

Abstract
In neuroendocrine PC12 cells, immature secretory granules (ISGs) mature through homotypic fusion and membrane remodeling. We present evidence that the ISG-localized synaptotagmin IV (Syt IV) is involved in ISG maturation. Using an in vitro homotypic fusion assay, we show that the cytoplasmic domain (CD) of Syt IV, but not of Syt I, VII, or IX, inhibits ISG homotypic fusion. Moreover, Syt IV CD binds specifically to ISGs and not to mature secretory granules (MSGs), and Syt IV binds to syntaxin 6, a SNARE protein that is involved in ISG maturation. ISG homotypic fusion was inhibited in vivo by small interfering RNA–mediated depletion of Syt IV. Furthermore, the Syt IV CD, as well as Syt IV depletion, reduces secretogranin II (SgII) processing by prohormone convertase 2 (PC2). PC2 is found mostly in the proform, suggesting that activation of PC2 is also inhibited. Granule formation, and the sorting of SgII and PC2 from the trans-Golgi network into ISGs and MSGs, however, is not affected. We conclude that Syt IV is an essential component for secretory granule maturation.
Introduction
In endocrine and neuroendocrine cells, bioactive molecules are packaged into nascent vesicles called immature secretory granules (ISGs), which bud from the TGN and are destined for regulated secretion (Arvan and Castle, 1998; Tooze, 1998). ISGs undergo a series of maturation steps, including acidification of the granule lumen, prohormone processing (Orci et al., 1987; Moore et al., 2002), AP-1–dependent removal of proteins (Dittie et al., 1997; Kuliawat et al., 1997; Klumperman et al., 1998), and ISG–ISG homotypic fusion (Urbé et al., 1998), to become mature secretory granules (MSGs). MSGs, which are also called large dense core vesicles, accumulate in cells until they undergo fusion with the plasma membrane by regulated exocytosis.
An in vitro fusion assay that reconstitutes ISG–ISG fusion has revealed that ISG homotypic fusion is dependent on NSF and α-SNAP (Urbé et al., 1998) and on the SNARE protein syntaxin 6 (Stx6), but not on Stx1 or SNAP-25 (Wendler et al., 2001). SNAREs are essential components of membrane fusion, but whether they are sufficient for fusion and/or ensuring targeting specificity remains under debate. Additional proteins, including the Rabs (Zerial and McBride, 2001) and the synaptotagmins (Syts; Chapman, 2002), may coordinate and regulate vesicle trafficking and fusion.
The Syts are a family of proteins characterized by a short lumenal NH2 terminus, one transmembrane region, and tandem C2A and C2B domains (Perin et al., 1991; Bai and Chapman, 2004). Currently, it is thought that Syts participate in the regulation of various steps during membrane fusion, primarily at the plasma membrane. Syt I, which was the first isoform identified (Matthew et al., 1981), is involved in calcium-dependent exocytosis (Fernandez-Chacon et al., 2001) and functions as the calcium sensor that stabilizes the opening of the fusion pore at the final steps of fusion (Wang et al., 2001), at the docking step (Chieregatti et al., 2002, 2004), and during vesicle recycling from the plasma membrane (Nicholson-Tomishima and Ryan, 2004). Syt I binds to the SNARE proteins Stx1 and SNAP-25 (Bennett et al., 1992; Schiavo et al., 1997), and this binding is thought to be important for the function of Syt I in membrane fusion.
A genomic analysis has identified 16 Syt isoforms in mammals (Craxton, 2004), so that, like SNAREs or Rabs, Syts constitute a large family of proteins, suggesting that they regulate multiple membrane events. In support of this, although still controversial, a differential distribution of Syt I, III, IV, and VII has been reported in neuroendocrine cells (Ibata et al., 2000; Sugita et al., 2001; Fukuda et al., 2004). Moreover, a recent study in Drosophila melanogaster showed that Syt isoforms localize to nonoverlapping subcellular compartments (Adolfsen et al., 2004).
Syt IV (Hilbush and Morgan, 1994) was characterized as an immediate early gene induced by depolarization in PC12 cells and rat brain (Vician et al., 1995). Syt IV knockout mice exhibit abnormalities in motor performance, suggesting a role in synaptic plasticity (Ferguson et al., 2000). The function of Syt IV in vesicular trafficking, however, remains unclear. Overexpressed Syt IV is sorted to MSGs upon NGF differentiation or forskolin treatment of PC12 cells, and is involved in the regulation of exocytosis (Fukuda et al., 2003; Wang et al., 2003; Fukuda and Yamamoto, 2004; Machado et al., 2004). Different studies have found contradictory localizations; Syt IV has been shown to colocalize with Syt I on synaptic vesicles and MSGs in PC12 cells (Ferguson et al., 1999), whereas others demonstrated that Syt IV has a juxtanuclear distribution (Ibata et al., 2000), is localized on ISGs and not MSGs in PC12 and AtT20 cells, and does not colocalize with Syt I (Eaton et al., 2000; Ibata et al., 2002; Fukuda et al., 2003).
The function of endogenous Syt IV in relation to its localization in nondifferentiated neuroendocrine cells has never been studied. In PC12 cells, Syt IV is found on ISGs, but not on MSGs, and we have investigated whether Syt IV could be involved in ISG–ISG homotypic fusion before its removal from MSGs. We show that the cytoplasmic domain (CD) of Syt IV, but not of Syt I, VII, or IX, inhibits ISG–ISG homotypic fusion in an in vitro homotypic fusion assay, and that this domain is recruited specifically to ISG membranes. The role of Syt IV in ISG homotypic fusion was confirmed in vivo after siRNA-mediated depletion of Syt IV. We also find that Syt IV binds Stx6, and that Syt IV interacts with Stx6 via both the C2A and C2B domains. We show in the in vitro homotypic fusion assay that addition of the Syt IV CD, together with an anti-Stx6 antibody, leads to an additive inhibition of fusion, and we speculate that Syt IV and Stx6 are involved in regulating different stages of ISG homotypic fusion. Furthermore, using the dominant-negative Syt IV CD, and by the reduction of Syt IV levels using siRNA, we demonstrate in vivo a reduction of secretogranin II (SgII) processing by prohormone convertase 2 (PC2). Finally, we show that in cells transfected with the dominant-negative Syt IV CD, PC2 is mostly in the unprocessed/inactive proform. Collectively, our data provide the first direct evidence that Syt IV is involved in an intracellular membrane fusion event and in the regulation of secretory granule maturation in neuroendocrine cells.
Results
Syt IV is localized to ISGs, and not MSGs, in PC12 cells
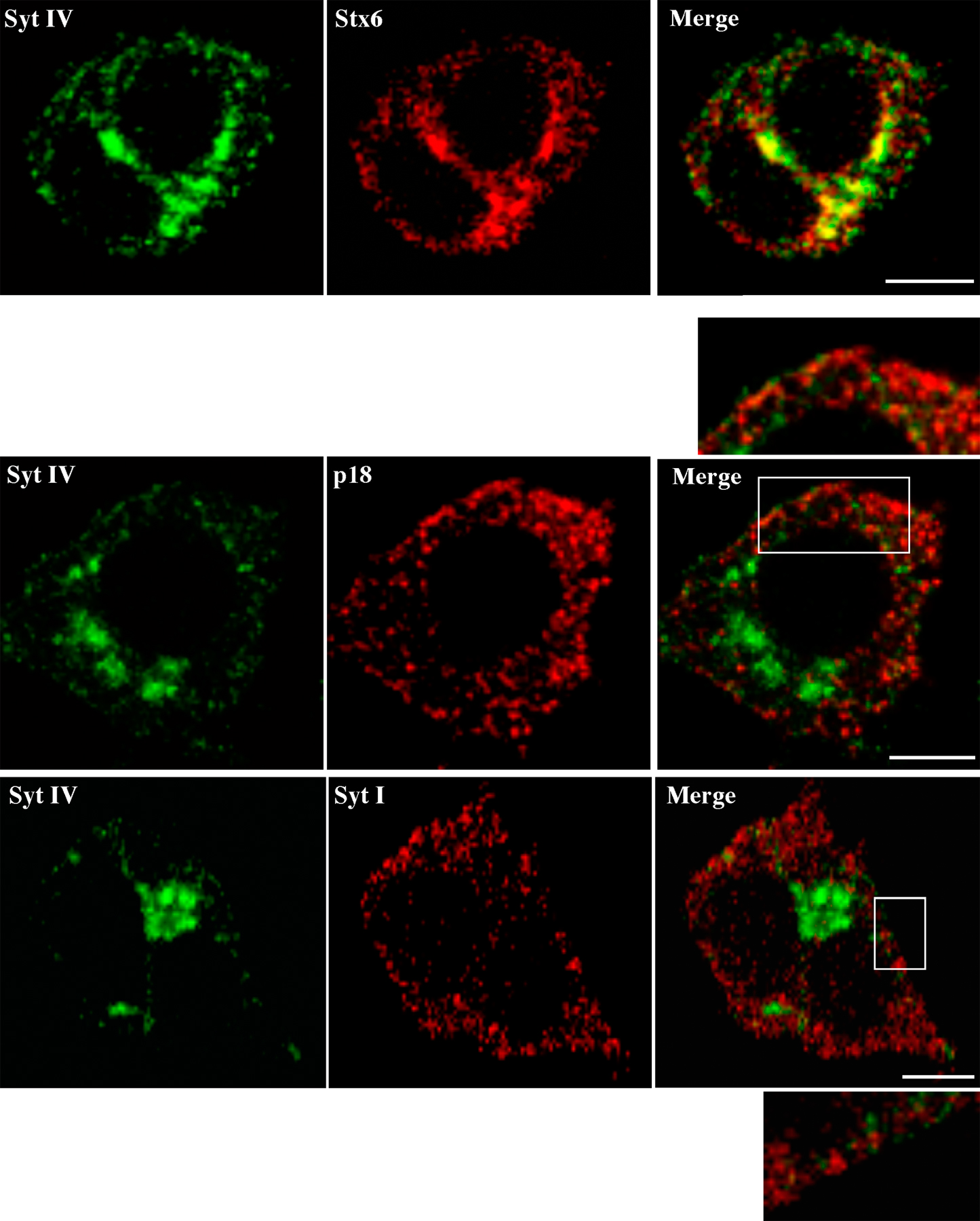
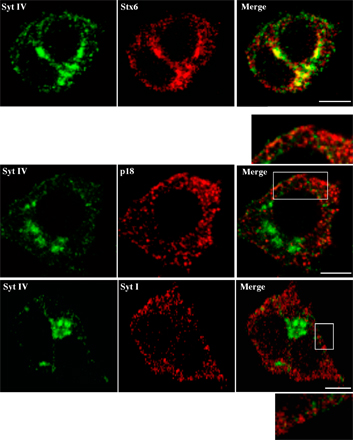
We first determined the distribution of Syt IV using subcellular fractionation techniques to isolate and separate ISGs and MSGs. PC12 cells were [35S]sulfate-labeled either for short periods (5 min pulse and 15 min chase) to label ISGs or overnight (1 h pulse, overnight chase) to label MSGs (Tooze et al., 1991). The postnuclear supernatant (PNS) from labeled cells was subjected to continuous velocity sucrose gradient fractionation and analysis. Syt IV was found mainly in the light ISG-containing fractions, as well as in Golgi-containing fractions at the bottom of the gradient (Fig. 1 A, top), in contrast to Syt I, which localizes mainly to MSGs and synaptic-like microvesicles (Perin et al., 1991) and is found mainly in the slightly heavier fractions (Fig. 1 A, bottom). The ISG and MSG fractions were further separated on discontinuous equilibrium sucrose gradients (Fig. 1, B and C). Syt IV was in the ISG-containing fractions, together with [35S]sulfate-labeled SgII and Stx6, which were previously shown to be on ISGs and not MSGs (Fig. 1 B; Wendler et al., 2001). However, Syt IV was not found in the MSG fractions, which were identified by the presence of [35S]sulfate-labeled SgII together with Syt I (Fig. 1 C, bottom). By indirect immunofluorescence in PC12 cells, Syt IV displayed a juxtanuclear distribution (Ibata et al., 2000) and colocalized with Stx6 (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200506163/DC1).
Figure 1.

Syt IV is found on the Golgi and ISGs in PC12 cells. (A) Fractions from a 0.3–1.2 M continuous velocity sucrose gradient loaded with a PNS from PC12 cells were collected and analyzed with anti–Syt IV (top) and Syt I (bottom) antibodies. Alternatively, PC12 cells were labeled with [35S]sulfate for 5 min and chased for 15 min (B) or labeled for 1 h and chased O/N (C) to label ISGs or MSGs, respectively, and then subjected to velocity centrifugation. (B and C) Fractions 1–4 (B) or 4–6 (C) from the velocity gradients were further separated on a 0.8–1.6 M discontinuous equilibrium sucrose gradient and analyzed. (B and C, top) Autoradiogram showing [35S]sulfate-labeled CgB (B) and SgII (B and C). (B and C, bottom) Western blot analysis using anti–Syt IV and anti-Stx6 antibodies (B) or anti–Syt IV and anti–Syt I antibodies (C). In B, the diffuse band between 120 and 90 kD is a heparan sulfate proteoglycan (Tooze and Huttner, 1990).
To confirm that Syt IV is not on MSGs, we used a PC12 cell line stably expressing PC2 (PC12/PC2 cells; Dittie and Tooze, 1995). In PC12/PC2 cells, PC2 cleaves SgII at several dibasic residues and produces a product of 18 kD (p18), which accumulates in MSGs and can be detected using an antibody that recognizes only p18, but not full-length or partially processed SgII. As shown in Fig. S1 B, a nonoverlapping distribution of Syt IV and p18 or Syt I was observed in PC12/PC2 cells. We conclude, in agreement with others (Eaton et al., 2000; Fukuda et al., 2003), that Syt IV is localized to ISGs, and not to MSGs, in PC12 cells.
Syt IV CD, but not Syt I, VII, or IX, inhibits in vitro ISG homotypic fusion
Because Syt IV is localized on ISGs, but not on MSGs, we hypothesized that it could play a role in granule maturation. In PC12 cells, ISGs undergo homotypic fusion during maturation, followed by excess membrane removal, most likely via AP-1–containing clathrin-coated vesicles. To test whether Syt IV is involved in the maturation of ISGs, we used an in vitro fusion assay that reconstitutes ISG homotypic fusion (Urbé et al., 1998). Addition of increasing amounts of the purified recombinant Syt IV CD into the complete fusion reaction resulted in a dose-dependent inhibition of ISG homotypic fusion by ∼40% (Fig. 2). Addition of the denatured CD had no effect on ISG–ISG fusion (unpublished data). In addition to Syt IV, Syt I, VII, and IX are the most abundant isoforms in PC12 cells, with Syt VII found at lower levels (Zhang et al., 2002; Wang et al., 2005). Furthermore, Syt I and IX are localized on MSGs and are involved in the regulation of calcium-dependent exocytosis in these cells (Elferink et al., 1993; Fukuda, 2002). Syt VII was localized either to the plasma membrane (Sugita et al., 2001) or to large dense core vesicles (Fukuda et al., 2004), and was also shown to be involved in the regulation of calcium-triggered fusion with the plasma membrane (Sugita et al., 2001; Fukuda et al., 2004; Wang et al., 2005).
Figure 2.

The CDs of Syt IV, but not of Syt I, VII, or IX, inhibit ISG homotypic fusion. An ISG homotypic fusion assay was performed as previously described by Urbé et al. (1998), which measures fusion by the production of p18 from [35S]sulfate-labeled SgII. The complete fusion reaction contains PC2 ISGs, PC12 [35S]sulfate-labeled PNS, and ATP. Syt IV (circle), Syt I (inverted triangle), Syt VII (triangle), or Syt IX (diamond) CDs were added at the indicated concentrations, on ice, and then the fusion assay was performed. After subtraction of the background from PC2-ISG minus reactions, the fusion was assayed by quantifying the amount of p18 produced and is presented as a percentage of the complete fusion reaction. The data represent the mean of three experiments done in duplicate. Error bars are the SEM. The addition of more Syt IV CD did not increase the inhibition beyond 50% (not depicted).
We asked if ISG homotypic fusion could be regulated by the other Syt isoforms present in PC12 cells. Purified Syt I, VII, and IX CDs, which were previously shown to inhibit exocytosis (Earles et al., 2001; Tucker et al., 2003), had no effect on ISG homotypic fusion (Fig. 2). We conclude that the inhibition of ISG–ISG fusion is specific for Syt IV and does not involve Syt I, VII, or IX.
Many membrane fusion events, including the fusion of secretory granules with the plasma membrane (Heinemann et al., 1994), require calcium. However, it has never been determined whether ISG homotypic fusion is calcium dependent. We found that the addition of increasing amounts of 1,2-bis (o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA) into the complete ISG–ISG fusion assay resulted in a dose-dependent inhibition of fusion of up to 95% (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200506163/DC1). The addition of equimolar BAPTA and calcium partially restored ISG–ISG fusion, and the addition of BAPTA alone after the fusion reaction had little effect. These results clearly demonstrate that ISG homotypic fusion is dependent on calcium. It was assumed that rat Syt IV is a calcium sensor because of the presence of the predicted calcium-binding residues in the C2B domain (Wang et al., 2003). However, a recent crystal structure revealed that changes in the orientation of the calcium-binding residues render the rat Syt IV C2B domain unlikely to bind calcium (Dai et al., 2004). Thus, it is likely that Syt IV is not the calcium sensor protein regulating ISG–ISG fusion and that another calcium-binding protein is regulating this process. Further work is required to clarify and confirm this speculation.
The Syt IV CD is recruited specifically to ISG membranes via protein components
Because the Syt IV CD was able to inhibit ISG homotypic fusion, we asked whether the site of inhibition resides on these membranes using an in vitro ISG-binding assay, which we have used to demonstrate ADP-ribosylation factor 1 and AP-1 binding to ISG membranes (Dittie et al., 1996; Austin et al., 2000). The addition of increasing amounts of Syt IV CD to ISGs showed a dose-dependent binding (Fig. 3 A), demonstrating that ISG membranes contain binding sites for Syt IV. No recombinant protein was detected in the pellet in the absence of ISGs, establishing that the binding is not a result of nonspecific aggregation of the protein. To test whether the binding is specific, 7 nM Syt IV CD was added to 50 μl ISGs or MSGs. The Syt IV CD bound to ISG, whereas no binding to MSG was observed (Fig. 3 B). In contrast, Syt I CD was able to bind both ISG and MSG membranes (Fig. 3 B). There was more binding of the Syt I CD to ISGs compared with MSGs, which might be attributed to the presence of synaptophysin-positive synaptic-like microvesicles in the ISG fraction (Regnier-Vigouroux et al., 1991). These results show that Syt IV binds specifically to components that are present on ISGs and absent from MSGs, and further suggest that the site of inhibition of ISG–ISG fusion by the Syt IV CD is on the ISG membrane.
Figure 3.

Syt IV CD is recruited to ISG membranes. (A) 20 nM of Syt IV CD was incubated for 30 min at 37°C with or without 50 μl PC12 ISGs, or increasing amounts of Syt IV CD were added to 50 μl ISGs in fusion buffer with ATP. The amount of Syt IV bound to ISG membranes was determined by immunoblotting with anti–Syt IV antibody and quantified using ImageJ software. The data are the mean of three experiments done in duplicate. Error bars are the SEM. (B) 7 nM Syt IV or Syt I CDs were preincubated with 50 μl ISGs or MSGs in fusion buffer with ATP. The amount of Syt IV or Syt I bound was determined as described in Materials and methods. (C) 50 μl ISGs were preincubated with increasing amounts of trypsin. Trypsin inhibitor was added before (*) or after the preincubation. Binding reactions including 18 nM Syt IV CD were performed as described in Materials and methods. The amount of Syt IV bound to ISGs was detected with an anti–Syt IV antibody (top). The blot was stripped and reprobed with an anti-SgII antibody (bottom).
In liposome-binding assays, some Syts such as Syt I and IX, but not Syt IV, are able to bind phospholipids via their C2 domains in a calcium–dependent manner (Hui et al., 2005). However, others showed that Syt IV is able to bind liposomes containing only negatively charged phospholipids (Fukuda et al., 1996). Although we do not know the lipid composition of ISG membranes, it is unlikely that they are composed solely of negatively charged phospholipids. To determine whether the recruitment of the Syt IV CD is mediated through protein or lipid components, we repeated the binding assays after pretreatment of ISG membranes with increasing amounts of trypsin, followed by the addition of trypsin inhibitor. We found that the recruitment of Syt IV CD to ISGs is trypsin sensitive (Fig. 3 C). When trypsin inhibitor was added before trypsin, the recruitment to ISG membranes of Syt IV was not reduced. The lumenal protein SgII was not degraded by the trypsin treatment (Fig. 3 C, bottom), demonstrating that ISGs remained intact during the assay. Although we cannot exclude the activation of phospholipases by trypsin, which would alter the composition of the membrane, our results, together with the recent confirmation of Syt IV's inability to bind phospholipids (Hui et al., 2005), suggest that protein components on ISGs are necessary for the recruitment of the Syt IV CD.
Syt IV interacts with the SNARE Stx6
Our results suggest that the Syt IV CD is able to inhibit ISG–ISG homotypic fusion via recruitment to ISGs. Syts bind to SNARE proteins to regulate membrane fusion. Syt I, for example, is able to bind Stx1 and SNAP-25 (Bai and Chapman, 2004), and it is thought that this binding is an important facet of the ability of Syt I to regulate exocytosis (Earles et al., 2001). We hypothesized that Syt IV may be part of a protein complex involved in an ISG homotypic fusion that contains Stx6, so we looked for an interaction between Stx6 and Syt IV. In GST pull-down assays from transfected human embryonic kidney 293 (HEK293) cell lysates, we found that myc-Stx6 binds to GST-Syt IV CD and not to GST alone (Fig. 4 A). Furthermore, we were able to coimmunoprecipitate Syt IV with Stx6 from PC12 cells, showing that endogenous Syt IV and Stx6 interact (Fig. 4 B). Next, to test whether Syt IV binds directly to Stx6, GST-Syt IV CD was incubated with increasing amounts of recombinant Stx6 CD. Syt IV CD was able to interact directly with Stx6 in a dose-dependent manner (Fig. 4 C). Together, these results suggest that Syt IV and Stx6 are part of the same protein complex.
Figure 4.

Syt IV interacts with Stx6. (A) Cell lysates from HEK cells transfected with myc-Stx6 were incubated with GST-Syt IV CD or GST bound to glutathione beads. (B) PC12 cells were lysed in TNTE buffer and incubated with 5 or 10 μg of anti-Stx6 antibody overnight at 4°C, and subsequently bound to protein G beads. Immunoprecipitates and 2.5% of the input were analyzed by immunoblotting with anti–Syt IV antibody. (C) Direct binding of Stx6 to Syt IV in vitro. 3 μg of GST-Syt IV CD bound to glutathione beads was incubated with the indicated amounts of the Stx6 CD in binding buffer overnight, and bound protein was analyzed by immunoblotting with anti-Stx6 antibody 3D10. (D) The Syt IV–Stx6 interaction in vitro is independent of Ca2+. Binding assays were performed as in C, using 1 μM Stx6 CD, supplemented with either 1 mM CaCl2 or 2 mM EGTA.
Previous data showed that Syt VIII, which is another isoform that was initially characterized as a calcium binding–deficient Syt, was able to interact with Stx2 in a calcium-dependent manner (Hutt et al., 2005). Therefore, we tested the possibility that Syt IV, which was also thought not to bind calcium (Dai et al., 2004), binding to Stx6 might be promoted by calcium. We tested the binding of GST-Syt IV CD with recombinant Stx6 CD in the presence of either 2 mM EGTA or 1 mM Ca2+. Syt IV binding to Stx6 was similar in all conditions (Fig. 4 D), suggesting that the interaction is calcium independent.
Furthermore, to investigate which Syt IV domain is required for binding to Stx6, we performed coimmunoprecipitation experiments with lysates prepared from HEK293 cells expressing myc-Stx6 and either HA-Syt IV-C2A, HA-Syt IV-C2B, or HA-Syt IV CD. HA-Syt IV CD could coimmunoprecipitate myc-Stx6 (Fig. 5, A and B), as could both HA-Syt IV- C2A and HA-Syt IV-C2B domains (Fig. 5 C). Similarly, myc-Stx6 could coimmunoprecipitate both HA-Syt IV-C2A and HA-Syt IV-C2B domains (Fig. 5 D). This result demonstrates that both domains are involved in the binding to Stx6. The binding efficiency of the individual Syt IV domains to myc-Stx6 was lower compared with the binding efficiency of the complete CD, suggesting that the C2A and C2B domains cooperate to bind Stx6, as is the case for Syt I binding to SNAREs (Bai and Chapman, 2004).
Figure 5.

Syt IV interacts with Stx6 via both C2A and C2B domains. (A and B) HEK293 cells were cotransfected with HA-Syt IV CD and myc-Stx6. Lysates from untransfected and transfected cells were used for immunoprecipitation with anti-HA (+) or beads alone (−; A) and with anti-myc antibody or beads alone (B). Immunoprecipitates and 10% of total input were analyzed with anti-myc (A) or anti-HA (B) antibodies by immunoblotting. (C and D) Lysates from HEK293 cells cotransfected with either HA-Syt IV C2A or C2B domain and myc-Stx6 were subjected to immunoprecipitation with either anti-myc antibody or beads alone (C), blotted with anti-HA antibody or anti-HA antibody or beads alone (D), and then blotted with anti-myc antibody.
Syt IV and Stx6 inhibition of ISG homotypic fusion is additive
After our findings that Syt IV binds Stx6 and that both are involved in ISG–ISG fusion, we investigated the effect of inhibitory reagents in the homotypic fusion assay. Our previous results (Wendler et al., 2001), along with the results in Fig. 2, show that the inhibition by each reagent individually was at most 50%. Combining reagents in the fusion assay increased inhibition of ISG homotypic fusion to 80% (Fig. 6 A). The addition of anti-Stx1 antibody, which was previously shown to have no effect on fusion, combined with the Syt IV CD, had no additional effect. Likewise, the addition of anti-Stx6 antibody with Syt I CD had no additional effect, demonstrating that the additive inhibitory effect is specific for Syt IV and Stx6 (Fig. 6 B). Preincubation of the fusion reaction with anti-Stx6 antibody for 30 min on ice before the addition of Syt IV CD (and vice versa) resulted in the same degree of inhibition of ISG homotypic fusion as when the two components were added simultaneously (unpublished data).
Figure 6.

Syt IV CD and Stx6 antibody have an additive inhibitory effect on ISG homotypic fusion. (A) A complete fusion reaction was incubated with 9 μM Syt IV CD and 10 μg anti-Stx6 antibody, or Syt IV CD and anti-Stx6 antibody. Fusion reactions were performed, treated, and analyzed (see Materials and methods). p18 signals were quantified using ImageJ analysis software, and the background signal measured in the absence of PC2 ISGs was subtracted. The data is the mean of three independent experiments done in duplicate. Error bars are the SEM. (B) A complete fusion reaction was incubated with 9 μM Syt IV CD, 10 μg anti-Stx6 antibody, 9 μM Syt I CD, 10 μg anti-Stx1 antibody, Syt IV CD and anti-Stx1 antibody, 9 μM Syt I CD and 10 μg anti-Stx6 antibody, or 10 μg anti-Stx6 antibody and 9 μM Syt IV CD. p18 signals were quantified as described in A.
Syt IV is required for ISG fusion and SgII processing in vivo
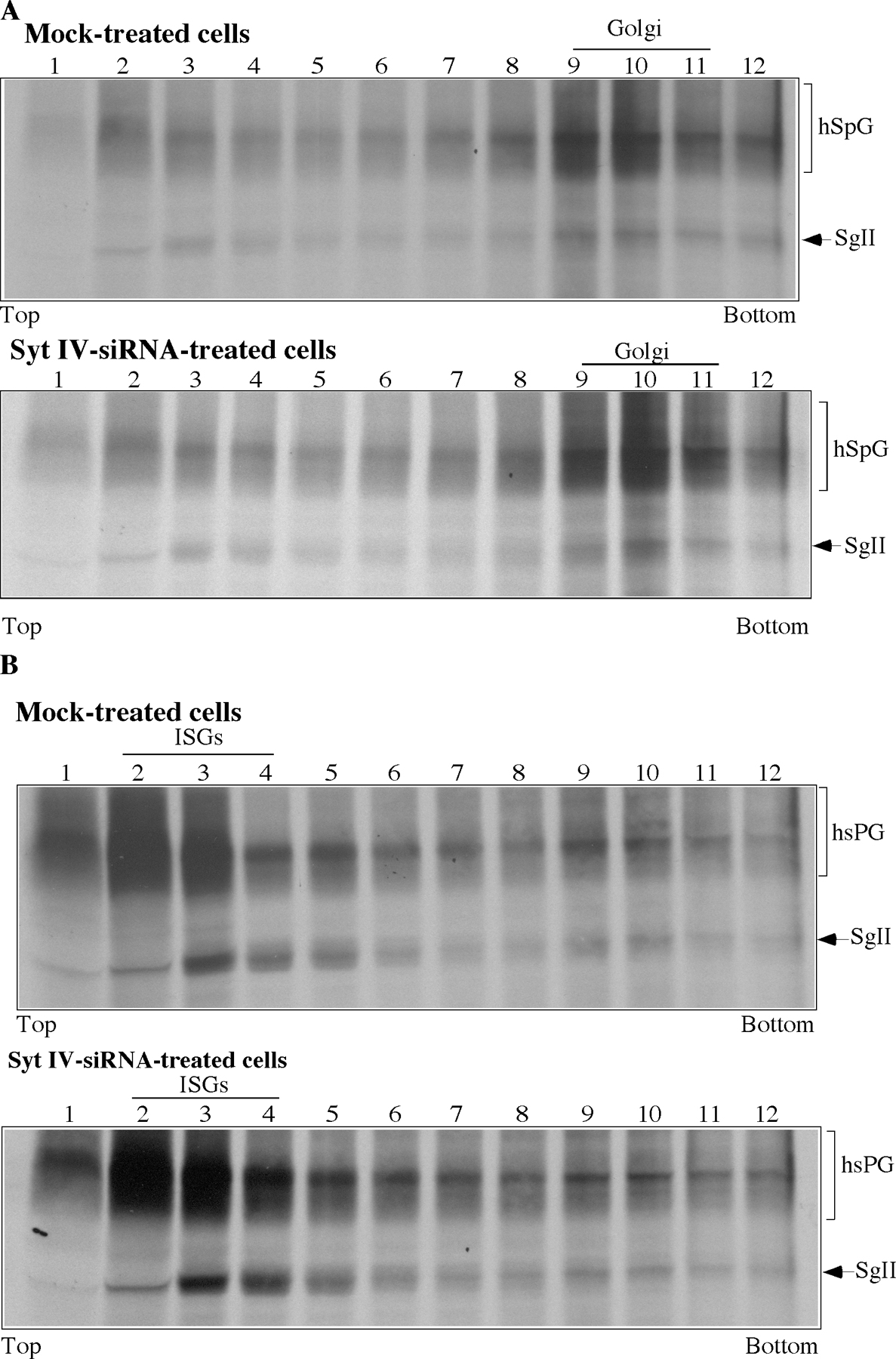
To test directly the role of Syt IV in ISG maturation in vivo, and in homotypic fusion in particular, we depleted Syt IV from PC12/PC2 cells using siRNA and asked if ISG–ISG fusion was affected. Using a Syt IV–specific antibody, we observed a reduction of up to 95% in Syt IV levels in >80% of the siRNA-treated PC12/PC2 cells by both indirect immunofluorescence and immunoblotting, whereas Syt I distribution and levels remained unchanged in the absence of Syt IV (Fig. 7, A and B). ISG maturation in vivo is characterized by an increase in the size of ISGs on velocity-controlled sucrose gradients that occurs between 15 and 75 min after ISGs are formed from the TGN (Tooze et al., 1991). This increase in size reflects ISG–ISG fusion (Urbé et al., 1998). Therefore, we tested whether Syt IV was required for the fusion of ISGs in vivo by assaying for an increase in the size of ISGs (in particular the [35S]sulfate-labeled granule protein SgII in the ISGs) during a chase of up to 60 min (Fig. 7 C). As shown in Fig. 7 (D and E), depletion of Syt IV resulted in an inhibition of the time-dependent size increase of ISGs.
Figure 7.

siRNA-mediated depletion of Syt IV results in inhibition of ISG fusion. PC12/PC2 cells were either transfected with 50 nM Syt IV siRNA or mock-transfected with transfection reagents alone. (A) 72 h after transfection, cells were labeled with anti–Syt IV (green) or anti–Syt I (blue). Images were taken using a Zeiss LSM510 confocal microscope. Bars, 20 μm. (B) 10 μg mock- or siRNA-treated cell lysates were solubilized and analyzed with anti–Syt IV and anti–Syt I antibodies. (C) Scheme for the pulse-chase labeling protocol and analysis of ISG size used in D and E, as well as in Fig. 8 A (Tooze et al., 1991). (D) 72 h after mock or Syt IV siRNA treatment, PC12 cells were labeled, harvested, and analyzed by velocity-controlled sucrose gradient fractionation. Fractions 1–12 were analyzed from gradients loaded with PNS prepared from mock- or siRNA-treated cells after a 60-min chase. In mock-treated cells, SgII is found in ISGs sedimenting in fractions 2–6, whereas in Syt IV siRNA-treated cells ISGs containing SgII are found in fractions 1–4. (E) Quantitation of the position of SgII in fractions 1–6 from velocity gradients loaded with cells treated as in D and chased for 30 and 60 min. The average of two experiments is shown.
During secretory granule maturation in endocrine or neuroendocrine cells, SgII, like many other secretory granule proteins and prohormones, is processed by PC2 in a pH-dependent reaction, which starts in the TGN and continues into the MSG (Urbé et al., 1997). We investigated whether PC2 processing of SgII was affected by the inhibition of fusion because of the absence of Syt IV by observing the processing of newly synthesized SgII in Syt IV siRNA-treated PC12/PC2 cells. In PC12/PC2 cells, after exit from the TGN and during the chase period, SgII is processed at several sites by PC2 to generate 38-, 28-, and 18-kD (p18) products (Dittie and Tooze, 1995). This processing can be detected after a short pulse of [35S]sulfate to label the TGN pool, followed by a chase with excess sulfate. In the absence of Syt IV, we observe reduced processing of the full-length 86-kD SgII (Fig. 8 A). The extent of this inhibition was determined from the ratio of p86 to p38 (Fig. 8 B). Before 30 min of chase, very little processing is detectable in this pulse-chase analysis, whereas maximum inhibition occurs between 45 and 60 min of chase, when in the mock-treated cells the rate of processing is highest. We conclude from these results that in PC12 cells Syt IV is required for fusion of ISGs and efficient processing of SgII.
Figure 8.

Syt IV depletion inhibits SgII processing in PC12/PC2 cells. (A) Syt IV siRNA– or mock-transfected PC12/PC2 cells were pulse-labeled with [35S]sulfate for 5 min and chased for the indicated length of time. Cells were lysed in TNTE and a heat-stable fraction was prepared and analyzed by SDS-PAGE gel and autoradiography. Arrowheads indicate the position of the SgII-processing products p38 and p18. (bottom) The same gel presented in the top image, but at higher exposure. (B) Quantification of the ratio of p38 and p86 using ImageJ. Three separate experiments were analyzed and the average is shown. Error bars are the SEM. P < 0.02, paired t test.
The Syt IV CD inhibits SgII processing and maturation of PC2
Next, we investigated the role of Syt IV in ISG maturation in vivo using the Syt IV CD, in particular, to determine if the reduction in SgII processing was a direct result of an inhibition of maturation or an alteration in PC2 sorting. As mentioned in the previous section, SgII in PC12/PC2 cells is processed to p18. p18 can be detected with an anti-p18 antibody recognizing only p18 and not larger SgII precursors. We used wild-type PC12 cells transfected with PC2 to eliminate the background signal from p18 stored in MSGs existing before addition of the dominant-negative HA-Syt IV CD, and asked if the appearance and accumulation of p18 in MSGs was affected by expression of the Syt IV CD. Transfection of the HA-Syt IV CD together with PC2 into PC12 cells resulted in an inhibition of p18 accumulation, whereas transfection of the Syt I CD together with PC2 or of PC2 alone, had no effect on p18 appearance (Fig. 9 A). Quantification of the intensity of the p18 signal in an equivalent number of cells cotransfected with HA-Syt IV CD and PC2 revealed a threefold decrease in p18 in these cells compared with cells transfected with PC2 alone or with Syt I CD and PC2 (Fig. 9 B).
Figure 9.

Syt IV CD inhibits PC2 maturation and PC2-dependent SgII processing. (A) PC12 cells were cotransfected with HA-Syt IV CD and PC2, HA-Syt I CD and PC2, or PC2 alone. The cells were fixed and labeled with rat anti-HA (green), rabbit anti-PC2 (blue), and mouse anti-p18 (red). (B) Transfected cells were individually selected and the mean of intensity of each channel was measured. Columns represent average of the mean of intensity of each channel. Error is the SEM, where n = 22 cells for all conditions. (C) PC12 cells were cotransfected with HA-Syt IV CD and PC2. Cells positive for both HA-Syt IV and PC2 were FACS-sorted by labeling with anti-HA antibody (Alexa Fluor 488) and anti-PC2 antibody (Alexa Fluor 647). As a control, PC12/PC2 cells labeled for PC2 were FACS-sorted using identical conditions. 150,000 HA-Syt IV+/PC2+ cells and 300,000 PC12/PC2 cells were loaded on an SDS-PAGE gel and immunoblotted with the anti-PC2 antibody. 5 μg of a PC12 cell lysate was used as a negative control for the PC2 antibody. (D) PC12 cells cotransfected with HA-Syt IV CD and PC2 were fixed and labeled with anti-HA, anti-PC2, and chromogranin B (CgB) antibodies. Bars, 5 μm.
Because we observed a decrease in SgII processing using the Syt IV CD, we tested whether excess full-length Syt IV increased p18 levels. Using FACS analysis, there was no difference in p18 in untransfected PC12/PC2 cells or cells transfected with full-length Syt IV or I (unpublished data). We speculate that the lack of a stimulatory effect on maturation by excess Syt IV may be attributable to a limitation of Syt IV effectors that are necessary for granule maturation, and which may also need to be overexpressed. We have also tested whether the effect of Syt IV on p18 production is calcium-dependent by mutating the first and second aspartate residues within the C2B domain to asparagine (D318/324N); these mutations were previously shown to block glutamate release in glial cells (Zhang et al., 2004). Expression of this double mutant or full-length Syt IV together with PC2 did not affect p18 production in PC12 cells (unpublished data), suggesting that the dominant-negative effect on SgII processing observed with HA Syt IV CD is independent of calcium.
PC2 is found as a 75-kD precursor in the TGN, which is then endoproteolytically cleaved to the 65-kD mature form in a pH-dependent manner (Zhou et al., 1999). PC2 activation depends on propeptide cleavage in the presence of bound 7B2, which is a member of the granin family shown to behave as a chaperone for pro-PC2 (Mbikay et al., 2001). We asked if inhibition of SgII processing by the dominant-negative Syt IV CD might be a result of failure to activate PC2. Therefore, we looked at PC2 in PC12 cells cotransfected with the HA-Syt IV CD and PC2. To do this, the doubly transfected cells and PC12/PC2 cells were FACS-sorted using anti-HA and anti-PC2 antibodies. In the doubly transfected cells expressing HA-Syt IV, there was a higher ratio of pro-PC2 to PC2 than in PC12/PC2 cells (Fig. 9 C). The processing defect of pro-PC2 is not a result of its failure to enter granules because in the doubly transfected cells PC2 colocalized with the MSG marker chromogranin B (CgB; Fig. 9 D). Furthermore, when PC12 cells expressing the HA-Syt IV CD were examined by EM after immunolabeling with an anti-HA antibody, compared with wild-type cells, the Golgi was not fragmented and secretory granule formation was not perturbed in these cells (unpublished data).
Discussion
In this study, we used a specific anti–Syt IV antibody (Ibata et al., 2000) and subcellular fractionation approaches in undifferentiated PC12 cells to demonstrate that Syt IV is found on ISGs and is absent from MSGs, confirming and extending previous studies (Eaton et al., 2000; Fukuda et al., 2003). The CD of Syt IV inhibited in vitro ISG homotypic fusion, suggesting that Syt IV is involved in secretory granule maturation. siRNA depletion demonstrated that Syt IV is required for ISG–ISG fusion in vivo. A previous study has shown that microinjection of recombinant Syt IV in endocrine β cells did not effect calcium-evoked insulin secretion (Gut et al., 2001). Similarly, catecholamine release from “cracked” PC12 cells was not changed by the addition of recombinant Syt IV (Tucker et al., 2003). Collectively, these data suggest that the ISG-localized Syt IV is not a regulator of calcium-triggered exocytosis in undifferentiated PC12 cells, but, rather, is involved in regulating ISG–ISG membrane fusion.
Interestingly, we found that the MSG-localized isoforms Syt I, IX, and VII did not have any effect on the regulation of ISG–ISG fusion. Our data are in agreement with studies showing that Syt IV displays a different distribution to Syt I, VII, and IX in PC12 cells (Ibata et al., 2000; Fukuda, 2004). Moreover, Syt IV lacks the ability to heterooligomerize with Syt I (Osborne et al., 1999; Fukuda and Mikoshiba, 2000) and does not copurify with Syt IX (Fukuda, 2004). Similarly, Syt VII was found to form complexes with Syt I and IX, but not with Syt IV (Fukuda et al., 2004; Wang et al., 2005). Syt I, VII, and IX were shown to be involved in calcium-regulated exocytosis in PC12 cells, leading to the idea that these isoforms regulate different membrane fusion events from those involving Syt IV.
The in vitro binding assay showed that the Syt IV CD is recruited to ISG membranes, which suggests that the site of inhibition is on these membranes. Syt IV CD bound specifically to ISGs and this binding is strictly via protein components, as the recruitment was completely inhibited after the pretreatment of ISG membranes with trypsin. Our experiments also confirm the data showing that Syt IV, in contrast to Syt I, is not a phospholipid-binding isoform (Dai et al., 2004). The proposal that the Syt IV CD functions on the membrane concurs with data on the mode of inhibition of the Syt I CD in vesicle fusion with the plasma membrane (Earles et al., 2001; Zhang et al., 2002), where the association of the added Syt I CD with Stx1 and SNAP-25 inhibited the assembly of the endogenous Syt I–SNARE complexes. In the same way, the CD of Syt IV might bind to ISG SNAREs, thereby inhibiting the formation of an endogenous Syt IV–ISG SNARE complex, leading to inhibition of ISG–ISG fusion.
The recruitment of the Syt IV CD to ISG membranes, as well as its inhibitory effect on fusion, led us to ask if Syt IV and Stx6 colocalize and interact. Immunostaining of PC12 cells with anti-Stx6 and anti–Syt IV antibodies showed colocalization mainly in the juxtanuclear area, which is unlikely to be endosomes because, unlike Stx6 (Bock et al., 1997), Syt IV is not found on endosomes (Fukuda and Yamamoto, 2004). The immunoprecipitation of endogenous Syt IV with Stx6, and direct binding of the recombinant proteins to each other in vitro, suggested that these two proteins are in the same complex and could be regulating the same fusion event. We found that both Syt IV-C2A and C2B domains bind Stx6, and that they most likely cooperate to bind to Stx6. Syt I was also found to bind to SNAP-25 and Stx1 through its C2A and C2B domains (Chapman et al., 1996; Gerona et al., 2000), and both domains are thought to cooperate to regulate fusion (Earles et al., 2001). It would be interesting to investigate whether an individual domain or both domains are required for ISG homotypic fusion. Unfortunately, because of the insolubility of the GST-C2A and C2B domains, we could not investigate whether individual domains have an inhibitory effect. It is unlikely that the formation of a Syt IV–Stx1–SNAP-25 complex (Wang et al., 2003; Rickman et al., 2004) is involved in ISG fusion, as ISG fusion is not sensitive to the addition of botulinum neurotoxins (Wendler et al., 2001).
To understand the mechanism by which Syt IV is regulating ISG homotypic fusion, and, more specifically, whether inhibition with the Syt IV CD is synergistic with the inhibition by Stx6 antibodies, we added both reagents to the fusion assay, which resulted in an additive inhibition of ISG–ISG fusion. One explanation for this result is that the fusion-competent ISGs are at different stages when isolated, and, therefore, some of the ISGs will be past the stage blocked by one or the other reagent. Our results support the notion that Syt IV and Stx6 function at different stages, rather than synergistically at the same stage of ISG–ISG fusion. We found that the binding of Syt IV CD to ISG membranes was not inhibited by the addition of anti-Stx6 antibody (and vice versa; unpublished data). This result suggests that the Syt IV–binding site in Stx6 is different from the epitope for the anti-Stx6 antibody. Our data cannot exclude the possibility that Syt IV and Stx6, although in the same complex, could function at different steps during tethering, docking, and fusion of ISGs.
Most importantly, we were able to demonstrate a role for Syt IV in the maturation of ISGs in vivo. First, using siRNA depletion of Syt IV we found that Syt IV was required for ISG fusion and for efficient processing of newly synthesized SgII by PC2. Second, the dominant-negative Syt IV CD inhibited SgII processing by exogenous PC2 in PC12 cells. The greatest reduction in processing occurred when the processing rate was maximal in untreated controls at 45–60 min. This correlates with the t 1/2 of 45 min for fusion and maturation of ISGs in PC12 cells. Recently, Kuliawat et al. (2004) showed that expression of a soluble form of Stx6 in INS-1 β cells slowed proinsulin processing and concluded that this reduction in processing was caused by an indirect effect on biosynthetic traffic. The inhibition of SgII processing that we observe is probably a direct result of an inability to activate PC2 in the maturing ISGs because PC2 was found mainly in the unprocessed proform in cells transfected with the dominant-negative Syt IV CD. We have been unable to test PC2 activation after siRNA treatment because of a lower than usual transfection efficiency after siRNA treatment, which precludes expressing PC2 in the Syt IV–deficient PC12 cells. As PC2 autocatalytic activation is dependent on acidification of the granules (Lamango et al., 1999), which occurs during the maturation from ISGs to MSGs, we speculate that the reduction in pro-PC2 processing could be caused by a failure to acidify the ISGs. Although the mechanism of secretory granule acidification is not known, it has been proposed to occur after an increase in the density of the vacuolar H+-ATPase, as well as a decrease in the H+ permeability (Wu et al., 2001). Perturbation of the vacuolar H+-ATPase has also been shown to reduce processing in Xenopus laevis intermediate pituitary cells (Schoonderwoert et al., 2000). We propose that the Syt IV CD, through an inhibition of homotypic fusion, which may also limit membrane remodeling, may inhibit acidification. Further biochemical and morphological analyses will be used to study the regulation of the secretory granule pH in vitro, and should provide further information on how secretory granule biogenesis occurs.
Materials and methods
Cells and reagents
PC12 cells (clone 251) and PC12/PC2 cells stably expressing PC2 were described previously (Dittie and Tooze, 1995). HEK293A cells were purchased from Invitrogen, BAPTA was obtained from Calbiochem, and [35S]sulfate was purchased from GE Healthcare. All other reagents were obtained from Sigma-Aldrich.
Antibodies
Polyclonal anti–Syt IV antibody was a gift from M. Fukuda (Institute of Physical and Chemical Research Institute, Saitama, Japan). Monoclonal anti-p18 and polyclonal anti-Stx6 antibody were previously described (Dittie and Tooze, 1995; Wendler et al., 2001). Anti-PC2 antibody was a gift from B. Eipper (University of Connecticut, Farmington, CT). Monoclonal anti-Stx6 antibodies were purchased from BD Biosciences and Abcam PVC (3D10), and monoclonal anti–Syt I antibodies were obtained from Synaptic Systems GmbH. Mouse anti-HA and anti-myc antibodies were developed in-house (Cancer Research UK, London, UK). Rat anti-HA was obtained from Roche, and rabbit anti-myc was obtained from Abcam PVC.
Constructs and protein expression
GST-Syt I CD was obtained from G. Schiavo (Cancer Research UK). GST-Syt VII and GST-Syt IX CDs were obtained from M. Fukuda. Rat Syt IV CD (aa 37–425), Syt IV-C2A domain (aa 150–261), and Syt IV-C2B domain (aa 281–245) were amplified by RT-PCR from RNA extracted from PC12 cells. GST-Syt IV CD was made by inserting the DNA into the SmaI–NotI sites of pGEX-2T expression vector, and the expressed proteins were purified using glutathione beads (GE Healthcare). To eliminate negatively charged bacterial contaminants, the GST fusion proteins were further purified on a size exclusion column, and then on a cation exchange column (Ubach et al., 2001). HA-Syt IV CD and HA-C2A and HA-C2B domains were inserted into pcDNA 3.1 (Invitrogen).
Labeling and subcellular fractionation of PC12 cells
PNS was prepared from [35S]sulfate-labeled PC12 cells and then loaded successively on a continuous velocity gradient, followed by a discontinuous equilibrium sucrose gradient to separate ISGs and MSGs, as previously described (Tooze et al., 1991; Dittie et al., 1996).
In vitro ISG–ISG homotypic fusion and binding assays
ISG–ISG homotypic fusion was performed as previously described (Urbé et al., 1998). In brief, complete fusion reactions are comprised of the following: 100 μl [35S]sulfate-labeled PNS from PC12 cells, 10 μl ISGs purified from PC12/PC2 cells, an ATP-regenerating system and purified recombinant Syt IV, I, VII, or IX CDs, affinity-purified polyclonal anti-Stx6 antibody, or BAPTA as indicated in the figure legends. Fusion was carried out for 30 min, followed by the processing reaction for 90 min at 37°C. The product of PC2 cleavage of SgII, which is 35S-labeled p18, was immunoprecipitated and subjected to SDS-PAGE and autoradiography. The amount of p18 was quantified using ImageJ (National Institutes of Health) analysis software. Binding assays were performed as previously described (Dittie et al., 1996). The amount of bound Syt IV or I CDs was detected using anti–Syt IV and anti–Syt I antibodies, and was quantified using ImageJ analysis software.
Transfections, indirect immunofluorescence, and FACS sorting of PC12 cells
PC12 cells were seeded on poly-d-lysine–coated coverslips and transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Cells were fixed 24 h after transfection with 3% paraformaldehyde, permeabilized with 0.2% Triton X-100, and stained with the appropriate primary and secondary antibodies diluted in PBS with 0.2% gelatin. Images were acquired by confocal microscopy using a confocal microscope and software (LSM510; Carl Zeiss MicroImaging, Inc.). For the quantification of p18 levels, confocal images were taken with identical acquisition parameters and the mean of intensity of p18 was measured in individual cells using Photoshop 7.0 software (Adobe). To obtain a homogenous population of cells expressing both HA-Syt IV and PC2, or PC2 alone, FACS-sorting was used. Transfected cells were fixed and stained the same as the PC12 cells, and sorted using the Moflo FACS sorter (DakoCytomation). 150,000 HA/PC2-positive cells (starting from 2 × 108 cells) and 300,000 PC2-positive cells were pelleted, lysed in sample buffer, and subjected to SDS-PAGE analysis.
siRNA treatment, ISG–ISG fusion, and SgII processing
A SMARTpool of four siRNAs duplexes (Dharmacon) against rat Syt IV (1: 5′-GAAGAAAGCAUUUGUGGUG-3′; 2: 5′-GGAGACAAAUUGCUAAGUG-3′; 3: 5′-CAACAAGACUCCUCCAUAC-3′; and 4: 5′-UAAAGGAGUUGAUAUCUAC-3′) was used at a final concentration of 50 nM. Duplex 3 gave comparable results when used alone. Transfection of siRNAs in PC12 cells was performed using Dharmafect 2 (Dharmacon) in DME-containing full serum without antibiotics for 24 h, followed by a second treatment 24 h later. 3 d after transfection, cells were either fixed for immunofluorescence, lysed in sample buffer, or subjected to pulse-chase labeling.
To monitor SgII processing, PC12/PC2 cells were used. To control for sorting and budding from the TGN, PC12 cells were used. Pulse-chase [35S]sulfate-labeling of PC12 cells and analysis of SgII were performed as previously described (Urbé et al., 1997).
GST pull-down experiments and coimmunoprecipitation
GST or GST-Syt IV CD bound to glutathione beads was incubated with Triton X-100–solubilized lysates from myc-Stx6–transfected HEK293 cells in binding buffer (25 mM Hepes-KOH, pH 7.2, 25 mM MgOAc, and 0.5% Triton X-100). Proteins bound to beads, and 10% of the input were subjected to SDS-PAGE and immunoblotting with anti-myc antibodies.
For immunoprecipitation, HEK293 cells were transfected as described above, lysed in TNTE buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 0.3% Triton X-100, and 5 mM EDTA), and incubated with anti-HA, anti-Myc coupled to protein G Sepharose beads (GE Healthcare), or with beads alone. For coimmunoprecipitation of endogenous Syt IV with Stx6, TNTE lysates from PC12 cells were incubated with monoclonal anti-Stx6 antibody (Abcam PVC) and protein G beads. Immunoprecipitates were analyzed by SDS-PAGE, followed by immunoblotting with anti-HA, anti-myc, or anti–Syt IV antibody.
Online supplemental material
Fig. S1 shows by indirect immunofluorescence that Syt IV colocalizes with Stx6, but not with p18, in PC12/PC2 cells. Fig. S2 shows that BAPTA inhibits ISG homotypic fusion. Fig. S3 shows that Golgi morphology and secretory granule biogenesis are normal in cells treated with Syt IV-siRNA. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200506163/DC1.
Supplementary Material
Acknowledgments
We thank Dr. G. Schiavo for advice and reagents and J. Tooze for critically reading the manuscript. In addition, we thank the EM Unit for help with EM and the members of the Tooze laboratory for support.
Abbreviations used in this paper: BAPTA, 1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid; CD, cytoplasmic domain; HEK, human embryonic kidney; ISG, immature secretory granule; MSG, mature secretory granule; PC2, prohormone convertase 2; PNS, postnuclear supernatant; SgII, secretogranin II; Stx, syntaxin; Syt, synaptotagmin.
References
- Adolfsen, B., S. Saraswati, M. Yoshihara, and J.T. Littleton. 2004. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. J. Cell Biol. 166:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvan, P., and D. Castle. 1998. Sorting and storage during secretory granule biogenesis: looking backward and looking forward. Biochem. J. 332:593–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin, C., I. Hinners, and S.A. Tooze. 2000. Direct and GTP-dependent interaction of ADP-ribosylation factor 1 with clathrin adaptor protein AP-1 on immature secretory granules. J. Biol. Chem. 275:21862–21869. [DOI] [PubMed] [Google Scholar]
- Bai, J., and E.R. Chapman. 2004. The C2 domains of synaptotagmin–partners in exocytosis. Trends Biochem. Sci. 29:143–151. [DOI] [PubMed] [Google Scholar]
- Bennett, M.K., N. Calakos, and R.H. Scheller. 1992. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 257:255–259. [DOI] [PubMed] [Google Scholar]
- Bock, J.B., J. Klumperman, S. Davanger, and R.H. Scheller. 1997. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol. Biol. Cell. 8:1261–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, E.R. 2002. Synaptotagmin: a Ca(2+) sensor that triggers exocytosis? Nat. Rev. Mol. Cell Biol. 3:498–508. [DOI] [PubMed] [Google Scholar]
- Chapman, E.R., S. An, J.M. Edwardson, and R. Jahn. 1996. A novel function for the second C2 domain of synaptotagmin. Ca2+-triggered dimerization. J. Biol. Chem. 271:5844–5849. [DOI] [PubMed] [Google Scholar]
- Chieregatti, E., J.W. Witkin, and G. Baldini. 2002. SNAP-25 and synaptotagmin 1 function in Ca2+-dependent reversible docking of granules to the plasma membrane. Traffic. 3:496–511. [DOI] [PubMed] [Google Scholar]
- Chieregatti, E., M.C. Chicka, E.R. Chapman, and G. Baldini. 2004. SNAP-23 functions in docking/fusion of granules at low Ca2+. Mol. Biol. Cell. 15:1918–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craxton, M. 2004. Synaptotagmin gene content of the sequenced genomes. BMC Genomics. 5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai, H., O.H. Shin, M. Machius, D.R. Tomchick, T.C. Sudhof, and J. Rizo. 2004. Structural basis for the evolutionary inactivation of Ca(2+) binding to synaptotagmin 4. Nat. Struct. Mol. Biol. 11:844–849. [DOI] [PubMed] [Google Scholar]
- Dittie, A.S., and S.A. Tooze. 1995. Characterization of the endopeptidase PC2 activity towards secretogranin II in stably transfected PC12 cells. Biochem. J. 310:777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittie, A.S., N. Hajibagheri, and S.A. Tooze. 1996. The AP-1 adaptor complex binds to immature secretory granules from PC12 cells, and is regulated by ADP-ribosylation factor. J. Cell Biol. 132:523–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittie, A.S., L. Thomas, G. Thomas, and S.A. Tooze. 1997. Interaction of furin in immature secretory granules from neuroendocrine cells with the AP-1 adaptor complex is modulated by casein kinase II phosphorylation. EMBO J. 16:4859–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earles, C.A., J. Bai, P. Wang, and E.R. Chapman. 2001. The tandem C2 domains of synaptotagmin contain redundant Ca2+ binding sites that cooperate to engage t-SNAREs and trigger exocytosis. J. Cell Biol. 154:1117–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton, B.A., M. Haugwitz, D. Lau, and H.P. Moore. 2000. Biogenesis of regulated exocytotic carriers in neuroendocrine cells. J. Neurosci. 20:7334–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elferink, L.A., M.R. Peterson, and R.H. Scheller. 1993. A role for synaptotagmin (p65) in regulated exocytosis. Cell. 72:153–159. [DOI] [PubMed] [Google Scholar]
- Ferguson, G.D., D.M. Thomas, L.A. Elferink, and H.R. Herschman. 1999. Synthesis degradation, and subcellular localization of synaptotagmin IV, a neuronal immediate early gene product. J. Neurochem. 72:1821–1831. [DOI] [PubMed] [Google Scholar]
- Ferguson, G.D., S.G. Anagnostaras, A.J. Silva, and H.R. Herschman. 2000. Deficits in memory and motor performance in synaptotagmin IV mutant mice. Proc. Natl. Acad. Sci. USA. 97:5598–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon, R., A. Konigstorfer, S.H. Gerber, J. Garcia, M.F. Matos, C.F. Stevens, N. Brose, J. Rizo, C. Rosenmund, and T.C. Sudhof. 2001. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 410:41–49. [DOI] [PubMed] [Google Scholar]
- Fukuda, M. 2002. Vesicle-associated membrane protein-2/synaptobrevin binding to synaptotagmin I promotes O-glycosylation of synaptotagmin I. J. Biol. Chem. 277:30351–30358. [DOI] [PubMed] [Google Scholar]
- Fukuda, M. 2004. RNA interference-mediated silencing of synaptotagmin IX, but not synaptotagmin I, inhibits dense-core vesicle exocytosis in PC12 cells. Biochem. J. 380:875–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda, M., and K. Mikoshiba. 2000. Calcium-dependent and -independent hetero-oligomerization in the synaptotagmin family. J. Biochem. (Tokyo). 128:637–645. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., and A. Yamamoto. 2004. Effect of forskolin on synaptotagmin IV protein trafficking in PC12 cells. J. Biochem. (Tokyo). 136:245–253. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., T. Kojima, and K. Mikoshiba. 1996. Phospholipid composition dependence of Ca2+-dependent phospholipid binding to the C2A domain of synaptotagmin IV. J. Biol. Chem. 271:8430–8434. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., E. Kanno, Y. Ogata, C. Saegusa, T. Kim, Y.P. Loh, and A. Yamamoto. 2003. Nerve growth factor-dependent sorting of synaptotagmin IV protein to mature dense-core vesicles that undergo calcium-dependent exocytosis in PC12 cells. J. Biol. Chem. 278:3220–3226. [DOI] [PubMed] [Google Scholar]
- Fukuda, M., E. Kanno, M. Satoh, C. Saegusa, and A. Yamamoto. 2004. Synaptotagmin VII is targeted to dense-core vesicles and regulates their Ca2+-dependent exocytosis in PC12 cells. J. Biol. Chem. 279:52677–52684. [DOI] [PubMed] [Google Scholar]
- Gerona, R.R., E.C. Larsen, J.A. Kowalchyk, and T.F. Martin. 2000. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J. Biol. Chem. 275:6328–6336. [DOI] [PubMed] [Google Scholar]
- Gut, A., C.E. Kiraly, M. Fukuda, K. Mikoshiba, C.B. Wollheim, and J. Lang. 2001. Expression and localisation of synaptotagmin isoforms in endocrine beta-cells: their function in insulin exocytosis. J. Cell Sci. 114:1709–1716. [DOI] [PubMed] [Google Scholar]
- Heinemann, C., R.H. Chow, E. Neher, and R.S. Zucker. 1994. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+. Biophys. J. 67:2546–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbush, B.S., and J.I. Morgan. 1994. A third synaptotagmin gene, Syt3, in the mouse. Proc. Natl. Acad. Sci. USA. 91:8195–8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui, E., J. Bai, P. Wang, M. Sugimori, R.R. Llinas, and E.R. Chapman. 2005. Three distinct kinetic groupings of the synaptotagmin family: candidate sensors for rapid and delayed exocytosis. Proc. Natl. Acad. Sci. USA. 102:5210–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt, D.M., J.M. Baltz, and J.K. Ngsee. 2005. Synaptotagmin VI and VIII and syntaxin 2 are essential for the mouse sperm acrosome reaction. J. Biol. Chem. 280:20197–20203. [DOI] [PubMed] [Google Scholar]
- Ibata, K., M. Fukuda, T. Hamada, H. Kabayama, and K. Mikoshiba. 2000. Synaptotagmin IV is present at the Golgi and distal parts of neurites. J. Neurochem. 74:518–526. [DOI] [PubMed] [Google Scholar]
- Ibata, K., T. Hashikawa, T. Tsuboi, S. Terakawa, F. Liang, A. Mizutani, M. Fukuda, and K. Mikoshiba. 2002. Non-polarized distribution of synaptotagmin IV in neurons: evidence that synaptotagmin IV is not a synaptic vesicle protein. Neurosci. Res. 43:401–406. [DOI] [PubMed] [Google Scholar]
- Klumperman, J., R. Kuliawat, J.M. Griffith, H.J. Geuze, and P. Arvan. 1998. Mannose 6–phosphate receptors are sorted from immature secretory granules via adaptor protein AP-1, clathrin, and syntaxin 6–positive vesicles. J. Cell Biol. 141:359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliawat, R., J. Klumperman, T. Ludwig, and P. Arvan. 1997. Differential sorting of lysosomal enzymes out of the regulated secretory pathway in pancreatic β-cells. J. Cell Biol. 137:595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuliawat, R., E. Kalinina, J. Bock, L. Fricker, T.E. McGraw, S.R. Kim, J. Zhong, R. Scheller, and P. Arvan. 2004. Syntaxin-6 SNARE involvement in secretory and endocytic pathways of cultured pancreatic beta-cells. Mol. Biol. Cell. 15:1690–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamango, N.S., E. Apletalina, J. Liu, and I. Lindberg. 1999. The proteolytic maturation of prohormone convertase 2 (PC2) is a pH-driven process. Arch. Biochem. Biophys. 362:275–282. [DOI] [PubMed] [Google Scholar]
- Machado, H.B., W. Liu, L.J. Vician, and H.R. Herschman. 2004. Synaptotagmin IV overexpression inhibits depolarization-induced exocytosis in PC12 cells. J. Neurosci. Res. 76:334–341. [DOI] [PubMed] [Google Scholar]
- Matthew, W.D., L. Tsavaler, and L.F. Reichardt. 1981. Identification of a synaptic vesicle–specific membrane protein with a wide distribution in neuronal and neurosecretory tissue. J. Cell Biol. 91:257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbikay, M., N.G. Seidah, and M. Chretien. 2001. Neuroendocrine secretory protein 7B2: structure, expression and functions. Biochem. J. 357:329–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, H.P., J.M. Andresen, B.A. Eaton, M. Grabe, M. Haugwitz, M.M. Wu, and T.E. Machen. 2002. Biosynthesis and secretion of pituitary hormones: dynamics and regulation. Arch. Physiol. Biochem. 110:16–25. [DOI] [PubMed] [Google Scholar]
- Nicholson-Tomishima, K., and T.A. Ryan. 2004. Kinetic efficiency of endocytosis at mammalian CNS synapses requires synaptotagmin I. Proc. Natl. Acad. Sci. USA. 101:16648–16652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orci, L., M. Ravazzola, M.J. Storch, R.G. Anderson, J.D. Vassalli, and A. Perrelet. 1987. Proteolytic maturation of insulin is a post-Golgi event which occurs in acidifying clathrin-coated secretory vesicles. Cell. 49:865–868. [DOI] [PubMed] [Google Scholar]
- Osborne, S.L., J. Herreros, P.I. Bastiaens, and G. Schiavo. 1999. Calcium-dependent oligomerization of synaptotagmins I and II. Synaptotagmins I and II are localized on the same synaptic vesicle and heterodimerize in the presence of calcium. J. Biol. Chem. 274:59–66. [DOI] [PubMed] [Google Scholar]
- Perin, M.S., N. Brose, R. Jahn, and T.C. Sudhof. 1991. Domain structure of synaptotagmin (p65). J. Biol. Chem. 266:623–629. [PubMed] [Google Scholar]
- Regnier-Vigouroux, A., S.A. Tooze, and W.B. Huttner. 1991. Newly synthesized synaptophysin is transported to synaptic-like microvesicles via constitutive secretory vesicles and the plasma membrane. EMBO J. 10:3589–3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman, C., M. Craxton, S. Osborne, and B. Davletov. 2004. Comparative analysis of tandem C2 domains from the mammalian synaptotagmin family. Biochem. J. 378:681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiavo, G., G. Stenbeck, J.E. Rothman, and T.H. Sollner. 1997. Binding of the synaptic vesicle v-SNARE, synaptotagmin, to the plasma membrane t-SNARE, SNAP-25, can explain docked vesicles at neurotoxin-treated synapses. Proc. Natl. Acad. Sci. USA. 94:997–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonderwoert, V.T., J.C. Holthuis, S. Tanaka, S.A. Tooze, and G.J. Martens. 2000. Inhibition of the vacuolar H+-ATPase perturbs the transport, sorting, processing and release of regulated secretory proteins. Eur. J. Biochem. 267:5646–5654. [DOI] [PubMed] [Google Scholar]
- Sugita, S., W. Han, S. Butz, X. Liu, R. Fernandez-Chacon, Y. Lao, and T.C. Sudhof. 2001. Synaptotagmin VII as a plasma membrane Ca(2+) sensor in exocytosis. Neuron. 30:459–473. [DOI] [PubMed] [Google Scholar]
- Tooze, S.A. 1998. Biogenesis of secretory granules in the trans-Golgi network of neuroendocrine and endocrine cells. Biochim. Biophys. Acta. 1404:231–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze, S.A., and W.B. Huttner. 1990. Cell-free protein sorting to the regulated and constitutive secretory pathways. Cell. 60:837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooze, S.A., T. Flatmark, J. Tooze, and W.B. Huttner. 1991. Characterization of the immature secretory granule, an intermediate in granule biogenesis. J. Cell Biol. 115:1491–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, W.C., J.M. Edwardson, J. Bai, H.J. Kim, T.F. Martin, and E.R. Chapman. 2003. Identification of synaptotagmin effectors via acute inhibition of secretion from cracked PC12 cells. J. Cell Biol. 162:199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubach, J., Y. Lao, I. Fernandez, D. Arac, T.C. Sudhof, and J. Rizo. 2001. The C2B domain of synaptotagmin I is a Ca2+-binding module. Biochemistry. 40:5854–5860. [DOI] [PubMed] [Google Scholar]
- Urbé, S., A.S. Dittie, and S.A. Tooze. 1997. pH-dependent processing of secretogranin II by the endopeptidase PC2 in isolated immature secretory granules. Biochem. J. 321:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbé, S., L.J. Page, and S.A. Tooze. 1998. Homotypic fusion of immature secretory granules during maturation in a cell-free assay. J. Cell Biol. 143:1831–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vician, L., I.K. Lim, G. Ferguson, G. Tocco, M. Baudry, and H.R. Herschman. 1995. Synaptotagmin IV is an immediate early gene induced by depolarization in PC12 cells and in brain. Proc. Natl. Acad. Sci. USA. 92:2164–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, C.T., R. Grishanin, C.A. Earles, P.Y. Chang, T.F. Martin, E.R. Chapman, and M.B. Jackson. 2001. Synaptotagmin modulation of fusion pore kinetics in regulated exocytosis of dense-core vesicles. Science. 294:1111–1115. [DOI] [PubMed] [Google Scholar]
- Wang, C.T., J.C. Lu, J. Bai, P.Y. Chang, T.F. Martin, E.R. Chapman, and M.B. Jackson. 2003. Different domains of synaptotagmin control the choice between kiss-and-run and full fusion. Nature. 424:943–947. [DOI] [PubMed] [Google Scholar]
- Wang, P., M.C. Chicka, A. Bhalla, D.A. Richards, and E.R. Chapman. 2005. Synaptotagmin VII is targeted to secretory organelles in PC12 cells, where it functions as a high-affinity calcium sensor. Mol. Cell. Biol. 25:8693–8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendler, F., L. Page, S. Urbe, and S.A. Tooze. 2001. Homotypic fusion of immature secretory granules during maturation requires syntaxin 6. Mol. Biol. Cell. 12:1699–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, M.M., M. Grabe, S. Adams, R.Y. Tsien, H.P. Moore, and T.E. Machen. 2001. Mechanisms of pH regulation in the regulated secretory pathway. J. Biol. Chem. 276:33027–33035. [DOI] [PubMed] [Google Scholar]
- Zerial, M., and H. McBride. 2001. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2:107–117. [DOI] [PubMed] [Google Scholar]
- Zhang, Q., M. Fukuda, E. Van Bockstaele, O. Pascual, and P.G. Haydon. 2004. Synaptotagmin IV regulates glial glutamate release. Proc. Natl. Acad. Sci. USA. 101:9441–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X., M.J. Kim-Miller, M. Fukuda, J.A. Kowalchyk, and T.F. Martin. 2002. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 34:599–611. [DOI] [PubMed] [Google Scholar]
- Zhou, A., G. Webb, X. Zhu, and D.F. Steiner. 1999. Proteolytic processing in the secretory pathway. J. Biol. Chem. 274:20745–20748. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}