

Abstract
In eukaryotes, the multivesicular body (MVB) sorting pathway plays an essential role in regulating cell surface protein composition, thereby impacting numerous cellular functions. Vps4, an ATPase associated with a variety of cellular activities, is required late in the MVB sorting reaction to dissociate the endosomal sorting complex required for transport (ESCRT), a requisite for proper function of this pathway. However, regulation of Vps4 function is not understood. We characterize Vta1 as a positive regulator of Vps4 both in vivo and in vitro. Vta1 promotes proper assembly of Vps4 and stimulates its ATPase activity through the conserved Vta1/SBP1/LIP5 region present in Vta1 homologues across evolution, including human SBP1 and Arabidopsis thaliana LIP5. These results suggest an evolutionarily conserved mechanism through which the disassembly of the ESCRT proteins, and thereby MVB sorting, is regulated by the Vta1/SBP1/LIP5 proteins.
Introduction
The endosomal system coordinates intracellular protein trafficking between several subcellular compartments, including the Golgi, plasma membrane, and lysosome. A variety of sorting reactions confer distinct outcomes onto endosomal membrane proteins: recycling to an earlier compartment, residence within a compartment, or degradation in the lysosome. Delivery of endosomal membrane proteins to the lumen of the hydrolytic lysosome is the result of a sorting event that occurs during the formation of multivesicular bodies (MVBs; for reviews see Katzmann et al., 2002; Gruenberg and Stenmark, 2004; Babst, 2005). MVB formation occurs when the limiting membrane of the endosome invaginates and buds into its own lumen, bringing with it a subset of the proteins residing therein, including endocytosed, activated cell surface receptors. Heterotypic fusion of the MVB with the lysosome results in the delivery of the cargo-containing intralumenal MVB vesicles to the degradative activities sequestered within the lysosomal lumen. The sorting of proteins to the intralumenal vesicles of an MVB is not limited to endocytic cargoes. A subset of cargo directly sent from the Golgi apparatus is actively sorted into these vesicles without transiting the cell surface, typically referred to as biosynthetic MVB cargo (for review see Katzmann et al., 2002). In addition to facilitating delivery of proteins to the lysosome for degradation or processing, the MVB machinery participates in the budding of retroviruses from the host cell (for review see Morita and Sundquist, 2004). Thus, understanding the mechanisms governing MVB sorting is important with regards to processes as diverse as the modulation of cell surface–receptor signaling and the budding of retroviruses.
For cellular homeostasis to be maintained, entry into the degradative MVB pathway must be tightly regulated, and the machinery mediating this sorting event is highly conserved between yeast and man (for reviews see Katzmann et al., 2002; Raiborg et al., 2003; Babst, 2005). Recent progress toward our understanding of this process has been greatly facilitated by genetic and biochemical studies in yeast. Posttranslational modification of endosomal proteins with ubiquitin serves as a signal for their inclusion into intralumenal vesicles during MVB formation (for review see Katzmann et al., 2002). Endosomal membrane proteins that have been covalently modified with ubiquitin are sorted into the MVB pathway via the action of the class E vacuolar protein sorting (Vps) proteins. The majority of the class E Vps proteins are subunits of protein complexes called ESCRTs (endosomal sorting complexes required for transport; for reviews see Katzmann et al., 2002; Raiborg et al., 2003; Babst, 2005). Class E vps mutants display the phenotype of missorting MVB cargoes to the limiting membrane of the vacuole and the class E compartment rather than the vacuolar lumen, indicating a requisite role for these factors in the function of the MVB pathway (Odorizzi et al., 1998).
The biochemical activities of the ESCRTs and their accessory proteins have begun to be characterized. At present, the best defined are the cargo-recognition activities of Vps27/Hrs and ESCRT-I, both of which bind ubiquitinated cargoes and direct them into the MVB pathway in a coordinated manner (Katzmann et al., 2001; Bilodeau et al., 2002; Katzmann et al., 2003). Genetic evidence places ESCRT-II function downstream of ESCRT-I, where it plays a critical role in the assembly of ESCRT-III (Babst et al., 2002b). Appropriate ESCRT-III assembly is required for the recruitment of additional factors involved in the deubiquitination of MVB cargoes before their inclusion into intralumenal vesicles (e.g., Doa4 and Bro1; Amerik et al., 2000; Luhtala and Odorizzi, 2004). Additionally, ESCRT-III recruits the AAA-ATPase (ATPase associated with a variety of cellular activities) Vps4 to the MVB sorting site for dissociation of the ESCRTs at a step late in the sorting reaction (Babst et al., 1998). Loss of Vps4 ATPase activity results in an accumulation of ESCRT proteins on endosomal compartments and concurrent loss of MVB sorting (Babst et al., 1998, 2002a). Although ESCRT-III has been shown to recruit Vps4 to the site of MVB sorting, no regulators of its ATPase activity have been described to date.
Vps4 is the only class E Vps protein that has a readily measurable enzymatic activity, the hydrolysis of ATP. In the absence of ATP, Vps4 exist as a dimer. ATP binding by Vps4 results in its homooligomerization into a complex of ∼440 kD. ATP hydrolysis by the Vps4 oligomer, required for the release of the ESCRTs from the endosomal membrane, returns Vps4 to its inactive dimeric state (Babst et al., 1998). Spatial and temporal regulation of this Vps4 cycle is critical for the function of the MVB sorting reaction, as this represents a potential mechanism by which to modulate flux through the pathway. We provide evidence that Vta1 is a positive regulator of Vps4 in vivo. This finding is supported by biochemical analyses indicating Vta1 directly binds to Vps4 and stimulates its ATPase activity in vitro. Additionally, we define a 30-residue COOH-terminal sequence highly conserved in yeast Vta1, mammalian SBP1 (SKD1 binding protein 1), and plant LIP5 as a new region (Vta1/SBP1/LIP5 [VSL]) necessary and partially sufficient for stimulation of Vps4 ATPase activity. These findings implicate Vta1 as a novel regulatory component of the MVB machinery that contributes to the temporal and spatial activation of Vps4. Conservation of the VSL region suggests that this is an evolutionarily conserved mechanism by which Vta1, SBP1, and LIP5 regulate the MVB sorting reaction.
Results
Vta1 positively modulates Vps4 function in vivo
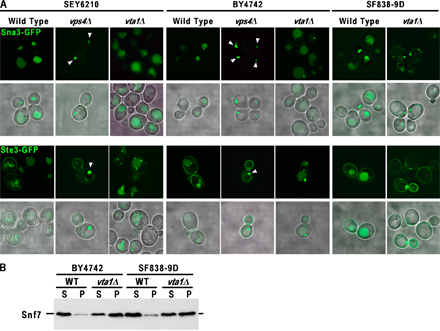
The VTA1 (Vps twenty associated 1) ORF (YLR181c) was identified as required for proper function of the MVB pathway during the course of genetic analyses using the Research Genetics (BY4742) deletion collection (Fig. 1 and Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200508166/DC1; unpublished data). Consistent with this observation, two recent reports have implicated Vta1 in the function of the MVB pathway, but the function of this protein was not elucidated (Yeo et al., 2003; Shiflett et al., 2004). To better define the role Vta1 plays in the MVB pathway, endosomal MVB cargoes originating from either the Golgi or plasma membrane were analyzed in cells lacking Vta1 (vta1Δ) as compared with wild-type cells or cells lacking the class E protein Vps4 (vps4Δ; Fig. 1 A and Fig. S1). Generation of VTA1 deletion strain in our standard genetic background (SEY6210) revealed a significantly weaker phenotype than was observed in the BY4742 background. For this reason, MVB sorting phenotypes were examined in SEY6210, BY4742, and SF838-9D genetic backgrounds to document any variation in the vta1Δ phenotype.
Figure 1.

MVB sorting defect in vta1Δ is distinct from the known class E vps mutant. (A) The MVB cargo GFP-CPS was visualized by fluorescent and brightfield microscopy in living cells corresponding to wild type, vta1Δ, or the known class E vps mutant vps4Δ in three distinct genetic backgrounds. Arrowheads highlight the class E compartment in vps4Δ cells. (B) Synthetic genetic interaction between vta1Δ and vps4tsf. Ste2-GFP was visualized by fluorescent microscopy in vta1Δ, vps4tsf, and vta1Δ vps4tsf genetic backgrounds at permissive temperature. (C) Vta1 is required for efficient release of the ESCRT-III subunits Snf7 and Vps24 from endosomal membranes. Differential centrifugation was performed on wild-type, vps4Δ, vta1Δ, and vps4Δ vta1Δ cell extracts. The resulting soluble (S) and pellet (P) fractions were probed with anti-Snf7 or anti-Vps24 antibodies.
The proper sorting of GFP-tagged MVB cargo proteins is scored by their localization within the lumenal space of the vacuole, as opposed to its limiting membrane. Using the biosynthetic MVB cargo GFP-tagged carboxypeptidase S (CPS) to visualize this process, the vast majority of the fusion protein was localized to the vacuole lumen in wild-type cells (Fig. 1 A). In contrast to cells lacking Vps4, the MVB sorting pathway was only partially defective in cells lacking Vta1, as indicated by the presence of GFP-CPS in the lumen as well as the limiting membrane of the vacuole. In addition, vta1Δ cells lacked the characteristic class E compartment that was readily observed in vps4Δ cells (Fig. 1 A and Fig. S1 A, arrowheads). One possible explanation for the defects seen in CPS sorting would be a defect in its ubiquitin modification. This was addressed by immunoprecipitation of CPS from wild-type and vta1Δ cells, followed by anti-ubiquitin Western blotting. This analysis revealed no defect in ubiquitin modification of CPS in vta1Δ cells (unpublished data). The trafficking of an endocytic MVB cargo (Ste3-GFP) or an ubiquitin-independent biosynthetic MVB cargo (Sna3-GFP) was also examined. As expected, both Ste3- and Sna3-GFP delivery to the vacuole lumen was perturbed in a vps4Δ strain (Fig. S1). Depending on the genetic background, the severity of the phenotype observed with loss of Vta1 function varied; vta1Δ cells in the SEY6210 background displayed a phenotype largely indistinguishable from wild type, whereas the other backgrounds displayed more severe missorting phenotypes (Fig. S1 A). Additional tests were performed to address hallmarks of the class E phenotype, including secretion of carboxypeptidase Y and the processing of CPS to its mature form; these analyses again revealed a distinct difference between vps4Δ and vta1Δ cells, with vta1Δ cells exhibiting wild-type to intermediate phenotypes (Fig. S1). These results indicate that endosomal function in general, and MVB sorting in particular, is not dramatically perturbed in the vta1Δ mutant in all genetic backgrounds, in contrast to previously characterized class E vps mutants. This suggests that Vta1 is likely not a core component of the class E Vps machinery but rather represents a potential modulator of ESCRT function.
In vitro, Vta1 has been demonstrated to bind the AAA-ATPase Vps4 (Yeo et al., 2003). Similarly, the murine homologue of Vta1, SBP1, has also been shown to bind the murine homologue of Vps4, SKD1 (Fujita et al., 2004). These observations, together with the Vta1 localization data presented in Fig. 2, led us to examine whether Vta1 impacted the Vps4-dependent MVB sorting reaction in vivo. To uncover a synthetic genetic interaction, vta1Δ was combined with a previously described temperature-sensitive allele of Vps4 (Babst et al., 1997) and the trafficking of Ste2-GFP was analyzed in the double mutant as well as in vta1 and vps4tsf single-mutant cells at permissive temperature. Ste2-GFP was delivered to the lumen of the vacuole in both vta1Δ and vps4tsf cells (Fig. 1 B), an indication of efficient function of the MVB pathway. However, the vta1Δ vps4tsf double mutant accumulated Ste2-GFP in exaggerated endosomal compartments with no obvious vacuolar signal, indicative of defective MVB sorting. The synthetic genetic interaction suggests that Vta1 acts with Vps4 in the MVB pathway in vivo.
Figure 2.

Interactions of Vta1 and Vps4 in vivo. (A) Vta1 requires Vps4 for endosomal association. Fluorescence microscopy was performed on wild-type, vps4Δ, and vps4E233Q cells expressing Vta1-GFP and the endosomal marker DsRed-FYVE. Arrowheads indicate endosomes in wild-type cells or class E compartments in vps4Δ and vps4E233Q. Dashed blue lines are used to outline cells. Bar, 5 μM. (B) Quantification of endosomal association of Vta1 in wild type, vps4Δ, and vps4E233Q. Differential centrifugation was performed on extracts made from wild-type, vps4Δ, and vps4E233Q cells expressing Vta1-HAand probed with anti-HA antibody. Immunoreactive species were quantitated and graphed as the percentage present in soluble and membrane fractions. This experiment was performed twice, and representative data is shown from one of the experiments. (C) Vta1 is not required for endosomal localization of Vps4. Immunofluorescence was performed to visualize the subcellular localization of Vps4 and Snf7 in either vps4Δ or vps4Δ vta1Δ cells expressing Vps4E233Q-HA. Bar, 5 μm.
The endosomal association of ESCRT-III subunits (Vps20, Snf7, Vps2, and Vps24) is exquisitely sensitive to the activity of Vps4. In wild-type cells, only 30% (or less) of these subunits are endosome associated because of the ESCRT-III disassembly activity of Vps4. In cells lacking Vps4, >90% of ESCRT-III subunits are localized to endosomal membranes (Babst et al., 2002a). To determine whether Vta1 and Vps4 act together at a common point in the MVB pathway, the impact of Vta1 function on ESCRT-III membrane association was examined. Subcellular fractionation was performed on wild-type, vps4Δ, vta1Δ, and vps4Δ vta1Δ cells, and the resulting fractions were probed for the ESCRT-III subunits Snf7 and Vps24 (Fig. 1 C). Consistent with previous results, Snf7 and Vps24 were largely soluble in wild-type lysates and largely membrane associated in the vps4Δ lysates. In contrast, vta1Δ cells exhibit an intermediate phenotype, with Snf7 and Vps24 equally distributed between the soluble and membrane fractions. Interestingly, the genetic backgrounds that displayed more severe MVB sorting defects also revealed increased membrane association of ESCRT-III subunits with deletion of VTA1 (Fig. S1 B). The double-mutant fractionation profile is indistinguishable from the profile observed for the vps4Δ cells (Fig. 1 C). These findings are consistent with Vta1 stimulating ESCRT-III release via Vps4 and suggest that Vps4 function is partially compromised when Vta1 function is lost.
Vps4 is required for recruitment of Vta1 to the MVB sorting reaction
To analyze Vta1 localization, a Vta1-GFP fusion protein expressed from an integrated VTA1-GFP gene was analyzed by fluorescence microscopy. Identification of phosphatidylinositol-3-phosphate (PtdIns[3]P)–positive endosomal structures was facilitated by coexpressing a DsRed-FYVE (PtdIns[3]P binding domain defined by Fab1/YDR313c/Vps27/EEA1) chimera (Katzmann et al., 2003). Extensive colocalization of Vta1-GFP with the DsRed-FYVE reporter was observed in wild-type cells, with an additional cytoplasmic signal (Fig. 2 A). This colocalization indicates that Vta1 associates primarily with endosomal membranes, consistent with previous interpretations (Shiflett et al., 2004) and supportive of a role for Vta1 in MVB protein sorting. All known class E Vps proteins accumulate on the class E compartment in vps4Δ cells, as Vps4 is required to dissociate the ESCRT complexes (Katzmann et al., 2001, 2003; Babst et al., 2002a,b). Surprisingly, although vps4Δ cells displayed aberrant class E structures (visualized with the DsRed-FYVE chimera), Vta1-GFP did not display obvious colocalization with these structures (Fig. 2 A). Instead, Vta1-GFP displayed an increased cytoplasmic distribution and membrane associations that were distinct from the class E compartment. To more quantitatively address this altered localization, subcellular fractionation was performed with cells expressing a functional, chromosomally integrated HA-tagged form of Vta1. Fractions were probed with anti-HA or marker antibodies, and immunoreactive species were quantitated. In wild-type cells, ∼40% of the Vta1 is present in the membrane fraction (Fig. 2 B). In contrast, Vta1-HA membrane association was decreased to ∼10% in the vps4Δ strain (Fig. 2 B). Thus, in contrast to previously described ESCRT subunits, Vps4 appears to be required for Vta1 association with the class E compartment. At present, the nature of the structures with which Vta1-GFP associates in the vps4Δ cells is unclear. However, as seen in wild-type cells, a portion of the Vta1-GFP does not colocalize with DsRed-FYVE and is associated with unique structures. It would appear that loss of Vps4 function causes a shift in distribution toward these PtdIns(3)P-negative structures. One interpretation of these observations is that Vta1 may associate with the endosomes undergoing MVB formation via Vps4 itself.
To test this idea, the ATP hydrolysis–defective form of Vps4 (Vps4E233Q), previously shown to accumulate on endosomal membranes (Babst et al., 1998), was used to examine the impact on Vta1-GFP endosomal localization. Visualization of vps4Δ cells expressing Vps4E233Q revealed extensive colocalization of Vta1-GFP with DsRed-FYVE–positive structures (Fig. 2 A), consistent with a role for Vps4 itself in the recruitment of Vta1 to endosomal structures. Similarly, subcellular fractionation revealed that Vta1-HA membrane association was increased to ∼65% in the context of the Vps4E233Q mutant (Fig. 2 B). These data indicate that Vps4 plays a role in appropriate Vta1 localization to the MVB sorting reaction and suggest that the recruitment of Vta1 occurs through a direct association with Vps4. In contrast, Vta1 is not required for Vps4 association with the MVB sorting machinery because Vps4E233Q localization is indistinguishable in VTA1 and vta1Δ cells (Fig. 2 C). Vta1 is clearly not essential for Vps4 function because vta1Δ cells do not phenocopy vps4Δ cells in all respects. These data implicate Vta1 as a positive modulator of Vps4 in vivo.
Vta1 promotes Vps4 oligomerization
To elucidate the mechanism by which Vta1 impacts Vps4 function, biochemical characterization of Vta1 was initiated. Consistent with published data, we found that both Vta1 from yeast cell extract (Shiflett et al., 2004) and bacterially expressed Vta1 elute from a gel filtration column in the range of 300 kD, in spite of a predicted molecular mass of ∼40 kD (Fig. 3 A). Together, these observations suggest that Vta1 forms a homomeric complex. The subtle difference in size observed between bacterially produced Vta1 and yeast Vta1 may be due to the sizes of the epitopes used in each case (His6 for bacteria vs. 3× HA for yeast); alternatively, Vta1 homooligomers may be associated with other factors in yeast extracts.
Figure 3.

Vta1 homooligomerization and heterooligomerization with Vps4. (A) Vta1 exists as a homooligomer in solution. Sephacryl S-200 gel filtration analyses were performed on yeast extracts or bacterial extracts, and Vta1-HA or His6-Vta1 was visualized with appropriate antibodies. (B) Vta1 and Vps4 form a higher order protein complex in the presence of ATP. Purified recombinant Vta1 and Vps4E233Q were subjected to Superose 6 gel filtration analysis under a variety of conditions. UV traces from the fast protein liquid chromatography runs, wherein the y axis corresponds to absorbance at 280 nm and the x axis corresponds to the elution volume, are presented to summarize results. The peak of the 1-MD complex was subjected to SDS-PAGE and Coomassie blue staining, displayed in the inset.
Molecular mass determination by gel filtration can be misleading, as the shape of the molecule affects the measurement. Analytical ultracentrifugation was therefore used to determine the precise molecular mass of the His6-Vta1 homooligomer. Varied concentrations of Vta1 protein were subjected to equilibrium centrifugation, and the resulting data indicated a native molecular mass for Vta1 of 82 kD, suggesting that Vta1 forms a dimer (theoretical molecular mass of His6-Vta1 is 40.9 kD). To ensure that during the analytical centrifugation experiment Vta1 did not change its oligomeric state, gel filtration was performed after equilibrium centrifugation. The Vta1 sample again eluted in a mass range of 300 kD, demonstrating that the Vta1 complex remained stable during centrifugation (unpublished data). Thus, the discrepancy between the gel filtration data and the ultracentrifugation data indicate that the Vta1 dimer has a very large Stoke's radius and is likely to be rod shaped.
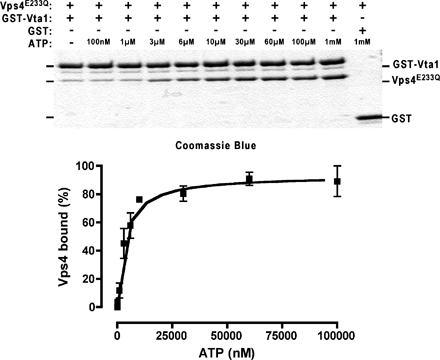
To examine the direct association of Vps4 and Vta1, purified, bacterially expressed proteins were subjected to gel filtration chromatography using a Superose 6 column in the presence of either ATP or ADP (Fig. 3 B). Vps4 bound to ATP forms a homooligomeric complex of ∼500 kD. The ATPase activity of wild-type Vps4 prevents resolution of this complex by gel filtration, requiring the use of the ATPase-defective mutant Vps4E233Q (Fig. 3 B; Babst et al., 1998). Consistent with previous results, in the presence of ADP, Vps4E233Q fractionates predominantly as a dimer. Dimeric Vta1 complex migrates at a slightly smaller apparent mass than the 440-kD marker and the oligomeric Vps4E233Q complex; once again, the presumptive rodlike structure of Vta1 is likely the explanation for this unexpected fractionation pattern of the Vta1 dimer. The addition of equimolar amounts of Vta1 to the Vps4E233Q plus ATP sample promoted formation of a Vta1–Vps4(ATP) complex that eluted at nearly 1 MD (Fig. 3 B); moreover, Coomassie blue staining of the 1-MD complex fractions revealed an apparent Vta1/Vps4 ratio of ∼1:1 (Fig. 3 B, inset; silver staining yielded identical results [not depicted]). Further addition of Vta1 (threefold molar excess) did not substantially affect the amount of complex formed, suggesting that the ∼1:1 stoichiometry represents a Vta1-saturated complex.
Formation of this 1-MD complex was dependent on ATP because the Vta1 + Vps4E233Q ADP sample did not form this high–molecular mass species (Fig. 3 B). This is in apparent contrast to previously reported results wherein an interaction between Vps4 and Vta1 was found to be independent of the nucleotide-bound state of Vps4 (Yeo et al., 2003). However, although we detect a weak association between Vta1 and Vps4E233Q in GST pull-down assays in the absence of ATP, the near stoichiometric association of GST-Vta1 and Vps4E233Q required ATP (Fig. 3 B and Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200508166/DC1). Although ATP is required for stoichiometric binding, the Vps4 NH2-terminal region is dispensable for Vta1–Vps4 complex formation (Fig. 3 B). These observations suggested that in the presence of ATP Vta1 binds to either the AAA or COOH-terminal region Vps4, consistent with published two-hybrid data (Yeo et al., 2003).
One interpretation of the data presented in Fig. 3 B is that Vta1 might function as an assembly factor that supports the oligomerization of ATP-bound Vps4 dimers into the active, higher order oligomeric form of Vps4. If this were true, addition of Vta1 might help assemble multimeric Vps4-ATP when it is too dilute to form complexes by itself. To test this model, we used gel filtration to analyze the size of ATP-bound Vps4E233Q at low concentration (10-fold lower than in Fig. 3 B) in the presence or absence of Vta1 (Fig. 4 A). Western blot analysis of the resulting fractions using anti-Vps4 antiserum revealed that Vps4E233Q elutes from the column in the mass range of the Vps4 dimer (∼100 kD) at this low-protein concentration. In contrast, in the presence of Vta1, Vps4E233Q is found in fractions corresponding to the 1-MD mass range, indicating that the binding of Vta1 to Vps4 stabilizes the higher oligomeric form of the ATPase.
Figure 4.

Vta1 stimulates Vps4 ATPase activity. (A) Vta1 stimulates Vps4 assembly into its ATPase-competent form. A low concentration of Vps4 was subjected to Superose 6 gel filtration analyses (containing ATP as in Fig. 3 B) in the presence or absence of Vta1, and immunoreactive species were detected by Western blotting with anti-Vps4 antisera. (B) ATPase activity of Vps4 in the presence of Vta1, measured as a function of Vta1 concentration. (C) ATPase activity of Vps4 in the presence of Vta1, measured as a function of Vps4 concentration. Values in both B and C are presented as specific activity per Vps4 molecule. Prism 4 software was used to fit data points to a curve with nonlinear regression.
Vta1 stimulates Vps4 ATPase activity
Oligomerization of Vps4 is required for the hydrolysis of the bound ATP (Babst et al., 1998). Vta1 induces oligomerization of Vps4 and therefore would be predicted to increase the ATP hydrolysis rate of Vps4. To test this hypothesis, the ATPase activity of Vps4 was measured (ADP/min/Vps4 molecule) in the presence and absence of Vta1. The addition of Vta1 to Vps4 (100 nM or 1.5 μM) stimulated Vps4 ATPase activity in a concentration-dependent manner, with half-maximal stimulation at ∼2 μM Vta1 (1.6–2.8 μM; Fig. 4 B); however, Vps4 ATPase activity stimulation was not observed with the addition of either BSA (not depicted) or certain truncated forms of Vta1 (Figs. 6 D, 7 C, and 8 B), and Vta1 exhibited no ATPase activity by itself (not depicted). Consistent with previous results, Vps4 ATPase activity was strongly dependent on Vps4 protein concentration, with 100 nM Vps4 exhibiting activity of 5 ADP/min/Vps4 and 1.5 μM Vps4 exhibiting activity of 30 ADP/min/Vps4 (Fig. 4 B; Babst et al., 1998). The protein concentration–dependent activity of Vps4 shown in Fig. 4 C can be described by the Michaelis-Menten equation with a Km of 1.1 μM, similar to the previously reported Km of 0.6 μM (Babst et al., 1998). The addition of either 1 or 6 μM Vta1 resulted in a decrease of the Vps4 Km to 118 and 69 nM, respectively. In addition, the Vmax increased from the previously reported 45 to 71 ADP/min/Vps4 with 6 μM Vta1. The lower Vps4 Km values observed in the presence of Vta1 are consistent with the model that Vta1 promotes the oligomerization of Vps4 dimers. However, the increase in the Vmax of Vps4 suggests that Vta1 not only promotes the oligomerization of Vps4 but may also affect other aspects of the hydrolysis kinetics.
Figure 6.

Domain analysis of Vta1. (A) Cells expressing Vta1-GFP were transformed with plasmids expressing PrA, PrA-Vta1, or mutant versions thereof; native lysates were generated; PrA fusion proteins were isolated using IgG Sepharose; and material was analyzed by Western blotting with anti-GFP antibody. (B) In vivo analysis of Vta1–Vps60 interactions. Cells were transformed with a plasmid expressing an HA-tagged form of Vps60, along with PrA-Vta1 constructs; native lysates were generated; PrA fusions were isolated using IgG Sepharose; and material was analyzed by Western blotting with anti-HA antibody. (C) In vitro analysis of Vta1–Vps4 interactions. Purified Vps4E233Q was incubated with GST or GST fusion proteins corresponding to Vta1 or mutant forms thereof; glutathione Sepharose was used as an affinity isolation step; and material was visualized by Western blotting with Vps4 or Coomassie blue staining. (D) ATPase activity of 0.6 μM Vps4 in the presence of mutated forms of Vta1. n = 3.
Figure 7.

An evolutionarily conserved motif in the COOH terminus of Vta1 homologues is necessary for Vps4 binding and stimulation of ATPase activity. (A) Alignment of the VSL region from Vta1 and homologues. Amino acids that have been altered are indicated by asterisks. (B) Vps4 binding by VSL point mutants. Purified Vps4E233Q was incubated with purified GST, GST-Vta1, or the indicated point mutant thereof; glutathione Sepharose was used as an affinity isolation step; and bound material was analyzed by Coomassie staining or anti-Vps4 Western blotting. (C) Stimulation of Vps4 ATPase activity by VSL point mutants. Vps4 ATPase activity was measured in the conditions indicated and normalized to wild-type Vta1 to calculate relative stimulation. n = 3. Error bars indicate SEM. (D) Effect of VSL point mutations on MVB sorting reaction. GFP-CPS was visualized in vta1Δ cells (BY4742) expressing the indicated form of Vta1. (E) Mutations in the VSL region perturb ESCRT-III recycling. Differential centrifugation was performed on vta1Δ cells expressing the indicated form of Vta1. The resulting soluble (S) and pellet (P) fractions were probed with anti-Snf7 antibody. White lines indicate that intervening lanes have been spliced out.
Figure 8.

The VSL domain of Vta1 and SBP1 is sufficient for Vps4 binding. (A) Purified Vps4E233Q was incubated with purified GST, GST-Vta1, GST-Vta1ΔC40, GST-VSLVta1, GST-SBP1, or GST-VSLSBP1; glutathione Sepharose was used as an affinity isolation step; and material was visualized by staining with Coomassie blue. (B) ATPase activity of Vps4 is stimulated by the VSL domain of Vta1. This experiment was performed twice, and representative data is presented from one of those experiments. (C) ATPase activity of Vps4 is stimulated by hSBP1 and its VSL domain. n = 4. Error bars indicate SEM.
Domain analysis of Vta1
To further dissect the mechanism of Vta1 action, a structure–function analysis was initiated. The COILS program (Lupas et al., 1991) identifies three potential coiled-coil regions in Vta1—residues 34–63, residues 231–263, and the COOH-terminal 30 amino acids (Fig. 5). To analyze the involvement of these regions in Vta1 activity, we generated alleles lacking residues 1–68 (Vta1ΔN68), 37–68 (Vta1Δ37–68), 224–330 (Vta1ΔC106), or 290–330 (Vta1ΔC40). These alleles, as well as wild-type Vta1, were expressed in yeast as COOH-terminal fusions to PrA (IgG binding units of protein A from Staphylococcus aureus). Whereas PrA-Vta1 could complement the CPS sorting defects observed in the BY4742 vta1Δ strain, none of the Vta1 truncations could restore VTA1 function (Fig. 5 and not depicted), although equivalent expression was observed (Fig. 6 A). This result indicates that both the COOH- and NH2-terminal coiled-coil domains are important for Vta1 function in vivo.
Figure 5.

Coiled-coil domains of Vta1 are required for function. (A) Schematic representation of Vta1 and mutant forms. Putative coiled-coil domains are indicated, as is the VSL region. (B) In vivo analysis of Vta1 coiled-coil mutants. BY4742 cells deleted for VTA1 were transformed with PrA-VTA1 chimeras as indicated, and MVB sorting was addressed by visualization of GFP-CPS.
To examine homodimerization, PrA-Vta1 alleles were expressed in yeast harboring a VTA1-GFP allele at the genomic VTA1 locus. Cell lysates were generated under nondenaturing conditions, PrA-Vta1 fusions were affinity purified with IgG Sepharose and resolved by SDS-PAGE, and Vta1-GFP and PrA-Vta1 were detected by Western blotting with anti-GFP monoclonal antibody. As expected, Vta1-GFP copurified with full-length PrA-Vta1, consistent with the formation of a Vta1 homodimer (Fig. 6 A). Deletion of the last 106 or 40 residues of Vta1 was sufficient to abolish the association with Vta1-GFP. Consistent with this observation, purified Vta1ΔC40 and Vta1ΔC106 behaved as apparent monomers when subjected to Superose 6 gel filtration analyses (Fig. 3 B and not depicted). Together, these results imply that the COOH-terminal 40 amino acids of Vta1 are required for homooligomerization. In contrast, the PrA-Vta1ΔN68 and PrA-Vta1Δ37–68 proteins were competent to bind Vta1-GFP; thus, the NH2-terminal coiled-coil domain is not required for Vta1 self-assembly.
Vta1 has been reported to associate with the class E Vps proteins Vps60/Mos10 and Vps20 (Yeo et al., 2003; Shiflett et al., 2004). Although the gene name (Vps twenty associated 1) implies that Vta1 binds Vps20, we have been unable to detect any interaction between Vps20 and Vta1 under a variety of conditions (unpublished data). In contrast, the association of full-length Vta1 with Vps60 was readily detectable with this PrA-based copurification approach (Fig. 6 B). Although Vta1ΔC40 was unable to homodimerize, the PrA fusion protein retained the ability to bind Vps60 (Fig. 6 B). In contrast, deletion of NH2-terminal residues 1–68 or 37–68 abolished the Vta1–Vps60 interaction. These findings implicate the NH2-terminal coiled-coil region as mediating the Vta1 association with Vps60.
To examine domains of Vta1 required for association with Vps4, the various forms of Vta1 were expressed in bacteria as NH2-terminal GST fusions. GST pull-down assays were performed with purified Vps4E233Q in the presence of 1 mM ATP and the various forms of Vta1. Vps4–Vta1 associations were analyzed by Coomassie blue staining and Western blotting (Fig. 6 C). Whereas full-length GST-Vta1 was capable of binding Vps4E233Q, deletion of the last 40 residues of Vta1 abolished interaction with Vps4E233Q. This finding indicates that COOH-terminal portion of Vta1 is required for both Vta1 dimerization and Vps4 binding, although the absence of Vps4 binding could be a secondary consequence of a defect in Vta1 dimerization. In contrast to Vta1ΔC40, the NH2-terminal deletions (Vta1ΔN68 and Vta1Δ37–68) are still competent to bind Vps4 (Fig. 6 C).
To further address the relevance of the Vta1–Vps4 association, the GST-Vta1ΔN68 and GST-Vta1Δ37–68 proteins were examined for their abilities to stimulate Vps4 ATPase activity. To minimize the amount of fusion protein required, we used a Vps4 ATPase assay that included glutathione beads and the conjugated proteins to supply the reactions with the desired amount of Vta1 protein. Using this assay with Vta1 as well as GST-Vta1, we were able to demonstrate that the GST tag and the glutathione beads do not interfere with Vta1 enhancement of Vps4 ATPase activity (Fig. 6 D). The GST-Vta1ΔN68 and GST-Vta1Δ37–68 fusion proteins were able to stimulate the ATPase activity of Vps4 to levels comparable to those of wild-type Vta1. In contrast, deletion of the COOH-terminal 40 residues abolished the ability of Vta1 to stimulate Vps4. These findings indicate that the Vta1 NH2-terminal coiled-coil region is required for interaction with Vps60 but not for homodimerization, nor binding and activation of Vps4. In contrast, the COOH-terminal region (in particular, residues 290–330) is required for Vta1 homodimerization, Vps4 binding, and Vps4 activation but is dispensable for Vps60 binding. These results suggest that Vta1 binds Vps60 and -4 independently. Consistent with this model, we have observed the association of PrA-Vta1 and Vps60-HA in lysates generated from vps4Δ strains (unpublished data), and we have reconstituted the Vta1–Vps4 complex in vitro without Vps60 (Fig. 3). However, given that Vta1 truncations that fail to bind Vps60 also fail to complement (Fig. 5), we cannot exclude the possibility that the Vps60–Vta1 interaction modulates Vta1 stimulation of Vps4 in vivo.
The VSL region binds and activates Vps4
The Vta1 COOH-terminal 40 residues are required for Vta1 homodimerization and Vps4 binding and stimulation. Although no defined domains have been identified in Vta1, this portion of Vta1 is highly conserved in homologous proteins in yeast, plants, and mammals. In particular, the last 30 residues of Vta1, mammalian SBP1, and plant LIP5 (Fig. 7 A) exhibit very significant sequence conservation. Based on these representative members, we have named this sequence motif the VSL region. The spacing of leucine and small hydrophobic residues in conjunction with the requirement of this VSL region in Vta1 dimerization suggests that the VSL region may form a coiled-coil structure, although this has not yet been confirmed experimentally. Other residues of note include the conserved charged residues at positions 2, 5, 14, 15, and 25 and the conserved aromatic residues at positions 6 and 13 (Fig. 7 A).
To examine the role of this VSL region in Vps4 binding and stimulation, site-directed mutagenesis was used to alter anticipated surface residues, including Lysines 299, 302, and 322 and Serine 306. Mutants were then analyzed for function both in vivo and in vitro and revealed a range of defects. PrA-tagged forms of each mutant were expressed in cells harboring Vta1-GFP to analyze the ability of these proteins to dimerize; all four mutants were able to copurify Vta1-GFP to levels indistinguishable from wild type (unpublished data), indicating that dimerization is unaffected by these mutations. GST-tagged forms of each mutant were expressed in bacteria and purified for use in Vps4 association and activation assays. Although homodimerization was unaffected, mutation of Lysines 299 and 302 and, to a lesser extent, Serine 306 disrupted binding to Vps4E233Q in the GST pull-down assay (Fig. 7 B). In contrast, Lysine 322 was largely dispensable for the Vta1–Vps4 interaction. In the Vps4 activity assay (Fig. 7 C), GST-Vta1K322A exhibited Vps4 ATPase stimulation comparable to wild type; GST-Vta1S306A displayed a modest reduction in Vps4 stimulation, consistent with the partial defect in Vps4 binding; and the GST-Vta1K299A and GST-Vta1K302A mutants were most compromised for Vps4 stimulation. Thus, Vps4 stimulation deficits correlated with Vps4 binding. Analysis of in vivo function also corresponded with these in vitro phenotypes. Vta1K302A and Vta1K299A exhibited the least Vps4 stimulation in vitro and similarly displayed the most severe GFP-CPS sorting and ESCRT-III recycling defects when expressed in vta1Δ cells (Fig. 7, D and E). Vta1S306A displayed more subtle defects in vivo, and Vta1K322A was indistinguishable from wild type. Together, these data demonstrate a correlation between the abilities of Vta1 to bind Vps4 via the VSL region, to stimulate Vps4 ATPase activity, and to effect efficient function of the MVB pathway. Moreover, these results demonstrate that the VSL region is directly involved in Vps4 binding in addition to being required for Vta1 dimerization.
To determine whether the VSL region is sufficient for Vps4 binding and activation, the last 40 residues of Vta1 (290–330) were fused to the COOH terminus of GST (GST-VSLVta1). This bacterially expressed protein was then used in GST pull-down assays as previously described for GST-Vta1. As can be seen in Fig. 8 A, GST-VSLVta1 is capable of binding Vps4E233Q, similarly to full-length Vta1, whereas GST-Vta1ΔC40 cannot. This result indicates that the VSL region is both necessary and sufficient for Vta1 association with Vps4. To address whether the VSL region is capable of stimulating Vps4 activity, GST-VSLVta1 was used in Vps4 ATPase assays. Whereas GST or GST-Vta1ΔC40 addition did not stimulate the ATPase activity of Vps4, the addition of GST-VSLVta1 significantly stimulated Vps4 activity at this concentration from 3 to ∼10 ADP/min/Vps4 (P = 0.03; unpublished data). We also performed ATPase assays with the purified 40-mer VSL peptide (VSLVta1 sans GST) and found that the Vta1 VSL region displayed concentration-dependent Vps4 stimulation (Fig. 8 B). Although this activation was less pronounced than stimulation by full-length Vta1, this result indicates that the Vta1 VSL region is both necessary and partly sufficient for Vps4 activation.
Murine SBP1 has been demonstrated to bind the Vps4 orthologue SKD1 (Fujita et al., 2004). Therefore, human SBP1 (hSBP1) and the VSL region of hSBP1 (VSLSBP1) were also examined for Vps4 binding and stimulation to determine whether the human VSL region exhibited comparable activity. Full-length hSBP1 or the last 41 residues of SBP1 (267–307) were fused to the COOH terminus of GST (GST-SBP1 and -VSLSBP1) and used in pull-down assays with Vps4. GST-VSLSBP1 and -SBP1 were capable of binding Vps4E233Q (Fig. 8 C). Moreover, examination of Vps4 ATPase stimulation indicated that SBP1 and the VSLSBP1 region were also capable of enhancing yeast Vps4 ATPase activity (P = 0.0002 and P < 0.0001; Fig. 8 C) to an extent similar to the VSLVta1 region. This result suggests a conserved mechanism by which the VSL region can stimulate Vps4.
Discussion
Vps4 is an AAA-ATPase whose activity is required for proper function of the MVB sorting pathway (for review see Babst, 2005). As a family, the AAA-ATPases function in a variety of cellular processes through a similar mechanism of action. AAA-ATPases use the energy from ATP hydrolysis to induce conformational changes in other proteins or protein complexes, ultimately causing unfolding or dissociation of the substrate protein (for review see Hanson and Whiteheart, 2005). The function of Vps4 would appear to be the removal or disassembly of the ESCRT machinery at a late step within the MVB sorting reaction (for review see Babst, 2005). Loss of Vps4 function results in accumulation of ESCRT machinery on the endosomal membrane and concurrent dysfunction of the MVB sorting pathway (Babst et al., 1997, 1998). The mammalian homologue of Vps4, SKD1, is believed to play an equivalent role in mammalian cells (Bishop and Woodman, 2000; Yoshimori et al., 2000). The pivotal role for Vps4 in the later steps of MVB cargo sorting highlights the importance of understanding how its ATPase activity is regulated during this complicated sorting event. In the present study, we have demonstrated that Vta1 binds directly to Vps4 through a conserved region, resulting in stimulation of Vps4 ATPase activity and potentiating the recycling of ESCRT proteins. The mechanism by which Vta1 stimulates Vps4 function appears to be evolutionarily conserved, underscoring its importance in the context of the MVB sorting reaction.
We have demonstrated that Vta1 plays an important role in stimulating Vps4 activity for maximal function in vivo. Our data indicate that Vps4 is still recruited to the endosome in vta1Δ cells and that the basal Vps4 activity is sufficient to maintain some level of MVB sorting. The positive regulatory role of Vta1 might explain the phenotypic variations observed upon deletion of VTA1 in different genetic backgrounds (Yeo et al., 2003; Shiflett et al., 2004). It is possible that in alternative genetic backgrounds, the basal Vps4 activity is not sufficient to escape the class E phenotype. Additionally, changes in environmental/growth conditions may increase flux through the endosomal system, which could place a bigger burden on the MVB pathway, specifically Vps4. If Vps4 function was already compromised by loss of Vta1 function, this defect could be amplified under increased endosomal flux. We also observed that there is some level of cargo-specific MVB sorting defects occurring in vta1Δ cells. Together, these data suggest that Vta1 functions as a positive regulator of Vps4 function, thereby impacting the function of the MVB pathway.
Model for Vps4 activation by Vta1
Vps4 exists as a dimer in its ADP-bound state, whereas ATP binding induces assembly into a homooligomeric complex (Babst et al., 1998). The precise makeup of this complex is not entirely clear; although Vps4 appears to assemble into a decamer (Babst et al., 1998), the majority of characterized AAA-ATPases appear to exist as ring structures with sixfold symmetry (for review see Hanson and Whiteheart, 2005). Regardless of the precise composition, Vps4 assembly into this higher order complex correlates with ATPase activity with a previously reported Vmax of 45 ADP/min/Vps4 (Babst et al., 1998). ATP hydrolysis by the Vps4 complex results in a return to the dimeric, ADP-bound state (Babst et al., 1998). Hence, the relationship between oligomeric state and ATPase activity results in a cycle of oligomerization, ATP hydrolysis, and dissociation of the Vps4 complex. The nucleotide-binding state of Vps4 also regulates its subcellular localization and substrate interactions in vivo. ADP-bound Vps4 localizes to the cytoplasm, whereas the ATP-bound form localizes to MVBs (Babst et al., 1998). ATP hydrolysis is required for removal of ESCRTs (Babst et al., 2002a,b; Katzmann et al., 2001, 2003) and the return of Vps4 to its dimeric, soluble state (Babst et al., 1998).
In the present studies, we have used in vivo and in vitro studies to demonstrate that Vta1 is a positive regulator of Vps4. We propose that this activation is the result of two distinct functions of the Vta1 protein. First, Vta1 is capable of stabilizing the steady-state levels of the ATPase-competent homooligomeric form of Vps4. This is supported by the observation that Vta1 stimulates oligomeric assembly of Vps4 at concentrations normally too low to support this. This assembly correlates with increased ATPase activity of low Vps4 concentrations in the presence of Vta1, resulting in a lower Km of the enzymatic reaction. Although a conserved region of Vta1 (the VSL region) is required for this activity, this region does not appear to be sufficient to drive maximal Vps4 activity. A second activating mechanism appears to be required for full potentiation, as Vta1 also increases the Vmax of Vps4, suggesting that Vta1 is directly stimulating ATP hydrolysis of the Vps4 oligomer in addition to promoting increased steady-state levels of the oligomer itself. It is possible, for instance, that binding of Vta1 to the Vps4 oligomer induces cooperativity among the Vps4 subunits, resulting in a more efficient reaction. Further experimentation is required to complete our understanding of Vta1–Vps4 stimulation.
Three functional regions have been identified in Vps4: an NH2-terminal microtubule interacting and trafficking region (Ciccarelli et al., 2003), a single AAA domain in the central portion of the protein, and a COOH-terminal region. The NH2-terminal microtubule interacting and trafficking region is dispensable for Vps4 dimerization, ATP-dependent oligomerization, and ATP hydrolysis in vitro; however, Vps4ΔN fails to complement in vivo and fails to colocalize with endosomally localized ESCRT-III subunits (Babst et al., 1998). These observations suggest that the NH2-terminal region of Vps4 is a substrate binding region that interacts with endosome-localized ESCRT-III. In contrast, loss of the COOH-terminal region results in a monomeric form of Vps4 that is unable to hydrolyze ATP or carry out the MVB sorting reaction in vivo (unpublished data). This suggests that this COOH-terminal region is critical for dimerization and subsequent higher order assembly of Vps4. We have demonstrated that Vta1 stimulates or stabilizes the Vps4 oligomeric state, and others have demonstrated that Vps4 residues 360–406 (the β domain) can bind Vta1 (Scott et al., 2005). We therefore speculate that the Vta1 stimulation of Vps4 occurs in part through binding the Vps4 COOH-terminal region.
These observations can be used to construct a model for Vps4 structure and function and Vta1 stimulation (Fig. 9). The basic structure of Vps4 appears to be a dimer. Upon ATP binding, Vps4 oligomerizes into a higher order oligomer. It is reasonable to propose that in vivo Vps4-ATP dimers bind to the substrate ESCRT-III on the endosomal membrane via the NH2-terminal coiled-coil domain. In the absence of Vta1, the interaction of the NH2-terminal region with its substrate (ESCRT-III) appears to be sufficient to permit oligomerization of Vps4 into its ATP hydrolysis–competent state, and as such Vps4 function is sufficient to permit MVB sorting at some level. However, in the wild-type setting, the present dataset indicates that optimal Vps4 assembly and ATPase activity requires Vta1. Hydrolysis of ATP by the Vta1–Vps4 complex results in a conformational change of the substrate binding region that leads to disassembly of ESCRT factors. In the ADP-bound state, the ATPase-competent Vta1–Vps4 complex is unstable and dissociates.
Figure 9.

Model for Vta1–Vps4 function in MVB sorting. ATPase activity of Vps4 requires the assembly of ATP-bound Vps4 dimers into an oligomeric form. The ATPase function of Vps4 is required for proper function of the MVB sorting pathway. In wild-type cells, assembly of oligomeric Vps4 and associated ATPase activity is stimulated by Vta1. This Vta1–Vps4 oligomer associates with the endosomal membrane via ESCRT-III subunits to potentiate the dissociation of ESCRTs. In vta1Δ cells, Vps4 function is partially compromised and ESCRT membrane association is partially stabilized.
Type 2 AAA proteins, such as NSF and p97, form stable hexameric complexes. In contrast, type 1 AAA-ATPases, such as Vps4 and katanin, have dynamic oligomeric structures, and assembly seems to play an important regulatory role in their function. In the case of Vps4, the assembly step is at least in part controlled by Vta1. We suggest that the use of assembly factors similar to Vta1 might be a common mechanism by which type 1 AAA-ATPases are regulated. The existence of the evolutionarily conserved VSL region within Vta1 homologues suggests that this may represent a conserved mechanism by which Vps4/SKD1 proteins are stimulated. Consistent with this notion, the human homologue of Vta1, SBP1, was capable of binding to Vps4 and stimulating its ATPase activity, as were peptide fragments corresponding to the VSL regions from Vta1 and SBP1. However, additional portions of Vta1 appear to participate in activating Vps4, as the VSLVta1 peptide stimulates Vps4 to only a portion of the level seen with full-length Vta1. We therefore suggest that the VSL region represents a conserved interaction site, whereas additional contact points are required for further stabilizing or activating the ATPase-competent Vps4 complex.
Although additional portions of Vta1 are likely to facilitate Vps4 activation, we have identified the Vta1 NH2 terminus (residues 1–68) as largely dispensable for Vps4 binding and stimulation. However, these residues are required for Vta1 function in vivo. These observations indicate that Vta1 may be involved in other reactions in addition to Vps4 stimulation or, alternatively, that Vta1 activity may be regulated via interactions through the NH2-terminal region; at present, we favor the second model. For example, Vta1 binds to Vps60 through this region, and this may represent another level of regulation. Vps4 lies at a critical juncture in the MVB pathway regulating release of ESCRT components from the endosomal membrane, and we have demonstrated that Vta1 directly stimulates Vps4 activity. The interaction of other proteins with Vta1 may serve to indirectly regulate Vps4 and thus modulate flux through the MVB pathway. We therefore suggest that the VSL family is a critical nexus modulating flux through the MVB pathway from plants to yeast to man.
Materials and methods
Plasmid construction and yeast strains
The VTA1 ORF was amplified from yeast genomic DNA and cloned into the BamHI and SalI sites of the pET28b expression vector (Novagen) to generate pET28Vta1. pET28Vta1Δ37–68, pET28Vta1ΔN68, pET28Vta1ΔC40, and pET28Vta1ΔC100 were constructed by PCR amplifying the relevant regions of VTA1 and cloning into the BamHI and SalI sites of pET28b. hSBP1 was amplified from a human breast cDNA library (a gift from C. Mendelson, University of Texas Southwestern, Dallas, TX) and cloned into the BamHI and SalI sites of pGST-parallel1 (pGST–hSBP1). VSL-region coding sequences of Vta1 and SBP1 were synthesized and cloned into the BamHI and SalI sites of pGST-parallel1 (pGST-VSLVta1 and pGST-VSLSBP1). All cloned PCR products and oligonucleotides were sequenced to exclude unexpected mutations. Inserts from pET28Vta1 constructs were then subcloned into the BamHI and SalI sites of pGST-parallel1 and pPrA416, the yeast expression vector pGPD416 containing an NH2-terminal Protein A tag (this study).
The following yeast strains were used in this study: SEY6210 (MATα leu2-3,112 ura3-52 his3-Δ200 trp1-Δ901 lys2-801 suc2-Δ9 [Robinson et al., 1988]), SEY6210.1 (SEY6210; MATa [Wendland et al., 1998]), MBY4 (SEY6210; vps4Δ∷TRP1 [Babst et al., 1997]), JPY48 (SEY6210; vta1Δ∷HIS3 [this study]), JPY47 (SEY6210.1; vta1Δ∷HIS3 [this study]), JPY50 (MBY4; vta1Δ∷HIS3 [this study]), JPY42 (SEY6210; VTA1-GFP∷HIS3 [this study]), JPY43 (MBY4; VTA1-GFP∷HIS3 [this study]), JPY46 (SEY6210; VTA1-HA∷HIS3 [this study]), and JPY45 (MBY4; vta1Δ∷HIS3 [this study]). BY4742 strains were obtained from Open Biosystems. SF838-9D strains were a gift from R. Piper (University of Iowa, Iowa City, IA).
Protein expression and purification
Protein expression was performed in the HMS174 DE3 bacterial strain at 25°C for 14–20 h with 0.5 mM IPTG. His6-fusion proteins were purified by Ni+2-affinity chromatography (5 ml HiTrap Chelating FF or Ni-NTA resin), treated with thrombin (optional), incubated with ATP to dissociate chaperones, subjected to anion exchange chromatography (5 ml HiTrap Q FF or Bioscale Q2 column), and resolved on a Superose 6 10/30 column (optional). GST fusion proteins were purified by glutathione-affinity chromatography (glutathione Sepharose 4B resin) including treatment with ATP to dissociate chaperones and elution of the Vta1 protein or peptide by cleavage with His6TEV; His6TEV was then removed with Ni-NTA resin. Alternatively, GST fusion proteins were used in pull-down assays and ATPase assays as fusion proteins still bound to the glutathione Sepharose 4B resin. Vps4 was purified as previously described (Babst et al., 1998).
Biochemical analyses
GST pull-down experiments were performed as previously described (Katzmann et al., 2001), with the following modifications: ATPase buffer (see ATPase assay) plus 0.05% Tween 20 was used with varying concentrations of ATP as needed, and Vps4E233Q was used at 6 μg/ml. Visualization of bound material was accomplished by Coomassie staining or Western blotting with anti-Vps4 antisera. Protein A purification was performed as in Katzmann et al. (2001), with slight modifications. 5 OD600 units of cells were spheroplasted, lysed in PBS plus 0.05% Tween 20 (PBST), cleared, and incubated with IgG Sepharose 6 Fast Flow in spin microcolumns for 60 min. Bound material was visualized with monoclonal HA.11 (Covance) or monoclonal anti-GFP (CLONTECH Laboratories, Inc.). Gel filtration analysis was performed as previously described (Babst et al., 2002b). Analysis of CPS and carboxypeptidase Y transport to the vacuole by pulse-chase immunoprecipitation was performed as previously described (Babst et al., 2002a). Vta1 equilibrium centrifugation was performed in an Optima XL-1 (Beckman Coulter) centrifuge at concentrations of 0.5 and 2 mg/ml in PBS.
Subcellular localization
Subcellular fractionation was performed as previously described (Babst et al., 2002a). Microscopy was performed on living cells using a fluorescence microscope (Nikon) fitted with FITC and rhodamine filters and a digital camera (Coolsnap HQ; Photometrix), and images were deconvolved using Delta Vision software (Applied Precision, Inc.).
ATPase assay
Measurement of Vps4 ATPase activity was performed as previously described (Babst et al., 1998). To ensure accurate calculations of activity, experiments were performed in a manner such that substrate (ATP) was not limiting and product inhibition was not observed. α-[32P]ATP was combined with cold nucleotide to yield a 10-mM ATP × Ci/mole mix. Vps4 (100 nM–1.5 μM) was combined with Vta1 proteins (100 nM–30 μM) in ATPase reaction buffer (0.1 M KOAc, 20 mM Hepes, and 5 mM MgOAc, pH 7.5) in a total of 18 μl at 30°C. Reactions were initiated by the addition of ATP to 1 mM. 1-μl samples were removed at various time points after ATP addition (5, 10, 15, and 20 min for low Vps4 concentrations and 40 s, 1 min 20 s, and 2 min for high Vps4 concentrations) and resolved by thin-layer chromatography using precoated PEI Cellulose TLC glass plates (Merck) and developing buffer (0.75M KPO4, pH 3.5). Plates were dried and exposed to phosphoimager screens for 4–12 h. Screens were processed using the Storm 840 system (GE Healthcare), and ADP and ATP signal was quantitated using ImageQuant software package (GE Healthcare). For analysis of GST fusion proteins, samples (lysate and glutathione beads) were washed extensively with PBST and ATPase buffer. Residual buffer was aspirated with a 30-gauge needle, and 500 nM Vps4 was added in a total of 18 μl ATPase reaction buffer. ATP addition, time-point collection, and sample processing was then performed as described for the untagged proteins. Data was analyzed with Excel (Microsoft) to determine ATP hydrolysis rates and Prism 4 (GraphPad) to determine kinetic and statistical parameters.
Online supplemental material
Fig. S1 shows variation of vta1Δ phenotypes in distinct genetic backgrounds. Table S1 show that the vta1Δ phenotype is distinct from the vps4Δ phenotype. Fig. S2 shows that ATP is required for stoichiometric Vta1–Vps4 interaction in vitro. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200508166/DC1.
Supplementary Material
Acknowledgments
We thank Drs. Chris Burd and Bruce Horazdovsky for critical evaluation of the manuscript and members of the Katzmann and Horazdovsky Laboratories helpful discussions.
This work has been supported by grant RO1 GM 73024-1 from the National Institutes of Health (to D.J. Katzmann) and grant 0530210N from the American Heart Association (to M. Babst).
Abbreviations used in this paper: CPS, carboxypeptidase S; ESCRT, endosomal sorting complex required for transport; hSPB1, human SPB1; MVB, multivesicular body; PtdIns(3)P, phosphatidylinositol-3-phosphate; Vps, vacuolar protein sorting; VSL, Vta1/SBP1/LIP5.
References
- Amerik, A.Y., J. Nowak, S. Swaminathan, and M. Hochstrasser. 2000. The Doa4 deubiquitinating enzyme is functionally linked to the vacuolar protein-sorting and endocytic pathways. Mol. Biol. Cell. 11:3365–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst, M. 2005. A protein's final ESCRT. Traffic. 6:2–9. [DOI] [PubMed] [Google Scholar]
- Babst, M., T.K. Sato, L.M. Banta, and S.D. Emr. 1997. Endosomal transport function in yeast requires a novel AAA-type ATPase, Vps4p. EMBO J. 16:1820–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst, M., B. Wendland, E.J. Estepa, and S.D. Emr. 1998. The Vps4p AAA ATPase regulates membrane association of a Vps protein complex required for normal endosome function. EMBO J. 17:2982–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst, M., D.J. Katzmann, E.J. Estepa-Sabal, T. Meerloo, and S.D. Emr. 2002. a. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev. Cell. 3:271–282. [DOI] [PubMed] [Google Scholar]
- Babst, M., D.J. Katzmann, W.B. Snyder, B. Wendland, and S.D. Emr. 2002. b. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev. Cell. 3:283–289. [DOI] [PubMed] [Google Scholar]
- Bilodeau, P.S., J.L. Urbanowski, S.C. Winistorfer, and R.C. Piper. 2002. The Vps27p Hse1p complex binds ubiquitin and mediates endosomal protein sorting. Nat. Cell Biol. 4:534–539. [DOI] [PubMed] [Google Scholar]
- Bishop, N., and P. Woodman. 2000. ATPase-defective mammalian VPS4 localizes to aberrant endosomes and impairs cholesterol trafficking. Mol. Biol. Cell. 11:227–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccarelli, F.D., C. Proukakis, H. Patel, H. Cross, S. Azam, M.A. Patton, P. Bork, and A.H. Crosby. 2003. The identification of a conserved domain in both spartin and spastin, mutated in hereditary spastic paraplegia. Genomics. 81:437–441. [DOI] [PubMed] [Google Scholar]
- Fujita, H., Y. Umezuki, K. Imamura, D. Ishikawa, S. Uchimura, A. Nara, T. Yoshimori, Y. Hayashizaki, J. Kawai, K. Ishidoh, et al. 2004. Mammalian class E Vps proteins, SBP1 and mVps2/CHMP2A, interact with and regulate the function of an AAA-ATPase SKD1/Vps4B. J. Cell Sci. 117:2997–3009. [DOI] [PubMed] [Google Scholar]
- Gruenberg, J., and H. Stenmark. 2004. The biogenesis of multivesicular endosomes. Nat. Rev. Mol. Cell Biol. 5:317–323. [DOI] [PubMed] [Google Scholar]
- Hanson, P.I., and S.W. Whiteheart. 2005. AAA+ proteins: have engine, will work. Nat. Rev. Mol. Cell Biol. 6:519–529. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., M. Babst, and S.D. Emr. 2001. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 106:145–155. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., G. Odorizzi, and S.D. Emr. 2002. Receptor downregulation and multivesicular-body sorting. Nat. Rev. Mol. Cell Biol. 3:893–905. [DOI] [PubMed] [Google Scholar]
- Katzmann, D.J., C.J. Stefan, M. Babst, and S.D. Emr. 2003. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J. Cell Biol. 162:413–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhtala, N., and G. Odorizzi. 2004. Bro1 coordinates deubiquitination in the multivesicular body pathway by recruiting Doa4 to endosomes. J. Cell Biol. 166:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas, A., M. Van Dyke, and J. Stock. 1991. Predicting coiled coils from protein sequences. Science. 252:1162–1164. [DOI] [PubMed] [Google Scholar]
- Morita, E., and W.I. Sundquist. 2004. Retrovirus budding. Annu. Rev. Cell Dev. Biol. 20:395–425. [DOI] [PubMed] [Google Scholar]
- Odorizzi, G., M. Babst, and S.D. Emr. 1998. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 95:847–858. [DOI] [PubMed] [Google Scholar]
- Raiborg, C., T.E. Rusten, and H. Stenmark. 2003. Protein sorting into multivesicular endosomes. Curr. Opin. Cell Biol. 15:446–455. [DOI] [PubMed] [Google Scholar]
- Robinson, J.S., D.J. Klionsky, L.M. Banta, and S.D. Emr. 1988. Protein sorting in Saccharomyces cerevisiae: isolation of mutants defective in the delivery and processing of multiple vacuolar hydrolases. Mol. Cell. Biol. 8:4936–4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott, A., H.Y. Chung, M. Gonciarz-Swiatek, G.C. Hill, F.G. Whitby, J. Gaspar, J.M. Holton, R. Viswanathan, S. Ghaffarian, C.P. Hill, and W.I. Sundquist. 2005. Structural and mechanistic studies of VPS4 proteins. EMBO J. 24:3658–3669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiflett, S.L., D.M. Ward, D. Huynh, M.B. Vaughn, J.C. Simmons, and J. Kaplan. 2004. Characterization of Vta1p, a class E Vps protein in Saccharomyces cerevisiae. J. Biol. Chem. 279:10982–10990. [DOI] [PubMed] [Google Scholar]
- Wendland, B., S.D. Emr, and H. Riezman. 1998. Protein traffic in the yeast endocytic and vacuolar protein sorting pathways. Curr. Opin. Cell Biol. 10:513–522. [DOI] [PubMed] [Google Scholar]
- Yeo, S.C., L. Xu, J. Ren, V.J. Boulton, M.D. Wagle, C. Liu, G. Ren, P. Wong, R. Zahn, P. Sasajala, et al. 2003. Vps20p and Vta1p interact with Vps4p and function in multivesicular body sorting and endosomal transport in Saccharomyces cerevisiae. J. Cell Sci. 116:3957–3970. [DOI] [PubMed] [Google Scholar]
- Yoshimori, T., F. Yamagata, A. Yamamoto, N. Mizushima, Y. Kabeya, A. Nara, I. Miwako, M. Ohashi, M. Ohsumi, and Y. Ohsumi. 2000. The mouse SKD1, a homologue of yeast Vps4p, is required for normal endosomal trafficking and morphology in mammalian cells. Mol. Biol. Cell. 11:747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}