

Abstract
Diacylglycerol kinases (DGKs) inhibit diacylglycerol (DAG) signaling by phosphorylating DAG. DGK-1, the Caenorhabditis elegans ortholog of human neuronal DGKθ, inhibits neurotransmission to control behavior. DGK-1, like DGKθ, has three cysteine-rich domains (CRDs), a pleckstrin homology domain, and a kinase domain. To identify DGK domains and amino acid residues critical for terminating DAG signaling in vivo, we analyzed 20 dgk-1 mutants defective in DGK-1-controlled behaviors. We found by sequencing that the mutations included nine amino acid substitutions and seven premature stop codons that impair the physiological functions of DGK-1. All nine amino acid substitutions are in the second CRD, the third CRD, or the kinase domain. Thus, these domains are important for the termination of DAG signaling by DGK-1 in vivo. Seven of the substituted amino acid residues are present in all human DGKs and likely define key residues required for the function of all DGKs. An ATP-binding site mutation expected to inactivate the kinase domain retained very little physiological function, but we found two stop codon mutants predicted to truncate DGK-1 before its kinase domain that retained significantly more function. We detected novel splice forms of dgk-1 that can reconcile this apparent conflict, as they skip exons containing the stop codons to produce DGK-1 isoforms that contain the kinase domain. Two of these isoforms lack an intact pleckstrin homology domain and yet appear to have significant function. Additional novel isoform(s) account for all of the DGK-1 function necessary for one behavior, dopamine response.
Diacylglycerol kinases (DGKs)1 phosphorylate diacylglycerol (DAG), thus attenuating signaling by this important second messenger (1, 2). DAG activates protein kinase C to control numerous cellular processes mediated by neurotransmitters, growth factors, and hormones (3). DAG also activates the synaptic vesicle priming protein UNC-13 (4, 5) to control neurotransmission and certain transient receptor potential cation channels (6). In humans, nine DGK isozymes have been identified (DGKα, β, γ, δ, η, ε, ζ, ι, and θ), but their physiological functions remain largely unknown (1, 2).
C. elegans DGK-1 provides a genetically tractable model for elucidating the physiological functions of diacylglycerol kinases. DGK-1 is expressed in neurons and is 38% identical to the human brain enzyme DGKθ (7). Genetic analysis of DGK-1 has shown that it depletes DAG generated by Gαo signaling in response to the neurotransmitters dopamine and serotonin (7, 8). Because the synaptic vesicle priming protein UNC-13 requires DAG to promote neurotransmission, this depletion of DAG by DGK-1 results in reduced neurotransmission (7). Loss of DGK-1 leads to increased neurotransmission and, thus, strong behavioral defects, including defects in dopamine-controlled locomotion behavior and defects in serotonin-controlled egg-laying behavior (7, 8). Similar behavioral defects are also caused by loss of C. elegans Gαo, a neural G protein, due to an increase of UNC-13 at synaptic release sites (7). Furthermore, the loss of DGK-1 suppresses the behavioral defects caused by the overexpression of constitutively active Gαo (9). These genetic data have led to the hypothesis that Gαo signaling may activate DGK-1 to deplete DAG levels.
All human DGKs have a kinase domain and two or three cysteine-rich domains (CRDs) (1, 2). Most DGK isozymes contain additional distinct conserved domains. The diversity of DGKs is further increased by the fact that in six DGK isozymes alternative splicing has been shown to produce isoforms with altered domain structures (10–15). DGK-1, like its human ortholog DGKθ, has three CRDs, a pleckstrin homology (PH) domain, and a kinase domain (7, 16).
The functions of the various conserved domains in DGKs remain poorly understood. Their roles have been examined by transgenically expressing DGKs in yeast, COS7, or other cells and assessing the effects of mutations on enzymatic activity and subcellular localization (17–20). The CRDs are similar to the DAG-binding domains of protein kinase C, suggesting that the CRDs might bind DAG and present it for phosphorylation (21). Consistent with this hypothesis, mutating CRDs results in the dramatic loss of in vitro kinase activity (17–20). However, it appears that not all CRDs are able to bind DAG (22), and the CRDs of some DGK isozymes are not absolutely required for in vitro kinase activity (19, 23). Another proposed function of the CRDs is based on the observation that certain DGK isozymes translocate to plasma membranes to act on DAG generated in response to extracellular signals, and in DGKζ the CRDs are required for such translocation (18). PH domains are, in general, involved in protein-protein or lipid-protein interactions (24). The roles of PH domains in DGKs remain unclear, though the DGKδ PH domain has been shown to bind phospholipids (25) and to be required for translocation to the plasma membrane (13). The kinase (“catalytic”) domain has a weak similarity to phosphofructokinases (26), and for two isozymes the expression of this domain alone was sufficient to produce diacylglycerol kinase activity (19, 23). However, the levels of activity produced were lower than those of the full-length enzymes, suggesting that other domains also contribute to catalysis.
We carried out a structure/function analysis of C. elegans DGK-1 in its physiological setting. By analyzing a panel of dgk-1 mutant strains generated in genetic screens for animals defective in DGK-1 controlled behaviors, we found that the second CRD, the third CRD, and the kinase domain of DGK-1 are important for attenuating DAG signaling in vivo. Seven conserved amino acid residues in these domains are critical for DGK-1 function and likely define key residues required for the function of all DGKs in vivo. Our analysis also led us to detect novel DGK-1 isoforms that arise from alternative splicing and to show that these isoforms have significant physiological functions.
EXPERIMENTAL PROCEDURES
Construction of Plasmids and Recombinant Protein Purification
The dgk-1a cDNA from pKP137 (7) was subcloned into pFastBacHTb (Invitrogen) to generate a baculovirus expressing N-terminally His-tagged DGK-1 (His-DGK-1), which was purified from baculovirus-infected High Five (Invitrogen) insect cell extracts using nickel-nitrilotri-acetic acid agarose affinity chromatography (Qiagen). DNA encoding amino acids 633–950 of DGK-1a was cloned into pET19b (Novagen) to express an N-terminally His-tagged DGK-1 kinase domain (KD) fragment, His-KD, in BL21(DE3) cells (Novagen). His-KD was purified in the presence of 8 M urea.
C. elegans Alleles, Transgenes, and Culture
C. elegans strains used for behavioral assays were cultured at 20 °C under standard conditions (27), except where otherwise noted. All 20 dgk-1 alleles were outcrossed four times to the wild-type strain (Bristol N2) before behavioral analysis. vs8, vs9, vs24, and vs55 are dgk-1 alleles obtained from a screen for mutants that lay eggs at an early developmental stage (28). vs67 and vs71 are dgk-1 alleles obtained from a screen for mutants resistant to paralysis by dopamine (8). sy424, sy425, sy426, sy428, sy429, sy435, sy436, sy442, sy453, sy454, sy455, sy456, and sy512 are dgk-1 alleles obtained from a screen for suppressors of the locomotion defect in animals expressing constitutively active GOA-1(Q205L) from a transgene (9, 29). These suppressor alleles were separated from the transgene during the outcrosses. The gonad of dgk-1(nu62) null mutant animals was injected with a mix of 10 ng/μl myo-2-gfp coinjection marker (gift from A. Fire, Stanford University) and 10 ng/μl test construct pAJ4, pAJ6, or pAJ8 to generate transgenic lines.
C. elegans Protein Extracts and Fractionation
The C. elegans strains used for Western blotting and biochemical experiments were grown in liquid culture at 20 °C as mixed stage populations. Worms were purified by floatation on 30% sucrose and transferred to lysis buffer (50 mM HEPES pH 7.4, 1 mM EDTA, 150 mM NaCl, 10% glycerol, 1 μM dithiothreitol, 1 μM phenylmethylsulfonyl fluoride, 1 μg/val leupeptin, and 1 μg/ml pepstatin) and lysed by passing three times through a French press. Debris was removed by centrifugation at 2,000 rpm in an IEC clinical centrifuge. The resulting total (T) lysates were flash-frozen in liquid nitrogen and stored at −80 °C. Protein concentrations were determined by Bradford analysis (Bio-Rad). When required, total lysates were fractionated before flash-freezing into soluble and pellet fractions by centrifugation at 100,000 × g for 45 min.
Antibodies and Western Blotting
Anti-DGK-1 antibodies were affinity purified using purified His-KD as described (30). Proteins were separated by SDS-PAGE and then blotted onto nitrocellulose filters. The anti-β-tubulin antibody was E7 (developed by Michael Klymkowsky and obtained from the Developmental Studies Hybridoma Bank, University of Iowa). Western blots were developed using chemiluminescence detection reagents (Pierce) and BioMax MR film. The Western blots in Fig. 3 are representative of four blots with at least two independently prepared extracts per genotype. The mobility on SDS-PAGE of the proteins studied was 110 kDa for His-DGK-1 and DGK-1 and 55 kDa for (β-tubulin. In DGK-1 Western blots, the presence of nonspecific bands prevented detection of any possible DGK-1 products of lower molecular mass than the full-length 110-kDa protein.
Fig. 3. Effects of dgk-1 mutations on C. elegans behavior and the DGK-1 protein.
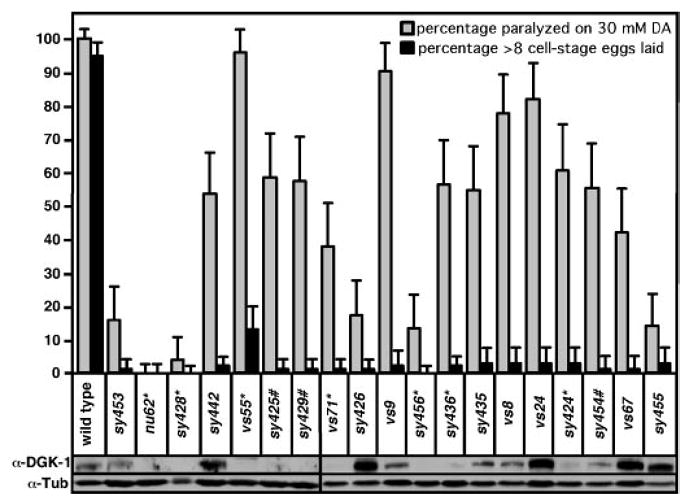
Two behaviors were measured, namely paralysis induced by the neurotransmitter dopamine (DA) and the laying of late stage (>8 cell) eggs. For dopamine-induced paralysis n = 50, and for the laying of late stage eggs n = 100. Error bars indicate 95% confidence interval. Western blots detecting full-length DGK-1 and the loading control tubulin (α-Tub) in total protein extracts of the wild-type and mutants are also shown. In the egg-laying assay, vs55 was the only mutant with significantly greater function than the null mutant nu62 (p < 0.0001). In the dopamine response assay, every mutant except sy428 showed significantly greater function than nu62 (p < 0.002).
Behavioral Assays
Proportion of eggs laid at a late developmental stage was measured as in Bany et al. (31) using animals that were aged 36 h past their fourth larval stage. We measured susceptibility to paralysis by dopamine using a modification of the method in Chase et al. (8) by placing animals on agar plates containing 30 mM dopamine and determining the fraction of animals paralyzed (unable to move in a 1-min interval). The error bars on all measures of behavior denote 95% confidence intervals for a single proportion calculated using Wilson’s estimates, and p values for comparison of two proportions were calculated using the proportion of pooled values (32). We analyzed five independent transgenic lines for each test construct to test susceptibility to paralysis by dopamine (number of animals assayed per line, > 40). dgk-1(sy512) was strongly defective in both behaviors tested but had no mutations in the dgk-1 coding regions and no detectable DGK-1 protein on a Western blot (data not shown).
In Vitro DGK-1 Activity Assay
The DGK-1 activity assay was adapted from that developed for its human ortholog, DGKθ (16). 125 ng of purified His-DGK-1 in 20 μl (or an equivalent volume of buffer) was added to 100 mM HEPES, pH 8, 10 mM MgCl2, 0.7 mM dithiothreitol, 2 mM n-octyl β-D-glucopyranoside (Sigma), 40 μM L-phosphatidylserine (Avanti Polar Lipids), and either 40 μM 1,2-dioleoyl-sn-glycerol (Avanti Polar Lipids) or the substrate indicated (40 μM) in Table I to a final volume of 246 μl 2 mM [γ-32P]ATP at 55 × 104 dpm/pmol in 4 μl was added to start the reaction. The reaction was stopped after 20 min at 25 °C by adding 500 μl of 0.5 M EDTA. The lipids were extracted using 2 ml of a 2:1 mixture of CHC13/CH3OH, and either the radioactive product in 100 μl of the extracted liquid was quantitated using scintillation counting or the extracted liquid was dried down to ~10 μl and separated by thin layer chromatography. The reaction product, phosphatidic acid (PA), was visualized as a radioactive spot after thin layer chromatography using BioMax MR film. For reference, 5 μg of PA (Avanti Polar Lipids) was also separated on the same thin layer chromatography plate and detected using phosphomolybdic acid (Sigma). For an assay of native DGK activity in soluble lysates of C. elegans, two independent lysates were prepared from each strain analyzed, and 100 μg of protein was assayed as described above, except that the specific activity of radiolabeled ATP was increased 4-fold, and the concentration of the diacylglycerol substrate was increased 2-fold.
Table I. Activity of DGK-1 fusion protein expressed in insect cells.
30 h after insect cell infection with a baculovirus encoding MBP or MBP-DGK-1, soluble fractions of cell lysates were assayed for DAG kinase activity with the substrates indicated. Assays were done in triplicate.
| Substrate | Substrate phosphorylation
|
|
|---|---|---|
| MBP | MBP-DGK-1 | |
| nmol/mg/min | ||
| 1,2-Dioleoyl-sn-glycerol | 0.06 ± 0.003 | 1.9 ± 0.03 |
| 1-Stearoyl-2-arachidonoyl-sn-glycerol | 0.02 ± 0.002 | 2.1 ± 0.04 |
| 1,3-Dioleoyl-sn-glycerol | 0.006 ± 0.0004 | 0.08 ± 0.002 |
| 1-Oleoyl-rac-glycerol | 0.005 ± 0.0006 | 0.01 ± 0.0003 |
Sequence Similarity
Genomic sequences spanning exons 1–14 of dgk-1a from C. elegans and Caenorhabditis briggsae were compared by NCBI-BLAST using the default settings, and regions of extended similarity (>7 bp) outside of and contiguous to known exons were noted. Amino acid sequences were compared using ClustalW and Megalign (Lasergene). The GenBank™ accession numbers of the sequences used were NM_075790 (DGK-1, REFSEQ), NM_001347 (DGKθ, REFSEQ), NM_001345 (DGKα, REFSEQ), NM_003648 (DGKδ, REFSEQ), NM_003647 (DGKε, REFSEQ), and U51477 (DGKζ).
Splice Form Analysis
Reverse transcriptase (RT) reactions were performed using a primer complementary to exon 10 of DGK-1. Single-stranded cDNA products were then used as templates in a PCR with various 5′-primers and a 3′-primer that overlapped the RT primer. No products were detected when RT-PCR reactions were attempted with 5′-primers in the region between splice site ii and putative splice site iii.
RESULTS
DGK-1 Phosphorylates DAG in Vitro and Is Predominantly a Soluble Enzyme
Because there has been little molecular characterization of DGK-1, we first examined some of the properties of this enzyme. We expressed His6 affinity-tagged recombinant DGK-1 in baculovirus-infected insect cells and used nickel affinity chromatography to obtain a substantially purified enzyme (Fig. 1A). Purified His-DGK-1 phosphorylated the diacylglycerol 1,2-dioleoyl-sn-glycerol in vitro to make PA (Fig. 1B). The specific activity of this protein was 80 nmol/mg/min (data not shown), which is ~135-fold less than that of the purified rat DGK-1 ortholog in a comparable in vitro assay (33). We also purified a maltose binding protein (MBP) affinity-tagged DGK-1 fusion protein that had a low specific activity similar to that of the His-DGK-1 protein. To test whether this relatively low specific activity may be due to C. elegans DGK-1 having a different substrate specificity than the mammalian enzyme, we examined the ability of crude cell lysates expressing MBP-DGK-1 to phosphorylate different species of diacylglycerols and monoacylglycerols (Table I). We found that DGK-1 prefers 1,2-diacyl-sn-glycerols to 1,3-diacyl-sn-glycerols and 1-monoacylglycerols, similarly to its mammalian ortholog (16). We found that the low activity of purified DGK-1 can be explained by ~95% of the protein being present as inactive aggregates (supplemental Fig. 1). When monomeric MBP-DGK-1 was separated from the aggregates by gel filtration, its specific activity was ~7,500 nmol/min/mg, comparable with that of the rat DGK-1 ortholog in a similar in vitro assay (33). Purified monomeric MBP-DGK-1 or His-DGK-1 protein isolated by gel filtration, however, aggregated over time (data not shown), precluding a detailed biochemical analysis of the recombinant protein. This instability of recombinant purified DGK-1 fusion proteins may be due to a lack of either post-translational modifications or cofactors that are present in the native DGK-1 protein in C. elegans.
Fig. 1. In vitro activity of recombinant DGK-1 and the fractionation of native DGK-1.
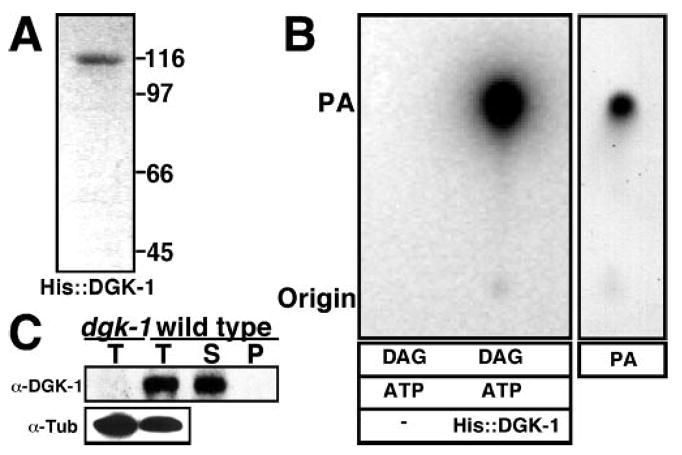
A, Coomassie-stained gel of recombinant His-tagged DGK-1 purified from insect cells. A single band is detected at ~110 kDa. B, in vitro DGK activity assay. Left, autoradiograph of a thin layer chromatogram showing PA generated upon phosphorylation of a DAG (1,2-dioleoyl-sn-glycerol) by purified recombinant His-DGK-1 with [γ-32P]ATP (ATP). His-DGK-1 (His::DGK-1) (125 ng) or no enzyme was added to the reaction as indicated. Right, chemically stained reference PA separated on the same thin layer chromatography plate. The positions of the origin and the PA spot are indicated. C, DGK-1 Western blot of fractionated C. elegans extracts. Total (T), soluble (S), and pellet (P) fractions of C. elegans extracts are shown. α-DGK-1, anti-DGK-1 Western blot. α-Tub, anti-tubulin Western used to verify similar loading of total extracts. dgk-1 denotes protein extract from dgk-1(nu62) null mutant worms. DGK-1 is a predominantly soluble protein.
To analyze native DGK-1 in C. elegans, we developed a DGK-1 antibody directed against part of the kinase domain. On Western blots, this antibody detected a single specific band (~110 kDa) in wild-type worm extracts that was absent in dgk-1 null mutant extracts (Fig. 1C). This band had a similar mobility in SDS-PAGE as purified His-DGK-1 (Fig. 1A). We pelleted membranes from C. elegans extracts by centrifugation at 100,000 × g for 45 min and found that the native DGK-1, like its mammalian ortholog DGKθ (33), remained in the soluble fraction (Fig. 1C) although one of its substrates, diacylglycerol, is expected to be present in the pellet fraction with membranes. Wild-type soluble protein extracts had a measurable albeit low level of DGK activity (0.003 ± 0.0008 nmol/mg/min). The DGK activity present in soluble protein extracts from dgk-1 null mutants (0.002 ± 0.0014 nmol/mg/min) was not significantly lower than the low DGK activity present in wild-type protein extracts. Thus DGK-1 is likely not the major soluble DGK activity in C. elegans, which encodes four other DGKs in its genome (1). In conclusion, DGK-1 is a predominantly soluble enzyme that, like DGKθ (34), likely translocates to membranes to inactivate the second messenger DAG by phosphorylation.
DGK-1 May Terminate DAG Signaling Independent of Gαo Signaling
Genetic analysis in C. elegans has led to the hypothesis that DGK-1 is activated by the Gαo G protein GOA-1 (9). We evaluated this hypothesis. We failed to detect translocation of DGK-1 to membranes in response to either expression of activated GOA-1 from a transgene (supplemental Fig. 2A, available in the on-line version of this article) or genetic mutations that alter GOA-1 signaling (supplemental Fig. 2B). We also failed to pull down DGK-1 from C. elegans extracts using purified, affinity-tagged GOA-1 whether the G protein was in its active state or not (supplemental Fig. 1C). Thus, we found no evidence that GOA-1 signaling affects DGK-1, suggesting that DGK-1 may act independently of GOA-1 to attenuate neural DAG signaling.
dgk-1 Mutants Define 19 Molecular Lesions That Compromise DGK-1 Function in Vivo
To identify DGK domains and amino acid residues critical for attenuating neural DAG signaling in vivo, we analyzed 20 dgk-1 mutants that were previously isolated based on defects in DGK-1-controlled behaviors (7–9, 28). We sequenced the dgk-1 coding regions from genomic DNA for each mutant and identified molecular lesions in 19 of the 20 mutants (Fig. 2A, and supplemental Table I). These molecular lesions included nine missense mutations, seven stop codon mutations, and three mutations that affect dgk-1 splice sites (Fig. 2B). No mutations were detected in the dgk-1 coding region of one mutant, dgk-1(sy512), which probably carries a mutation in the promoter or the regulatory regions of dgk-1 that were not sequenced. The nu62 (7) and sy428 mutations both result in stop codons very early in the DGK-1 open reading frame and therefore likely represent null alleles.
Fig. 2. Molecular lesions that compromise DGK-1 function in vivo.
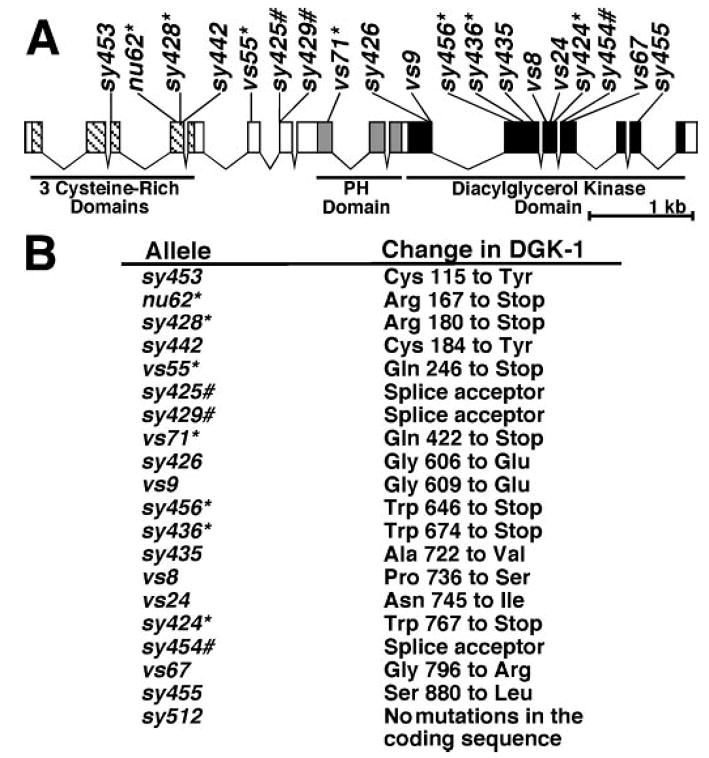
A, locations of dgk-1 mutations that compromise DGK-1 function in vivo. Exons of dgk-1a are indicated with boxes. Hatches, gray boxes, and black boxes represent sequences encoding cysteine-rich domains, the PH domain, and the kinase domain, respectively. Mutations are indicated with their allele names. Stop codons are denoted with an asterisk (*), and splicing mutations are indicated with a pound sign (#). B, the molecular lesions in the DGK-1 protein corresponding to the mutations shown in panel A. The amino acid numbers are with respect to DGK-1a.
In C. elegans, Gαq signals to generate DAG and thereby promotes egg laying and opposes the paralysis induced by exogenously applied dopamine (8, 35). DGK-1 functions to attenuate DAG signaling, and mutants lacking DGK-1 show enhanced Gαq signaling, i.e. hyperactive egg laying and resistance to dopamine-induced paralysis (7, 8). We measured the impact of molecular lesions on DGK-1 function in vivo by quantifying these behaviors in our panel of dgk-1 mutants (Fig. 3). We first measured defects in egg-laying behavior. Whereas wild-type animals retain fertilized eggs for >2 h and lay 95% of their eggs at late developmental stages (>8 cells), dgk-1(nu62) null mutants retain fertilized eggs for a shorter time and lay all of their eggs prematurely (before the 8-cell stage) (28). The percentage of late-stage eggs laid can be used as a measure of the residual DGK-1 function present in mutants. All dgk-1 mutants tested, except dgk-1(vs55), were almost completely defective for egg laying and laid <5% of their eggs at late developmental stages (Fig. 3). dgk-1(vs55) was the weakest dgk-1 mutant and retained significant DGK-1 function by this measure (p < 0.0001).
We also measured the susceptibility of the various mutants to the paralytic effect of dopamine. Whereas wild-type worms were 100% paralyzed when placed on agar medium containing 30 mM dopamine, dgk-1(nu62) null mutants were 0% paralyzed (Fig. 3). The remaining mutants showed different susceptibilities to paralysis, ranging from 96% paralyzed in dgk-1(vs55) animals to 4% paralyzed in dgk-1(sy428). Thus, dopamine-induced paralysis allows a range of levels of DGK-1 function to be detected in the mutants, possibly because lower levels of DGK-1 function are required for dopamine response than for egg laying. In principle, DGK-1 might function differently in egg laying than in dopamine-induced paralysis so that certain molecular lesions could result in defects in one behavior but not in the other. However, the mutant least defective in dopamine-induced paralysis, dgk-1(vs55), was also the least defective in egg laying. Furthermore, all missense mutants that were strongly defective in dopamine-induced paralysis (<30% paralyzed, e.g. dgk-1(sy453) and dgk-1(sy426)) were also strongly defective in egg laying (<4% of the eggs laid at late developmental stages). Hence, there was no evidence for molecular lesions that differentially affected the two behaviors.
Seven Missense Mutations Identify Conserved Amino Acid Residues Crucial for DGK-1 Function
Missense mutants can be used to identify amino acid residues important for the in vivo function of DGK-1. However, defects in some missense mutants may not reflect a role for the affected amino acid residue in enzymatic activity but may be due to misfolding of the mutant protein. Because misfolding can lead to protein degradation, we tested if there was detectable DGK-1 protein in extracts of dgk-1 mutants (Fig. 3, bottom). Whereas all stop codon mutants had little if any detectable full-length DGK-1 protein (Fig. 3, bottom), we found that all missense mutants had detectable full-length DGK-1 protein. In Fig. 3, some missense mutant extracts appear to have more DGK-1 than wild-type extracts, but this variation in the Western blot signal was not reproducible (data not shown). Thus, the missense mutations likely do not destabilize the DGK-1 protein, and they potentially define the amino acid residues of DGK-1 required for function of the folded protein.
All nine missense mutations are in the second CRD, the third CRD, or the kinase domain. To examine the conservation of the mutated amino acid residues, we performed a multiple sequence alignment of the affected domains (Fig. 4). Whereas most DGKs have only two CRDs, DGK-1 and its mammalian ortholog DGKθ have three CRDs, among which the second and third are most similar to the CRDs of other DGKs (21) and are therefore shown in our alignment.
Fig. 4. DGK-1 missense mutations and alignment with human DGKs.
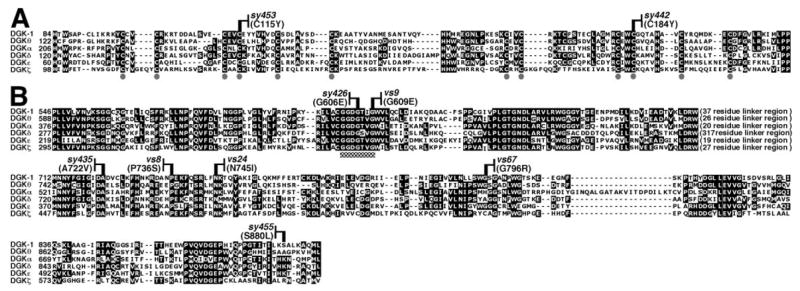
A, multiple sequence alignment of the second and third DGK-1 CRDs, with CRDs from one member of each human DGK subfamily (1). Gray circles identify the six conserved cysteines in each CRD (21). B, alignment of kinase domains. Checkered box denotes the ATP binding motif. A variable length non-conserved linker region is indicated. Allele names and molecular lesions of dgk-1 missense mutants are indicated above the alignments. Amino acids identical in three or more sequences are in black boxes. Seven of the nine DGK-1 missense mutations are in residues conserved in all human DGKs.
Two missense mutations affected the CRDs. In dgk-1(sy453) and dgk-1(sy442), the mutant DGK-1 protein made has conserved Cys residues replaced by Tyr residues in the second and third CRDs, respectively (Fig. 4A). These mutants have no detectable function in the egg-laying assay but do show reduced albeit significant function in the dopamine response assay (Fig. 3). Thus, the second and third CRDs are important for the physiological functions of DGK-1.
Seven missense mutations affected the kinase domain. We identified two mutations, sy426 and vs9, in the conserved Gly-Glu-Gly-Xaa-Xaa-Gly ATP binding motif of the DGK kinase domain that change the second and third conserved Gly of this motif to Glu, respectively (1) (Fig. 4B). In other DGKs, changes in the amino acid residue corresponding to that changed by sy426 (Fig. 4) abolished in vitro kinase activity (1, 36). We found three additional mutations (sy435, vs8, and vs24) in the kinase domain that change amino acid residues that are absolutely conserved among all DGKs (Fig. 4B). Finally, we found two mutations, vs67 and sy455, in the kinase domain that alter amino acid residues that were not conserved among all DGKs and may represent residues that are important for the function of only a subset of known DGKs. The mutation sy455 alters a Ser residue that might be phosphorylated because it is located in a possible protein kinase C phosphorylation site (37), and the human DGK-1 ortholog DGKθ can be phosphorylated in vitro by protein kinase C (1).
All of the kinase domain missense mutations severely reduced DGK-1 function in the egg-laying assay and also reduced DGK-1 function to varying extents in the dopamine response assay, indicating that the kinase domain is important for the physiological functions of DGK-1. We note that none of these missense mutations produced defects as severe as those in the dgk-1 null mutants nu62 and sy428. In addition, three stop codon mutations in the kinase domain (sy456, sy436, and sy424) also produced defects less severe than those in the null mutants (Fig. 3). The residual function present in all kinase domain mutants was only evident in the dopamine response assay, which is apparently more sensitive than the egg-laying assay. These results suggest that the kinase domain of DGK-1 may not be absolutely required for all of its physiological functions.
Two Stop Codon Mutants Predicted to Lack a Kinase Domain Retain Significant Function in Vivo
Two early stop codon mutants, vs55 and vs71, are predicted to produce truncated DGK-1 containing the CRDs but lacking the kinase domain. They retain significantly more function than the ATP-binding site mutant sy426 (Fig. 3). Indeed, vs55 mutants have greater function in the dopamine response assay than any kinase domain mutant and are actually indistinguishable from the wild-type in this assay (p > 0.999). These results raise the following question. How can a mutation that completely eliminates the kinase domain perturb function less than any mutation within the kinase domain? One possibility is that vs55 and vs71 actually do not completely eliminate the kinase domain. This could occur if there are alternative splice forms of dgk-1 that skip the exons containing the vs55 and vs71 stop codons to produce novel DGK-1 isoforms that include the kinase domain
New dgk-1 Splice Forms May Explain Residual Function in dgk-1(vs55) and dgk-1(vs71) Stop Codon Mutants
To detect the proposed novel splice forms, we used RT-PCR to amplify cDNAs from the region surrounding the two mutations. We found four different splice forms named dgk-1a, dgk-1b, dgk-1c, and dgk-1d (Fig. 5A). dgk-1a and dgk-1b have been reported earlier (7) and differ in using alternative splice donors (i and ii) from exon 5. dgk-1c and dgk-1d are novel splice forms identified in this study that use splice donors i or ii from exon 5, include exon 6, and then skip exons 7 and 8. Because vs71 lies in exon 8, these two novel splice forms are undisrupted in the dgk-1(vs71) mutant. The DGK-1c and DGK-1d isoforms do contain the kinase domain and thus may account for the significant DGK-1 function in the dgk-1(vs71) mutant (Fig. 3).
Fig. 5. Detection of alternative splice forms of dgk-1.
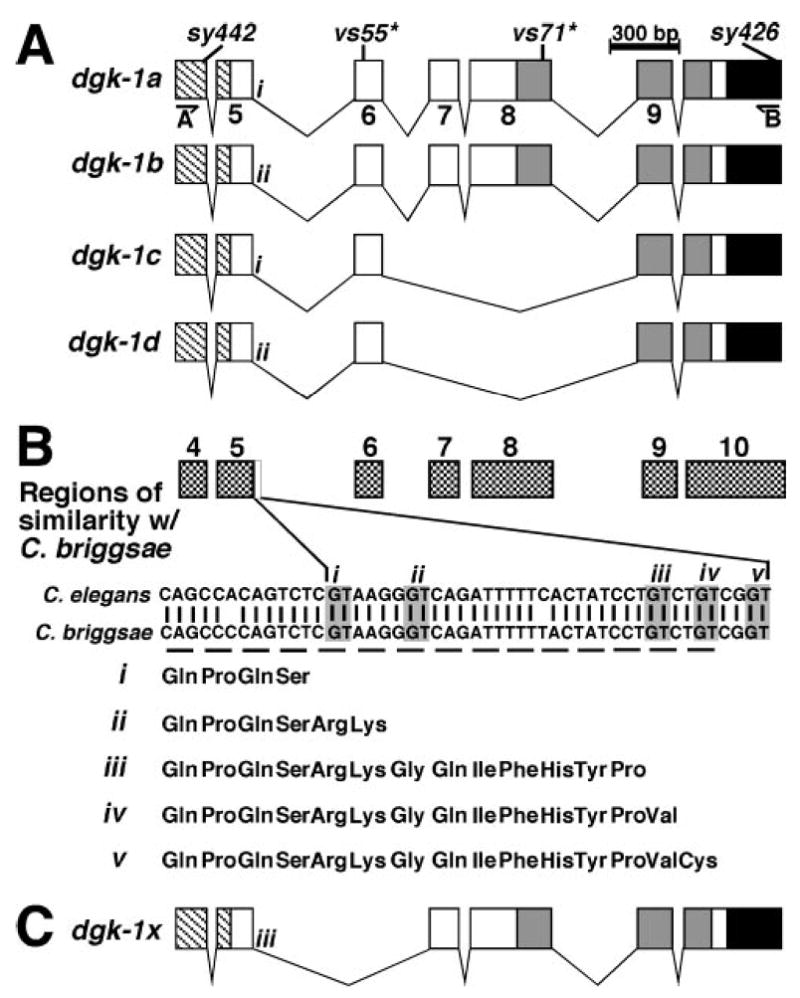
A, schematics showing splice forms of dgk-1 identified experimentally. Exons 4 through 10 of dgk-1a are schematized, with conserved domain coding sequences shaded and the mutations indicated as in Fig. 2A. Positions of the outer primers used for RT-PCR are indicated (A and B underneath harpoons). Different splice donor sites after exon 5 are indicated with either i or ii. B, comparison of C. elegans and C. briggsae sequences to detect possible novel exons of dgk-1. The regions of sequence similarity between C. elegans and C. briggsae are schematized by boxes that are aligned with the exon schematics in panel A. Checkered boxes correspond to the exons of dgk-1a as numbered. A region of similarity beyond exon 5 of dgk-1a is indicated with a white box and an expanded view showing the C. elegans and C. briggsae sequences. Conserved GT dinucleotides of potential splice donors are shown in gray boxes and are numbered i–v. The amino acids encoded by the potential exon 5 extensions are indicated below the DNA sequence. C, schematic showing a potential dgk-1 splice form that skips exon 6 (which contains vs55) and would make a DGK-1 isoform with a kinase domain.
We did not detect any DGK-1 isoforms that could similarly account for the significant function in dgk-1(vs55) (Fig. 3 and data not shown). To determine whether such isoforms might exist, we compared the genomic sequence of C. elegans dgk-1 with that of the related species, C. briggsae (38), to identify possible alternative splice forms (Fig. 5B). Coding sequences, and not introns, are highly conserved between these two species. Outside of the known dgk-1 coding sequences, we found only a single region of similarity >7 bp. This 30-bp region extended 3′ of previously known splice donors from exon 5 (Fig. 5B) and can extend the coding potential of exon 5 through the use of additional splice donors. A similar analysis was used to successfully identify an analogous exon extension allowing the subsequent detection of a novel splice form of the programmed cell death gene ced-4 (39). Three conserved splice donor sites within the potential extension of dgk-1 exon 5 (Fig. 5B, iii–v) could allow splicing to downstream exons, skipping the vs55 mutation to make functional DGK-1 proteins with kinase domains. For example, use of splice donor iii would require skipping exon 6 (which contains vs55) to maintain the dgk-1 reading frame. By splicing instead to exon 7, a protein with the kinase domain could be made (Fig. 5C).
It is possible that such a splice form is made at low levels and, hence, eluded detection. Alternatively, the dgk-1(vs55) mutant may produce a highly functional truncated protein lacking the kinase domain and containing only the three CRDs. To distinguish between these two possibilities, we evaluated the requirement for the kinase domain in the dgk-1 (vs55) mutant (Fig. 6). We constructed a transgene, pAJ4, containing the wild-type dgk-1 gene and its regulatory regions (Fig. 6A). This transgene was able to restore DGK-1 function in the dgk-1 null mutant dgk-1 (nu62) to wild-type levels (Fig. 6B). An analogous transgene, pAJ6, with the vs55 stop codon mutation (Fig. 6A) was also able to restore significant DGK-1 function to the dgk-1 null mutant (Fig. 6B). This was as expected, because the dgk-1(vs55) mutant animals showed significant DGK-1 function (Fig. 3). We reasoned that if pAJ6 confers function by producing DGK-1 isoforms containing the kinase domain but lacking the vs55 stop codon because of alternative splicing, we should be able to inactivate the kinase domain of these isoforms by adding the ATP-binding site mutation sy426 to pAJ6. Alternatively, if pAJ6 confers function by producing a truncated DGK-1 protein lacking the kinase domain, then adding the sy426 mutation to pAJ6 should have no effect. We constructed the transgene pAJ8, containing both the vs55 and sy426 mutations (Fig. 6A). pAJ8 had no detectable function (Fig. 6B). Thus the dgk-1(vs55) mutant likely makes a functional DGK-1 isoform with a kinase domain due to alternative splicing.
Fig. 6. Use of transgenes to test if a kinase domain is required for dgk-1(vs55) function.

A, schematics showing the transgenes used. Thick black line indicates the dgk-1 promoter region (~4.1 kb). Arrow-head represents the start and direction of transcription. The dgk-1 gene, the sequences encoding conserved domains of DGK-1, and the vs55 and sy426 mutations are schematized as in Fig. 2A. Thick white line indicates the 3′-untranslated region (UTR) of dgk-1. B, effect of transgene expression on susceptibility to paralysis by dopamine (DA). The genetic backgrounds and transgenes used are indicated. Error bars indicate 95% confidence interval.
DISCUSSION
In Vivo Structure/Function Analysis
The physiological functions of human DGKs are largely unknown. The readout of function that has been used in earlier structure/function studies of DGKs has involved transgenically expressing altered enzymes in cultured cells and measuring localization and/or in vitro kinase activity (17–20). In contrast, we carried out a structure/function study of the C. elegans DGK enzyme, DGK-1, by measuring defects in its physiological functions using animals in which the endogenous enzyme was altered by mutations.
In a structure/function study of human DGKθ (ortholog of DGK-1), virtually all alterations of the enzyme led to complete abolishment of in vitro activity (20). It is possible that the various altered DGKθ enzymes tested had differing levels of activity, all of which were below the detection limit of the in vitro assay used. We measured the behavioral defects in dgk-1 mutants using two DGK-1-controlled behaviors, egg laying and dopamine-induced paralysis. Most of the mutants were indistinguishable by the egg-laying assay, as they showed no DGK-1 function by this measure. However, by measuring susceptibility to the paralytic effects of dopamine, we were able to finely discriminate between the defects due to the various molecular lesions of the DGK-1 protein, possibly because this assay, unlike the egg-laying assay, allowed detection of even very low levels of DGK-1 function.
Conserved Domains and Amino Acid Residues Critical for the in Vivo Function of DGK-1 and Possibly All DGKs
The CRDs of DGKs are characterized by a series of six conserved cysteine residues (Fig. 4A, gray circles) (21). sy453 and sy442 change the fourth conserved Cys in the second and the third CRDs of DGK-1, respectively, indicating that these CRDs are important for DGK-1 function in vivo. Because we did not find any missense mutations in the first CRD of DGK-1 within our panel of mutants that had been selected for defects in DGK-1 controlled behaviors, it is possible that this domain is not essential for the behaviors measured. Consistent with this suggestion, in a structure/function study of DGKθ the only altered enzyme that retained significant in vitro activity was an N-terminal truncation that removed the first CRD (20).
The kinase domain of DGKs is distantly related to that of phosphofructokinases (26). Based on the structure of bacterial phosphofructokinase (40) and on sequence alignments, it has been suggested that DGK kinase domains consist of two conserved subdomains, a DGKc (catalytic) subdomain that binds ATP and a DGKa (accessory) subdomain that binds DAG (19).
The DGKc subdomain contains a conserved Gly-Glu-Gly-Xaa-Xaa-Gly ATP binding motif. Mutation of the second Gly in this motif has been shown to result in defective DGKs that cannot phosphorylate DAG in vitro (1, 36). Consistent with this finding, we found that a mutation that alters the corresponding residue of DGK-1 caused severe defects in both behaviors assayed (sy426 in Figs. 3 and 4). We note that the sy426 mutant retained a low but measurable amount of function in the dopamine response assay, suggesting that kinase activity may not be absolutely required for all of the DGK-1 function in vivo. Mutating the third Gly residue in the ATP binding motif has a less dramatic effect on DGK-1 function in vivo (vs9 in Figs. 3 and 4). This may be explained by the fact that the third Gly is not conserved in phosphofructokinases (19) and, thus, may not be essential for binding ATP.
Within a 34-amino acid stretch of the DGKa subdomain of DGK-1 we found three mutations that alter residues conserved among all DGKs (sy435, vs8, and vs24 in Figs. 3 and 4). Thus, these mutations identify amino acid residues in a novel motif that may be required for DAG binding by all DGKs.
Physiological Role of DGK Isoforms Generated through Alternative Splicing
We found two early stop codon mutants (vs55 and vs71) that were expected to produce a DGK-1 protein truncated before the kinase domain retained greater function than the ATP-binding site mutant sy426 (Fig. 3). The genes encoding six human DGK isozymes (DGKβ, γ, δ, ι, η, and ζ) are alternatively spliced to generate novel isoforms (10–15), and we hypothesized that the significant function retained in vs55 and vs 71 was possibly due to unidentified dgk-1 splice forms that skip these stop codon mutations through alternative splicing. We identified two such splice forms that skip the exon containing the vs71 mutation to make two novel DGK-1 isoforms lacking part of the PH domain but containing the kinase domain. These DGK-1 isoforms likely account for the function seen in the dgk-1(vs71) mutant. Because dgk-1(vs71) had no detectable function in the egg-laying assay and only affects DGK-1 isoforms containing an intact PH domain, these isoforms must play the major role in regulating egg laying. In contrast, the significant function of dgk-1(vs71) in the dopamine response assay suggests that the DGK-1 isoforms lacking an intact PH domain play a significant role in dopamine response, because these isoforms are unaffected by vs71.
The vs55 stop codon mutant was indistinguishable from the wild-type in the dopamine response assay and also retained significant function in the egg-laying assay. Using transgenes containing both the vs55 stop codon mutation and an inactivating mutation in the downstream kinase domain, we showed that the exon containing the vs55 mutation is likely skipped to generate functional DGK-1 isoform(s) containing a kinase domain. We were unable to detect such splice form(s) by RT-PCR, suggesting these forms are of low abundance. It is possible that these splice form(s) are expressed only in a small subset of cells in the animal, such as the motor neurons that respond to dopamine (8). Thus, our genetic approach has allowed us to deduce the presence of physiologically important DGK-1 isoform(s) whose very low abundance might have otherwise prevented their discovery by molecular approaches.
Supplementary Material
Acknowledgments
We thank Yvonne Hajdu-Cronin, Paul Sternberg, I. Amy Bany, and Daniel L. Chase for dgk-1 mutants that were used in this study. Some mutants used in this work were provided by the Caenorhabditis Genetics Center, which is funded by the National Institutes of Health National Center for Research Resources (NCRR). We also thank Daniel L. Chase for making the immunogen used for generating the anti-DGK-1 antibody.
Footnotes
This work was supported by a Leukemia and Lymphoma Society Scholar award (to M. R. K.) and by grants from the National Institutes of Health.
The on-line version of this article (available at http://www.jbc.org) contains further information on this subject in the form of supplemental experimental procedures, references, two figures, and a table.
The abbreviations used are: DGK, diacylglycerol kinase; CRD, cysteine-rich domain; DAG, diacylglycerol; MBP, maltose-binding protein; PA, phosphatidic acid; PH, pleckstrin homology; RT, reverse transcriptase.
References
- 1.van Blitterswijk WJ, Houssa B. Cell Signal. 2000;12:595–605. doi: 10.1016/s0898-6568(00)00113-3. [DOI] [PubMed] [Google Scholar]
- 2.Kanoh H, Yamada K, Sakane F. J Biochem. 2002;131:629–633. doi: 10.1093/oxfordjournals.jbchem.a003144. [DOI] [PubMed] [Google Scholar]
- 3.Nishizuka Y. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- 4.Maruyama IN, Brenner S. Proc Natl Acad Sci U S A. 1991;88:5729–5733. doi: 10.1073/pnas.88.13.5729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richmond JE, Davis WS, Jorgensen EM. Nat Neurosci. 1999;2:959–964. doi: 10.1038/14755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 7.Nurrish S, Ségalat L, Kaplan JM. Neuron. 1999;24:231–242. doi: 10.1016/s0896-6273(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 8.Chase DL, Pepper JS, Koelle MR. Nat Neurosci. 2004;7:1096–1103. doi: 10.1038/nn1316. [DOI] [PubMed] [Google Scholar]
- 9.Hajdu-Cronin YM, Chen WJ, Patikoglou G, Koelle MR, Sternberg PW. Genes Dev. 1999;13:1780–1793. doi: 10.1101/gad.13.14.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kai M, Sakane F, Imai S-i, Wada I, Kanoh H. J Biol Chem. 1994;269:18492–18498. [PubMed] [Google Scholar]
- 11.Ding L, Bunting M, Topham MK, McIntyre TM, Zimmerman GA, Prescott SM. Proc Natl Acad Sci U S A. 1997;94:5519–5524. doi: 10.1073/pnas.94.11.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caricasole A, Bettini E, Sala C, Roncarati R, Kobayashi N, Caldara F, Goto K, Terstappen GC. J Biol Chem. 2002;277:4790–4796. doi: 10.1074/jbc.M110249200. [DOI] [PubMed] [Google Scholar]
- 13.Sakane F, Imai S-i, Yamada K, Murakami T, Tsushima S, Kanoh H. J Biol Chem. 2002;277:43519–43526. doi: 10.1074/jbc.M206895200. [DOI] [PubMed] [Google Scholar]
- 14.Murakami T, Sakane F, Imai S-i, Houkin K, Kanoh H. J Biol Chem. 2003;278:34364–34372. doi: 10.1074/jbc.M301542200. [DOI] [PubMed] [Google Scholar]
- 15.Ito T, Hozumi Y, Sakane F, Saino-Saito S, Kanoh H, Aoyagi M, Kondo H, Goto K. J Biol Chem. 2004;279:23317–23326. doi: 10.1074/jbc.M312976200. [DOI] [PubMed] [Google Scholar]
- 16.Houssa B, Schaap D, van der Wal J, Goto K, Hisatake K, Yamakawa A, Shibata M, Takenawa T, van Blitterswijk WJ. J Biol Chem. 1997;272:10422–10428. doi: 10.1074/jbc.272.16.10422. [DOI] [PubMed] [Google Scholar]
- 17.Nagaya H, Wada I, Jia YJ, Kanoh H. Mol Biol Cell. 2002;13:302–316. doi: 10.1091/mbc.01-05-0255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Santos T, Carrasco S, Jones DR, Mérida I, Eguinoa A. J Biol Chem. 2002;277:30300–30309. doi: 10.1074/jbc.M200999200. [DOI] [PubMed] [Google Scholar]
- 19.Abe T, Lu X, Jiang Y, Boccone CE, Qian S, Vattem KM, Wek RC, Walsh JP. Biochem J. 2003;375:673–680. doi: 10.1042/BJ20031052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Los AP, van Baal J, de Widt J, Divecha N, van Blitterswijk WJ. Biochim Biophys Acta. 2004;1636:169–174. doi: 10.1016/j.bbalip.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Houssa B, van Blitterswijk WJ. Biochem J. 1998;331:677–680. doi: 10.1042/bj3310677u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shindo M, Irie K, Masuda A, Ohigashi H, Shirai Y, Miyasaka K, Saito N. J Biol Chem. 2003;278:18448–18454. doi: 10.1074/jbc.M300400200. [DOI] [PubMed] [Google Scholar]
- 23.Sakane F, Kai M, Wada I, Imai S-i, Kanoh H. Biochem J. 1996;318:583–590. doi: 10.1042/bj3180583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw G. BioEssays. 1996;18:35–46. doi: 10.1002/bies.950180109. [DOI] [PubMed] [Google Scholar]
- 25.Takeuchi H, Kanematsu T, Misumi Y, Sakane F, Konishi H, Kikkawa U, Watanabe Y, Katan M, Hirata M. Biochim Biophys Acta. 1997;1359:275–285. doi: 10.1016/s0167-4889(97)00109-2. [DOI] [PubMed] [Google Scholar]
- 26.Labesse G, Douguet D, Assairi L, Gilles AM. Trends Biochem Sci. 2002;27:273–275. doi: 10.1016/s0968-0004(02)02093-5. [DOI] [PubMed] [Google Scholar]
- 27.Brenner S. Genetics. 1974;77:71–94. doi: 10.1093/genetics/77.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bany IA. PhD thesis. Yale University; New Haven, CT: 2004. Genetic and Cellular Analysis of the Inhibition of Egg Laying in Caenorhabditis elegans. [Google Scholar]
- 29.Mendel JE, Korswagen HC, Liu KS, Hajdu-Cronin YM, Simon MI, Plasterk RHA, Sternberg PW. Science. 1995;267:1652–1655. doi: 10.1126/science.7886455. [DOI] [PubMed] [Google Scholar]
- 30.Chase DL, Patikoglou GA, Koelle MR. Curr Biol. 2001;11:222–231. doi: 10.1016/s0960-9822(01)00071-9. [DOI] [PubMed] [Google Scholar]
- 31.Bany IA, Dong MQ, Koelle MR. J Neurosci. 2003;23:8060–8069. doi: 10.1523/JNEUROSCI.23-22-08060.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moore DS, McCabe GP. Introduction to the Practice of Statistics. 4. W. H. Freeman and Company; New York: 2003. [Google Scholar]
- 33.Kato M, Takenawa T. J Biol Chem. 1990;265:794–800. [PubMed] [Google Scholar]
- 34.Walker AJ, Draeger A, Houssa B, van Blitterswijk WJ, Ohanian V, Ohanian J. 2001;353:129–137. [PMC free article] [PubMed] [Google Scholar]
- 35.Brundage L, Avery L, Katz A, Kim UJ, Mendel JE, Sternberg PW, Simon MI. Neuron. 1996;16:999–1009. doi: 10.1016/s0896-6273(00)80123-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Topham MK, Bunting M, Zimmerman GA, McIntyre TM, Blackshear PJ, Prescott SM. Nature. 1998;394:697–700. doi: 10.1038/29337. [DOI] [PubMed] [Google Scholar]
- 37.Newton AC. J Biol Chem. 1995;270:28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 38.Stein LD, Bao Z, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwalla A, Clarke L, Clee C, Coghlan A, Coulson A, D’Eustachio P, Fitch DH, Fulton LA, Fulton RE, Griffiths-Jones S, Harris TW, Hillier LW, Kamath R, Kuwabara PE, Mardis ER, Marra MA, Miner TL, Minx P, Mullikin JC, Plumb RW, Rogers J, Schein JE, Sohrmann M, Spieth J, Stajich JE, Wei C, Willey D, Wilson RK, Durbin R, Waterston RH. PLoS Biol. 2003;1:166–192. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shaham S, Horvitz RH. Cell. 1996;56:201–208. doi: 10.1016/s0092-8674(00)80092-6. [DOI] [PubMed] [Google Scholar]
- 40.Shirakihara Y, Evans PR. J Mol Biol. 1988;204:973–994. doi: 10.1016/0022-2836(88)90056-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.