

Abstract
Role of Src kinases in acute lymphoblastic leukaemia has been recently demonstrated in leukaemia mouse model. Retained activation of Src kinases by the BCR-ABL oncoprotein in leukaemic cells following inhibition of BCR-ABL kinase activity by imatinib indicates that Src activation by BCR-ABL is independent of BCR-ABL kinase activity and provides an explanation for reduced effectiveness of the BCR-ABL kinase activity inhibitors in Philadelphia chromosome-positive acute lymphoblastic leukaemia. Simultaneous inhibition of kinase activity of both BCR-ABL and Src kinases results in long-term survival of mice with acute lymphoblastic leukaemia. Leukaemic stem cells exist in acute lymphoblastic leukaemia, and complete eradication of this group of cells would provide a curative therapy for this disease.
Keywords: Src kinases, acute lymphoblastic leukaemia (ALL), BCR-ABL oncogene, signaling pathway, tyrosine kinase inhibitor, leukaemic stem cell
1. Introduction
The human Philadelphia (Ph) chromosome arises from a translocation between chromosomes 9 and 22, and results in formation of the chimeric and constitutively activated BCR-ABL tyrosine kinase. Ph+ leukaemias include chronic myeloid leukaemia (CML) and B-cell acute lymphoblastic leukaemia (B-ALL). CML often begins with a chronic phase and can progress to a terminal blastic phase, in which either acute myeloid or acute B-lymphoid leukemia develops. Some Ph+ leukemia patients have B-ALL as their initial clinical appearance. The BCR-ABL tyrosine kinase inhibitor imatinib mesylate (Gleevec) is the standard of care for Ph+ leukemia. Imatinib induces a remarkable hematologic response in chronic phase CML patients (Druker et al., 2001). However, imatinib does not completely eliminate BCR-ABL-expressing leukemic cells (Graham et al., 2002; Marley, Deininger, Davidson, Goldman, & Gordon, 2000), and patients develop drug resistance (Gorre et al., 2001). Imatinib prolongs survival of mice with BCR-ABL-induced CML (Hu et al., 2004; Wolff & Ilaria, 2001), but does not cure the disease (Hu et al., 2004). Regardless, current therapeutic efforts have focused on targeting BCR-ABL kinase activity using kinase inhibitors. It is generally believed that shutting down the kinase activity of BCR-ABL will completely inhibit its functions, leading to inactivation of its downstream signaling pathways. However, our obtained evidence suggests this is not the case. We find that Src kinases remain active following imatinib inhibition of BCR-ABL kinase activity in leukaemic cells (Hu et al., 2006). In addition, signaling molecules/pathways utilized by BCR-ABL to induce these leukaemias are not identical, although these two distinct diseases can be caused by the same BCR-ABL oncogene. This idea is proposed based on our recent finding that some Src kinases are required for proliferation of BCR-ABL-expressing B-lymphoid cells but not myeloid cells in mice (Hu et al., 2004). This finding is of important clinical implication, because it suggests that different therapeutic strategies are needed for treating chronic phase CML and B-ALL in patients. In this review, I will briefly summarize our current understanding of the signaling pathways that are activated by BCR-ABL, with an emphasis on the activation of Src kinases in leukaemia development in mice. This review will also emphasize our idea that sole inhibition of BCR-ABL kinase activity by kinase inhibitors is insufficient to shut down all BCR-ABL downstream signaling pathways, as BCR-ABL also activates, independently of its kinase activity, Src kinases and perhaps other signaling pathways. It should be pointed out that results discussed in this article are obtained mainly from studies in mice, and that my focus is on activation of Src kinase by BCR-ABL kinase other than other kinases or mechanisms.
2. BCR-ABL signaling and Src kinases
BCR-ABL activates multiple signaling pathways, including Ras, MAPK, STAT, JNK/SAPK, PI-3 kinase, NF-kB and c-MYC (Sawyers, 1997). BCR-ABL functions also link to apoptotic pathways (Amarante-Mendes et al., 1998; Dubrez et al., 1998; Goetz, van der Kuip, Maya, Oren, & Aulitzky, 2001; Honda & Hirai, 2001; Jonuleit et al., 2000; Majewski et al., 1999; McGahon et al., 1995; Neshat, Raitano, Wang, Reed, & Sawyers, 2000; Parada et al., 2001; Sanchez-Garcia & Martin-Zanca, 1997; Skorski et al., 1997; Skorski et al., 1996) and Src family kinases (Danhauser-Riedl, Warmuth, Druker, Emmerich, & Hallek, 1996; Lionberger, Wilson, & Smithgall, 2000; Warmuth et al., 1997). Src family kinases are a group of structurally related non-receptor protein tyrosine kinases. In mouse, there are at least eight family members (Blk, Fgr, Fyn, Hck, Lck, Lyn, c-Src, and Yes), and these kinases contain four Src homology domains but diverge in the unique domain (Lowell & Soriano, 1996). BCR-ABL interects with Src kinases. BCR-ABL coimmunoprecipitates with and activates Lyn and Hck in myeloid cell lines (Danhauser-Riedl et al., 1996; Warmuth et al., 1997). Hck binds directly to BCR-ABL through its three SH domains and distal portion of the C-terminal tail, and is required for transformation of the myeloid leukaemia cell line to IL-3 independency by BCR-ABL (Lionberger et al., 2000). Src kinases may be also linked to BCR-ABL function indirectly through other signaling molecules or pathways such as protein tyrosine phosphatases (Bruecher-Encke, Griffin, Neel, & Lorenz, 2001; Tauchi et al., 1994). In particular, Lyn and SHP-1 (SH2-containing tyrosine phosphotase-1) are functionally related (Daigle, Yousefi, Colonna, Green, & Simon, 2002; Gardai et al., 2002), and Lyn could regulate BCR-ABL function through SHP-1. Src family kinases are also functionally linked to Btk (Bruton’s tyrosine kinase)(Park et al., 1996; Rawlings et al., 1996), and we have observed that Btk is involved in BCR-ABL leukemogenesis (unpublished data).
There are many indirect evidences that indicate an association between Src kinases and hematologic malignancies, as summarized previously (Li, 2005). (Burnett et al., 1991; Ernould, Ferry, Barret, Genton, & Boutin, 1994; Fischer et al., 1989; Lynch et al., 1993; Myers et al., 1995; O’Connor, Torigoe, Reed, & Santoli, 1992; Tilbrook et al., 2001; Torigoe, O’Connor, Santoli, & Reed, 1992; Uckun et al., 1995; Waki et al., 1994; Willman et al., 1991; Yamaguchi et al., 1997). Direct evidence for the roles of Src kinases in leukaemogenesis is inadequate; recently, we provided convincing evidence that Src kinases are required for proliferation of leukaemic cells in mice with ALL (Hu et al., 2004; Hu et al., 2006).
Critical role of Src kinases in proliferation of BCR-ABL-transformed B-lymphoid cells but not myeloid cells in mice
It is unclear whether specific oncogenes use cell type-specific signaling networks to induce particular types of cancer (Deininger, 2004). If this were true, it would help us to determine the choices of available therapeutic strategies and to develop new and effective therapies. Using gene knockout mice, we have found that three Src kinases (Lyn, Hck, and Fgr) are required for proliferation of BCR-ABL-expressing pre-B cells but not myeloid progenitor cells (Hu et al., 2004), although these Src kinases are activated by BCR-ABL in these myeloid cells. This finding is important, as it suggests that Src kinases are better targets for treating ALL but not chronic phase CML, although possible roles for these kinases in regulation of CML stem cells and the transition from chronic phase CML to lymphoid blast crisis. It should be pointed out that our study does not exclude a role for other Src family kinases (rather than Lyn, Hck, and Fgr) in proliferation of BCR-ABL-trandformed myeloid cells in CML mice. It is possible that cell type-specific signaling represents a common mechanism in cancer development. If so, identification of the unique signaling network involved in each type of cancer is critical for developing effective cancer therapies.
Activation of Src kinases by BCR-ABL is not dependent on its kinase activity and inhibition of Src kinases is critical to ALL therapy in mice
It is generally believed that the kinase activity of BCR-ABL is responsible for activation of all its downstream signaling pathways; thus, BCR-ABL kinase activity inhibitors should completely inhibit BCR-ABL functions and cure the disease. We find that Src kinases remain active following imatinib inhibition of BCR-ABL kinase activity in leukaemic cells (Hu et al., 2006). When we treated mice with ALL induced by BCR-ABL-T315I that is resistant to inhibition of BCR-ABL kinase activity by both imatinib (Gorre et al., 2001; Roumiantsev et al., 2002; Warmuth et al., 2003) and dasatinib (Shah et al., 2004), we observed that imatinib (which only inhibits BCR-ABL) had no therapeutic effect, whereas dasatinib (which inhibits both BCR-ABL and Src kinases) significantly prolonged survival of the mice (Hu et al., 2006). We have tested genetically whether Src kinases play a role in CML transition to lymphoid blast crisis using a serial transplantation assay (Pear et al., 1998). Mice were transplanted with BCR-ABL transduced bone marrow (BM) cells from either wild type or Lyn-/-Hck-/-Fgr-/- mice to induce CML, and BM cells from the CML mice were subsequently transferred into recipient mice. Strikingly, mice receiving wild type CML BM cells developed ALL, whereas none of the mice receiving Lyn-/-Hck-/-Fgr-/- CML BM cells developed this disease. These results indicate that CML transition to lymphoid blast crisis requires Src kinases. CML progression is associated with additional genetic changes including mutations in the tumor suppressor genes INK4a, pRB, and p53 (Feinstein et al., 1991; Sill, Goldman, & Cross, 1995; Towatari, Adachi, Kato, & Saito, 1991). Arf gene loss enhances oncogenicity of and limits imatinib response to BCR-ABL-induced ALL in mice (Williams, Roussel, & Sherr, 2006). A potential genetic interaction between Src kinases and those tumor suppressors needs to be investigated. Together, kinase activity-independent activation of Src kinases by BCR-ABL provides a new idea to help our understanding of signaling mechanisms involved in leukemia development.
Pro-B leukemic cells function as ALL stem cells, and inhibition of Src kinases may help prevent them from developing into ALL
All mice treated with dasatinib survived long time as long as the treatment continues (Hu et al., 2006), indicating that inhibition of Src kinases is critical to the treatment of ALL. We asked whether dasatinib could completely eradicate leukaemic cells in ALL mice and cure the mice. A small percentage of BCR-ABL-expressing cells (<1%) remained in peripheral blood of these mice, even after three months of dasatinib treatment. After treatment was stopped, BCR-ABL-expressing cells grew, but dropped again to less than 1% after the treatment resumed (Hu et al., 2006). However, these cells persisted in BM of the treated ALL mice, and were capable of transferring the same disease to secondary recipient mice (unpublished data). These results indicate that these residual leukaemic cells contain ALL stem cells and that continuous administration of dasatinib could prevent these residual cells from developing into fatal ALL, although this compound at the dose used did not completely kill these residual cells. These residual leukaemic cells are B220+/CD43+ pro-B cells and function as ALL stem cells after acquiring self-renewal capacity. Although inhibition of Src kinases by dasatinib did not completely eradicate B-ALL stem cells, preventing these cells from developing into lethal ALL suggests that Src inhibition may at least have a cytostatic effect on these stem cells. Our identification and isolation of ALL stem cells in mice provides a valuable system for studying biology of these stem cells and for developing new therapies to target these cells.
Conclusion
It becomes more and more clear that imatinib may not cure Ph+ leukemia due to the development of clinical drug resistance. Recently, three BCR-ABL kinase activity inhibitors, dasatinib (Shah et al., 2004), AP23464 (O’Hare et al., 2004), and AMN107 (Weisberg et al., 2005), have been shown to inhibit almost all imatinib-resistant BCR-ABL mutants, with an exception of the BCR-ABL-T315I mutant. The development of new BCR-ABL kinase activity inhibitors that are effective on identified and emerging drug-resistant BCR-ABL mutants has been a major focus in treatment of Ph+ leukemia, because it is believed that inhibition of BCR-ABL kinase activity would completely suppress BCR-ABL functions. We have obtained opposing evidence that imatinib-inhibited BCR-ABL can still activates Src kinases, which play a critical role in the development of BCR-ABL-induced ALL. This Src pathway would help leukaemic cells to survive treatment with BCR-ABL kinase activity inhibitors and eventually allow resistant BCR-ABL-T315I cells to grow out. The kinase activity-independent activation of Src kinases by BCR-ABL explains why Ph+ B-ALL is less sensitive than chronic phase CML to imatinib therapy and suggests that sole inhibition of BCR-ABL kinase activity by kinase inhibitors will not cure Ph+ leukaemia. Although the next generation of BCR-ABL kinase inhibitors aims at increasing drug potency or overriding imatinib resistance caused by kinase domain point mutations including BCR-ABL-T315I, to achieve a durable therapeutic effect in patients with Ph+ B-ALL and lymphoid blast crisis, Src kinases must be targeted. Dasatinib does not completely eradicate leukaemic stem cells in ALL mice, but targeting Src kinases helps achieve long-term control of the disease. Curative drug therapy of ALL would require targeting not only BCR-ABL kinase activity and Src-dependent pathways but also pro-B leukemic stem cells (Fig. 1). It will be critical to assess whether BCR-ABL-expressing pro-B cells serve as stem cells in patients with Ph+ B-ALL or lymphoid blast crisis CML. Moreover, identification of unknown pathways in leukaemic stem cells will be critical for developing curative therapies for Ph+ leukaemia.
Fig. 1. Roles of BCR-ABL kinase activity-independent Src pathway and leukaemic stem cells in ALL therapy.
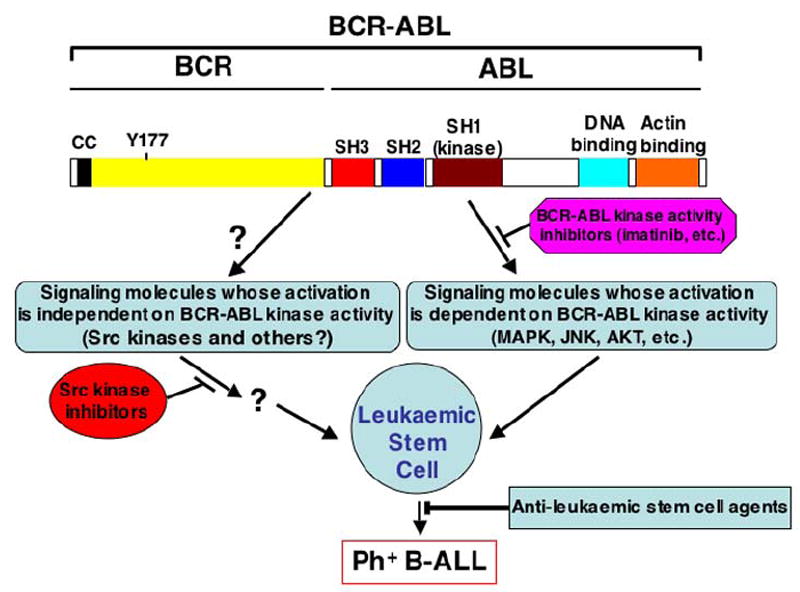
Because the activation of Src kinases is independent of BCR-ABL kinase activity, inhibition of BCR-ABL kinase activity by imatinib does not reduce BCR-ABL-stimulated Src activation. Inhibition of Src kinases may have a cytostatic effect on ALL stem cells, but these cells also survive through other unknown mechanisms. Therefore, simultaneous inhibition of functions of both BCR-ABL (by imatinib) and Src kinases (by an Src kinase inhibitor) and leukaemic stem cells (by an anti-stem cell agent to be developed) is needed for curative therapy of Ph+ B-ALL. cc: coiled-coil domain; Y177: tyrosine 177; SH: Src homology domain.
Acknowledgments
The author would like to thank Barbara Tennent for critically reading the manuscript; and P Cherry for the secretarial assistance. This work was supported by the grants from Main Cancer Foundation, Department of Defense, and National Cancer Institute.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amarante-Mendes GP, Naekyung Kim C, Liu L, Huang Y, Perkins CL, Green DR, et al. Bcr-Abl exerts its antiapoptotic effect against diverse apoptotic stimuli through blockage of mitochondrial release of cytochrome C and activation of caspase-3. Blood. 1998;91(5):1700–1705. [PubMed] [Google Scholar]
- Bruecher-Encke B, Griffin JD, Neel BG, Lorenz U. Role of the tyrosine phosphatase SHP-1 in K562 cell differentiation. Leukemia. 2001;15(9):1424–1432. doi: 10.1038/sj.leu.2402214. [DOI] [PubMed] [Google Scholar]
- Burnett RC, David JC, Harden AM, Le Beau MM, Rowley JD, Diaz MO. The LCK gene is involved in the t(1;7)(p34;q34) in the T-cell acute lymphoblastic leukemia derived cell line, HSB-2. Genes Chromosomes Cancer. 1991;3(6):461–467. doi: 10.1002/gcc.2870030608. [DOI] [PubMed] [Google Scholar]
- Daigle I, Yousefi S, Colonna M, Green DR, Simon HU. Death receptors bind SHP-1 and block cytokine-induced anti-apoptotic signaling in neutrophils. Nat Med. 2002;8(1):61–67. doi: 10.1038/nm0102-61. [DOI] [PubMed] [Google Scholar]
- Danhauser-Riedl S, Warmuth M, Druker BJ, Emmerich B, Hallek M. Activation of Src kinases p53/56lyn and p59hck by p210bcr/abl in myeloid cells. Cancer Res. 1996;56(15):3589–3596. [PubMed] [Google Scholar]
- Deininger M. Src kinases in Ph+ lymphoblastic leukemia. Nat Genet. 2004;36(5):440–441. doi: 10.1038/ng0504-440. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344(14):1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Dubrez L, Eymin B, Sordet O, Droin N, Turhan AG, Solary E. BCR-ABL delays apoptosis upstream of procaspase-3 activation. Blood. 1998;91(7):2415–2422. [PubMed] [Google Scholar]
- Ernould AP, Ferry G, Barret JM, Genton A, Boutin JA. Substrate phosphorylation capacities of the major tyrosine protein kinase from the human promyelocytic cell line, HL-60. Int J Pept Protein Res. 1994;43(5):496–504. doi: 10.1111/j.1399-3011.1994.tb00549.x. [DOI] [PubMed] [Google Scholar]
- Feinstein E, Cimino G, Gale RP, Alimena G, Berthier R, Kishi K, et al. p53 in chronic myelogenous leukemia in acute phase. Proc Natl Acad Sci U S A. 1991;88(14):6293–6297. doi: 10.1073/pnas.88.14.6293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Wendling F, Boulet I, Cocault L, Soula M, Fagard R, et al. Elevated level of p60c-src in virus-transformed murine megakaryocytic cell lines. Oncogene. 1989;4(7):901–905. [PubMed] [Google Scholar]
- Gardai S, Whitlock BB, Helgason C, Ambruso D, Fadok V, Bratton D, et al. Activation of SHIP by NADPH oxidase-stimulated Lyn leads to enhanced apoptosis in neutrophils. J Biol Chem. 2002;277(7):5236–5246. doi: 10.1074/jbc.M110005200. [DOI] [PubMed] [Google Scholar]
- Goetz AW, van der Kuip H, Maya R, Oren M, Aulitzky WE. Requirement for Mdm2 in the survival effects of Bcr-Abl and interleukin 3 in hematopoietic cells. Cancer Res. 2001;61(20):7635–7641. [PubMed] [Google Scholar]
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293(5531):876–880. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- Graham SM, Jorgensen HG, Allan E, Pearson C, Alcorn MJ, Richmond L, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325. doi: 10.1182/blood.v99.1.319. [DOI] [PubMed] [Google Scholar]
- Honda H, Hirai H. Model mice for BCR/ABL-positive leukemias. Blood Cells Mol Dis. 2001;27(1):265–278. doi: 10.1006/bcmd.2000.0374. [DOI] [PubMed] [Google Scholar]
- Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, et al. Requirement of Src kinases Lyn, Hck and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet. 2004;36(5):453–461. doi: 10.1038/ng1343. [DOI] [PubMed] [Google Scholar]
- Hu Y, Swerdlow S, Duffy TM, Weinmann R, Lee FY, Li S. Targeting multiple kinase pathways in leukemic prognitors and stem cells is essential for improved treatment of Ph+ leukemia in mice. Proc Natl Acad Sci U S A. 2006;103(45):16870–16875. doi: 10.1073/pnas.0606509103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonuleit T, van der Kuip H, Miething C, Michels H, Hallek M, Duyster J, et al. Bcr-Abl kinase down-regulates cyclin-dependent kinase inhibitor p27 in human and murine cell lines. Blood. 2000;96(5):1933–1939. [PubMed] [Google Scholar]
- Li S. Src kinases as targets for B cell acute lymphoblastic leukaemia therapy. Expert Opin Ther Targets. 2005;9(2):329–341. doi: 10.1517/14728222.9.2.329. [DOI] [PubMed] [Google Scholar]
- Lionberger JM, Wilson MB, Smithgall TE. Transformation of myeloid leukemia cells to cytokine independence by Bcr-Abl is suppressed by kinase-defective Hck. J Biol Chem. 2000;275(24):18581–18585. doi: 10.1074/jbc.C000126200. [DOI] [PubMed] [Google Scholar]
- Lowell CA, Soriano P. Knockouts of Src-family kinases: stiff bones, wimpy T cells, and bad memories. Genes Dev. 1996;10(15):1845–1857. doi: 10.1101/gad.10.15.1845. [DOI] [PubMed] [Google Scholar]
- Lynch SA, Brugge JS, Fromowitz F, Glantz L, Wang P, Caruso R, et al. Increased expression of the src proto-oncogene in hairy cell leukemia and a subgroup of B-cell lymphomas. Leukemia. 1993;7(9):1416–1422. [PubMed] [Google Scholar]
- Majewski M, Nieborowska-Skorska M, Salomoni P, Slupianek A, Reiss K, Trotta R, et al. Activation of mitochondrial Raf-1 is involved in the antiapoptotic effects of Akt. Cancer Res. 1999;59(12):2815–2819. [PubMed] [Google Scholar]
- Marley SB, Deininger MW, Davidson RJ, Goldman JM, Gordon MY. The tyrosine kinase inhibitor STI571, like interferon-alpha, preferentially reduces the capacity for amplification of granulocyte-macrophage progenitors from patients with chronic myeloid leukemia. Exp Hematol. 2000;28(5):551–557. doi: 10.1016/s0301-472x(00)00142-9. [DOI] [PubMed] [Google Scholar]
- McGahon AJ, Nishioka WK, Martin SJ, Mahboubi A, Cotter TG, Green DR. Regulation of the Fas apoptotic cell death pathway by Abl. J Biol Chem. 1995;270(38):22625–22631. doi: 10.1074/jbc.270.38.22625. [DOI] [PubMed] [Google Scholar]
- Myers DE, Jun X, Waddick KG, Forsyth C, Chelstrom LM, Gunther RL, et al. Membrane-associated CD19-LYN complex is an endogenous p53-independent and Bc1-2-independent regulator of apoptosis in human B-lineage lymphoma cells. Proc Natl Acad Sci U S A. 1995;92(21):9575–9579. doi: 10.1073/pnas.92.21.9575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neshat MS, Raitano AB, Wang HG, Reed JC, Sawyers CL. The survival function of the Bcr-Abl oncogene is mediated by Bad-dependent and -independent pathways: roles for phosphatidylinositol 3-kinase and Raf. Mol Cell Biol. 2000;20(4):1179–1186. doi: 10.1128/mcb.20.4.1179-1186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor R, Torigoe T, Reed JC, Santoli D. Phenotypic changes induced by interleukin-2 (IL-2) and IL-3 in an immature T-lymphocytic leukemia are associated with regulated expression of IL-2 receptor beta chain and of protein tyrosine kinases LCK and LYN. Blood. 1992;80(4):1017–1025. [PubMed] [Google Scholar]
- O’Hare T, Pollock R, Stoffregen EP, Keats JA, Abdullah OM, Moseson EM, et al. Inhibition of wild-type and mutant Bcr-Abl by AP23464, a potent ATP-based oncogenic protein kinase inhibitor: implications for CML. Blood. 2004;104(8):2532–2539. doi: 10.1182/blood-2004-05-1851. [DOI] [PubMed] [Google Scholar]
- Parada Y, Banerji L, Glassford J, Lea NC, Collado M, Rivas C, et al. BCR-ABL and interleukin 3 promote haematopoietic cell proliferation and survival through modulation of cyclin D2 and p27Kip1 expression. J Biol Chem. 2001;276(26):23572–23580. doi: 10.1074/jbc.M101885200. [DOI] [PubMed] [Google Scholar]
- Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, et al. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 1996;4(5):515–525. doi: 10.1016/s1074-7613(00)80417-3. [DOI] [PubMed] [Google Scholar]
- Pear WS, Miller JP, Xu L, Pui JC, Soffer B, Quackenbush RC, et al. Efficient and rapid induction of a chronic myelogenous leukemia-like myeloproliferative disease in mice receiving P210 bcr/abl-transduced bone marrow. Blood. 1998;92:3780–3792. [PubMed] [Google Scholar]
- Rawlings DJ, Scharenberg AM, Park H, Wahl MI, Lin S, Kato RM, et al. Activation of BTK by a phosphorylation mechanism initiated by SRC family kinases. Science. 1996;271(5250):822–825. doi: 10.1126/science.271.5250.822. [DOI] [PubMed] [Google Scholar]
- Roumiantsev S, Shah NP, Gorre ME, Nicoll J, Brasher BB, Sawyers CL, et al. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr-253 in the Abl kinase domain P-loop. Proc Natl Acad Sci U S A. 2002;99(16):10700–10705. doi: 10.1073/pnas.162140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Garcia I, Martin-Zanca D. Regulation of Bcl-2 gene expression by BCR-ABL is mediated by Ras. J Mol Biol. 1997;267(2):225–228. doi: 10.1006/jmbi.1996.0779. [DOI] [PubMed] [Google Scholar]
- Sawyers CL. Signal transduction pathways involved in BCR-ABL transformation. Baillieres Clin Haematol. 1997;10(2):223–231. doi: 10.1016/s0950-3536(97)80004-2. [DOI] [PubMed] [Google Scholar]
- Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- Sill H, Goldman JM, Cross NC. Homozygous deletions of the p16 tumor-suppressor gene are associated with lymphoid transformation of chronic myeloid leukemia. Blood. 1995;85(8):2013–2016. [PubMed] [Google Scholar]
- Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway. Embo J. 1997;16(20):6151–6161. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skorski T, Nieborowska-Skorska M, Wlodarski P, Perrotti D, Martinez R, Wasik MA, et al. Blastic transformation of p53-deficient bone marrow cells by p210bcr/abl tyrosine kinase. Proc Natl Acad Sci U S A. 1996;93(23):13137–13142. doi: 10.1073/pnas.93.23.13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauchi T, Feng GS, Shen R, Song HY, Donner D, Pawson T, et al. SH2-containing phosphotyrosine phosphatase Syp is a target of p210bcr-abl tyrosine kinase. J Biol Chem. 1994;269(21):15381–15387. [PubMed] [Google Scholar]
- Tilbrook PA, Palmer GA, Bittorf T, McCarthy DJ, Wright MJ, Sarna MK, et al. Maturation of erythroid cells and erythroleukemia development are affected by the kinase activity of Lyn. Cancer Res. 2001;61(6):2453–2458. [PubMed] [Google Scholar]
- Torigoe T, O’Connor R, Santoli D, Reed JC. Interleukin-3 regulates the activity of the LYN protein-tyrosine kinase in myeloid-committed leukemic cell lines. Blood. 1992;80(3):617–624. [PubMed] [Google Scholar]
- Towatari M, Adachi K, Kato H, Saito H. Absence of the human retinoblastoma gene product in the megakaryoblastic crisis of chronic myelogenous leukemia. Blood. 1991;78(9):2178–2181. [PubMed] [Google Scholar]
- Uckun FM, Evans WE, Forsyth CJ, Waddick KG, Ahlgren LT, Chelstrom LM, et al. Biotherapy of B-cell precursor leukemia by targeting genistein to CD19-associated tyrosine kinases. Science. 1995;267(5199):886–891. doi: 10.1126/science.7531365. [DOI] [PubMed] [Google Scholar]
- Waki M, Kitanaka A, Kamano H, Tanaka T, Kubota Y, Ohnishi H, et al. Antisense src expression inhibits U937 human leukemia cell proliferation in conjunction with reduction of c-myb expression. Biochem Biophys Res Commun. 1994;201(2):1001–1007. doi: 10.1006/bbrc.1994.1801. [DOI] [PubMed] [Google Scholar]
- Warmuth M, Bergmann M, Priess A, Hauslmann K, Emmerich B, Hallek M. The Src family kinase Hck interacts with Bcr-Abl by a kinase-independent mechanism and phosphorylates the Grb2-binding site of Bcr. J Biol Chem. 1997;272(52):33260–33270. doi: 10.1074/jbc.272.52.33260. [DOI] [PubMed] [Google Scholar]
- Warmuth M, Simon N, Mitina O, Mathes R, Fabbro D, Manley PW, et al. Dual-specific Src and Abl kinase inhibitors, PP1 and CGP76030, inhibit growth and survival of cells expressing imatinib mesylate-resistant Bcr-Abl kinases. Blood. 2003;101(2):664–672. doi: 10.1182/blood-2002-01-0288. [DOI] [PubMed] [Google Scholar]
- Weisberg E, Manley PW, Breitenstein W, Bruggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Williams RT, Roussel MF, Sherr CJ. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci U S A. 2006;103(17):6688–6693. doi: 10.1073/pnas.0602030103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willman CL, Stewart CC, Longacre TL, Head DR, Habbersett R, Ziegler SF, et al. Expression of the c-fgr and hck protein-tyrosine kinases in acute myeloid leukemic blasts is associated with early commitment and differentiation events in the monocytic and granulocytic lineages. Blood. 1991;77(4):726–734. [PubMed] [Google Scholar]
- Wolff NC, Ilaria RL., Jr Establishment of a murine model for therapy-treated chronic myelogenous leukemia using the tyrosine kinase inhibitor STI571. Blood. 2001;98(9):2808–2816. doi: 10.1182/blood.v98.9.2808. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Tanaka T, Waki M, Kitanaka A, Kamano H, Kubota Y, et al. Antisense src expression inhibits tyrosine phosphorylation of Shc and its association with Grb2 and Sos which leads to MAP kinase activation in U937 human leukemia cells. Leukemia. 1997;11(4):497–503. doi: 10.1038/sj.leu.2400605. [DOI] [PubMed] [Google Scholar]