

Abstract
We have been able to observe the dynamic interactions between a specific messenger RNA (mRNA) and its protein product in vivo by studying the synthesis and assembly of peripherin intermediate filaments (IFs). The results show that peripherin mRNA-containing particles (messenger ribonucleoproteins [mRNPs]) move mainly along microtubules (MT). These mRNPs are translationally silent, initiating translation when they cease moving. Many peripherin mRNPs contain multiple mRNAs, possibly amplifying the total amount of protein synthesized within these “translation factories.” This mRNA clustering is dependent on MT, regulatory sequences within the RNA and the nascent protein. Peripherin is cotranslationally assembled into insoluble, nonfilamentous particles that are precursors to the long IF that form extensive cytoskeletal networks. The results show that the motility and targeting of peripherin mRNPs, their translational control, and the assembly of an IF cytoskeletal system are linked together in a process we have termed dynamic cotranslation.
Introduction
Intermediate filament (IF) proteins are assembled into either homopolymer or heteropolymer 10-nm-diam cytoskeletal filaments in a complex multistep process (Strelkov et al., 2003). Pairs of protein chains interact in parallel and in register to form an α-helical coiled–coil dimer, which is the basic building block of IF. Little is known about the mechanisms responsible for dimer formation. However, it is known that in vitro dimers assemble into antiparallel tetramers that associate laterally to form unit-length filaments (ULFs). The ULFs are ∼60-nm-long and contain ∼32 protein chains. These anneal end-to-end to form ∼10-nm-diam IFs (Strelkov et al., 2003). Of these various IF structures, only small amounts of tetramer have been detected in soluble fractions of lysed cells (Soellner et al., 1985; Eriksson et al., 2004).
Type III IF proteins such as vimentin and peripherin exist in several states within cells, including nonfilamentous particles (Prahlad et al., 1998). Particles form short IF, or squiggles, which form the long IF that comprise the cytoskeletal networks of interphase cells. Although the composition of particles is unknown, they most likely contain essential IF building blocks such as dimers or ULFs. The assembly of particles into IF networks has been studied in spreading fibroblasts and differentiating nerve cells (Helfand et al., 2003a). During spreading, ∼70% of the particles move rapidly along MT in a kinesin- and dynein-dependent manner (Helfand et al., 2004). As spreading progresses, many particles are converted into polymerized IF. Similar particles are seen in other IF systems, including type IV neurofilaments (Prahlad et al., 1998) and the types I and II keratins in epithelial cells (Liovic et al., 2003).
Although it is obvious that nonfilamentous particles are precursors in the assembly pathway of cytoskeletal IF, little is known about their formation in cells. We show that in rat pheochromocytoma cells (PC12), a significant fraction of peripherin particles are assembled cotranslationally in a process that we have termed dynamic cotranslation. Evidence for dynamic cotranslation is derived from RNA FISH and the simultaneous live imaging of both peripherin mRNA and its protein product. Individual peripherin mRNA particles (messenger RNPs [mRNPs]) possess numerous copies of peripherin mRNA, suggesting a mechanism involving the coordinated synthesis of coiled–coil dimers, the building blocks of IF. The results provide important and novel insights into the linkages among the motile properties and targeting of mRNAs, their translational control, and the dynamic properties and assembly of IF proteins.
Results
Peripherin particles rapidly recover their fluorescence after photobleaching
The dynamic properties of IF particles were analyzed in GFP-peripherin–expressing PC12 cells by FRAP. The cells used in these experiments were optimized for particle formation by short-term (2–4 h) NGF treatment. At this time, there was a dramatic increase in peripherin expression in the form of particles (Fig. 1). Within 1 min after whole-cell photobleaching, fluorescent particles were visible (Fig. 1 A and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1), whereas filaments could not be detected for 5–10 min (not depicted; Helfand et al., 2003a). This rapid recovery suggested that some particles might be engaging in de novo synthesis of peripherin. We tested this possibility by photobleaching GFP-peripherin–expressing cells in the presence of cycloheximide to inhibit protein synthesis. Under these conditions, the fluorescence recovery of particles was not detected even after 10–15 min (Video 2).
Figure 1.

FRAP analysis of a PC12 cell expressing GFP-peripherin. (A) A series of images of a single PC12 cell transfected with GFP-peripherin for 24 h, followed by NGF treatment for 4 h. This cell was photobleached in its entirety, and recovery of fluorescence was monitored by time-lapse imaging for a period of 10 min at 10-s intervals (Video 1). The cell, which is in the early stage of neurite extension, is depicted before photobleaching in i (phase and fluorescence) and ii (fluorescence). Particles were not seen immediately after photobleaching (iii), but were seen within 1 min after bleaching (iv). This rapid appearance of fluorescent particles after whole-cell photobleaching is completely inhibited by cycloheximide, as demonstrated by identical time-lapse observations over a 10-min period (Video 2 and not depicted). (B) The majority of peripherin particles move rapidly through the cytoplasm along microtubule tracks (Video 1; Helfand et al., 2004) making it extremely difficult to determine whether an individual fluorescent particle could recover its fluorescence. Therefore, PC12 cells were transfected with GFP-peripherin for 24 h, treated with NGF for 2 h, and exposed to nocodazole to disassemble microtubules and inhibit peripherin particle motility. In this experiment, one half of the cell (prebleach image shown in v) was left unbleached, as shown in the 10-s postbleach image (vi), to ensure that particles were immobilized. Time-lapse observations demonstrated that fluorescent particles appeared within 1 min (vii) and that after 10 min (viii) ∼30% of the particles (n = 200) seen before photobleaching had recovered their fluorescence (Video 3). There was no significant increase in the number of particles that recovered after a longer recovery period of 30 min (not depicted). The 10-min postbleach image (viii) and the higher magnification image (ix) are displayed as an overlay between the prebleach (green) and the 10-min after bleach (red) images. Overlap between the green and the red appear yellow and represent particles that recovered their fluorescence. Not all the particles that recovered are yellow, but are red because of slight changes in particle position and shape over the 10-min observation period, leading to an incomplete overlap. Videos 1–3 are available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1. Bars, 10 μm.
We also sought to determine whether the same particles seen before photobleaching were the ones that recovered their fluorescence. However, because ∼70% of the peripherin particles in PC12 cells engage in rapid MT-dependent movements (Helfand et al., 2003a), it was necessary to treat cells with the MT inhibitor nocodazole to inhibit motility. Transfected cells that had been differentiated in NGF for 2 h were treated with nocodazole for 30 min. Under these conditions, individual particles could be identified both before and at time intervals after photobleaching. As a control in these experiments, only half of the cell was photobleached to provide an internal standard to ensure that particles were indeed immobilized (Fig. 1 B and Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1). Some particles recovered their fluorescence within 1 min after photobleaching, and, by 10 min, ∼30% of the immobilized particles (n = 200) in the photobleached half seen before photobleaching were fluorescent. No further increase in the number of fluorescent particles was detected for periods up to 30 min (not depicted).
Some peripherin particles are closely associated with peripherin mRNA
To determine whether peripherin particles were associated with peripherin mRNA, GFP-peripherin–expressing PC12 cells were fixed and processed for FISH within 2–4 h after the addition of NGF (Fig. 2 A). Biotinylated antisense probes were used to detect peripherin mRNA. Analysis of ∼1,800 randomly selected peripherin particles in the thin, peripheral regions of 20 cells revealed that ∼29% were closely associated with peripherin mRNAs, which were also seen as discrete particles (Fig. 2, A and D [green, peripherin particles; red, peripherin mRNA]; for split image of Fig. 2, A and D, see Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1). A close association was defined as a minimum of 1/4 overlap between diameters of the red and green signals (Fig. S2 shows a more detailed description of the association between peripherin particles and mRNA). To ensure that this association was not random, statistical tests were performed using a modification of a previously published procedure (Helfand et al., 2003a; see Materials and methods). The expected number of peripherin particles that could associate with the peripherin mRNA based on chance alone was calculated using this same 1/4 overlap criterion for each of the 20 cells analyzed. This calculation included the determination of the number and size of individual peripherin mRNA and protein particles, as well as the total area of the cytoplasmic region analyzed. The resulting data were subjected to a two-tailed t test with Bonferroni correction to determine if the differences between the actual and expected values were statistically significant (P < 0.006; see Materials and methods). We determined that the probability of the association seen between peripherin particles and mRNA, based on chance alone, was less than one in 3.7 × 108 (P ≅ 3.7e − 8). The association of endogenous peripherin particles with peripherin mRNA in untransfected cells was also determined by a combination of immunofluorescence and FISH. Approximately 1,000 particles were analyzed as above, and the analysis revealed that ∼30% were associated with peripherin mRNA, reflecting the similar association seen in transfected cells (Fig. S1, G and H).
Figure 2.

Peripherin mRNA association with peripherin particles. (A and D) PC12 cells were transfected with GFP-peripherin for 24 h, followed by exposure to NGF for 4 h, and processed for FISH to determine whether peripherin particles were associated with peripherin mRNA. Biotinylated antisense RNA probes were used for detection of peripherin mRNA (red). Peripherin protein is shown in green (Fig. S1, A and B, for split images). Approximately 29% of the peripherin particles (n = 1,776 particles in 20 cells) are associated with peripherin mRNA (D, arrows). An association was defined as a minimum of 1/4 overlap between the green and red signals, and the quantitation was performed in thin regions of the cytoplasm, not in the thick, juxtanuclear regions where potential for random overlap was higher because of the large amount of both peripherin mRNA and protein. (B and E) GFP-peripherin–transfected PC12 cells were treated with puromycin before processing for FISH with probes specific for peripherin mRNA. Peripherin protein particles are shown in green and peripherin mRNA is in red (Fig. S1, C and D, for split images). This treatment reduced the association between peripherin particles and peripherin mRNA to statistically insignificant levels (n = 750 peripherin particles in 15 cells). (C and F) As a control for the specificity of the association between peripherin mRNA and peripherin particles, we also determined whether actin mRNA was associated with peripherin particles. To this end, PC12 cells that were transfected as in A were processed for FISH using biotinylated antisense-RNA probes for actin mRNA. Unlike the overlapping distributions of peripherin particles and peripherin mRNA throughout the cytoplasm (A), the actin mRNA (C, red) appears to localize in cytoplasmic regions separate from the majority of peripherin protein particles (green; Fig. S1, E and F, for split images). There is no statistically significant association between actin mRNA and peripherin particles (F; n = 500 peripherin particles in 10 cells). (G) Because ∼70% of the peripherin particles move at any given time, it was of interest to determine whether particles associated with mRNA are motile. Time-lapse images of moving GFP-peripherin particles (n = 40) were captured (i–iii) and the cells were fixed with formaldehyde directly on the microscope stage. These cells (n = 15) were then processed for FISH to detect peripherin mRNA and the motile particles were relocated in the confocal microscope (iv). None of the moving particles (green; arrowheads) was associated with peripherin mRNA (red). In contrast, some stationary particles (asterisks) within the same microscope field of view were associated with peripherin mRNA. Fig. S1 and Videos 4 and 5 are available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1. Bars, 5 μm.
The protein synthesis inhibitor puromycin, which acts by releasing nascent protein chains from ribosomes (Yarmolinsky and de la Haba, 1959; Joklik and Becker, 1965), reduced the association between peripherin protein particles and mRNA from ∼29% to levels that were attributable to random overlap (Fig. 2, B and E; for split image of Fig. 2 B see Fig. S1, C and D; n = 15 cells; P ≅ 0.52).
As an additional control for the specificity of the interaction between peripherin particles and peripherin mRNA, we analyzed the association between peripherin particles and β-actin mRNA. Unlike the overlapping cytoplasmic distributions between peripherin particles and mRNA seen in Fig. 2 A, the majority of β-actin mRNA and peripherin particles appeared to reside in distinct regions of the cell (Fig. 2 C; for split image of Fig. 2 C see Fig. S1, E and F). No significant association could be detected based on the same statistical analyses performed for peripherin mRNA and particle association (Fig. 2 F; n = 10 cells; P ≅ 0.04).
As mentioned in the previous paragraphs, ∼70% of peripherin particles were motile and ∼30% were stationary (Helfand et al., 2003a). Therefore, we attempted to determine whether moving peripherin particles were associated with mRNA. Motile particles in GFP-peripherin–transfected PC12 cells that were grown on locator coverslips were observed by time-lapse imaging for 10–20 s (Video 4, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1), and the cells were then immediately fixed on the microscope stage. After fixation, the coverslip was processed for FISH and the appropriate microscope field was relocated (Fig. 2 G). In 15 different experiments involving 15 different cells, 40 moving particles were followed and none of these was associated with peripherin mRNA (Fig. 2 G, arrowheads). In contrast, particles in the same field of view, which were not moving during the observation period, were associated with mRNA (Fig. 2 G, asterisks). This result suggested that only stationary particles were engaged in protein synthesis (Video 5 shows footage of a different cell).
Peripherin particles and mRNPs move independently of each other
To determine the motile behavior of both peripherin particles and mRNAs, a system was developed that permitted the simultaneous imaging of both a specific mRNA and its translation product in live cells. A construct containing the enhanced CFP (ECFP)–peripherin coding sequence (CDS), followed by 24 MS2–binding repeats (R24; Fusco et al., 2003) and the peripherin 3′untranslated region (UTR), was introduced into PC12 cells along with a plasmid encoding YFP-MS2 (a bacteriophage MS2 coat protein fused to YFP). When the ECFP-peripherin-R24-3′UTR construct is transcribed, the R24 sequence in the mRNA forms 24 stem loops, each consisting of an identical sequence of 19 nucleotides. The YFP-MS2 protein binds to these stem-loop structures, thereby providing a visible reporter for locating and tracking the movements of these peripherin mRNAs. The translation product of this YFP-MS2–tagged peripherin mRNA is CFP-peripherin (Fig. 3 A).
Figure 3.

Simultaneous imaging of peripherin mRNA and its protein product in vivo. (A) A schematic representation of the system permitting the direct observation of both peripherin mRNA and its protein product. A construct containing the ECFP-peripherin CDS, followed by R24 and the peripherin 3′UTR, is introduced into PC12 cells along with a plasmid encoding for YFP-MS2 (a bacteriophage coat protein). When the ECFP-peripherin-R24-3′UTR construct is transcribed, the R24 sequence forms 24 stem loops, each consisting of an identical sequence of 19 nucleotides (1). The YFP-MS2 protein binds to these stem-loop structures, thereby providing a visible reporter for localizing and tracking the movements of peripherin mRNAs (2). The translation product of this YFP-MS2–tagged peripherin mRNA is CFP-peripherin (3). (B) A region of the cytoplasm of a living PC12 cell cotransfected with the pECFP-peripherin-R24-3′UTR and YFP-MS2 constructs for 24 h and subsequently treated with NGF for 4 h (i, fluorescence image; ii, phase image). Time-lapse imaging of this cell expressing both constructs showed rapid movements of YFP-MS2–tagged peripherin mRNA (red) and its protein product, CFP-peripherin (green), throughout the cell body as well as in early developing neurites (Video 6). (C) Higher magnification time-lapse observations (iii–v) of a PC12 cell cotransfected and treated with NGF as described in B reveal that peripherin mRNPs (red; arrowheads) move rapidly away from stationary peripherin protein particles (green; blue asterisks; Video 7). Movement of peripherin protein particles away from stationary mRNPs was not observed. Videos 6 and 7 are available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1. Bars: (B) 5 μm; (C) 1 μm.
Live imaging of PC12 cells expressing both constructs contained CFP-peripherin particles and YFP-peripherin mRNA, which also appeared as distinct particles or mRNPs (Fig. 3 B). Time-lapse imaging revealed rapid and independent movements of both peripherin mRNPs and protein particles. These were dispersed throughout the cytoplasm, including early outgrowing neurites (Video 6, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1; one frame of this movie is shown in Fig. 3 B). Approximately 80% of the peripherin mRNPs were motile, with an average speed of 0.42 ± 0.15 μm/s (n = 200 mRNPs in 5 cells), which is ∼1.5 times faster than the average rate of movement of peripherin particles (Helfand et al., 2003a). In no case could we detect mRNPs associated with protein particles moving together (n = 54 cells). In time-lapse movies, stationary mRNPs could be found in close association with stationary protein particles. In some cases, the mRNPs were seen to move rapidly away, leaving the peripherin particle behind (n = 20 mRNPs in 10 cells; Fig. 3 C and Video 7). The reverse situation, in which a protein particle moved away from a stationary mRNP, was never observed. Furthermore, treatment of cells with 10 μg/ml of the protein synthesis inhibitor puromycin for 30–60 min increased the fraction of motile mRNPs to ∼99% (n = 1,286 mRNPs in 6 cells). These findings lend further support to the idea that motile mRNPs are translationally silent.
Most of cellular peripherin mRNA exists in clusters
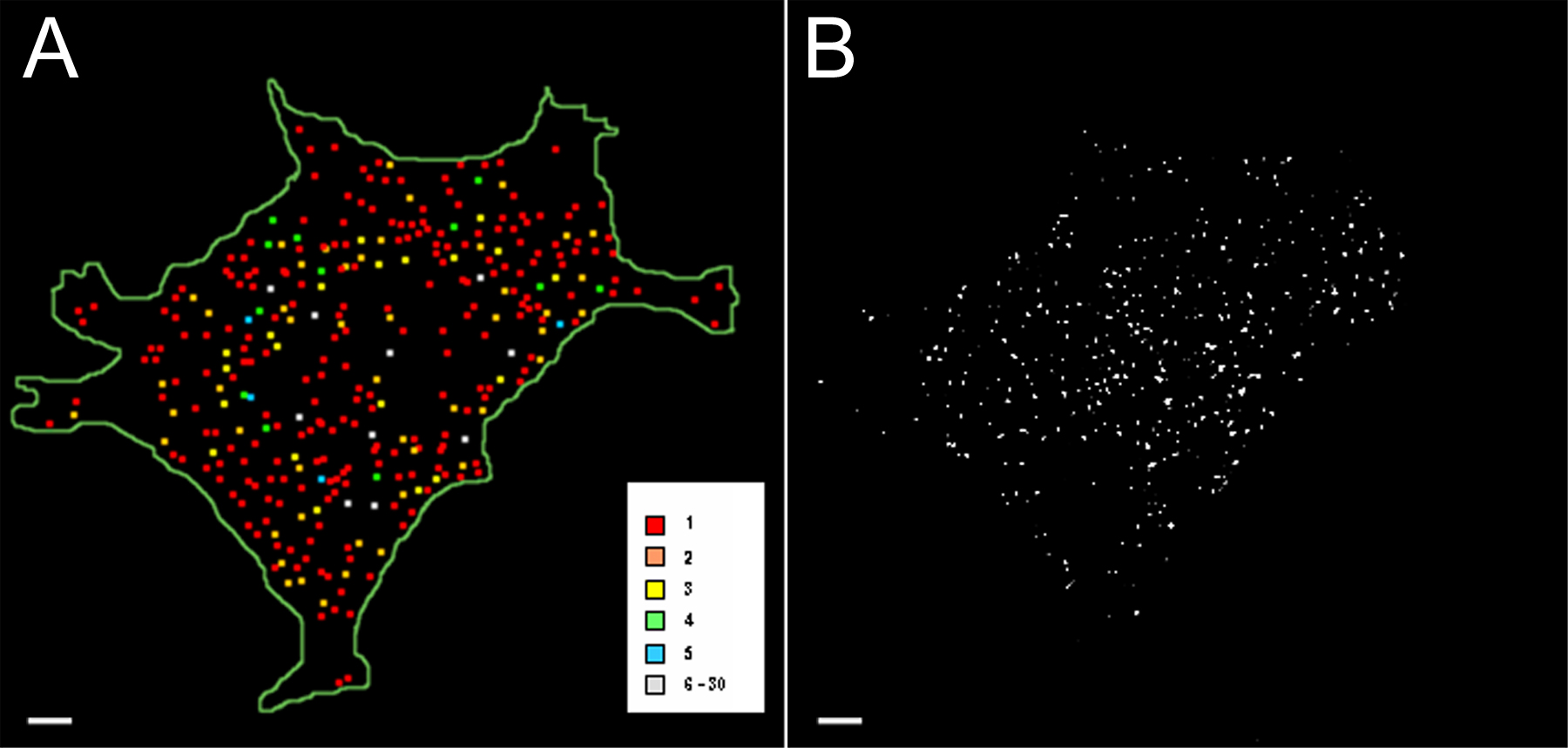
To evaluate the copy number of peripherin mRNA molecules in each mRNP, we used a quantitative FISH method, which is capable of detecting single RNA molecules (Femino et al., 1998; see Materials and methods). The results of this analysis (n = 7 cells) demonstrated that peripherin mRNPs contain one or more mRNAs (Fig. 4, A and B). The distribution of the mRNA clusters or mRNPs shows that ∼50% of the mRNPs contain one mRNA and the other ∼50% contain two or more peripherin mRNAs (Fig. 4 C). However, the ∼50% of mRNPs that contain one mRNA account for only ∼20% of the total peripherin mRNA. Therefore, ∼80% of the total peripherin mRNA exists in clusters of two or more mRNAs (Fig. 4 D).
Figure 4.

Quantification of peripherin mRNA. Cells transfected with ECFP-peripherin-R24-3′UTR construct for 24 h, followed by 4 h in NGF, were processed for double FISH with a Cy3-labeled probe to peripherin mRNA and a Cy5-labeled probe to the MS2-binding repeat region. This made it possible to quantitatively analyze peripherin mRNAs that could be detected in live cell observations using the MS2 tagging method. 3D Z-stacks of the Cy3 and Cy5 probe were obtained. These images were deconvolved, and the cells were analyzed by a single RNA molecule quantification method (Femino et al., 1998) to determine the number of RNA transcripts in each mRNP. (A) A color-coded map of the mRNPs containing 1–30 mRNA molecules in a single cell. The map is a compressed image of all the Z-slices. Low-level fluorescent signals caused by background noise and with values lower than that for a single probe (as in B) have been excluded. (B) A deconvolved image of a single Z-slice in the middle of the cell used for the color-coded map in A. (C) The distribution of the number of peripherin mRNA molecules per mRNP. The data show that ∼50% of the mRNPs contain one mRNA (red) and the other half contain two or more peripherin mRNAs displayed according to the other colors indicated. (D) Analyses of the distribution of total cellular peripherin mRNA show that the single mRNAs account for only 20% of the total peripherin mRNA; therefore, 80% of the total peripherin mRNA exists in clusters of two or more. Bars, 2 μm.
Mechanisms responsible for peripherin mRNA clustering
Type III IF proteins such as peripherin are known to self-assemble into higher order structures (Strelkov et al., 2003). This capacity for self-assembly may play a role in the clustering of peripherin mRNAs caused by the interactions between nascent protein chains. mRNA clustering may also be attributable to regulatory regions, such as the 3′UTR, within the mRNA molecules. An examination of the possible role of the nascent peripherin protein chains in mRNA clustering was initiated by inserting a stop codon (TAA) between the CFP and peripherin CDSs in the ECFP-peripherin-R24-3′UTR construct to inhibit peripherin synthesis. To verify the action of the stop codon, we transfected rat embryonic fibroblasts (Rat2) that do not express endogenous peripherin with the ECFP-TAA-peripherin-R24-3′UTR construct. Controls consisted of Rat2 cells transfected with ECFP-peripherin-R24-3′UTR. After a 24-h transfection, whole cell lysates were separated by SDS-PAGE and prepared for immunoblotting (Fig. 5 A). In cells expressing the control plasmid, both peripherin and GFP antibodies (known to react with CFP) recognized a product corresponding to the predicted size of the CFP-peripherin fusion protein, ∼81 kD. In cells expressing the construct with the TAA insertion, there was no detectable peripherin by immunoblotting and the GFP antibody instead recognized an ∼27-kD product, which is the predicted size of CFP (Fig. 5 A). Immunofluorescence analyses of cells transfected with ECFP-TAA-peripherin-R24-3′UTR showed no peripherin staining, and the CFP signal was diffuse and evenly distributed throughout the cytoplasm (unpublished data). In contrast, in cells transfected with the control plasmid, both the peripherin antibody staining and CFP fluorescence were filamentous and colocalized with the endogenous Rat2 vimentin network (unpublished data), suggesting that the fused peripherin protein was, as expected, coassembling with this type III IF protein (Herrmann and Aebi, 2000).
Figure 5.
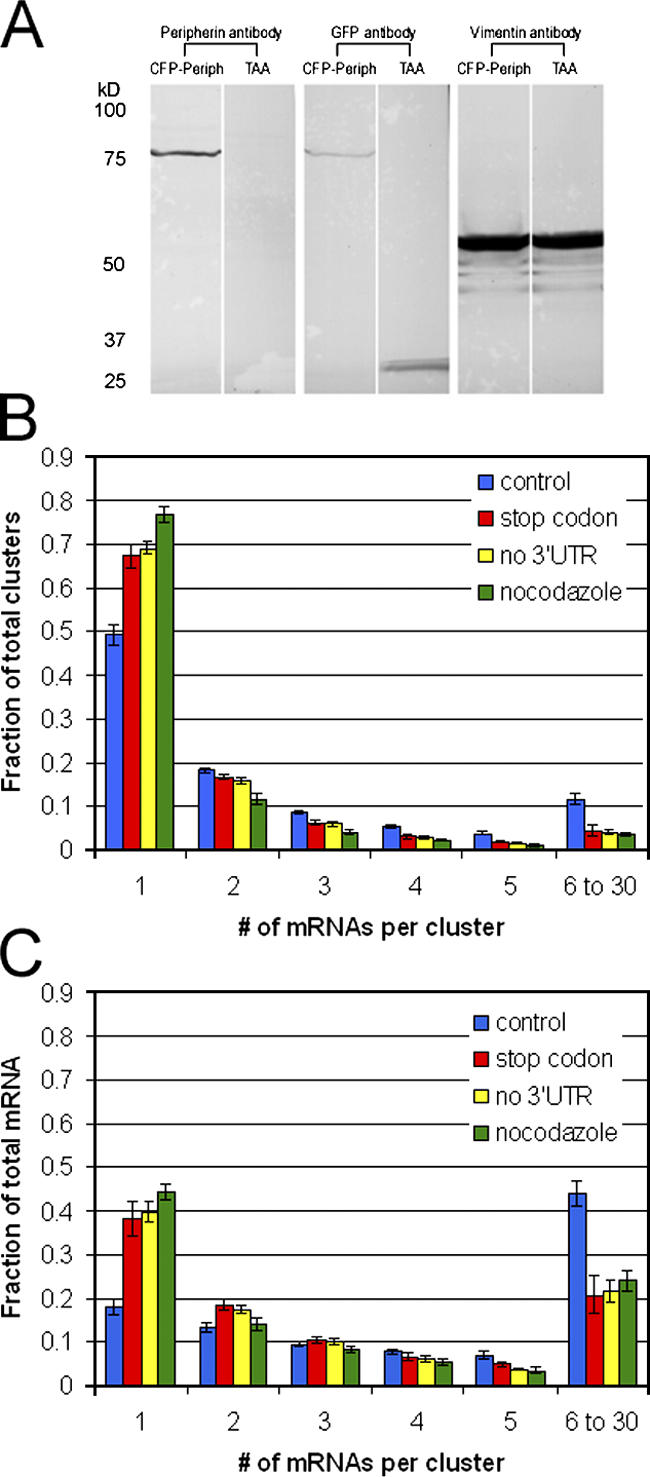
Insights into peripherin mRNA clustering. The relative roles of the nascent peripherin protein chain, the 3′UTR of peripherin mRNA, and MT in the formation of peripherin mRNA clusters were studied. (A) To determine the role of the nascent peripherin protein in mRNA clustering, a peripherin translation-null construct (ECFP-TAA-peripherin-R24-3′UTR) was created by inserting a stop codon (TAA) between the CFP and peripherin CDSs in the ECFP-peripherin-R24-3′UTR construct. To confirm the effectiveness of the inserted stop codon in preventing the translation of peripherin, Rat2 cells that are null for peripherin were transfected with either the ECFP-peripherin-R24-3′UTR construct as a control or the ECFP-TAA-peripherin-R24-3′UTR construct. After 24 h, whole cell lysates of these transfected Rat2 cells were separated by SDS-PAGE, transferred to nitrocellulose, and immuno-blotted with antibodies against CFP (GFP antibody) and peripherin. Vimentin, the endogenous Rat2 IF protein was used as a loading control. In cells expressing the control plasmid (CFP-Periph), both the peripherin and GFP antibodies recognized a product with a molecular weight of ∼81 kD, the predicted size of the CFP-peripherin fusion protein. In cells expressing the construct with the TAA insertion, there was no detectable peripherin by immunoblotting, and the GFP antibody recognized an ∼27-kD product, the predicted size of CFP. (B) A histogram comparing the distributions of peripherin mRNP clusters in control cells (blue bars, ECFP-peripherin-R24-3′UTR), ECFP-TAA-peripherin-R24-3′UTR–transfected cells (red bars), cells expressing the 3′UTR-null construct (yellow bars, ECFP-peripherin-R24), and control cells (ECFP-peripherin-R24-3′UTR) treated with nocodazole during NGF-induced differentiation (green bars). PC12 cells were transfected with the various constructs for 24 h, followed by NGF for 4 h. The cells were prepared for FISH and quantitatively analyzed as described in Fig. 4. Error bars depict the SEM. (C) A histogram comparing the distributions of total peripherin mRNA in control cells (blue bars, ECFP-peripherin-R24-3′UTR); ECFP-TAA-peripherin-R24-3′UTR transfected cells (red bars); cells expressing the 3′UTR-null ECFP-peripherin-R24 construct (yellow bars); and control cells (ECFP-peripherin-R24-3′UTR) treated with nocodazole.
The quantitative FISH method was used to analyze PC12 cells transfected with the ECFP-TAA-peripherin-R24-3′UTR construct for 24 h followed by NGF for 4 h. The results showed an ∼30% increase in the number of mRNPs containing single mRNAs (Fig. 5 B, red bars; n = 8 cells) compared with controls that were transfected with the ECFP-peripherin-R24-3′UTR construct (Fig. 5 B, blue bars). However, calculation of the total amount of peripherin mRNA in clusters of two or more still accounted for ∼60% of the total cellular peripherin mRNA (Fig. 5 C). These data suggest that mRNA clusters can form in the absence of peripherin translation and that the clustering is mediated to a certain extent by the mRNA itself.
To more specifically define the region of peripherin mRNA that might be involved in mRNA clustering, we tested the role of the 3′UTR by deleting this sequence from the original ECFP-peripherin-R24-3′UTR construct. Cells transfected with this construct and processed for FISH were quantitatively analyzed as described in the previous paragraph. The results were very similar to the stop codon experiment, showing an ∼30% increase in single mRNAs (Fig. 5 B, yellow bars; n = 20 cells). As with the stop codon construct, ∼60% of the total peripherin mRNA still formed clusters (Fig. 5 C).
To further examine the mechanisms underlying the peripherin mRNA clustering, PC12 cells expressing the ECFP-peripherin-R24-3′UTR construct were treated with nocodazole both before and during NGF treatment (see Materials and methods). Under these conditions, the absence of a fully polymerized MT system resulted in an ∼54% increase in the number of mRNPs containing single mRNAs (Fig. 5 B, green bars; n = 12 cells; Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1) and an ∼30% decrease in the fraction of total mRNA in clusters (Fig. 5 C).
Observing peripherin particle formation in vivo
The close associations found between some of the mRNPs and peripherin particles suggested a cause and effect relationship, indicating that the protein particle formation resulted from translation of the colocalized mRNA. Therefore, we sought to observe the generation of a protein particle in vivo. Cells were double transfected with ECFP-peripherin-R24-3′UTR and YFP-MS2 and treated with NGF for 4 h, at which time nocodazole was added to depolymerize MT (30 min; see Materials and methods). This procedure inhibited the rapid movements of the majority of both peripherin mRNPs and protein particles. This afforded us the opportunity to observe individual mRNPs over a period of time consistent with the synthesis of protein by time-lapse imaging. In several cases, an mRNP was seen closely associated with a weak, diffuse CFP signal. Within 30–180 s, the CFP fluorescence intensity increased significantly, ultimately appearing indistinguishable from other IF particles (Fig. 6 A and Video 8, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1).
Figure 6.

Observing peripherin particle formation in vivo. (A) A live cell cotransfected with both ECFP-peripherin-R24-3′UTR and YFP-MS2 for 24 h, followed by NGF treatment for 4 h, was treated with nocodazole for 30 min to depolymerize microtubules. This treatment inhibited the motility of both peripherin particles and mRNPs, thereby facilitating the observation of peripherin particle formation in association with mRNPs. Time-lapse imaging shows an mRNP (red) that appears to be engaged in protein synthesis (green; arrowhead) in a thin peripheral region of the cytoplasm (Video 8). The green signal appears first as a dim haze (i and ii) that becomes brighter and more distinct with time (iii), resulting in the formation of a typical IF particle by 180 s (iv). (B) The association of ribosomes with peripherin mRNPs. Cells expressing both ECFP-peripherin-R24-3′UTR and YFP-MS2 were processed for indirect immunofluorescence using an antibody against the S6 subunit of ribosomes. Approximately 70% of peripherin mRNPs (vi, light blue) associated with peripherin protein particles (v; green) were also associated with ribosomes (vii, red) as shown in the triple overlay in viii. (C) Distribution of peripherin particles. 29% of peripherin particles are associated with peripherin mRNA. Of this fraction, ∼70% are also associated with ribosomes. The remaining 30% may represent complexes in which the translation process has ceased and the ribosomes have dissociated. Video 8 is available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1. Bars, 5 μm.
The association of ribosomes with peripherin mRNPs
Our findings suggest that ∼30% of peripherin particles are cotranslationally assembled in close association with their mRNPs. Therefore, it was expected that ribosomes would be associated with these mRNP–peripherin particle complexes. An antibody directed against the S6 ribosomal subunit was used to determine whether ribosomes were associated with these complexes in cells expressing both YFP-MS2 and ECFP-peripherin-R24-3′UTR after treatment with NGF. Approximately 70% of the peripherin particles that associated with peripherin mRNPs also associated with ribosomes (Fig. 6, B and C). 10 cells were analyzed and the statistical significance of the associations was determined using the method described for peripherin particle and mRNA association, but modified for triple colocalizations (see Materials and methods). It should be noted that only ∼20% of peripherin mRNPs were associated with ribosomes and that the majority were associated with neither ribosomes nor peripherin particles.
Further evidence for mRNA clustering in IF composed of two types of proteins
Keratin IF are assembled from heterodimers containing a type I and II protein (Steinert et al., 1994). We analyzed HeLa cells expressing keratin 8 and 18 to determine if keratin mRNAs form clusters and whether these clusters contain both types of mRNA. In fully spread cells, keratin networks are more stable when compared with Type III IF networks such as peripherin (Yoon et al., 2001). Therefore, early spreading cells (after trypsinization and replating), in which the keratin network was more dynamic and undergoing active assembly (Yoon et al., 1998, 2001; unpublished data), were used for analysis. To detect the two types of keratin mRNAs in these spreading cells, a biotinylated oligonucleotide probe specific for K18 mRNA and a digoxigenin-conjugated probe specific for K8 mRNA were used for double FISH analysis (Fig. 7 A). Approximately 15% of the two types of mRNA signals appeared to colocalize or overlap extensively (Fig. 7 B; n = 8 cells; n ≅ 900 mRNPs; P ≅ 0.0004). The existence of hetero-mRNP clusters provides further evidence that IF mRNA clustering is a general phenomenon.
Figure 7.

Keratin 18 and 8 mRNA colocalize in a subpopulation of mRNPs. HeLa cells were trypsinized and replated for 2 h; then they were processed for double FISH to detect keratin 18 and 8 mRNA. (A) For keratin mRNA detection a biotinylated probe specific for keratin 18 mRNA (red) and a digoxigenin-labeled probe specific for keratin 8 mRNA (green) were used. (B) A ventral view of an early spreading HeLa cell hybridized with both K18 (red) and K8 probes (green). Approximately 15% of both types of mRNA signals appeared to colocalize or overlap completely (arrowheads; n = 8 cells; n ≅ 900 mRNPs; P ≅ 0.0004). Bar, 5 μm.
Discussion
The peripherin system of IFs in PC12 cells has provided a unique opportunity to observe the dynamic properties of a specific species of mRNA, along with its translation product. This is attributable to the finding that newly synthesized peripherin accumulates in close proximity to its mRNPs, which is demonstrated by the findings that ∼30% of GFP-peripherin particles rapidly recover their fluorescence after photobleaching in a cycloheximide-sensitive fashion. This rapid recovery of fluorescence shows that the folding and maturation time for GFP-peripherin is on the order of minutes. Although the folding and maturation process of GFP takes ∼1 h in bacteria and yeast (Rizzo and Piston, 2004), it has recently been shown that GFP cDNA injected into the nuclei of mammalian cells is expressed and fluoresces in the cytoplasm within 40 min (Dean, 2004). In addition, the refolding of extensively denatured GFP in vitro takes a few minutes, as determined by chromophore fluorescence (Enoki et al., 2004). Furthermore, our FISH analyses demonstrated that a similar percentage (∼29%) of peripherin particles was closely associated with peripherin mRNA and that this association was inhibited by puromycin. The loss of this association in the presence of puromycin shows that the relationship between peripherin mRNAs and protein particles depends on translation.
The interactions between peripherin particles and mRNA were directly visualized in living cells using the dual-transfection method developed for simultaneous imaging of both peripherin mRNA and its protein product. Time-lapse imaging shows active movement of both peripherin mRNPs and protein particles throughout the cytoplasm. However, the movements were independent of one another, and motile peripherin mRNPs were never seen associated with peripherin protein particles. Various mRNA species are known to move along both MT and microfilaments, powered by molecular motors such as kinesin, dynein, and myosin (for review see Tekotte and Davis, 2002). Specific examples of kinesin-dependent mRNA movement include the mRNA-encoding myelin basic protein in oligodendrocytes (Carson et al., 1997) and tau mRNA in axons of neuronal P19 cells (Aronov et al., 2002). In addition, the localization of several zygotically transcribed mRNAs in Drosophila melanogaster embryos have been shown to be dependent on cytoplasmic dynein (for review see Tekotte and Davis, 2002). Although it has been hypothesized that motile mRNAs are not capable of translational activity, this has never been observed in vivo. Our dual peripherin mRNA- and protein-imaging method provides direct evidence that moving mRNPs are translationally inactive. In those instances where stationary peripherin mRNPs were found in close association with peripherin particles, the mRNP invariably moved rapidly away, leaving the protein particle behind. The reverse situation was never observed. In further support of these observations, the translational inhibitor puromycin increased the fraction of motile mRNPs to ∼99%.
Visualizing the early stages of protein synthesis in association with peripherin mRNPs turned out to be extremely difficult because of the rapid motility of the majority of both mRNPs and protein particles along MT tracks. To observe the relatively small number of stationary mRNPs engaging in protein synthesis cells were treated with nocodazole. This disassembled MT and therefore stopped the movement of the vast majority of peripherin mRNPs not engaged in protein synthesis, as well as the particles not associated with mRNA. Under these conditions, we were able to observe peripherin particles forming in close association with those translation factories that had presumably formed before the disassembly of MT. In addition, ∼70% of the mRNPs associated with particles were also associated with ribosomes. The ∼30% that remained may represent mRNPs that have ceased the translational process and have already dissociated from ribosomes (Fig. 8 and Video 9, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1). Most likely, newly synthesized peripherin particles will at some point engage molecular motors and initiate motility or alternatively, remain in place, ultimately assembling into short IFs that appear to link in tandem to form long IFs (Fig. 8 and Video 9; Helfand et al., 2004). On the other hand, the departing mRNP may move to a new site of synthesis, associate with ribosomes, and begin another round of translation (Fig. 8 and Video 9).
Figure 8.

A model depicting dynamic cotranslation mRNA particles. mRNPs (red) containing one or more peripherin mRNA are moved along MT (blue) by molecular motors such as kinesin and dynein. These motile mRNPs are translationally silent. When these complexes stop moving they associate with ribosomes (yellow) and engage in translational activity. The regulation of mRNP motility could involve the inactivation of their associated motors, the uncoupling of these motors, or the dissociation from their MT tracks. Many mRNPs appear to contain two or more mRNAs. As the IF protein is synthesized from multiple IF mRNAs, the protein chains are cotranslationally assembled into higher order structures that appear as nonfilamentous particles (green). Once a critical concentration of protein is achieved, the ribosomes disengage, followed by the dissociation of the mRNPs, which can either reasssociate with molecular motors or activate preexisting motors and move rapidly away along MT. The newly synthesized IF particles could assemble into IF at their site of synthesis or recruit motors to begin their journey as IF precursors to different regions of the cytoplasm. Once they reach their targets, they assemble into short IF (squiggles) that link up in tandem to form longer filaments (Video 9, available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1; Helfand et al., 2003a).
Quantitative analyses of peripherin mRNPs demonstrate that ∼80% of total peripherin mRNA resides in clusters of two or more. Analyses of cells expressing the ECFP-TAA-Peripherin-R24-3′UTR construct, from which peripherin protein is not synthesized, demonstrate that ∼60% of the total mRNA remains in clusters of two or more. The reduction from ∼80 to ∼60% is mostly attributable to an ∼30% increase in mRNPs containing single mRNAs and an ∼60% decrease in the largest mRNPs containing 6–30 mRNAs, suggesting that interactions between the nascent peripherin protein chains may contribute to the formation of large mRNA clusters. There was no significant change in the number of mRNPs containing two to five mRNAs under these experimental conditions. In the absence of a 3′UTR, there was a similar decrease in clustered peripherin mRNPs with the most dramatic decrease again in the large 6–30 mRNA-containing clusters. There was also an ∼30% increase in mRNPs containing single mRNAs. The MS2-binding repeat region of the mRNA is most likely not involved in the mRNA clustering, as a previous work using the same repeat region conjugated to actin mRNA showed that the vast majority of the mRNA existed as single molecules (Shav-Tal et al., 2004). Treatment of cells with nocodazole resulted in the most dramatic decrease in peripherin mRNA clustering, suggesting that the mRNAs are not clustered by simple diffusion, but require a regulated mechanism involving MT and most likely MT-based motors. The partial inhibition of clustering in nocodazole-treated cells may be because of an actin microfilament–based clustering mechanism (Stebbings, 2001).
It has been suggested that the nonfilamentous peripherin particles contain dimers, which are the essential building blocks of IF, and perhaps higher order structures such as tetramers and ULFs (Chang and Goldman, 2004). Dimer assembly requires the formation of coiled–coil interactions between the α-helical–rich central rod domains of two parallel and precisely in register IF protein chains (Strelkov et al., 2003). The α-helical coiled–coil structure is one of the primary subunit oligomerization motifs in proteins, originally discovered in keratin IF by x-ray diffraction (McArthur, 1943). The mechanism responsible for coiled–coil dimer assembly remains unknown. The finding that the majority of peripherin mRNA is located in clusters of two or more suggests that the formation of the coiled–coil interactions between pairs of peripherin protein chains takes place cotranslationally, involving the coordinated synthesis of closely associated mRNAs. Interestingly, during muscle differentiation, myosin assembly intermediates containing myosin heavy chain coiled–coil dimers also appear as globular “foci,” similar in appearance to peripherin particles. These foci are also thought to form cotranslationally (Srikakulam and Winkelmann, 2004). The double FISH experiments showing the subpopulation of hetero-mRNPs containing both K8 and K18 mRNAs also support the possibility that the cotranslational assembly of IF dimers is a general phenomenon.
Our study provides evidence for the clustering of IF mRNAs and its physiological significance. Other mRNAs, such as β-actin, have been found to exist as single mRNAs, using the same quantitative FISH method that we have described (Fusco et al., 2004). Furthermore, whereas peripherin mRNPs are distributed throughout the cell, actin mRNPs are concentrated in certain regions such as the lamellipodia of moving cells. This targeting of actin mRNPs is most likely related to the maintenance of high concentrations of monomeric soluble actin required for the extensive actin polymerization that takes place in lamellipodia (Shestakova et al., 2001). In contrast, IF mRNPs can move to many sites distributed throughout the cell to initiate the synthesis and formation of insoluble, nonfilamentous precursors such as peripherin particles. This provides a cell with the capacity to control the de novo assembly of fully polymerized IF anywhere in its cytoplasm. This temporal and spatial control of IF polymerization may allow cells to locally regulate the distribution and various functions of IF, such as those involved in determining the mechanical properties of the cytoplasm and in signal transduction (Chang and Goldman, 2004).
The direct observation of a specific mRNP and its protein product in the same living cell provides a powerful tool for determining the mechanisms and the order of events involved in targeted protein synthesis. Collectively, the results of this work reveal a process called dynamic cotranslation, which has profound implications for translational control in vertebrate cells.
Materials and methods
Cell culture
Stock cultures of PC12 cells were maintained in complete medium (DME containing 10% calf serum and 1 mM sodium pyruvate) at 37°C. For studies of peripherin particles, cells from stock cultures were removed with trypsin-EDTA (Invitrogen), replated onto laminin-coated (Roche) coverslips, and cultured in differentiation medium (DM; DME containing 5% calf serum, 1 mM sodium pyruvate, and 30 ng/ml of NGF [Roche] for 2–4 h; Helfand et al., 2003a).
For live imaging of cells in nocodazole (whole cell FRAP experiments and observation of particle formation experiments), PC12 cells were transfected with the appropriate constructs for 24 h, differentiated in NGF for 2–4 h, and then treated with 10 μg/ml nocodazole (Sigma-Aldrich) for 30 min before imaging. For analysis of mRNA clustering in nocodazole, cells were transfected with the ECFP-peripherin-R24-3′UTR construct for 24 h and then treated with both NGF and nocodazole for 4 h before quantitative FISH analysis. For translation inhibition experiments, cells were treated with either 10 μg/ml cycloheximide (Calbiochem) or 10 μg/ml puromycin (Sigma-Aldrich) for time periods of 5 min to 1 h.
Rat2 cells were maintained in DME containing 10% fetal calf serum at 37°C. For immunofluorescence studies, cells were trypisinized and replated onto uncoated coverslips.
HeLa cells were maintained in DME containing 10% calf serum at 37°C. For FISH studies, cells were trypsinized and replated onto uncoated coverslips for 2 h before fixation.
Antibodies
Primary antibodies used in this study included rabbit anti-peripherin (Parysek et al., 1988), mouse anti-biotin (Jackson ImmunoResearch Laboratories), sheep anti-digoxigenin (Boehringer), mouse anti-S6 (a gift from D.L. Spector, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY, and from R. Traut, University of California, Davis, Davis, CA), mouse anti-GFP (Roche), and mouse anti-vimentin (clone V9; Sigma-Aldrich). FITC-, rhodamine-, and Cy5-conjugated goat anti–rabbit and anti–mouse and donkey anti–sheep IgG (Jackson ImmunoResearch Laboratories) antibodies were used as secondary antibodies for indirect immunofluorescence. For immunoblotting, peroxidase-conjugated goat anti–rabbit and anti–mouse secondary antibodies were used (Jackson ImmunoResearch Laboratories).
SDS-PAGE and immunoblotting
For immunoblotting experiments, 70% confluent 100-mm dishes of transfected Rat2 cells were prepared for SDS-PAGE analysis and transferred to nitrocellulose for immunoblotting (Helfand et al., 2003b). All antibody incubations were performed in PBS containing 5% nonfat dry milk (Sigma-Aldrich).
Immunofluorescence
For anti-biotin labeling after FISH, PC12 cells grown on laminin-coated coverslips were rinsed in PBS and fixed with 3.5% formaldehyde (Tousimis) at room temperature for 5 min. HeLa cells were spread for 2 h on uncoated coverslips before fixation with formaldehyde. After formaldehyde fixation, cells were permeabilized with 0.05% Triton X-100 for 5 min. This fixation and permeabilization method was also used for the immunolabeling of Rat2 cells. For S6 labeling, PC12 cells were permeabilized for 15 s in IF lysis buffer (PBS containing 0.6 M KCl, 5 mM EDTA, 5 mM EGTA, and protease inhibitors [1 mM PMSF, 1 mM tosyl-l-arginine methyl ester, and 1 mg/ml each of leupeptin, pepstatin, and aprotinin; Sigma-Aldrich]) containing 0.1% Triton X-100 (Prahlad et al., 1998) and fixed for 5 min in 3.5% formaldehyde (Tousimis). Cells were then washed with PBS and processed for indirect immunofluorescence, as previously described (Prahlad et al., 1998; Yoon et al., 1998). After staining, coverslips were washed in PBS and mounted on glass slides in gelvatol containing 100 mg/ml 1,4-diazabicyclo[2.2.2]octane (Sigma-Aldrich; Yoon et al., 1998). Images of fixed, stained preparations were taken with a microscope (LSM 510; Carl Zeiss MicroImaging, Inc.; Yoon et al., 1998).
Probes for FISH
For the detection of peripherin mRNA in PC12 cells, either a 2′O-methylated RNA oligonucleotide complementary to the 3′UTR region of rat peripherin mRNA (position 1,473–1,492) or a probe complementary to the coding region of peripherin mRNA (position 288–320) was used. Each probe was labeled at its 5′ end with either biotin or Cy3. A biotin-labeled RNA oligonucleotide probe complementary to rat β-actin mRNA (position 402–421) was used in control experiments. For quantitative FISH analyses, cells were simultaneously hybridized with a Cy5-labeled probe to a region near the MS2-binding sites (Fusco et al., 2003) and the Cy3-peripherin probe. For keratin double FISH experiments, a probe antisense to position 338–366 in human keratin 8 mRNA CDS and a probe antisense to position 227–261 of human keratin 18 mRNA CDS were used. The 5′ end of the K8 probe was labeled with digoxigenin and the K18 was labeled with biotin. All probes were obtained from Integrated DNA Technologies.
FISH
PC12 cells were transfected with either GFP-peripherin, ECFP-peripherin-R24-3′UTR, ECFP-TAA-peripherin-R24-3′UTR, or ECFP-peripherin-R24 for 24 h and maintained in DM on laminin-coated coverslips for 2–4 h, followed by fixation with 4% paraformaldehyde (in 1× PBS, pH 7.4) for 15 min. Cells were then rinsed in 1× PBS three times for 10 min each, followed by permeabilization with 0.5% Triton X-100 for 5 min on ice. Cells were washed twice for 10 min in PBS and then in 2× SSC for 10 min. Cells were hybridized in hybridization buffer (final concentration of 2× SSC, 10% dextran sulfate, 50% formamide, 1 mg/ml tRNA mix, and 1 ng/μl of RNA probe) overnight at 37°C. The hybridization was performed on a coverslip sandwiched between two pieces of parafilm that were sealed at their edges to prevent dehydration. For the keratin double FISH experiment, HeLa cells were trypsinized and replated for 2 h and then processed in the same manner as the PC12 cells. In some experiments, untransfected PC12 cells were first processed for immunofluorescence using peripherin antibodies to detect endogenous peripherin particles, followed by a second fixation and hybridization with peripherin mRNA probes. The next day, after hybridization, both transfected and nontransfected cells were washed twice in 2× SSC (50% formamide) for 30 min and then in 1× SSC for 15 min. Cells were then processed for indirect immunofluorescence using either mouse anti-biotin, sheep anti-digoxigenin, or both, followed by goat anti–mouse-FITC and donkey anti–sheep-rhodamine IgG for secondary detection. As a negative control for hybridization, cells were treated with RNase A (Sigma-Aldrich) before hybridization.
Image analysis for RNA quantification
Images were acquired with an epifluorescence microscope (model BX61; Olympus) with an internal focus motor and a 60×, 1.35 NA, oil objective (UPlanApo; Olympus) using a 100-W mercury arc lamp for illumination. Digital images were acquired using a camera (CoolSNAP HQ; Roper Scientific), and stacks of 26 images were taken with a Z-step size of 0.2 μm using IPLab software (Windows v3; Scanalytics) and filter sets for CFP, Cy3, and Cy5 (Chroma Technology Corp.). For quantification of RNAs all three-dimensional (3D) stacks were first deconvolved using Exhaustive Photon Reassignment (Scanalytics), which uses a quantitative, constrained iterative algorithm with an acquired point-spread function.
Single molecule mRNA identification and quantification was performed as previously described (Femino et al., 1998) using the Cy3-labeled probe to the peripherin message. This probe has only one binding site on the transcript and can therefore serve for quantification. Only cells hybridized with both the Cy3-peripherin and Cy5-MS2 probes were selected for quantitation, as these are the ones expressing the exogenous construct. The number of probes per particle was determined by calculating the total fluorescent intensity (TFI) of individual RNPs and dividing that value by the TFI per probe as follows: TFI per probe was calculated for the Cy3-peripherin probe by collecting images from serial probe dilutions. 5 μl of each probe dilution (ranging from 1.5 to 1.5 × 10−3 ng/μl) were placed between a coverslip and a slide, onto which 170-nm blue fluorescent beads had previously been dried. Using the beads as markers, the distance between the coverslip and slide was measured (in micrometers) and the center plane of the dilution was located. A range of interest (256 × 256 pixels) that excluded beads was identified, and a single image was captured at the center plane, using an exposure time identical to that used to capture cell images. This procedure was repeated three times for each dilution, each at a different location on the coverslip. The TFI per probe was then obtained by plotting the integrated fluorescence in the total imaged volume against the known number of molecules in that volume. The slope of the resulting curve represents the TFI per one fluorescent probe molecule and is further used in the calculations. In parallel, TFI per 3D RNA particle in the FISH images was calculated from deconvolved images with a script written for IPLab. To compensate for the deconvolution, this value was then divided by the number of planes in the point-spread function used by EPR. Each RNA now had an assigned TFI, which was then divided by the TFI/probe molecule calculated from the dilution curve, resulting in the number of RNA molecules in an imaged particle. Color map image volumes were generated, using a program written for IPLab, to visualize the number and spatial position of clusters of mRNA. The program identified mRNA clusters from the acquired 3D images, which were deconvolved using a threshold and routine segmentation algorithms. The total intensity of each object was calculated and used to assign a color code for each mRNA cluster that corresponded to the number of mRNAs contained in the cluster. Signals below those calculated for single mRNAs were excluded in the color map presentations.
Statistical analysis
The single FISH images (using either peripherin or actin probes) of GFP-peripherin–transfected cells were subjected to statistical tests using a modification of a previously published procedure (Helfand et al., 2003a). These tests were performed to make certain that the associations between peripherin particles and peripherin mRNPs were not random. One cytoplasmic region from each cell (n = 20 cells for peripherin FISH; n = 15 cells for peripherin FISH in puromycin; and n = 10 cells for actin FISH) was used for the statistical analysis of images captured by confocal microscopy. For the purposes of our calculations, we assumed that the particles and mRNA signals were circular and that any particles that showed >1/4 overlap (in radii) with an mRNA signal were defined as “associations.” The following formula was used to calculate the expected number of peripherin particles (E) that would coincide (>1/4 overlap) with either peripherin or actin mRNA based on chance alone:
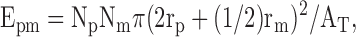 |
where Np is the total number of peripherin particles in a cytoplasmic region, Nm is the number of mRNPs (either peripherin- or actin-specific) in a cytoplasmic region, rp is the average radius of a peripherin particle, rm is the average radius of an mRNP, and AT is the total area of the cytoplasmic region analyzed. Measurements (radii and area) were performed using image analysis software (LSM510; Carl Zeiss MicroImaging, Inc.).
The number of associations actually observed was also determined for each image. This was defined as the number of peripherin particles that were observed to associate (by a minimum of 1/4 overlap) with peripherin or actin mRNA (n = 1,776, 750, and 500 randomly picked peripherin particles were analyzed for associations with mRNA in the peripherin FISH, peripherin FISH in puromycin, and actin FISH experiments, respectively). Finally, two-tailed t tests with Bonferroni correction were used to determine if the differences between the actual and expected values were statistically significant. P < 0.006 was defined as significant.
When the stringency of the association criteria was increased to a minimum of 1/2 overlap, the following formula was used to calculate the expected number of associations based on chance alone:
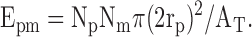 |
When the stringency of the association criteria was further increased to complete overlaps, the following formula was used to calculate the expected number of associations based on chance alone:
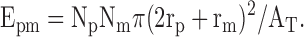 |
When the stringency of the association criteria was decreased to touching, the following formula was used to calculate the expected number of associations based on chance alone:
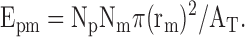 |
To test the significance of triple associations between YFP-MS2, CFP-peripherin, and ribosomal staining, the following formula was used:
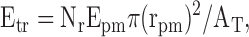 |
where Etr is the total number of ribosomes expected to be associated with both peripherin particles and mRNPs by chance alone, Epm is the expected number of peripherin particles that would associate with mRNPs by chance alone, Nr is the total number of ribosomes in a cytoplasmic region, rpm is the average radius of a peripherin particle-mRNP doublet, and AT is the total area of the cytoplasmic region analyzed. To determine the actual number of triplet associations observed. 10 cells were analyzed (167 peripherin particles that were associated with mRNPs were analyzed) and only extensive or complete overlaps were considered colocalizations.
For the keratin double FISH analysis, a complete overlap between the two mRNP signals was considered as a colocalization. To determine the expected number of colocalizations based on chance alone for these associations, the following formula was used:
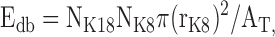 |
where Edb is the total number of K18 mRNPs expected to be associated with K8 mRNPs by chance alone, NK18 is the total number of K18 mRNPs in the cytoplasmic region analyzed, NK8 is the total number of K8 mRNPs in the cytoplasmic region analyzed, rK8 is the average radius of K8 mRNP signals, and AT is the total area of the cytoplasmic region analyzed. To determine the actual number of complete colocalizations between K18 and K8 mRNPs, eight cells (∼1,000 mRNPs for each type of mRNA) were analyzed.
Constructs used for transfection
The EGFP-C1-peripherin plasmid was constructed by amplifying rat peripherin cDNA (∼1.4 kb; provided by L. Parysek, University of Cincinnati, Cincinnati, OH) using PCR with primers that insert BamHI sites at the 5′ and 3′ ends. The resulting BamHI–BamHI fragment was subcloned into the BamHI site of pEGFP-C1 (CLONTECH Laboratories, Inc.). The ECFP-peripherin-R24-3′UTR plasmid was constructed by modifying the EGFP-C1-peripherin plasmid. 24 MS2-binding repeat sequences (∼1.3 kb) were fused to the 3′ end of the ECFP-peripherin CDS, followed by the 3′UTR (∼0.3 kb) of peripherin mRNA. The ECFP-TAA-peripherin-R24-3′UTR construct was created by inserting a TAA between the CFP and peripherin CDSs of the ECFP-peperipherin-R24-3′UTR construct. For the 3′UTR deletion construct (ECFP-peripherin-R24), this sequence was removed from the ECFP-peripherin-R24-3′UTR construct. YFP-MS2 was expressed from a plasmid with a CMV promoter ( Fusco et al., 2003).
Transfection
Constructs were introduced into either PC12 or Rat2 cells by electroporation (Yoon et al., 2001). After electroporation, PC12 cells were maintained in complete medium for 24 h. After 24 h, the transfected cells were trypsinized, replated onto laminin-coated coverslips, and maintained in DM for 2–4 h (which was optimal for visualizing peripherin particle synthesis) before imaging/analysis. Immunoblot analysis of GFP-peripherin–transfected PC12 cells shows that GFP-peripherin composes ∼25% of the total peripherin in an average transfected cell and that the exogenous peripherin incorporates into the endogenous IF network (Helfand et al., 2003a). Furthermore, transfected cells display a GFP-IF pattern that is indistinguishable from the endogenous IF in nontransfected cells prepared for indirect immunofluorescence (Helfand et al., 2003a).
Rat2 cells were also transfected by electroporation and plated on uncoated coverslips or tissue culture plates in Rat2 medium for ∼24 h before processing for immunofluorescence and immunoblotting.
Live cell imaging and analysis
Laminin-coated coverslips plated with transfected PC12 cells were assembled into a Focht Chamber System 2 (Bioptechs) for live cell imaging. The closed chamber system was calibrated to maintain cells at 37°C during microscopic examination. The culture medium used in these preparations was Leibovitz L-15 (Invitrogen) containing 5% calf serum, 1 mM sodium pyruvate, and 30 ng/ml NGF.
Time-lapse observations were made using a LSM510 confocal microscope as previously described (Yoon et al., 1998). Images were captured at 5–10-s intervals at a resolution of 512 × 512 dots per inch with an average scan time of 5 s. Images were collected for 20-s to 30-min time periods.
FRAP analyses were performed on GFP-peripherin–transfected PC12 cells using the LSM510 microscope as previously described (Yoon et al., 1998). For some experiments, the entire cell was photobleached and in other cases only half of the cell was bleached. In some experiments, differentiated cells (in DM for 2–4 h) were treated with 10 μg/ml nocodazole (Sigma-Aldrich) for 30 min or 10 μg/ml cycloheximide (Calbiochem) for 60 min before FRAP analysis.
To determine if peripherin mRNA was associated with motile or stationary peripherin protein particles, GFP-peripherin–expressing cells were plated on laminin-coated locator coverslips (Bellco Glass, Inc.) that were glued with Sylgard (Dow Corning Co.) to the bottom of a 35-mm dish that had an ∼2-cm-wide hole cut in it. A region of a cell containing motile particles was monitored by time-lapse microscopy for 20 s with 5-s intervals and then fixed directly on the microscope stage with formaldehyde. The dish was then removed from the microscope stage and processed for FISH. The particles imaged in live cells were relocated and imaged for peripherin mRNA labeling. 15 different cells were analyzed in this fashion.
The average rate of translocation of peripherin mRNPs (n = 200) in five cells was obtained by monitoring the total distance traveled by each RNA particle during the entire image capture sequence and dividing it by the capture time, as previously described (Yoon et al., 1998). To analyze the relationship between translation and mRNP motility, double transfected cells that were differentiated for ∼4 h in NGF were treated with 10 μg/ml puromycin (Sigma-Aldrich) for 30–60 min. Six cells and 1,286 mRNPs were analyzed for motility in puromycin. Each video ranged from 3–10 min and rapid movement or disappearance of an mRNP out of the focal plane was also categorized as moving.
Image processing
Gaussian blur, unsharp mask filters, and gamma adjustments have been applied to images shown in Figs. 1–3 , 6, and 7, and Fig. S1, using Photoshop software (Adobe) to decrease background noise.
Online supplemental material
Fig. S1 (A–F) shows split images of overlays shown in Fig. 2 (A–C). G and H are low and high magnification overlay images showing the distribution of endogenous peripherin particles and mRNA. Fig. S2 is a comparison of the average percentage of peripherin particles observed to associate with peripherin mRNA and the expected percentage based on chance alone, using different association criteria. Fig. S3 is a color-coded map showing peripherin mRNP distributions in a cell differentiated in the presence of nocodazole. A single deconvolved image from the primary image stack is also included. Videos 1–9 are of stills shown in Fig. 1 (A and B), Fig. 2 G, Fig. 3 (B and C), Fig. 6 A, and Fig. 8. Videos referred to in Fig. 1 A and another example of the experiment described in Fig. 2 G are shown as well. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200511033/DC1.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the technical support provided by Anuradha Nadimpalli and Shailesh M. Shenoy.
We would also like to acknowledge the support of a Method to Extend Research in Time award GM-36806-16 from the National Institute of General Medical Sciences and National Institutes of Health grants EB2060 and AR41480.
Abbreviations used in this paper: 3D, three-dimensional; CDS, coding sequence; DM, differentiation medium; ECFP, enhanced CFP; IF, intermediate filament; mRNP, messenger RNP; MT, microtubules; TFI, total fluorescent intensity; ULF, unit-length filament; UTR, untranslated region.
References
- Aronov, S., G. Aranda, L. Behar, and I. Ginzburg. 2002. Visualization of translated tau protein in the axons of neuronal P19 cells and characterization of tau RNP granules. J. Cell Sci. 115:3817–3827. [DOI] [PubMed] [Google Scholar]
- Carson, J.H., K. Worboys, K. Ainger, and E. Barbarese. 1997. Translocation of myelin basic protein mRNA in oligodendrocytes requires microtubules and kinesin. Cell Motil. Cytoskeleton. 38:318–328. [DOI] [PubMed] [Google Scholar]
- Chang, L., and R.D. Goldman. 2004. Intermediate filaments mediate cytoskeletal crosstalk. Nat. Rev. Mol. Cell Biol. 5:601–613. [DOI] [PubMed] [Google Scholar]
- Dean, D. 2004. Gene delivery by direct injection and facilitation of expression by mechanical stretch. In Live Cell Imaging. A Laboratory Manual. R.D. Goldman and D.L. Spector, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 51–66.
- Enoki, S., K. Saeki, K. Maki, and K. Kuwajima. 2004. Acid denaturation and refolding of green fluorescent protein. Biochemistry. 43:14238–14248. [DOI] [PubMed] [Google Scholar]
- Eriksson, J.E., T. He, A.V. Trejo-Skalli, A.S. Hrmala-Brasken, J. Hellman, Y.H. Chou, and R.D. Goldman. 2004. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117:919–932. [DOI] [PubMed] [Google Scholar]
- Femino, A.M., F.S. Fay, K. Fogarty, and R.H. Singer. 1998. Visualization of single RNA transcripts in situ. Science. 280:585–590. [DOI] [PubMed] [Google Scholar]
- Fusco, D., N. Accornero, B. Lavoie, S.M. Shenoy, J.M. Blanchard, R.H. Singer, and E. Bertrand. 2003. Single mRNA molecules demonstrate probabilistic movement in living Mammalian cells. Curr. Biol. 13:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco, D., E. Bertrand, and R.H. Singer. 2004. Imaging of single mRNAs in the cytoplasm of living cells. Prog. Mol. Subcell. Biol. 35:135–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand, B.T., P. Loomis, M. Yoon, and R.D. Goldman. 2003. a. Rapid transport of neural intermediate filament protein. J. Cell Sci. 116:2345–2359. [DOI] [PubMed] [Google Scholar]
- Helfand, B.T., M.G. Mendez, J. Pugh, C. Delsert, and R.D. Goldman. 2003. b. A role for intermediate filaments in determining and maintaining the shape of nerve cells. Mol. Biol. Cell. 14:5069–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfand, B.T., L. Chang, and R.D. Goldman. 2004. Intermediate filaments are dynamic and motile elements of cellular architecture. J. Cell Sci. 117:133–141. [DOI] [PubMed] [Google Scholar]
- Herrmann, H., and U. Aebi. 2000. Intermediate filaments and their associates: multi-talented structural elements specifying cytoarchitecture and cytodynamics. Curr. Opin. Cell Biol. 12:79–90. [DOI] [PubMed] [Google Scholar]
- Joklik, W.K., and Y. Becker. 1965. Studies on the genesis of polyribosomes. J. Mol. Biol. 13:496–510. [DOI] [PubMed] [Google Scholar]
- Liovic, M., M.M. Mogensen, A.R. Prescott, and E.B. Lane. 2003. Observation of keratin particles showing fast bidirectional movement colocalized with microtubules. J. Cell Sci. 116:1417–1427. [DOI] [PubMed] [Google Scholar]
- McArthur, I. 1943. Structure of alpha-keratin. Nature. 152:38–41. [Google Scholar]
- Parysek, L.M., R.L. Chisholm, C.A. Ley, and R.D. Goldman. 1988. A type III intermediate filament gene is expressed in mature neurons. Neuron. 1:395–401. [DOI] [PubMed] [Google Scholar]
- Prahlad, V., M. Yoon, R.D. Moir, R.D. Vale, and R.D. Goldman. 1998. Rapid movements of vimentin on microtubule tracks: kinesin-dependent assembly of intermediate filament networks. J. Cell Biol. 143:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo, M., and D. Piston. 2004. Fluorescent protein tracking and detection. In Live Cell Imaging. A Laboratory Manual. R.D. Goldman and D.L. Spector, editors. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. 3–23.
- Shav-Tal, Y., X. Darzacq, S.M. Shenoy, D. Fusco, S.M. Janicki, D.L. Spector, and R.H. Singer. 2004. Dynamics of single mRNPs in nuclei of living cells. Science. 304:1797–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova, E.A., R.H. Singer, and J. Condeelis. 2001. The physiological significance of β-actin mRNA localization in determining cell polarity and directional motility. Proc. Natl. Acad. Sci. USA. 98:7045–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soellner, P., R.A. Quinlan, and W.W. Franke. 1985. Identification of a distinct soluble subunit of an intermediate filament protein: tetrameric vimentin from living cells. Proc. Natl. Acad. Sci. USA. 82:7929–7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikakulam, R., and D.A. Winkelmann. 2004. Chaperone-mediated folding and assembly of myosin in striated muscle. J. Cell Sci. 117:641–652. [DOI] [PubMed] [Google Scholar]
- Stebbings, H. 2001. Cytsoskeleton-dependent transport and localization of mRNA. Int. Rev. Cytol. 211:1–31. [DOI] [PubMed] [Google Scholar]
- Steinert, P.M., A.C. North, and D.A. Parry. 1994. Structural features of keratin intermediate filaments. J. Invest. Dermatol. 103:19S–24S. [DOI] [PubMed] [Google Scholar]
- Strelkov, S.V., H. Herrmann, and U. Aebi. 2003. Molecular architecture of intermediate filaments. Bioessays. 25:243–251. [DOI] [PubMed] [Google Scholar]
- Tekotte, H., and I. Davis. 2002. Intracellular mRNA localization: motors move messages. Trends Genet. 18:636–642. [DOI] [PubMed] [Google Scholar]
- Yarmolinsky, M.B., and G.L. de la Haba. 1959. Inhibition by puromycin of amino acid incorporation into protein. Proc. Natl. Acad. Sci. USA. 45:1721–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, K.H., M. Yoon, R.D. Moir, S. Khuon, F.W. Flitney, and R.D. Goldman. 2001. Insights into the dynamic properties of keratin intermediate filaments in living epithelial cells. J. Cell Biol. 153:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, M., R.D. Moir, V. Prahlad, and R.D. Goldman. 1998. Motile properties of vimentin intermediate filament networks in living cells. J. Cell Biol. 143:147–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}