

Abstract
Mutations in the gene encoding SP-C (surfactant protein C; SFTPC) have been linked to interstitial lung disease (ILD) in children and adults. Expression of the index mutation, SP-CΔexon4, in transiently transfected cells and type II cells of transgenic mice resulted in misfolding of the proprotein, activation of endoplasmic reticulum (ER) stress pathways, and cytotoxicity. In this study, we show that stably transfected cells adapted to chronic ER stress imposed by the constitutive expression of SP-CΔexon4 via an NF-κB–dependent pathway. However, the infection of cells expressing SP-CΔexon4 with respiratory syncytial virus resulted in significantly enhanced cytotoxicity associated with accumulation of the mutant proprotein, pronounced activation of the unfolded protein response, and cell death. Adaptation to chronic ER stress imposed by misfolded SP-C was associated with increased susceptibility to viral-induced cell death. The wide variability in the age of onset of ILD in patients with SFTPC mutations may be related to environmental insults that ultimately overwhelm the homeostatic cytoprotective response.
Introduction
SP-C (surfactant protein C) is a transmembrane protein that is synthesized as a 191– or 197–amino acid proprotein by type II epithelial cells of the lung. Processing of the proprotein in the distal secretory pathway liberates a 35–amino acid peptide that is secreted into the airspaces as a component of pulmonary surfactant. Mutations in the gene encoding SP-C (SFTPC) have been associated with the development of sporadic and familial interstitial lung disease (ILD). In all cases to date, the mutation was present on only one allele consistent with a dominant-negative effect. The index mutation, a heterozygous base substitution of A for G at the first base of intron 4 (c.460 + 1 G→A), led to the internal deletion of 37 amino acids from the ER luminal domain, generating a truncated proprotein (SP-CΔexon4; Nogee et al., 2001). Two separate SFTPC mutations associated with ILD, SP-CL188Q and SP-CI73T, were detected in three kindreds (Thomas et al., 2002a; Chibbar et al., 2004; Cameron et al., 2005). The age of onset and penetrance of ILD varied markedly in all three kindreds.
Studies in transiently transfected cells suggested that the c.460 + 1 G→A mutation led to misfolding of the mutant proprotein, retention of SP-Cwt in the ER, activation of the unfolded protein response (UPR), and apoptosis (Bridges et al., 2003; Wang et al., 2003; Mulugeta et al., 2005). SP-CΔexon4 was also associated with cytotoxicity and lung dysmorphogenesis when expressed in type II cells of transgenic mice (Bridges et al., 2003). The UPR is activated by conditions that perturb ER homeostasis, including the accumulation of misfolded proteins (Schroder and Kaufman, 2005). This response encompasses translational and transcriptional changes within the cell to alleviate the stress and to promote restoration of ER homeostasis. A model for the time-dependent induction of the UPR has been proposed, suggesting that translational repression via PERK activation/eIF2α phosphorylation occurs first followed by the cleavage of ATF6, activation of IRE1/XBP-1, and expression of ATF6 and XBP-1 target genes (Yoshida et al., 2003). If ER homeostasis cannot be restored by these pathways or by the induction of adaptive responses, apoptosis may occur as a means of avoiding the untoward effects of cell necrosis. ER stress–induced apoptosis has been associated with induction of the transcription factor C/EBP homologous protein, activation of c-Jun amino-terminal kinase via IRE1, and activation of the ER stress–specific caspases 4 (human; Hitomi et al., 2004) and 12 (mouse; Nakagawa et al., 2000; Urano et al., 2000; Hetz et al., 2003; for review see Oyadomari and Mori, 2004). Although the effects of acute ER stress, which is imposed by xenotoxic agents such as thapsigargin and tunicamycin, are well established, little is known about the molecular pathways involved in adaptation to chronic ER stress imposed by a misfolded protein.
The variability in the age of onset and penetrance of disease in the SP-CL188Q and SP-CI73T pedigrees suggests that both genetic and environmental factors may influence the manifestation of lung disease. Based on the results of the aforementioned studies in human patients and transiently transfected cells, experiments were designed to test the hypotheses that (1) chronic ER stress imposed by misfolded SP-C promotes adaptation and cell survival and (2) adaptation increases susceptibility to environmental stress. Clonal cell lines stably expressing SP-CΔexon4 or SP-Cwt were generated to identify cytoprotective pathways that are associated with adaptation to the constitutive expression of misfolded SP-C and to assess the cytotoxic effects of environmental stress on adapted cells.
Results
Generation and characterization of stably transfected cell lines
To determine the molecular mechanisms underlying SP-CΔexon4-induced cytotoxicity, HEK293 cell lines stably expressing SP-Cwt or SP-CΔexon4 were generated. Multiple clonal lines were obtained for each construct, and two lines were chosen for subsequent experimentation based on equivalent expression of SP-C mRNA, which was initially assessed by RT-PCR (Table I) and subsequently confirmed by microarray analysis (Fig. 1 b). These cell lines were morphologically indistinguishable by light microscopy (Fig. 1 a) or electron microscopy (not depicted) and exhibited similar doubling rates (not depicted). Basal SP-C protein levels were assessed by Western blot analysis of cell lysates with an antibody directed against the NH2-terminal peptide of the proprotein (proSP-C), a region which is unaffected by the Δexon4 mutation. Despite equivalent mRNA levels, expression of the SP-CΔexon4 protein was barely detectable compared with SP-Cwt, which is consistent with rapid proteasome-dependent turnover of the mutant proprotein (Fig. 1 c).
Table I.
Transcriptional profiling reveals differential expression of genes associated with apoptosis in SP-CΔexon4 cells
| Fold change (microarray) |
Fold change (real-time PCR) |
Gene | Description | Functiona | NF-κB–binding sites |
|---|---|---|---|---|---|
| +4.8 | +4.2 | MYC | V-myc myelocytomatosis viral oncogene homologue (avian) |
Regulation of apoptosis | 1 |
| +2.3 | ND | SNCA | Synuclein, α | Anti | 1 |
| +2.1 | ND | PROK2 | Prokineticin 2 | Anti | 1 |
| +1.7 | +2.9 | IKBKG | Inhibitor of κ-light polypeptide gene enhancer in B cells, kinase γ |
Pro | 1 |
| +1.6 | ND | PBEF | Pre–B cell colony–enhancing factor 1 |
Anti | 1 |
| +1.6 | ND | BNIP1 | Bcl2/adenovirus E1B 19-kD interacting protein 1 |
Anti and pro | 2 |
| +1.5 | ND | MAPK14 | Mitogen-activated protein kinase 14 |
Anti | 2 |
| +1.5 | ND | MYB | V-myb myeloblastosis viral oncogene homologue (avian) |
Regulation of apoptosis | 1 |
| +2.1 | ND | PDCD8 | Programmed cell death 8 | Pro | 0 |
| +2.0 | ND | HRK | Harakiri, Bcl2-interacting protein |
Pro | 0 |
| +1.8 | ND | HTATIP2 | HIV-1 Tat interactive protein 2 | Anti | 0 |
| +1.7 | ND | BCAP31 | B cell receptor–associated protein 31 |
Apoptosis | 0 |
| +1.6 | ND | PLAGL1 | Pleiomorphic adenoma genelike 1 |
Pro | 0 |
| +1.6 | ND | BAX | Bcl2-associated X protein | Pro | 0 |
| +1.6 | ND | P8 | P8 protein | Pro | 0 |
| +1.6 | ND | BAG2 | Bcl2-associated athanogene 2 | Apoptosis | 0 |
| +1.6 | ND | MRPS30 | Mitochondria ribosomal protein S30 |
Apoptosis | 0 |
| +1.5 | ND | TNFRSF6 | TNF receptor superfamily, member 6 |
Anti | 0 |
| −3.0 | ND | SFRP1 | Secreted frizzled rp1 | Anti | 2 |
| −3.0 | ND | CD24 | CD24 antigen | Pro | 1 |
| −2.1 | ND | FLI1 | Friend leukemia virus integration 1 |
Cell death | 1 |
| −2.0 | ND | RUNX3 | Runt-related transcription factor 3 |
Apoptosis | 2 |
| −1.6 | ND | PSEN1 | Presenilin 1 | Anti | 1 |
| −1.6 | ND | CDC2L2 | Cell division cycle 2–like 2 | Apoptosis | 1 |
| −1.6 | −3.6 | DUSP6 | Dual specificity phosphatase 6 | Apoptosis | 1 |
| −3.0 | ND | TGF-β1 | Transforming growth factor β1 | Anti | 0 |
| −2.1 | ND | Bcl2 | B cell CLL/lymphoma 2 | Anti | 0 |
| −1.9 | ND | PHLDA2 | Pleckstrin homology-like DA2 | Apoptosis | 0 |
| −1.9 | ND | AKT1 | V-akt murine thymoma viral oncogene |
Anti | 0 |
| −1.7 | ND | TIA1 | RNA-binding protein | Apoptosis | 0 |
| −1.8 | ND | PTPN6 | Protein tyrosine phosphatase N6 | Apoptosis | 0 |
| −1.7 | ND | PRKCZ | Protein kinase C, ζ | Anti | 0 |
| −1.7 | ND | MCL1 | Bcl-2 related protein | Anti and pro | 0 |
| +1.7 | +2.3 | IRAK1 | IL-1 receptor-associated kinase 1 | NF-κB pathway | 0 |
The clonal cell lines expressing SP-Cwt, SP-CΔexon4, or empty vector were subjected to transcriptional profiling using the human U133 gene chip set from Affymetrix. Genes that were specifically increased or decreased in SP-CΔexon4 over both SP-Cwt and an empty vector control (i.e., mutant-specific changes) are reported as fold changes from microarray analysis (first column). Select targets were validated by real-time PCR (second column). The last column lists the number of putative NF-κB–binding sites located in the 5′ flanking sequence (1 kb) of the gene on the left. Data represent the mean of three analyses for each genotype.
Anti and pro designations signify anti- and pro-apoptosis; evidence of apoptosis is from the Gene Ontology database.
Figure 1.

Generation of clonal stably transfected cell lines. (a) Phase light micrographs of stably transfected HEK293 cell lines expressing SP-Cwt or SP-CΔexon4. (b) Expression of SP-C mRNA in the two cell lines. Error bars represent SD. (c) 30 μg of cell lysates were subjected to immunoblot analysis using antibody directed against the NH2 terminus of proSP-C to detect SP-Cwt (Mr = 21 k) and SP-CΔexon4 (Mr = 17 k). (d and e) Subcellular localization of SP-Cwt to lysosomes (d) and SP-CΔexon4 to ER (e) in clonal cell lines by confocal microscopy. Bars (a and d), 20 μm; (e) 10 μm.
SP-CΔexon4 protein was previously shown to be rapidly degraded in a proteasome-dependent manner and failed to be exported from the ER when transiently expressed in HEK293 cells (Bridges et al., 2003). To determine the subcellular localization of the SP-C variants in stably transfected cell lines, cells were stained with antibodies directed against proSP-C and LAMP-1 and analyzed by confocal microscopy. A bright, punctuate staining pattern was observed for proSP-C in SP-Cwt cells, whereas cells expressing SP-CΔexon4 showed faint, diffuse staining that was detected only in the presence of proteasome inhibitor (Fig. 1 d). SP-Cwt traffics to the lamellar body in type II cells but is redirected to the lysosome in cells that lack lamellar bodies (for reviews see Dell'Angelica et al., 2000; Weaver et al., 2002). Colocalization of SP-Cwt with the lysosomal marker LAMP-1 demonstrated efficient export of wild-type protein from the ER (Fig. 1 d). In contrast, SP-CΔexon4 exhibited a faint and diffuse staining pattern that colocalized with an anti-KDEL antibody, an ER marker (Fig. 1 e). These data in clonal cell lines, coupled with previous results in transiently transfected cells (Bridges et al., 2003), suggest that SP-Cwt is correctly folded and exported from the ER, whereas SP-CΔexon4 fails quality control and is rapidly degraded by the proteasome.
Transcriptional profiling reveals differential expression of genes associated with apoptosis in SP-CΔexon4 cells
Molecular pathways induced by the chronic expression of SP-CΔexon4 were identified by transcriptional profiling of SP-Cwt and SP-CΔexon4 clonal cell lines (the complete dataset can be found at http://www.ncbi.nlm.nih.gov/geo/; GenBank/EMBL/DDBJ accession no. GSE2980). Unexpectedly, known components of the UPR/ER-associated degradation (ERAD) pathways were not increased in cells that constitutively expressed SP-CΔexon4, which is in contrast to results in transiently transfected cells (Bridges et al., 2003). However, several genes associated with anti- and pro-apoptosis pathways were differentially expressed in the SP-CΔexon4 clonal cell line, including BAX and Bcl-2 (Table I, Fig. S1, and Table S1; available at http://www.jcb.org/cgi/content/full/jcb.200508016/DC1). Interestingly, two transcripts linked to the NF-κB pathway, interleukin-1 receptor-associated 1 (IRAK1) and the γ subunit of the Iκ-B kinase complex (IKBKG), were increased in the SP-CΔexon4 cells, which is consistent with alterations in the NF-κB signaling pathway. Analyses of the 5′ flanking sequences of apoptosis-associated genes revealed that ∼44% (15/34) contained putative NF-κB binding sites, suggesting that they may be direct targets of NF-κB.
To determine whether NF-κB activity was increased in SP-CΔexon4 cells, an NF-κB luciferase reporter construct was transiently transfected into the clonal cell lines. SP-CΔexon4 cells exhibited an 8.6-fold increase in basal NF-κB activity compared with cells stably transfected with empty vector or SP-Cwt (Fig. 2 a). Cotransfection of a stabilized, nonphosphorylatable form of I-κBα (super repressor [SR]) with the NF-κB reporter resulted in a dose-dependent decrease in luciferase activity in SP-CΔexon4 cells that was completely suppressed to levels detected in untreated SP-Cwt cells (Fig. 2 b). IκBα protein in SP-CΔexon4 cells was decreased compared with SP-Cwt cells, which is consistent with increased NF-κB activity (Fig. 2 c). Treatment of SP-CΔexon4 cells with SN50 peptide to inhibit the nuclear translocation of NF-κB resulted in cellular retraction and a significant increase in cell death, as indicated by propidium iodide (PI) staining (Fig. 3 a) and MTS reduction assay (Fig. 3 c). Treatment of SP-CΔexon4 cells with a control peptide, SN50mut, at equivalent doses and duration had no effect (Fig. 3 b). Cells expressing SP-Cwt were unaffected by SN50 or SN50mut treatment (not depicted). Treatment of SP-CΔexon4 cells with SN50 decreased basal NF-κB activity as measured by a NF-κB reporter (Fig. 3 d) and inhibited nuclear translocation of the p65 subunit of NF-κB (Fig. 3 e). Altogether, these results suggest that NF-κB, mediated in part by the p65 subunit, plays an important cytoprotective role in adaptation to the constitutive expression of SP-CΔexon4.
Figure 2.
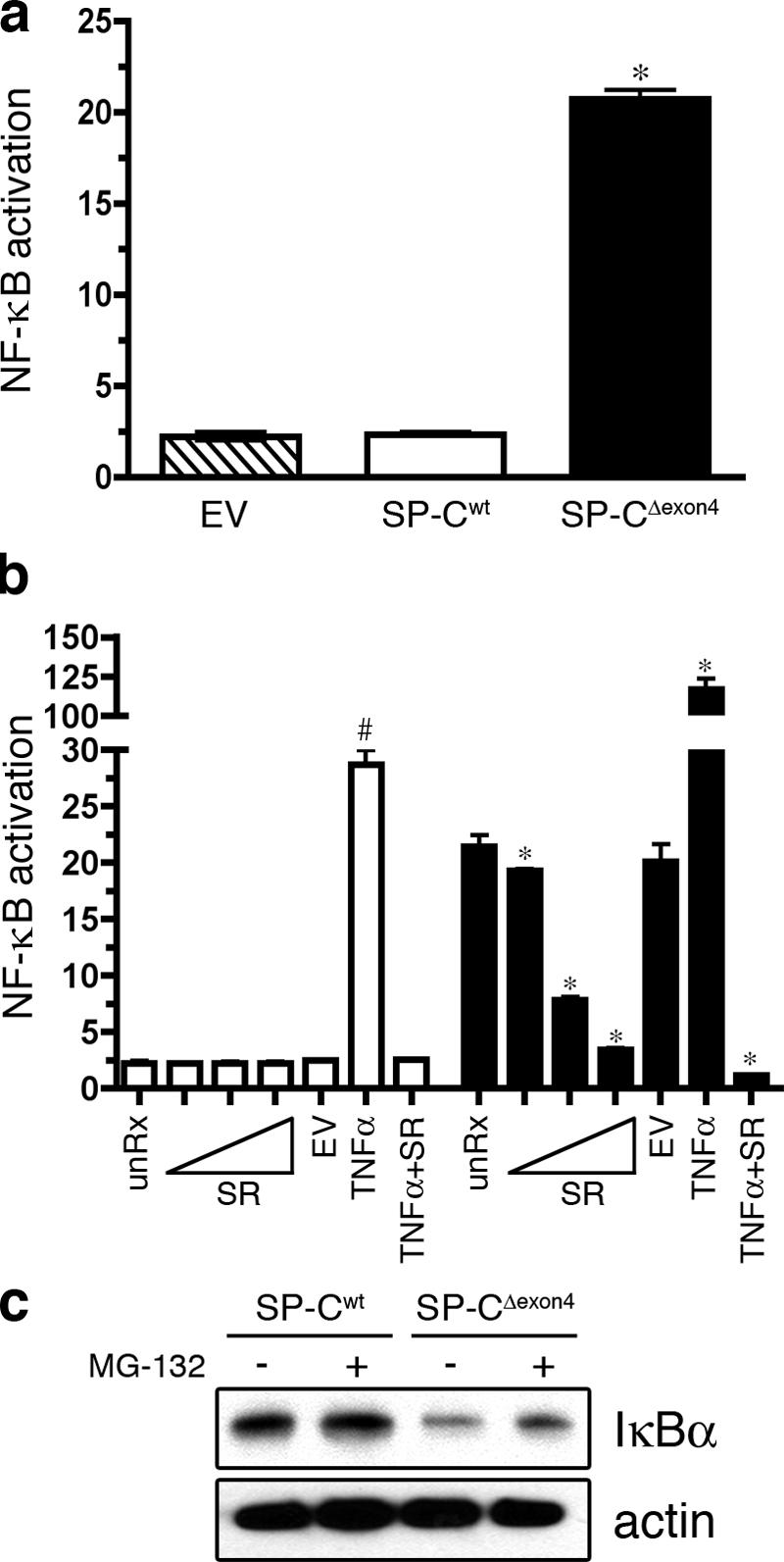
Basal NF-κB activity is increased in cells expressing SP-CΔexon4. (a) The clonal cell lines expressing empty vector (EV), SP-Cwt, or SP-CΔexon4 were transiently cotransfected with an NF-κB reporter construct (pELAM-Luc) and a plasmid encoding renilla luciferase (pRL-TK). Luciferase activity was read 48 h after transfection. Similar results were obtained with two additional NF-κB luciferase reporter constructs, including a minimal IL-8 promoter and a synthetic NF-κB promoter (not depicted). *, P < 0.05 compared with SP-Cwt. (b) SP-Cwt (open bars) or SP-CΔexon4 (closed bars) clonal cell lines were transiently transfected with pELAM-Luc, pRL-TK, and increasing amounts (10 pg–1 ng) of a plasmid encoding a super repressor of NF-κB activity (SR). EV indicates 10 ng of empty vector control. Groups with 10 ng/ml TNFα were treated for 12 h. Luciferase activity was assessed 36 h after transfection. Results are presented as means ± SD (error bars) of the ratio of firefly/renilla, expressed as relative luciferase units; each determination was performed in triplicate, and the experiment was repeated four times. *, P < 0.05 compared with SP-CΔexon4 untreated; #, P < 0 .05 compared with SP-Cwt unRx. (c) Immunoblot analysis of whole cell lysates using antibody directed against IκBα (top) or actin (bottom). Cells were treated with 5 μm MG-132 for 4 h before harvest.
Figure 3.

Inhibition of NF-κB nuclear translocation induces death of SP-CΔexon4 cells. (a and b) The clonal cell line expressing SP-CΔexon4 was treated with SN50, a cell-permeable peptide that inhibits the nuclear translocation of NF-κB (a), or a mutant SN50 peptide (SN50mut; b) at the indicated doses. Cells were stained with PI 24 h after the addition of peptide and phase (top), and fluorescent (bottom) images were captured directly from the culture plate. Bars, 50 μm. (c) Clonal cell lines expressing SP-Cwt or SP-CΔexon4 were treated with the SN50 peptide at indicated doses for 24 h, and cell death was quantitated by MTS assay. (d) The clonal cell line expressing SP-CΔexon4 was transiently cotransfected with the pELAM-Luc and pRL-TK vectors. Cells were treated with SN50 or SN50mut peptide 24 h after transfection at indicated doses. SR was cotransfected with pELAM-Luc and pRL-TK as positive controls. Luciferase activity was assessed 48 h after transfection. Results in c and d are presented as means ± SD (error bars); each determination was performed in triplicate, and the experiment was repeated three times. *, P < 0.05 compared with SP-CΔexon4 unRx; #, P < 0.05 compared with SP-Cwt unRx. (e) Immunoblot analysis of nuclear extracts with an anti-p65 antibody (top) or histone H2B (bottom). SP-CΔexon4 cells were treated with 50 μg/ml SN50 or SN50mut for 24 h before harvest. SP-Cwt cells were treated with 10 ng/ml TNFα for 6 h for positive control.
SP-CΔexon4 expression increases susceptibility to viral-induced death
The increased expression of pro-apoptotic transcripts in SP-CΔexon4 cells (Table I) suggested that adapted cells may be more susceptible to secondary stress. Infection with respiratory syncytial virus (RSV) preceded the onset of ILD in several patients carrying the SP-CL188Q mutation. To determine whether the expression of SP-CΔexon4 increased susceptibility to RSV, the clonal cells lines were infected at a multiplicity of infection (MOI) of 10 pfu/cell, and cell viability was assessed 24 h after infection. Although cell viability was minimally affected in SP-Cwt cells, as indicated by the low level of PI staining (Fig. 4 a [b and f]), the infection of cells expressing SP-CΔexon4 resulted in significant cell death (Fig. 4 a [d and h]). Western blot analysis demonstrated that SP-CΔexon4 protein accumulated in a manner directly correlated with the viral titer, whereas SP-Cwt protein levels were unaffected (Fig. 4 b). Several viruses, including adenovirus, influenza, and hepatitis B and C viruses, have been reported to induce ER stress/UPR pathways (Meyer et al., 1992; Pahl and Baeuerle, 1995; Pahl et al., 1996; Tardif et al., 2004). To determine whether RSV activated the UPR in cells stably expressing SP-C, an UPR element luciferase reporter construct was transiently transfected into cells expressing SP-CΔexon4 or SP-Cwt before infection with RSV (Fig. 4 c). Baseline levels of luciferase activity were similar between the cell lines, which is consistent with the microarray data, indicating that the UPR was not activated in cells stably expressing SP-CΔexon4. However, luciferase activity was increased in both cell lines after RSV infection, and the effect was exacerbated in SP-CΔexon4 cells (2.5-fold increase in SP-Cwt vs. 4.4-fold increase in SP-CΔexon4; Fig. 4 c). Furthermore, RSV-induced cell death appeared to be independent of NF-κB, as NF-κB activity was sustained in both RSV-infected SP-CΔexon4 and SP-Cwt cells (Fig. 4 d). Consistent with this finding, UPR activity was not induced in SP-CΔexon4 cells when NF-κB activity was inhibited with SN50 (Fig. 4 e). Collectively, these data indicate that RSV infection was associated with accumulation of the mutant proprotein, pronounced activation of the UPR, and increased cell death independent of NF-κB activity.
Figure 4.

SP-CΔexon4 increases susceptibility to RSV-induced cell death. (a) Cells expressing SP-Cwt (panels a, b, e, and f) or SP-CΔexon4 (panels c, d, g, and h) were infected with RSV at 10 MOI (panels b, f, d, and h). Cells were stained with propidium iodide (PI) 24 hr after infection, and phase (panels a–d) or fluorescent (panels e–h) images were captured directly from the culture plate. Bars, 50 μm. (b) Immunoblot analysis of cells expressing SP-Cwt or SP-CΔexon4 after infection with 0.1, 1, or 10 MOI RSV for 24 h. Immunoblots were probed with an anti–proSP-C antibody or an anti-actin antibody (load-ing control). (c) Clonal cell lines expressing SP-Cwt or SP-CΔexon4 were transiently transfected with a reporter construct consisting of five UPR elements driv-ing luciferase (UPRE-Luc) and pRL-TK. Cells were infected with 5 MOI RSV 24 h after transfection, and luciferase activity was assessed 24 h after infection. Results are presented as means ± SD (error bars) of the ratio of firefly/renilla, expressed as relative luciferase units; each determination was performed in triplicate, and the experiment was repeated four times. (d) Cell lines were transiently transfected with pELAM-Luc and pRL-TK vectors and infected with RSV at indicated doses 24 h after transfection. (e) SP-CΔexon4 cells were transiently transfected with the UPRE-Luc and pRL-TK vectors. Cells were treated with SN50 or SN50mut peptide 24 h after transfection at indicated doses. Cells were treated with 10 μg/ml tunicamycin for 12 h for posi-tive control. (d and e) Luciferase activity was assessed 48 h after transfection and normalized to renilla luciferase activity. Results are presented as means ± SD; each determination was performed in triplicate, and the experiment was repeated three times. Data indicate fold change relative to untreated control for each genotype. (c–e) #, P < 0.05 compared with SP-Cwt unRx; *, P < 0.05 compared with SP-CΔexon4 unRx; ♦, P < 0.05 compared with RSV-infected SP-Cwt.
Proteasome function is decreased in RSV-infected SP-CΔexon4 cells
Accumulation of mutant SP-C proprotein in RSV-infected SP-CΔexon4 cells suggested that proteasome function was inhibited. To assess the impact of RSV infection on proteasome activity, a proteasome reporter construct, pZsProSensor-1, was transiently transfected into the clonal cell lines. Cells were infected with 1–10 MOI of RSV and analyzed 24 h later by fluorescence microscopy and FACS analysis. Fluorescence in untreated SP-CΔexon4 cells (mean fluorescence intensity [MFI] = 305) was higher than that observed in untreated SP-Cwt cells (MFI = 156), indicating a modest inhibition of basal proteasome activity in the presence of the mutant proprotein (Fig. 5, a [e] vs. b [e]). Infection of SP-CΔexon4 cells with RSV resulted in a dose-dependent increase in fluorescence detected by microscopy and FACS (Fig. 5 a [e–h]). In contrast, fluorescence was not altered in SP-Cwt cells infected with RSV (Fig. 5 b [e–h]). Transfection efficiency was similar between the two cell lines, and the proteasome reporter was activated in SP-Cwt cells treated with MG-132 (Fig. 5 c). These results demonstrate that basal proteasome function is decreased in SP-CΔexon4 cells and that this perturbation is exacerbated during RSV infection.
Figure 5.

Proteasome function is decreased in RSV-infected SP-CΔexon4 cells. (a and b) The clonal cell lines expressing SP-CΔexon4 (a) or SP-Cwt (b) were transiently transfected with the proteasome sensor pZsProSensor-1 construct and infected with RSV at the indicated titers 24 h after transfection. Phase (top) and fluorescent (bottom) images were captured directly from the culture plate 24 h after infection. After image capture, cells were harvested and subjected to FACS analysis to determine MFI (reported in the top right corner of the bottom panels). (c) Clonal cells expressing SP-Cwt (panels a and c) or SP-CΔexon4 (panels b and d) were transfected with a plasmid encoding EGFP as a control for transfection efficiency (panels a and b) or with the pZsProSensor-1 vector in the presence of MG-132 (panels c and d) as a positive control. Cells were harvested and subjected to FACS analysis 24 h after transfection. Values for GFP+ cells and mean fluorescence intensity (MFI) are shown in the top right corner of each panel. Data is representative of three independent experiments. Bars, 50 μm.
SP-CΔexon4 sensitizes cells to proteasome inhibition and is associated with accumulation of mutant proprotein and activation of XBP-1
To determine whether decreased proteasome function induced ER stress and activation of apoptosis independently of RSV infection, the clonal cell lines were treated with the proteasome inhibitor MG-132. The SP-CΔexon4 cell line was exquisitely sensitive to proteasome inhibition. The majority of cells retracted and detached from the plate after treatment with 250 nM MG-132 for 18 h, and a more pronounced effect was detected at the 500 nM of concentration (Fig. 6 a [g and h]). SP-Cwt cells were minimally affected by similar treatment (Fig. 6 a [a–d]). Cell viability was decreased ∼10% in SP-Cwt cells and 50% in SP-CΔexon4 cells treated with 500 nM MG-132 (Fig. 6 b).
Figure 6.

SP-CΔexon4 sensitizes cells to proteasome inhibition. (a) SP-Cwt and SP-CΔexon4 clonal cell lines were plated at equal cell densities and treated with the proteasome inhibitor MG-132 at the indicated concentrations. Phase photographs of the cells were captured directly from the culture plate 18 h after treatment. Bars, 50 μm. (b) MTS assay of cells treated as indicated in a. Results are presented as means ± SD (error bars); each determination was performed in triplicate, and the experiment was repeated three times. *, P < 0.05 compared with SP-CΔexon4 unRx; #, P < 0.05 compared with SP-Cwt unRx. (c) Immunoblots of whole cell lysates from clonal cell lines expressing SP-Cwt or SP-CΔexon4 after MG-132 treatment. Immunoblots were probed with antibodies against proSP-C antibody or actin (loading control). (d) Clonal cell lines expressing SP-Cwt or SP-CΔexon4 were cultured in the presence (+) or absence (−) of 500 nM MG-132 for 18 h. Total RNA was isolated from cells and subjected to RT-PCR analysis with primers spanning the intron of unspliced XBP-1 mRNA. XBP-1(u) denotes unspliced, inactive XBP-1; XBP-1(s) denotes spliced, active XBP-1; products were separated on a 4% agarose gel.
To determine whether MG-132–induced cytotoxicity was associated with SP-CΔexon4 accumulation, immunoblot analysis was performed with a proSP-C antibody after MG-132 treatment. There was a dose-dependent increase in SP-CΔexon4 protein, whereas levels of the SP-Cwt protein were unaffected by MG-132 treatment (Fig. 6 c). Accumulation of unfolded proteins in the ER results in the activation of the IRE1–XBP-1 pathway (for review see Harding et al., 2002). Active XBP-1 was not detected in SP-Cwt cells after MG-132 treatment; in contrast, robust activation of XBP-1 was detected in SP-CΔexon4 cells after proteasome inhibition (Fig. 6 d). Collectively, these results demonstrate that proteasome inhibition resulted in dose-dependent cytotoxicity associated with the accumulation of SP-CΔexon4 and the activation of XBP-1.
Accumulation of SP-CΔexon4 is associated with apoptosis
To determine whether the accumulation of SP-CΔexon4 after proteasome inhibition was associated with apoptosis, cell lines were treated with MG-132 and stained with the fluorescent DNA-intercalating dye H33342 to assess nuclear architecture. Cells expressing SP-Cwt showed a diffuse, uniform staining of the nucleus typical of viable, healthy cells (Fig. 7 a, left panel and inset). Although the majority of cells expressing SP-CΔexon4 detached from the plate after MG-132 treatment and were excluded from this analysis, the cells that remained attached exhibited punctuate staining and nuclear condensation that is consistent with apoptosis (Fig. 7 a, right panel and inset). Furthermore, treatment of SP-CΔexon4–expressing HEK293 cells with MG-132 for 18 h resulted in the activation of caspase-3 (Fig. 7 b). Caspase-3 activation was not detected in SP-Cwt cells treated with MG-132, indicating that this event was specific for cells expressing mutant SP-CΔexon4 (Fig. 7 b). As previously reported, the expression of SP-CΔexon4 in the distal lung epithelium of transgenic mice resulted in cytotoxicity associated with lung dysmorphogenesis (Bridges et al., 2003). Immunohistochemistry was performed on fetal lung sections from a transgene-positive animal (Fig. 7 d, TG#2) and a wild-type littermate control (Fig. 7 d, WT) with an antibody that detects the cleaved isoform of caspase-3. Robust staining for active caspase-3 was detected in sloughed epithelial cells in the distal airway and isolated intact epithelial cells in the transgenic animal, whereas lung epithelial cells in the wild-type animal were negative (Fig. 7 d). Together, these results demonstrate that the accumulation of SP-CΔexon4, induced by proteasome inhibition in vitro or overexpression in vivo, was associated with the activation of apoptotic pathways. In contrast, RSV infection resulted in specific, extensive death of SP-CΔexon4–expressing cells (Fig. 4) without evidence of caspase-3 activation (Fig. 7 c).
Figure 7.

Accumulation of SP-CΔexon4 is associated with cell death. (a) SP-Cwt (left) and SP-CΔexon4 (right) clonal cell lines were plated at equal cell density on poly-d-lysine–coated coverslips and treated with 500 nM MG-132. 18 h after treatment, the cells that remained attached to the coverslips were fixed, permeabilized, and stained with H33342 dye to visualize the nuclear architecture. Higher magnification of one representative cell is shown in the insets. Bars, 50 μm; (insets), 10 μm. (b) Clonal cell lines expressing SP-Cwt or SP-CΔexon4 were cultured in the presence or absence of MG-132 at the indicated concentrations, harvested 18 h after treatment, and subjected to immunoblot analysis with a caspase-3 antibody that detects both the precursor and cleaved isoforms. (c) Clonal cell lines expressing SP-Cwt or SP-CΔexon4 were infected with RSV at the indicated plaque-forming units s for 24 h. Cell lysates were harvested and subjected to immunoblot analysis with a caspase-3 antibody. SP-CΔexon4 cells were treated with MG-132 overnight for a positive control. (d) Immunohistochemistry for cleaved caspase-3 on lung sections from wild-type (WT) or transgenic mice (TG#2) in which SP-CΔexon4 was expressed in the distal lung epithelium. Closed arrowheads represent cleaved caspase-3–positive cell debris, and open arrowheads represent red blood cells. Bars, 20 μm.
Discussion
The SP-CΔexon4 mutation was chosen for these experiments because of its link to ILD in human patients (Nogee et al., 2001, 2002), the severity of this mutation on the structure of the proprotein, and its association with cytotoxicity and lung dysmorphogenesis when expressed in transgenic mice (Bridges et al., 2003). Results of experiments performed in this study using stably transfected cells indicated that the SP-CΔexon4 cell line adapted to the constitutive expression of misfolded SP-C and that NF-κB played a pivotal role in the adaptive response. Infection of cells expressing SP-CΔexon4 with RSV resulted in enhanced cytotoxicity associated with decreased proteasome function, accumulation of the mutant proprotein, and cell death. Collectively, these results suggest that adaptation to chronic ER stress imposed by misfolded SP-C may promote resistance to ILD, whereas environmental insults, such as viral infection, may trigger the onset of disease in patients with mutations in SFTPC.
Although the effects of acute ER stress, imposed by xenotoxic agents such as thapsigargin and tunicamycin, are well established, little is known about the molecular pathways involved in adaptation to chronic ER stress imposed by a misfolded protein. The lack of a cytotoxic response coupled with the absence of ER stress induction in SP-CΔexon4 clonal cells suggested that these cells successfully adapted to the constitutive expression of misfolded SP-C. Several lines of evidence support this hypothesis. First, the level of SP-C mRNA in the SP-CΔexon4 cell line was comparable with that in the SP-Cwt cell line, thus excluding low expression of the mutant protein as a reason for the survival of SP-CΔexon4 cells. Second, in contrast to the wild-type protein, SP-CΔexon4 protein was barely detectable, indicating that the misfolded proprotein was recognized as terminally misfolded and rapidly degraded via the ERAD pathway. Furthermore, chronic expression of SP-CΔexon4 was associated with modestly reduced proteasome function that is consistent with near saturation of this degradative pathway. Third, components of the ER stress/UPR pathways were not transcriptionally up-regulated in the clonal SP-CΔexon4 cell line, which is in marked contrast to results in transiently transfected HEK293 cells (Bridges et al., 2003; Mulugeta et al., 2005). Fourth, chronic expression of SP-CΔexon4 was associated with differential expression of both pro- and anti-apoptotic genes, approximately half of which contained putative NF-κB–binding sites in their promoters. Lastly, the inhibition of NF-κB, which is known to promote cell survival in response to a variety of stresses, resulted in the death of SP-CΔexon4 cells. Collectively, these results are consistent with an NF-κB–dependent adaptive response to chronic ER stress imposed by the constitutive expression of SP-CΔexon4. Although the induction of other molecular pathways involved in cell survival may occur, the activation of NF-κB appears to be critical for survival in HEK293 cells that constitutively express SP-CΔexon4.
The role of NF-κB in the regulation of cytokine-stimulated proinflammatory gene expression is well established (Li and Verma, 2002). In this study, up-regulation of proinflammatory cytokines was not detected by microarray analyses; however, it is certainly possible that inflammation associated with lung injury and repair contributes to pathogenesis in vivo. NF-κB activity is also increased in response to the accumulation of transmembrane proteins in the ER, including the E3/19k protein of adenovirus (Pahl et al., 1996), middle human hepatitis B surface protein (Meyer et al., 1992), expression of major histocompatibility complex class 1 in the absence of β2-microglobin protein (Pahl et al., 1996), and p450 (Szczesna-Skorupa et al., 2004). In addition, transient expression of the misfolded Z variant of α1-antitrypsin in CHO and HEK293 cells resulted in NF-κB activation associated with decreased levels of IκBα and IκBβ (Lawless et al., 2004). Activation of NF-κB in response to the accumulation of newly synthesized membrane or misfolded secretory proteins is referred to as the ER overload response. Constitutive activation of NF-κB promotes survival of a wide range of cells, including B cells, hepatic cells, and cancer cells, and is a major target for cancer therapy (Kucharczak et al., 2003). NF-κB promotes cell survival, in part, by activating anti-apoptotic genes, including members of the inhibitor of apoptosis family, TRAF1 and TRAF2, and the Bcl2 homologues Bfl-1/A1 and Bcl-Xl (Kucharczak et al., 2003). Recent studies using tunicamycin and thapsigargin as stressor agents demonstrated an association between ER stress–induced PERK activation, eIF2α phosphorylation, and NF-κB activation (Jiang et al., 2003; Deng et al., 2004), effectively linking the UPR and the ER overload response. Although a definitive role for NF-κB activation in response to ER stress has not been established, it is thought to promote cell survival in this context.
Transient transfection of HEK293 cells with SP-CΔexon4 resulted in specific up-regulation of BiP, XBP-1, and HedJ1, which is consistent with the induction of UPR and ERAD to promote clearance of the misfolded protein and the alleviation of ER stress (Bridges et al., 2003; Mulugeta et al., 2005). In contrast, the constitutive expression of SP-CΔexon4 resulted in the differential expression of apoptosis-related genes with an apparent balance shifted toward NF-κB–dependent anti-apoptotic responses. Identification of environmental triggers that tip the balance toward apoptosis/cell death are clearly important for understanding disease pathogenesis in humans with SFTPC mutations. ILD is often associated with infection and/or inflammation (Vassallo, 2003; Noble and Homer, 2004). The marked variability in severity and age of onset of lung disease in the SP-CL188Q pedigrees suggested that environmental factors might be involved in triggering the onset of ILD (Thomas et al., 2002a; Chibbar et al., 2004). Consistent with this hypothesis, five individuals in the SP- CL188Q kindreds were diagnosed with viral infection before the manifestation of lung disease. Importantly, cells stably expressing SP-CΔexon4 were much more susceptible to viral-induced cell death, suggesting that infection may be an environmental trigger for ILD.
Although RSV infection in vivo is associated with epithelial cell damage and cellular desquamation, these cytopathic effects are primarily mediated by an augmented immune response initiated by the infected host cells rather than viral-induced cell death. In support of this hypothesis, mice depleted of CD4 and CD8 cells exhibited persistent viral replication without illness (Graham et al., 1991). Furthermore, RSV infection of cultured airway epithelial cells (MOI of ∼20; i.e., twice the dose used in this study) did not result in cytopathology (Zhang et al., 2002). Consistent with these findings, HEK293 cells constitutively expressing SP-Cwt exhibited relatively little cell death in response to RSV infection (Fig. 4 a). In marked contrast, RSV infection of cells expressing SP-CΔexon4 resulted in extensive cell death associated with proteasome inhibition and accumulation of mutant proprotein. In vivo, injury of alveolar epithelial cells is an early event in progressive pulmonary fibrosis. Impaired reepithelialization leads to sustained proliferation/activation of interstitial fibroblasts and myofibroblasts, resulting in the accumulation of extracellular matrix and, ultimately, fibrosis (Thannickal et al., 2004; Noble and Homer, 2005). The increased susceptibility of some individuals to severe RSV pneumonia may further contribute to variability in the severity and onset of the disease (Peebles and Graham, 2005).
Although RSV infection was specifically cytotoxic for SP-CΔexon4–expressing cells, the molecular pathway leading to cell death is not clear. Cell death did not appear to be apoptosis dependent, as caspase-3 cleavage was undetectable in the RSV-infected cells (Fig. 7 c). RSV has been shown to inhibit apoptosis of cultured epithelial cells via the activation of EGF receptor–extracellular signal-regulated kinase and PI3K–AKT signaling pathways, thereby promoting self-replication (Thomas et al., 2002b; Monick et al., 2005). In this study, RSV-induced cytotoxicity was associated with proteasome dysfunction, accumulation of the mutant proprotein SP-CΔexon4, and pronounced activation of the UPR. Induction of ER stress and proteasome inhibition in RSV-infected SP-CΔexon4 cells may be related, in part, to forced synthesis of viral membrane proteins, including the F, G, and SH proteins. UPR activation was higher in SP-CΔexon4 cells than in SP-Cwt cells, suggesting that the production of RSV membrane proteins superimposed on the expression of misfolded SP-CΔexon4 protein may overwhelm the degradative capacity of the proteasome, leading to accumulation of cytotoxic forms of SP-CΔexon4 and subsequent cell death. Therefore, it is possible that proteasome dysfunction and accumulation of SP-CΔexon4 drives the cell down a nonapoptotic cell death pathway in the presence of RSV. In contrast, accumulation of SP-CΔexon4 in transgenic mice and MG-132–treated cells (i.e., in the absence of RSV infection) resulted in caspase-3 activation, which is typical of apoptosis (Fig. 7 c; Bridges et al., 2003).
Based on the findings of this study, the following model is proposed. SP-Cwt protein is correctly folded and exported from the ER, whereas SP-CΔexon4 is terminally misfolded and degraded by a proteasome-dependent pathway. Constitutive expression of terminally misfolded SP-CΔexon4 results in chronic ER stress and an NF-κB–dependent cytoprotective response. Superimposition of a secondary stress, such as RSV infection, on the constitutive expression of SP-CΔexon4 leads to proteasome dysfunction, accumulation of SP-CΔexon4, and cytotoxicity. Accumulation of SP-CΔexon4 and cytotoxicity may contribute to the pathogenesis of ILD associated with mutations in SFTPC.
Materials and methods
Reagents
Recombinant TNFα protein was obtained from PeproTech. The MTS reduction assay kit and Dual-Luciferase Reporter Assay System were purchased from Promega; MTS reduction assays were performed in 96-well plates according to the manufacturer's protocol. MG-132, SN50, and control peptide (SN50mut) were obtained from EMD Biosciences.
SP-C cDNA constructs and generation of stably transfected cell lines
Full-length human wild-type SP-C (SP-Cwt) cDNA and SP-CΔexon4, generated as previously described (Bridges et al., 2003), were subcloned into pTRE2-Hyg (BD Biosciences) to generate SP-Cwt/pTRE2-Hyg and SP-CΔexon4/pTRE2-Hyg. To generate stably transfected cell lines, HEK293 Tet-Off cells (CLONTECH Laboratories, Inc.) were transiently transfected with SP-Cwt/pTRE2-Hyg or SP-CΔexon4/pTRE2-Hyg using LipofectAMINE 2000 (Invitrogen) in the presence of 250 μg/ml hygromycin B as the selection agent. Doxycycline was present in the media during the selection process to obtain regulatable lines (i.e., expression of SP-Cwt or SP-CΔexon4 was dependent on doxycycline withdrawal). However, all lines that were initially regulatable eventually reverted to constitutive expression of the transgene. Therefore, SP-Cwt or SP-CΔexon4 cells for this study were cultured in the absence of doxycycline and G418 (selection agent for the tTA cassette). Clonal colonies were isolated, amplified, and screened for integration of the transgene by RT-PCR using SP-C–specific primers and Western analyses using an antibody directed against the NH2-terminal peptide of proSP-C (Bridges et al., 2003).
RNA isolation, RT-PCR, and microarray analysis
Total RNA for RT-PCR and real-time PCR analysis was isolated using the acidified guanidinium method (Chomczynski and Sacchi, 1987), treated with DNase I (DNA free; Ambion), and reverse transcribed into cDNA using SuperScript II Reverse Transcriptase (Invitrogen). Forward and reverse primer sequences for human XBP-1 are 5′-GGACTTAAGACAGCGCTTGG-3′ and 5′-TGAGAGGTGCTTCCTCGATT-3′. PCR reactions for XBP-1 were performed for 35 cycles, and products were separated on 4% agarose gels. Verification of microarray data for selected genes was performed by real-time PCR analysis with a SmartCycler (Cepheid) using reaction conditions as previously described (Hyatt et al., 2004).
RNA samples for microarray analysis were prepared as previously described (DeFelice et al., 2003) and hybridized to the GeneChip Human Genome U133 Set (HG-U133A and HG-U133B; Affymetrix, Inc.) according to the manufacturer's protocol. Affymetrix Microarray Suite 5.0 was used to scan and quantitate the gene chips under default scan settings. Normalization was performed using the Robust Multichip Average model (Irizarry et al., 2003a,b). Data were further analyzed using Significance Analysis Of Microarrays (Tusher et al., 2001) and Genespring 7.2 (Silicon Genetics). Detection of differential expression was performed using random permutation and Welch's approximate t test for mutant and control groups at P ≤ 0.01, false discovery rate ≤ 10%, a minimal of twofold changes in absolute ratio, and a minimum of two present calls by Affymetrix algorithm in three samples with the relative higher expression. Gene ontology analysis was performed using the database for annotation, visualization, and integrated discovery (Dennis et al., 2003). Potential protein–protein interactions were identified using PathwayAssist (Ariadne Genomics). NF-κB–binding sites (GGGACTTTCC) were scanned in −2-kb promoter regions of all differentially expressed genes from microarray analysis using MatInspector (Genomatix), allowing a maximum of one mismatch.
Cell culture and Western analysis
HEK293 stable cell lines were propagated as previously described (Bridges et al., 2003). Cell lysate preparation, protein standardization, and Western analysis were performed as previously described (Bridges et al., 2003). Nuclear extracts were isolated as previously described (Odoms et al., 2004). Antibody sources are as follows: anti–proSP-C (Vorbroker et al., 1995), anti–caspase-3 (Cell Signaling), antihistone H2B (Imgenex), anti-p65 subunit of NF-κB (Santa Cruz Biotechnology, Inc.), and anti-actin (Bridges et al., 2003).
Confocal fluorescence
Cells for confocal microscopy were prepared as previously described (Conkright et al., 2001). Primary antibodies included a polyclonal anti–proSP-C (Bridges et al., 2003), a monoclonal anti-LAMP1/CD107A (Research Diagnostics, Inc.), and a monoclonal anti-KDEL (StressGen Biotechnologies). Secondary antibodies included anti–rabbit FITC-conjugated and anti–mouse Texas red–conjugated secondary antibodies (Jackson ImmunoResearch Laboratories). Vectashield Hardset (Vector Laboratories) was used as the mounting medium. Fluorescence was visualized on a confocal microscope (LSM510; Carl Zeiss MicroImaging, Inc.) using FITC/Texas red filters and a 40× NA 1.3 objective.
Fluorescence and phase microscopy of live cells was performed on a microscope (LX70; Olympus) equipped with a UV lamp, FITC/Texas red filters, and a 20× NA 0.40 objective. Images were captured with a digital camera (MagnaFire; Optronics) using MagnaFire software (Optronics). For cell death assays, 2 μl of 1 mg/ml PI solution was added to live cells for 10 min at room temperature before visualization.
Flow cytometry
HEK293 cells were detached from the culture plate 24 h after RSV infection/transient transfection by treatment with trypsin/EDTA, washed once with PBS, and resuspended in FACS buffer consisting of PBS with 0.1% FBS and 0.05% sodium azide. Cell-associated fluorescence was measured on a FACScalibur flow cytometer using CellQuest software (BD Biosciences). For each sample, 20,000 events were acquired.
Immunohistochemistry
Immunohistochemistry was performed on mouse fetal lung samples that were previously described (Bridges et al., 2003) using an antibody that detects the cleaved form of caspase-3 (R&D Systems) at a 1:5,000 dilution. Localization of antigen–antibody complexes was performed as previously described (Bridges et al., 2003).
RSV production and infection protocol
The A2 strain of RSV was amplified, purified, and quantitated in Hep2 as previously described (LeVine et al., 1999). Clonal cell lines were infected with RSV using a previously established protocol for adenoviral infection (Bridges et al., 2003). For studies in which proteasome activity was assessed after RSV infection, the proteasome sensor plasmid pZsProSensor-1 (CLONTECH Laboratories, Inc.) was transfected into the cells using LipofectAMINE 2000 24 h before RSV infection. The pZsProSensor-1 plasmid encodes a destabilized GFP consisting of amino acids 422–461 of the degradation domain of mouse ornithine decarboxylase protein fused to the COOH terminus of the naturally occurring reef coral Zoanthus species protein (ZsGreen). This fusion protein is rapidly degraded by the proteasome in an ubiquitin-independent manner.
Luciferase constructs and assays
The NF-κB–dependent ELAM-1 promoter-driven firefly luciferase plasmid (pELAM-luc) was obtained from M.J. Fenton (University of Maryland, Baltimore, MA). The p5xATF6GL3 plasmid (also known as the UPR element luciferase reporter) was obtained from R. Prywes (Columbia University, New York, NY). The IκB SR expression plasmid was obtained from R. Hay (University of St. Andrews, St. Andrews, Scotland). The pRL-TK plasmid was purchased from Promega. 400 ng of firefly luciferase construct was cotransfected with 50 ng pRL-TK using LipofectAMINE 2000 (Invitrogen). Cells were harvested 48 h after transfection, and luciferase activity was quantified using the Dual-Luciferase Assay system in a Berthold multitube luminometer. Data are plotted as the ratio of firefly/renilla activity to correct for transfection efficiency among samples.
Statistics
Data were analyzed with InStat version 3.0 (GraphPad Software). Values are presented as means ± SD. Multiple comparisons were made by analysis of variance between groups using the Tukey-Kramer multiple comparisons test, and paired samples were analyzed by t tests. In both cases, statistical significance was defined as P < 0.05 or less.
Online supplemental material
Fig. S1 is a model of functional associations between differentially expressed genes in SP-CΔexon4 cells associated with apoptosis using PathwayAssist software. Table SI lists descriptive information of apoptosis-associated genes from the model in Fig. S1. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200508016/DC1.
Supplementary Material
Acknowledgments
The authors would like to thank Chen Xia Duan, Mei Wang, Senad Divanovic, Al Senft, Mukund Raghavan, and Xiaofei Shangguan for technical assistance and/or advice.
This work is supported by National Institutes of Health awards PO1-HL61646 and P50-HL56387 to T.E. Weaver.
Abbreviations used in this paper: ERAD, ER-associated degradation; ILD, interstitial lung disease; MFI, mean fluorescence intensity; MOI, multiplicity of infection; PI, propidium iodide; RSV, respiratory syncytial virus; SP-C, surfactant protein C; SR, super repressor; UPR, unfolded protein response.
References
- Bridges, J.P., S.E. Wert, L.M. Nogee, and T.E. Weaver. 2003. Expression of a human surfactant protein C mutation associated with interstitial lung disease disrupts lung development in transgenic mice. J. Biol. Chem. 278:52739–52746. [DOI] [PubMed] [Google Scholar]
- Cameron, H.S., M. Somaschini, P. Carrera, A. Hamvas, J.A. Whitsett, S.E. Wert, G. Deutsch, and L.M. Nogee. 2005. A common mutation in the surfactant protein C gene associated with lung disease. J. Pediatr. 146:370–375. [DOI] [PubMed] [Google Scholar]
- Chibbar, R., F. Shih, M. Baga, E. Torlakovic, K. Ramlall, R. Skomro, D.W. Cockcroft, and E.G. Lemire. 2004. Nonspecific interstitial pneumonia and usual interstitial pneumonia with mutation in surfactant protein C in familial pulmonary fibrosis. Mod. Pathol. 17:973–980. [DOI] [PubMed] [Google Scholar]
- Chomczynski, P., and N. Sacchi. 1987. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162:156–159. [DOI] [PubMed] [Google Scholar]
- Conkright, J.J., J.P. Bridges, C.L. Na, W.F. Voorhout, B. Trapnell, S.W. Glasser, and T.E. Weaver. 2001. Secretion of surfactant protein C, an integral membrane protein, requires the N-terminal propeptide. J. Biol. Chem. 276:14658–14664. [DOI] [PubMed] [Google Scholar]
- DeFelice, M., D. Silberschmidt, R. DiLauro, Y. Xu, S.E. Wert, T.E. Weaver, C.J. Bachurski, J.C. Clark, and J.A. Whitsett. 2003. TTF-1 phosphorylation is required for peripheral lung morphogenesis, perinatal survival, and tissue specific gene expression. J. Biol. Chem. 278:35574–35583. [DOI] [PubMed] [Google Scholar]
- Dell'Angelica, E.C., C. Mullins, S. Caplan, and J.S. Bonifacino. 2000. Lysosome-related organelles. FASEB J. 14:1265–1278. [DOI] [PubMed] [Google Scholar]
- Deng, J., P.D. Lu, Y. Zhang, D. Scheuner, R.J. Kaufman, N. Sonenberg, H.P. Harding, and D. Ron. 2004. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24:10161–10168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis, G., Jr., B.T. Sherman, D.A. Hosack, J. Yang, W. Gao, H.C. Lane, and R.A. Lempicki. 2003. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 4:P3. [PubMed] [Google Scholar]
- Graham, B.S., L.A. Bunton, P.F. Wright, and D.T. Karzon. 1991. Role of T lymphocyte subsets in the pathogenesis of primary infection and rechallenge with respiratory syncytial virus in mice. J. Clin. Invest. 88:1026–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H.P., M. Calfon, F. Urano, I. Novoa, and D. Ron. 2002. Transcriptional and translational control in the mammalian unfolded protein response. Annu. Rev. Cell Dev. Biol. 18:575–599. [DOI] [PubMed] [Google Scholar]
- Hetz, C., M. Russelakis-Carneiro, K. Maundrell, J. Castilla, and C. Soto. 2003. Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J. 22:5435–5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi, J., T. Katayama, Y. Eguchi, T. Kudo, M. Taniguchi, Y. Koyama, T. Manabe, S. Yamagishi, Y. Bando, K. Imaizumi, et al. 2004. Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and A{β}-induced cell death. J. Cell Biol. 165:347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, B.A., X. Shangguan, and J.M. Shannon. 2004. FGF-10 induces SP-C and Bmp4 and regulates proximal-distal patterning in embryonic tracheal epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 287:L1116–L1126. [DOI] [PubMed] [Google Scholar]
- Irizarry, R.A., B.M. Bolstad, F. Collin, L.M. Cope, B. Hobbs, and T.P. Speed. 2003. a. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry, R.A., B. Hobbs, F. Collin, Y.D. Beazer-Barclay, K.J. Antonellis, U. Scherf, and T.P. Speed. 2003. b. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 4:249–264. [DOI] [PubMed] [Google Scholar]
- Jiang, H.Y., S.A. Wek, B.C. McGrath, D. Scheuner, R.J. Kaufman, D.R. Cavener, and R.C. Wek. 2003. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol. Cell. Biol. 23:5651–5663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucharczak, J., M.J. Simmons, Y. Fan, and C. Gelinas. 2003. To be, or not to be: NF-kappaB is the answer–role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene. 22:8961–8982. [DOI] [PubMed] [Google Scholar]
- Lawless, M.W., C.M. Greene, A. Mulgrew, C.C. Taggart, S.J. O'Neill, and N.G. McElvaney. 2004. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J. Immunol. 172:5722–5726. [DOI] [PubMed] [Google Scholar]
- LeVine, A.M., J. Gwozdz, J. Stark, M. Bruno, J. Whitsett, and T. Korfhagen. 1999. Surfactant protein-A enhances respiratory syncytial virus clearance in vivo. J. Clin. Invest. 103:1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q., and I.M. Verma. 2002. NF-kappaB regulation in the immune system. Nat. Rev. Immunol. 2:725–734. [DOI] [PubMed] [Google Scholar]
- Meyer, M., W.H. Caselmann, V. Schluter, R. Schreck, P.H. Hofschneider, and P.A. Baeuerle. 1992. Hepatitis B virus transactivator MHBst: activation of NF-kappa B, selective inhibition by antioxidants and integral membrane localization. EMBO J. 11:2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monick, M.M., K. Cameron, J. Staber, L.S. Powers, T.O. Yarovinsky, J.G. Koland, and G.W. Hunninghake. 2005. Activation of the epidermal growth factor receptor by respiratory syncytial virus results in increased inflammation and delayed apoptosis. J. Biol. Chem. 280:2147–2158. [DOI] [PubMed] [Google Scholar]
- Mulugeta, S., V. Nguyen, S.J. Russo, M. Muniswamy, and M.F. Beers. 2005. A surfactant protein C precursor protein BRICHOS domain mutation causes endoplasmic reticulum stress, proteasome dysfunction, and caspase 3 activation. Am. J. Respir. Cell Mol. Biol. 32:521–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa, T., H. Zhu, N. Morishima, E. Li, J. Xu, B.A. Yankner, and J. Yuan. 2000. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 403:98–103. [DOI] [PubMed] [Google Scholar]
- Noble, P.W., and R.J. Homer. 2004. Idiopathic pulmonary fibrosis: new insights into pathogenesis. Clin. Chest Med. 25:749–758. [DOI] [PubMed] [Google Scholar]
- Noble, P.W., and R.J. Homer. 2005. Back to the future: historical perspective on the pathogenesis of idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 33:113–120. [DOI] [PubMed] [Google Scholar]
- Nogee, L.M., A.E. Dunbar, S.E. Wert, F. Askin, A. Hamvas, and J.A. Whitsett. 2001. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N. Engl. J. Med. 344:573–579. [DOI] [PubMed] [Google Scholar]
- Nogee, L.M., A.E. Dunbar, S. Wert, F. Askin, A. Hamvas, and J.A. Whitsett. 2002. Mutations in the surfactant protein C gene associated with interstitial lung disease. Chest. 121:20S–21S. [DOI] [PubMed] [Google Scholar]
- Odoms, K., T.P. Shanley, and H.R. Wong. 2004. Short-term modulation of interleukin-1beta signaling by hyperoxia: uncoupling of IkappaB kinase activation and NF-kappaB-dependent gene expression. Am. J. Physiol. Lung Cell. Mol. Physiol. 286:L554–L562. [DOI] [PubMed] [Google Scholar]
- Oyadomari, S., and M. Mori. 2004. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11:381–389. [DOI] [PubMed] [Google Scholar]
- Pahl, H.L., and P.A. Baeuerle. 1995. Expression of influenza virus hemagglutinin activates transcription factor NF-kappa B. J. Virol. 69:1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahl, H.L., M. Sester, H.G. Burgert, and P.A. Baeuerle. 1996. Activation of transcription factor NF-κB by the adenovirus E3/19K protein requires its ER retention. J. Cell Biol. 132:511–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles, R.S., Jr., and B.S. Graham. 2005. Pathogenesis of respiratory syncytial virus infection in the murine model. Proc. Am. Thorac. Soc. 2:110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder, M., and R.J. Kaufman. 2005. The mammalian unfolded protein response. Annu. Rev. Biochem. 74:739–789. [DOI] [PubMed] [Google Scholar]
- Szczesna-Skorupa, E., C.D. Chen, H. Liu, and B. Kemper. 2004. Gene expression changes associated with the endoplasmic reticulum stress response induced by microsomal cytochrome P450 overproduction. J. Biol. Chem. 279:13953–13961. [DOI] [PubMed] [Google Scholar]
- Tardif, K.D., K. Mori, R.J. Kaufman, and A. Siddiqui. 2004. Hepatitis C virus suppresses the IRE1-XBP1 pathway of the unfolded protein response. J. Biol. Chem. 279:17158–17164. [DOI] [PubMed] [Google Scholar]
- Thannickal, V.J., G.B. Toews, E.S. White, J.P. Lynch III, and F.J. Martinez. 2004. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 55:395–417. [DOI] [PubMed] [Google Scholar]
- Thomas, A.Q., K. Lane, J. Phillips III, M. Prince, C. Markin, M. Speer, D.A. Schwartz, R. Gaddipati, A. Marney, J. Johnson, et al. 2002. a. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 165:1322–1328. [DOI] [PubMed] [Google Scholar]
- Thomas, K.W., M.M. Monick, J.M. Staber, T. Yarovinsky, A.B. Carter, and G.W. Hunninghake. 2002. b. Respiratory syncytial virus inhibits apoptosis and induces NF-kappa B activity through a phosphatidylinositol 3-kinase-dependent pathway. J. Biol. Chem. 277:492–501. [DOI] [PubMed] [Google Scholar]
- Tusher, V.G., R. Tibshirani, and G. Chu. 2001. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA. 98:5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urano, F., X. Wang, A. Bertolotti, Y. Zhang, P. Chung, H.P. Harding, and D. Ron. 2000. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 287:664–666. [DOI] [PubMed] [Google Scholar]
- Vassallo, R. 2003. Viral-induced inflammation in interstitial lung diseases. Semin. Respir. Infect. 18:55–60. [DOI] [PubMed] [Google Scholar]
- Vorbroker, D.K., S.A. Profitt, L.M. Nogee, and J.A. Whitsett. 1995. Aberrant processing of surfactant protein C (SP-C) in hereditary SP-B deficiency. Am. J. Physiol. 268:L647–L656. [DOI] [PubMed] [Google Scholar]
- Wang, W.J., S. Mulugeta, S.J. Russo, and M.F. Beers. 2003. Deletion of exon 4 from human surfactant protein C results in aggresome formation and generation of a dominant negative. J. Cell Sci. 116:683–692. [DOI] [PubMed] [Google Scholar]
- Weaver, T.E., C.L. Na, and M. Stahlman. 2002. Biogenesis of lamellar bodies, lysosome-related organelles involved in storage and secretion of pulmonary surfactant. Semin. Cell Dev. Biol. 13:263–270. [DOI] [PubMed] [Google Scholar]
- Yoshida, H., T. Matsui, N. Hosokawa, R.J. Kaufman, K. Nagata, and K. Mori. 2003. A time-dependent phase shift in the Mammalian unfolded protein response. Dev. Cell. 4:265–271. [DOI] [PubMed] [Google Scholar]
- Zhang, L., M.E. Peeples, R.C. Boucher, P.L. Collins, and R.J. Pickles. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 76:5654–5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}