pairs was determined using the Pearson correlation coefficient (r).
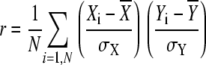 |
where N = number of expression samples, X = expression level for gene X in ith sample, and Y = expression level for gene Y in ith sample −1 ≤ r ≤ 1.
High positive r values indicate a correlation of expression patterns, while low negative r values indicate anticorrelation. To generate an accurate representation of gene expression, we combined four AtGenExpress compendia (hormone, pathogen, stress, and tissue; see Supplemental Data for NASCArrays sample identifiers for the data sets in each compendium) into one large multi-data-set compendium containing 945 data sets in total. The Arabidopsis Genome Initiative (AGI) number to ATH1 probe set lookup was performed with a table from TAIR called affy25k_array_elements-2006-01-06.txt. r values were binned into 0.1 r unit bins to generate a distribution. To determine whether the interolog coexpression distribution was enriched in pairs that exhibited high correlation coefficients, we performed a two-sample Kolmogorov-Smirnov test on the interolog and random distributions. Random distributions were generated by randomly generating 20,000 protein pairs from Arabidopsis from within the collection of interacting proteins as predicted in this article, from any of the AGI IDs on the ATH1 GeneChip, or from any AGI ID on the ATH1 GeneChip such that the topology of this random set matched that of our predicted interactome in terms of hubbiness. For all of these random sets r values were computed and subsequently binned into 0.1 r unit bins. These statistics were generated using the R programming language Statistics package, which includes a built-in function ks.test (the R Project for Statistical Computing: The R Reference Index—Kolmogorov-Smirnov Tests; http://www.r-project.org/). This function computed a P value as illustrated in Marsaglia et al. (2003).
In attempt to find a Gold Standard to compare the interolog pairs' coexpression, we looked at the coexpression between pairs of confirmed Arabidopsis PPIs from the BIND database (Bader et al., 2003). However, these findings were not conclusive due to the smaller quantity of confirmed PPIs as compared to the interolog dataset. As well, it appears as though not all confirmed PPIs in Arabidopsis are strongly coexpressed, and, therefore, this comparison was omitted. Again, it is not an absolute requirement that interacting proteins exhibit coexpression, as one member may be constitutively expressed while another is induced under a specific condition. Indeed, we noticed that if we examined the r values for interolog pairs for the individual AtGenExpress compendia (e.g. the Schmid et al., 2005 developmental map compendium versus the Kilian et al., 2007 abiotic stress compendium) it is quite often the case that the pairs are much less correlated in their expression patterns in one compendium than in another.
Supplemental Data
The following materials are available in the online version of this article.
Supplemental Figure S1. RNA splicing network expanded by predicted interactions.
Supplemental Figure S2. RHO-RAB network expanded by predicted interactions.
Supplemental Figure S3. Homeobox network expanded by predicted interactions.
Supplemental Figure S4. Distribution and construction of the CV.
Supplemental Figure S5. Analysis of hub size.
Supplemental Table S1. The Arabidopsis predicted interactome.
Supplemental Table S2. Sources of microarray expression data.
Supplemental Table S3. Resolution of conflicting localizations in SUBA.
Supplemental Materials and Methods S1. Enrichment analysis.
Supplemental File S1. Interactome network file in Cytoscape format.
Supplemental File S2. Interactome network file in Osprey format.
Supplementary Material
Acknowledgments
The authors thank Joel Bader for helpful discussions on the statistical treatment.
This work was supported by funds from the Australian Research Council Centre of Excellence Program (to A.H.M. and N.O.) and A.H.M. is funded as an Australian Research Council Australian Professorial Fellow. N.J.P. and R.A. are supported by grants from the Natural Sciences and Engineering Research Council of Canada. The Botany Array Resource was funded by a grant from Genome Canada/Ontario Genome Institute.
The author responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described in the Instructions for Authors (www.plantphysiol.org) is: Matt Geisler (mgeisler@plant.siu.edu).
Some figures in this article are displayed in color online but in black and white in the print edition.
The online version of this article contains Web-only data.
Open Access articles can be viewed online without a subscription.
References
- Bader GD, Betel D, Hogue CWV (2003) BIND: the biomolecular interaction network database. Nucleic Acids Res 31 248–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Donaldson I, Wolting C, Ouellette BFF, Pawson T, Hogue CWV (2001) BIND—the biomolecular interaction network database. Nucleic Acids Res 29 242–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhyay S, Sharan R, Ideker T (2006) Systematic identification of functional orthologs based on protein network comparison. Genome Res 16 428–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batada NN, Reguly T, Breitkreutz A, Boucher L, Breitkreutz BJ, Hurst LD, Tyers M (2006) Stratus not altocumulus: a new view of the yeast protein interaction network. PLoS Biol 4 1720–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj N, Lu H (2005) Correlation between gene expression profiles and protein-protein interactions within and across genomes. Bioinformatics 21 2730–2738 [DOI] [PubMed] [Google Scholar]
- Birney E, Andrews TD, Bevan P, Caccamo M, Chen Y, Clarke L, Coates G, Cuff J, Curwen V, Cutts T, et al (2004) An overview of ensembl. Genome Res 14 925–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitkreutz BJ, Stark C, Tyers M (2003) Osprey: a network visualization system. Genome Biol 4 R22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KB, Jurisica I (2005) Online predicted human interaction database. Bioinformatics 21 2076–2082 [DOI] [PubMed] [Google Scholar]
- Carter CJ, Bednarek SY, Raikhel NV (2004) Membrane trafficking in plants: new discoveries and approaches. Curr Opin Plant Biol 7 701–707 [DOI] [PubMed] [Google Scholar]
- Cornell M, Paton N, Oliver S (2004) A critical and integrated view of the yeast interactome. Comp Funct Genomics 5 382–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EC, Philips MR (2006) Spatial segregation of Ras signaling—new evidence from fission yeast. Cell Cycle 5 1936–1939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curwen V, Eyras E, Andrews TD, Clarke L, Mongin E, Searle SMJ, Clamp M (2004) The ensembl automatic gene annotation system. Genome Res 14 942–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezso Z, Oltvai ZN, Barabasi AL (2003) Bioinformatics analysis of experimentally determined protein complexes in the yeast Saccharomyces cerevisiae. Genome Res 13 2450–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley TPJ, Svenja Hester S, Shadforth IP, Runions J, Weimar T, Hanton SL, Griffin JL, Bessant C, Brandizzi F, Hawes C, et al (2006) Mapping the Arabidopsis organelle proteome. Proc Natl Acad Sci USA 103 6518–6523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Wall DP, Eisen MB (2004) Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci USA 101 9033–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi TK, Zhong J, Mathivanan S, Karthick L, Chandrika KN, Mohan SS, Sharma S, Pinkert S, Nagaraju S, Periaswamy B, et al (2006) Analysis of the human protein interactome and comparison with yeast, worm and fly interaction datasets. Nat Genet 38 285–293 [DOI] [PubMed] [Google Scholar]
- Ge H, Church GM, Vidal M (2001) Correlation between transcriptome and interactome mapping data from Sacchromyces cerevisiae. Nat Genet 29 482–486 [DOI] [PubMed] [Google Scholar]
- Giot L, Bader JS, Brouwer C, Chaudhuri A, Kuang B, Li Y, Hao YL, Ooi CE, Godwin B, Vitols E, et al (2003) A protein interaction map of Drosophila melanogaster. Science 302 1727–1736 [DOI] [PubMed] [Google Scholar]
- Hanisch D, Zien A, Zimmer R, Lengauder T (2002) Co-clustering of biological networks and gene expression data. Bioinformatics 18 S145–S154 [DOI] [PubMed] [Google Scholar]
- Heazlewood JL, Tonti-Filippini J, Verboom RE, Millar AH (2005) Combining experimental and predicted datasets for determination of the subcellular location of proteins in Arabidopsis. Plant Physiol 139 598–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heazlewood JL, Verboom RE, Tonti-Filippini J, Small I, Millar AH (2007) SUBA: the Arabidopsis subcellular database. Nucleic Acids Res 35 D213–D218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Weinl S, Batistic O, D'Angelo C, Bornberg-Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and exemplary data analysis of UV-B light, drought and cold stress responses. Plant J 50 347–363 [DOI] [PubMed] [Google Scholar]
- Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, et al (2006) Global landscape of protein complexes in the yeast Saccaromyces cerevisiae. Nature 440 637–643 [DOI] [PubMed] [Google Scholar]
- Lam BCH, Sage TL, Bianchi F, Blumwald E (2001) Role of SH3 domain-containing proteins in clathrin-mediated vesicle trafficking in Arabidopsis. Plant Cell 13 2499–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Jin JB, Song JH, Min MK, Park DS, Kim YW, Hwang IH (2002) The intermolecular interaction between the PH domain and the C-terminal domain of Arabidopsis dynamin-like 6 determines lipid binding specificity. J Biol Chem 277 31842–31849 [DOI] [PubMed] [Google Scholar]
- Lehner B, Fraser AG (2004) A first-draft human protein-interaction map. Genome Biol 5 R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leister D (2003) Chloroplast research in the genomic age. Trends Genet 19 47–56 [DOI] [PubMed] [Google Scholar]
- Li S, Armstrong CM, Bertin N, Ge H, Milstein S, Boxem M, Vidalain P-O, Han JJ, Chesneau A, Hao T, et al (2004) A map of the interactome network of the metazoan C. elegans. Science 303 540–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsaglia G, Tsang WW, Wang J (2003) Evaluating Kolmogorov's distribution. J Stat Softw 8 1–4 [Google Scholar]
- Miller JP, Lo RS, Ben-Hur A, Desmarais C, Stagljar I, Noble WS, Fields S (2005) Large-scale identification of yeast integral membrane protein interactions. Proc Natl Acad Sci USA 102 12123–12128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien KP, Remm M, Sonnhammer ELL (2005) Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33 D476–D480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm C, Sonnhammer E (2001) Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol 314 1041–1052 [DOI] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N, et al (2005) Towards a proteome-scale map of the human protein-protein interaction network. Nature 437 1173–1178 [DOI] [PubMed] [Google Scholar]
- Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU (2005) A gene expression map of Arabidopsis thaliana development. Nat Genet 37 501–506 [DOI] [PubMed] [Google Scholar]
- Shannon P, Markeil A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13 2498–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DZ, Ade J, Frye CA, Innes RW (2005) Regulation of plant defense responses in Arabidopsis by EDR2, a PH and START domain-containing protein. Plant J 44 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toufighi K, Brady SM, Austin R, Ly E, Provart NJ (2005) The botany array resource: e-northerns, expression angling, and promoter analyses. Plant J 43 153–163 [DOI] [PubMed] [Google Scholar]
- Tzafrir I, Pena-Muralla R, Dickerman A, Berg M, Rogers R, Hutchens S, Sweeney TC, McElver J, Aux G, Patton D, et al (2004) Identification of genes required for embryo development in Arabidopsis. Plant Physiol 135 1206–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, et al (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403 623–627 [DOI] [PubMed] [Google Scholar]
- Uhrig JF (2006) Protein interaction networks in plants. Planta 224 771–778 [DOI] [PubMed] [Google Scholar]
- Williams EJB, Bowles DJ (2006) Coexpression of neighboring genes in the genome of Arabidopsis thaliana. Genome Res 14 1060–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Ma MH, Brown KR, Geisler M, Li L, Tzeng E, Jia CYH, Jurisica I, Li S (2007) Systematic identification of SH3 domain-mediated human protein-protein interactions by peptide array target screening. Proteomics 7 1775–1785 [DOI] [PubMed] [Google Scholar]
- Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JJ, Bertin N, Chung S, Vidal M, Gerstein M (2004) Annotation transfer between genomes: protein–protein interologs and protein-DNA regulogs. Genome Res 14 1107–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}