

Abstract
The ubiquitin–proteasome pathway has been implicated in synaptic development and plasticity. However, mechanisms by which ubiquitination contributes to precise and dynamic control of synaptic development and plasticity are poorly understood. We have identified a PDZ domain containing RING finger 3 (PDZRN3) as a synapse-associated E3 ubiquitin ligase and have demonstrated that it regulates the surface expression of muscle-specific receptor tyrosine kinase (MuSK), the key organizer of postsynaptic development at the mammalian neuromuscular junction. PDZRN3 binds to MuSK and promotes its ubiquitination. Regulation of cell surface levels of MuSK by PDZRN3 requires the ubiquitin ligase domain and is mediated by accelerated endocytosis. Gain- and loss-of-function studies in cultured myotubes show that regulation of MuSK by PDZRN3 plays an important role in MuSK-mediated nicotinic acetylcholine receptor clustering. Furthermore, overexpression of PDZRN3 in skeletal muscle of transgenic mice perturbs the growth and maturation of the neuromuscular junction. These results identify a synapse-associated E3 ubiquitin ligase as an important regulator of MuSK signaling.
Introduction
The formation of synapses requires a complex interchange of signals between presynaptic nerve terminals and postsynaptic cells. This is best illustrated at the mammalian neuromuscular junction (NMJ), where a signaling cascade mediating postsynaptic differentiation has been well characterized (Colledge and Froehner, 1998; Sanes and Lichtman, 2001; Burden, 2002; Bezakova and Ruegg, 2003; Song et al., 2006). At the center of this signaling cascade are agrin, a proteoglycan derived from the terminals of presynaptic motoneurons (Nitkin et al., 1987; Bowe and Fallon, 1995), and MuSK, a muscle-specific receptor tyrosine kinase activated by agrin (Jennings et al., 1993; Valenzuela et al., 1995). Activation of MuSK leads to the assembly of the postsynaptic complex, and genetic studies in mice have shown that both agrin and MuSK are required for the formation of the NMJ (DeChiara et al., 1996; Gautam et al., 1996; Burgess et al., 1999). In particular, MuSK is required for all aspects of postsynaptic differentiation, including the initial agrin and nerve-independent clustering of nicotinic acetylcholine receptors (AChRs) (Lin et al., 2001; Yang et al., 2001; Willmann and Fuhrer, 2002), thus functioning as the major organizer of synapse formation at the NMJ. It is less clear how the activity of MuSK is regulated to ensure proper development and homeostasis of synapses.
Increasing evidence suggests that protein ubiquitination plays an important role in regulating synaptic development, maintenance, and plasticity (DiAntonio and Hicke, 2004). Protein ubiquitination is mediated by the sequential actions of three enzymes: an E1 ubiquitin activating enzyme, E2 ubiquitin conjugating enzyme, and E3 ubiquitin ligase. Of these, E3 ubiquitin ligases directly bind substrates and render substrate specificity to the ubiquitination reaction, thus acting as key modulators of the ubiquitin system. Consistent with this idea, genetic studies in Drosophila and Caenorhabditis elegans have demonstrated the importance of E3 ubiquitin ligases in synaptic development and function. Most notably, mutations of highwire, a putative E3 ubiquitin ligase in Drosophila, or its orthologue in C. elegans (rpm-1), lead to aberrant presynaptic development (Schaefer et al., 2000; Wan et al., 2000; Zhen et al., 2000; DiAntonio et al., 2001). Recent studies in C. elegans also suggest that E3 ubiquitin ligases play an important role in regulating the surface abundance of glutamate receptors at the postsynaptic membrane (Juo and Kaplan, 2004; van Roessel et al., 2004). Although ample evidence supports the role of protein ubiquitination in synaptic development and plasticity in vertebrates (Chapman et al., 1992; Serdaroglu et al., 1992; Colledge et al., 2003; Patrick et al., 2003; Zhao et al., 2003), the specific molecular mechanisms underlying these effects remain to be elucidated.
In the present study, we report the functional characterization of PDZRN3, a protein containing both RING and PDZ domains, as a synapse-associated E3 ubiquitin ligase at the mammalian NMJ. PDZRN3 (PDZ domain containing RING finger 3) was named based on its sequence similarity to PDZRN1 and 2 (Katoh and Katoh, 2004). It has also been named LNX3 and SEMCAP3 based on its sequence similarity to LNX1/LNX2 (Ligand-of-Numb protein X) and SEMCAP1/SEMCAP2 (M-SemF cytoplasmic domain-associated protein), respectively. A very recent study showed that PDZRN3 is expressed in muscle (Ko et al., 2006). In culture, the expression of PDZRN3 is increased during differentiation of myoblasts to myotubes and may play a role in myoblast fusion (Ko et al., 2006). We find that PDZRN3 mRNA is enriched in the synaptic region of the muscle and that PDZRN3 protein is concentrated at the NMJ. Coimmunoprecipitation shows that PDZRN3 interacts with MuSK in heterologous cells and in myotubes, and that this interaction is enhanced by agrin stimulation. Functionally, PDZRN3 promotes ubiquitination of MuSK and down-regulates cell surface levels of MuSK through its E3 ubiquitin ligase domain. Both gain- and loss-of-function studies in cultured myotubes reveal an important role for PDZRN3 in regulating agrin-induced AChR clustering. Furthermore, transgenic overexpression of PDZRN3 in vivo perturbs the growth and maturation of the NMJ. Our findings demonstrate an important role for PDZRN3 in regulating the growth and maturation of the NMJ.
Results
PDZRN3 interacts with MuSK
PDZ domain–mediated protein–protein interactions play important roles at synapses (Kim and Sheng, 2004). Many synaptic proteins at the mammalian NMJ, including MuSK, ErbB2, neuregulin-1, and Eph receptors, contain conserved PDZ binding motifs at the C termini (Torres et al., 1998), suggesting that PDZ domain proteins may also play a role at the NMJ. Our previous yeast two-hybrid screen using the last 25 amino acids of erbB2 and neuregulin-1 as baits identified Lnx1 as an ErbB2-interacting, PDZ domain–containing E3 ubiquitin ligase that is exclusively expressed in perisynaptic Schwann cells at the NMJ (Young et al., 2005). We then searched databases for additional RING and PDZ domain–containing proteins that may play a role at the NMJ and identified PDZRN3 as a potential candidate. Structurally, PDZRN3 (also called SEMCAP3 and LNX3; Katoh and Katoh, 2004; Ko et al., 2006) contains a RING domain, a zinc finger domain, one (PDZRN3A) or two (PDZRN3B) PDZ domains depending on alternative splicing, and a PDZ domain binding motif (TTV) at the C terminus (Fig. 1 A). Northern blot analysis indicates that there are two species of PDZRN3 mRNA with molecular sizes of 5.5 and 4.6 kb. Both splicing variants are highly expressed in skeletal and cardiac muscle of adult mice. Lower levels of expression are also seen in the brain, spinal cord, kidney, and lung (Fig. 1 B). Examination of mRNA expression by in situ hybridization of the diaphragm shows that PDZRN3 mRNA is expressed throughout muscle fibers with a distinct enrichment at the central region of the diaphragm muscle where synapses and synaptic nuclei are localized (Fig. 1 C), suggesting that PDZRN3 mRNA is preferentially transcribed by synaptic nuclei and thus may play a role at the NMJ.
Figure 1.

Interaction of MuSK with PDZRN3. (A) Protein domain structure of PDZRN3. Two splicing variants were identified. The major form of PDZRN3 (PDZRN3A) contains a RING finger domain (RF), a zinc finger domain (ZF), a PDZ domain (PDZ-1), and a PDZ binding motif (TTV). The longer form (PDZRN3B) contains an additional PDZ domain (PDZ-2). (B) Northern blot shows two species of PDZRN3 mRNA with molecular sizes of 5.5 and 4.6 kb. PDZRN3 is highly expressed in skeletal and cardiac muscle. Lower expression is also seen in the spinal cord, brain, kidney, and lung. The blot was re-probed with elongation factor-1α (EF-1α) as a loading control. (C) Wholemount in situ hybridization of diaphragm from P7 mice reveals that PDZRN3 mRNA is concentrated at the central region of the muscle, suggesting that PDZRN3 mRNA is preferentially transcribed by synaptic nuclei. Lower level of PDZRN3 mRNA is also expressed throughout the muscle fibers. (D) PDZRN3 interacts with MuSK in transfected COS-7 cells. PDZRN3A and MuSK were cotransfected into COS-7 cells. Immunoprecipitations (IP) were performed using an anti-PDZRN3 antibody and immunoblotted (IB) with an anti-MuSK antibody. MuSK and PDZRN3A were coimmunoprecipitated. Lysate lanes represent 10% of the input for coimmunoprecipitation. (E) A control experiment shows that Cbl, a RING-type E3 ubiquitin ligase, does not coimmuniprecipitate with MuSK when coexpressed in COS-7 cells. (F) PDZRN3 and MuSK form a complex in myotubes. Lysates from cultured C2C12 myotubes were immunoprecipitated with an anti-MuSK antibody and blotted with an anti-PDZRN3 antibody. Lysate lanes represent 5% of the input for coimmunoprecipitation. (G) Agrin stimulation enhances the interaction of PDZRN3 and MuSK. Lysates of cultured C2C12 myotubes with or without agrin stimulation were immunoprecipitated with an anti-MuSK antibody and blotted with an anti-PDZRN3 antibody. An increased amount of PDZRN3 was coimmunoprecipitated with MuSK from agrin-stimulated myotubes. Lysate lanes represent 5% of the input for coimmunoprecipitation. (H) The PDZ domain of PDZRN3 is required for its interaction with MuSK. Lysates from COS-7 cells expressing MuSK and either PDZRN3A, PDZRN3A lacking the RING and zinc finger domains (PDZRN3AΔRZ), or PDZRN3A lacking the PDZ domain (PDZRN3AΔPDZ) were immunoprecipitated with an anti-PDZRN3 antibody and blotted with an anti-MuSK antibody. Deletion of the PDZ domain from PDZRN3A completely abolished its interaction with MuSK. (I) The PDZ domain binding motif of MuSK is not necessary for its binding to PDZRN3. MuSK lacking the PDZ domain binding motif (deletion of the last three amino acids; MuSKΔ3) was coexpressed with PDZRN3A in COS-7 cells and coimmunoprecipitation was performed with an anti-PDZRN3 antibody. (J and K) Deletion mapping suggests that in the absence of the PDZ binding motif a large portion of the kinase domain of MuSK mediates the interaction with PDZRN3 (J). Only the transmembrane domain and the cytoplasmic domain are shown in the diagram. JXT = juxtamembrane domain. However, the presence of the PDZ binding motif (TTV) in the deletion construct of MuSK (ΔC2+TTV) restores its ability to interact with PDZRN3 (K), indicating that the PDZ binding motif of MuSK is sufficient but not necessary for its interaction with PDZRN3.
Because MuSK contains a conserved PDZ binding motif at its C terminus (Torres et al., 1998) we tested whether PDZRN3 interacts with MuSK by coexpressing MuSK and PDZRN3A in COS-7 cells. As shown in Fig. 1 D, MuSK can be specifically coimmunoprecipitated with PDZRN3A, suggesting a direct interaction between MuSK and PDZRN3A. As a control, we found that Cbl, a well-characterized RING-type E3 ubiquitin ligase (Levkowitz et al., 1999), does not interact with MuSK under the same conditions (Fig. 1 E). To test whether PDZRN3 interacts with MuSK under physiological conditions, we immunoprecipitated MuSK from cultured myotubes and found that PDZRN3 was coimmunoprecipitated with MuSK, indicating that they form a complex in myotubes (Fig. 1 F). Moreover, agrin stimulation significantly increased the association of MuSK and PDZRN3 in cultured myotubes (Fig. 1 G), suggesting a dynamic regulation of PDZRN3 and MuSK interaction.
To determine which domain(s) in the PDZRN3 protein mediates the interaction with MuSK, we made deletion constructs of PDZRN3A that lacked either the RING and zinc finger domains or the PDZ domain. When coexpressed with MuSK in COS-7 cells, PDZRN3A lacking the RING and zinc finger domains retained the ability to interact with MuSK, whereas PDZRN3A lacking the PDZ domains failed to interact with MuSK (Fig. 1 H). This suggests that the PDZ domain in PDZRN3A is required for its interaction with MuSK. However, deletion of the PDZ binding motif of MuSK only partially decreased its interaction with PDZRN3A (Fig. 1 I), suggesting that the PDZ domains of PDZRN3A may interact with internal sequences as well as the C-terminal motif on the cytoplasmic domain of MuSK. To further dissect the interaction of MuSK with PDZRN3A, we coexpressed full-length PDZRN3A with a series of deletion constructs of MuSK lacking the PDZ binding motif (last 3 amino acids). Coimmunoprecipitation data showed that, in the absence of the PDZ binding motif, a large portion of the kinase domain of MuSK is required for binding to PDZRN3A (Fig. 1 J). However, inclusion of the terminal PDZ binding motif (TTV) in deletion constructs of MuSK is sufficient to restore the binding with PDZRN3A (Fig. 1, J and K), indicating that the PDZ binding motif of MuSK is sufficient but not necessary for its binding to PDZRN3A in a heterologous system.
PDZRN3 proteins are concentrated postsynaptically at the NMJ
To examine the localization of PDZRN3 proteins, we generated a rabbit polyclonal antibody against the N-terminal RING domain (PDZRN3-10B) and a guinea pig polyclonal antibody against an internal unique sequence (PDZRN3-3B). Both affinity-purified antibodies specifically recognized a single band from C2C12 myotube lysates which corresponded to the size of the shorter PDZRN3 isoform (PDZRN3A in Fig. 1 A) from transfected COS-7 cells (Fig. 2, A and B), suggesting that PDZRN3A is the predominant form in myotubes. Consistent with the Northern blot result, PDZRN3 proteins are detectable in skeletal muscle, heart, spinal cord, and brain (Fig. 2 C).
Figure 2.

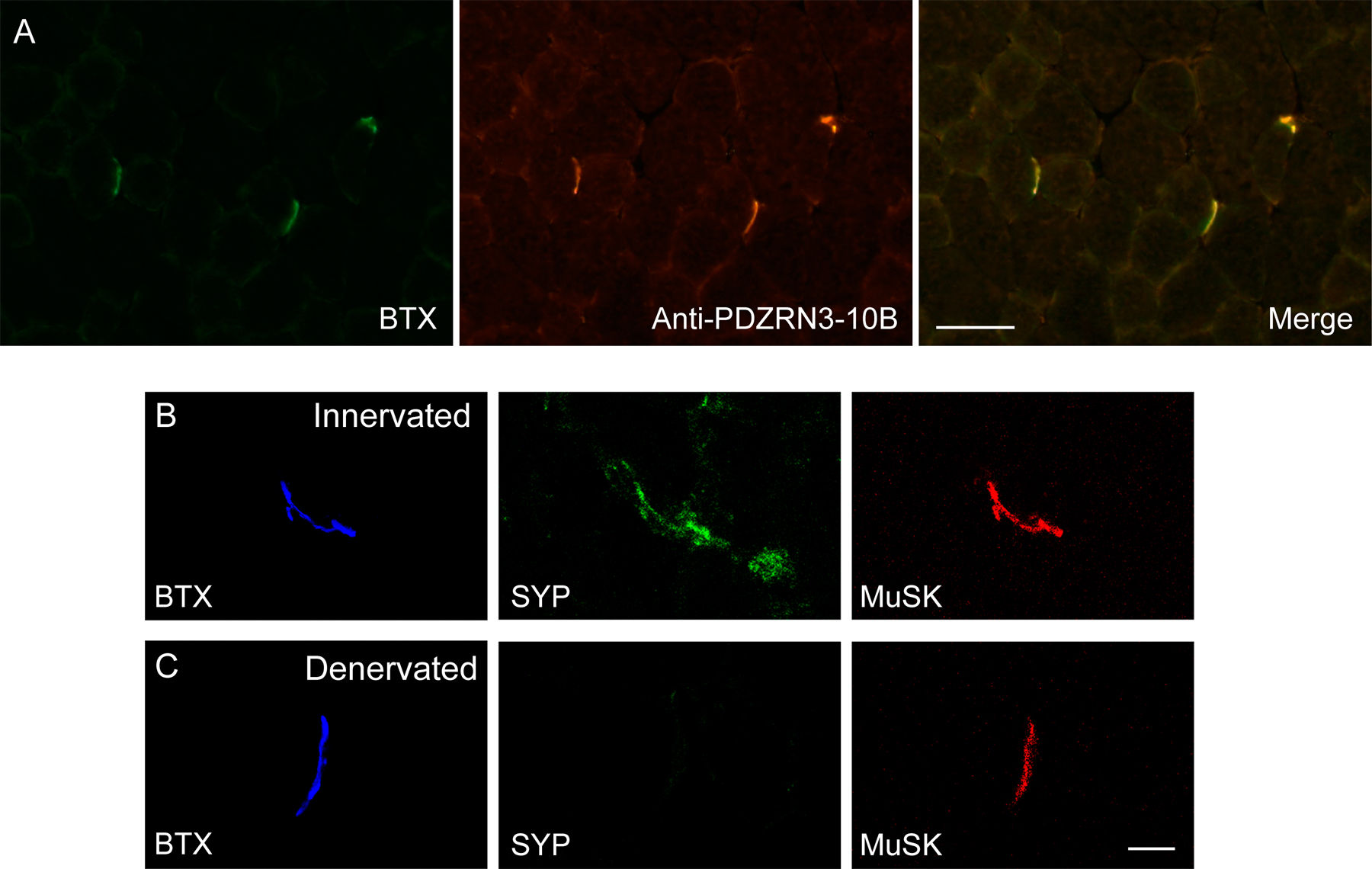
PDZRN3 is localized postsynaptically at the NMJ. (A and B) Characterization of a rabbit (PDZRN3-10B) and a guinea pig (PDZRN3-3B) antibody against PDZRN3. Both affinity-purified antibodies recognize a single band in myotube lysate corresponding to the size of PDZRN3A transiently expressed in COS-7 cells. (C) A Western blot shows that PDZRN3 protein is expressed in the skeletal muscle, heart, brain, and spinal cord. (D and E) Cross sections of adult mouse muscle were co-stained with α-bungarotoxin (BTX; D and E), which binds to AChRs, and either PDZRN3-10B antibody (D′) or PDZRN3-3B antibody (E′). PDZRN3 precisely colocalizes with AChRs at the NMJ (D′′ and E′′). (F) The synaptic staining of PDZRN3 is blocked by incubating the antibody with the immunizing antigen. (G and H) PDZRN3 localizes postsynaptically. Cross sections from innervated (G) or denervated (H) muscles were triple stained with α-BTX for postsynaptic AChRs (blue; G and H), anti-synaptophysin for presynaptic nerve terminals (green; G′and H′) and PDZRN3 (red; G′′ and H′′). 5 d after denervation, presynaptic terminals had degenerated (H′), but PDZRN3 staining persisted (H′′). (I) Developmental profile of PDZRN3 expression at the NMJ. Cross sections of muscles were prepared from mice at embryonic day (E) 14.5, 16.5, 18.5, postnatal day (P) 3, 7, and 21, and co-stained with α-BTX and PDZRN3. PDZRN3 staining first appears at E16.5. Bars, 10 μm.
To determine the localization of PDZRN3 proteins in the neuromuscular system, we performed coimmunostaining of adult mouse muscle with anti-PDZRN3 antibodies and α-bungarotoxin (α-BTX), which specifically binds nicotinic acetylcholine receptors (AChRs). Both anti-PDZRN3 antibodies stain the NMJ brightly with a pattern that precisely matches the staining of AChRs with α-BTX (Fig. 2; D–D′′, E–E′′). The staining is specific as it can be blocked by preincubation of the antibodies with cognate antigens (Fig. 2, F–F′′). To determine whether PDZRN3 is localized postsynaptically, we denervated hindlimb muscles in adult mice by transection of the sciatic nerve on one side of the body, and compared the expression of PDZRN3 in denervated and non-denervated muscles using triple labeling of α-BTX, anti-synaptophysin, and anti-PDZRN3. 5 d after nerve resection, presynaptic nerve terminals had degenerated as revealed by the lack of staining for the presynaptic vesicle protein synaptophysin, whereas staining for AChRs (Fig. 2, H–H′) and MuSK (Fig. S1 [available at http://www.jcb.org/cgi/content/full/jcb.200610060/DC1]; Bowen et al., 1998) remained. PDZRN3 staining persisted at the denervated NMJ and colocalized with AChR staining (Fig. 2, H–H′′), indicating that PDZRN3 is localized at the postsynaptic site of the NMJ, consistent with a potential role in regulating MuSK signaling.
The developmental profile of PDZRN3 expression at the NMJ was examined by immunostaining of muscle sections from various developmental stages. PDZRN3 staining at the NMJ first appears at approximately embryonic day (E) 16.5 (Fig. 2 I), a time point when NMJs have just formed. As the NMJ grows and matures, the staining intensity and complexity of PDZRN3 increases in concert with that of AChRs (Fig. 2 I), a developmental profile common to known postsynaptic regulators at the NMJ, such as MuSK and rapsyn (Noakes et al., 1993; Bowen et al., 1998).
PDZRN3 promotes MuSK ubiquitination
The presence of a classic RING domain with conserved key cysteine and histidine residues in the PDZRN3 protein suggests that PDZRN3 likely functions as a RING-type E3 ubiquitin ligase. To test this, we used an in vitro ubiquitination assay to examine whether the RING domain of PDZRN3 contains E3 ubiquitin ligase activity. The RING domain of PDZRN3 was fused to GST and expressed in Escherichia coli. Glutathione bead-purified GST fusion proteins were tested for self-ubiquitination in the presence of added E1 and E2 enzymes. Currently, more than two dozen E2 enzymes have been identified, and they show varying degrees of specificity when interacting with E3s (Kumar et al., 1997). Our initial tests of various E2s showed that the RING domain of PDZRN3 exhibits ubiquitin ligase activity in the presence of E1 and the UbcH5 family of E2 enzymes (Fig. 3 A). Consistent with this finding, GST pull-down assays showed that PDZRN3 directly interacts with the UbcH5B ubiquitin conjugating enzyme (Fig. 3 B). Furthermore, mutation of either one of the two key cysteines in the RING structure completely abolished the ubiquitin ligase activity of PDZRN3 (Fig. 3 C). Together, these data demonstrate that PDZRN3 is a catalytically active RING-type E3 ubiquitin ligase.
Figure 3.

PDZRN3 is an E3 ubiquitin ligase and promotes MuSK ubiquitination. (A) The RING domain of PDZRN3 was fused to GST and purified from E. coli. In vitro self-ubiquitination assays were performed in the presence of ubiquitin, E1, and various E2s. Self-ubiquitination of RING-GST (arrow) was only seen in the presence of the UbcH5 family of E2s. The arrowhead indicates the non-ubiquitinated RING-GST fusion protein. (B) GST pull-down experiment shows a direct interaction between UbcH5B and PDZRN3. GST or GST-UbcH5B proteins were immobilized on glutathione-sepharose beads. Whole cell lysates from PDZRN3 transfected COS-7 cells were incubated with GST or GST-UbcH5B beads and proteins were eluted. Western blot analysis shows that PDZRN3 was pulled down by GST-UbcH5B beads. (C) In vitro self-ubiquitination assay shows that mutation of the key cysteine residues in the RING domain of PDZRN3 (C18A and C38A) completely abolishes its ubiquitin ligase activity. The blot was first probed with an anti-FLAG antibody for the FLAG-tagged GST-RING proteins (top panel), then blotted with an anti-ubiquitin antibody to confirm the ubiquitination (bottom panel). (D) PDZRN3 promotes MuSK ubiquitination. MuSK was coexpressed with PDZRN3A, PDZRN3A lacking the RING domain (PDZRN3AΔRING) or PDZRN3A lacking the PDZ domains (PDZRN3AΔPDZ) in COS-7 cells. MuSK was immunoprecipitated with an anti-MuSK antibody and analyzed by Western blot with an anti-ubiquitin antibody to detect ubiquitination of MuSK. Only wild-type PDZRN3A promotes MuSK ubiquitination. (E) Coexpression of PDZRN3 and TrkB does not lead to increased ubiquitination of TrkB. TrkB was coexpressed with PDZRN3A in COS-7 cells. Coimmunoprecipitation was performed with an anti-TrkB antibody and blotted with an anti-ubiquitin antibody. (F) MuSK ubiquitination is increased upon agrin stimulation. Lysates from agrin-treated (1 nM for 2 h) and nontreated cultured myotubes were immunoprecipitated with an anti-MuSK antibody and blotted with an anti-ubiquitin antibody (top panel). The blot was reprobed with an anti-MuSK antibody to show the immunoprecipitated MuSK (bottom panel).
We next tested whether PDZRN3 promotes the ubiquitination of MuSK. MuSK was cotransfected with PDZRN3A, PDZRN3AΔRING (PDZRN3A lacking the RING domain), or PDZRN3AΔPDZ (lacking the PDZ domains) into cultured COS-7 cells. Transfected cells were harvested in the presence of 2% SDS and incubated at 4°C for 1 h to minimize nonspecific association of proteins with MuSK. MuSK was immunoprecipitated with anti-MuSK antibodies and analyzed by Western blot for ubiquitination using an anti-ubiquitin antibody. In the absence of PDZRN3A, MuSK from transfected COS-7 cells showed low levels of ubiquitination (Fig. 3 D). When MuSK was coexpressed with PDZRN3A, the levels of MuSK ubiquitination were significantly increased (Fig. 3 D). Deletion of the RING domain, which contains the E3 ubiquitin ligase activity, or the PDZ domain, which is required for the interaction with MuSK, completely abolished the effects of PDZRN3A on the ubiquitination of MuSK (Fig. 3 D). On the other hand, coexpression of PDZRN3A and TrkB, a functionally unrelated receptor tyrosine kinase, did not lead to increased ubiquitination of TrkB (Fig. 3 E). These data strongly suggest that MuSK is a specific substrate of PDZRN3 ubiquitin ligase activity.
To determine whether endogenous MuSK in myotubes is ubiquitinated, we immunoprecipitated MuSK from agrin-treated and nontreated myotubes and probed for MuSK ubiquitination using an anti-ubiquitin antibody. In the absence of agrin stimulation, a low level of MuSK ubiquitination was observed. However, the level of ubiquitination of MuSK was significantly increased upon agrin stimulation (Fig. 3 F). Together with the results showing the enhancement of MuSK and PDZRN3 interaction upon agrin stimulation, these data suggest that the ubiquitnation of MuSK is dynamically regulated and PDZRN3 may play an important role in this regulation.
Regulation of cell surface levels of MuSK by PDZRN3
To determine the functional consequences of MuSK-PDZRN3 interaction, we tested whether PDZRN3 directly regulates the level of MuSK on the cell surface. We coexpressed PDZRN3A and MuSK in COS-7 cells and measured cell surface levels of MuSK by membrane nonpermeable biotinylation of surface proteins and subsequent quantification of biotinylated MuSK by Western blot. Coexpression of PDZRN3A with MuSK significantly reduced the surface level of MuSK (Fig. 4, A and B). This effect was seen with both wild-type and constitutively kinase-active MuSK. Interestingly, PDZRN3A did not down-regulate the surface level of kinase-dead MuSK, which retains partial ability to interact with PDZRN3A (Fig. 4 C), suggesting that the effect of PDZRN3A on surface MuSK levels depends on the kinase activity of MuSK. This is consistent with our observation that agrin stimulation, which activates MuSK kinase activity, enhances the MuSK-PDZRN3 interaction and MuSK ubiquitination. This is also consistent with other studies showing that E3 ubiquitin ligases are often activated by the kinase activity of the receptor tyrosine kinases they regulate (Levkowitz et al., 1999; Miyake et al., 1999; Marmor and Yarden, 2004).
Figure 4.

PDZRN3 regulates the surface levels of MuSK. (A and B) PDZRN3 down-regulates surface levels of MuSK in COS-7 cells. PDZRN3A was coexpressed with wild-type (WT), kinase-active (KA), or kinase-dead (KD) MuSK in COS-7 cells and the surface levels of MuSK were determined by biotinylation assays followed by Western blot of MuSK. Coexpression of PDZRN3A leads to reduced surface levels of wild-type and kinase-active MuSK, but not kinase-dead MuSK. Transferrin receptors (TfR) were used as a control for surface biotinylation and GAPDH was used as a control of cell number. Bar graphs in B show the quantification data (mean ± SEM, n = 6). *, P < 0.05; **, P < 0.01. (C) Kinase-dead MuSK retains its ability to interact with PDZRN3. PDZRN3A and MuSK were cotransfected into COS-7 cells and immunoprecipitations were performed using an anti-PDZRN3 antibody. Both kinase-active (KA) and kinase-dead (KD) MuSK were coimmunoprecipitated with PDZRN3A. (D) PDZRN3A construct lacking the RING domain (PDZRN3AΔRING) was coexpressed with MuSK and the surface level of MuSK was determined as described above. Deletion of the RING domain abolishes the effect of PDZRN3A on the down-regulation of MuSK surface levels. (E and F) Relative endocytosis of MuSK in the presence and absence of PDZRN3A. The initial surface (E, left panel) and subsequent endocytosed MuSK (E, right panel) were determined by biotinylation assays, and relative endocytosis was calculated as the ratio of endocytosed/initial surface MuSK (F; mean ± SEM, n = 6). Coexpression of PDZRN3A with MuSK significantly increased the endocytosis of MuSK. *, P < 0.05.
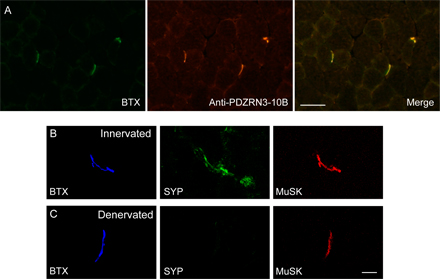
Because the RING domain contains the ubiquitin ligase activity, we examined whether the RING domain of PDZRN3 is required for the down-regulation of the surface level of MuSK using a PDZRN3A construct lacking the RING domain. As shown in Fig. 4 D, deletion of the RING domain abolishes the effect of PDZRN3 on the surface level of MuSK, suggesting that the E3 ubiquitin ligase activity of PDZRN3 plays a critical role in this regulation. To test the specificity of the effect of PDZRN3 on MuSK surface expression, we coexpressed PDZRN3A with erbB2, another receptor tyrosine kinase present at the NMJ. We found that PDZRN3A did not affect the surface level of erbB2 (Fig. S2 A, available at http://www.jcb.org/cgi/content/full/jcb.200610060/DC1). Moreover, the surface level of MuSK is not down-regulated by Cbl (Fig. S2 B), a RING-type E3 ubiquitin ligase that has been shown to down-regulate the signaling of several receptor tyrosine kinases such as PDGF and EGF receptors (Levkowitz et al., 1999; Miyake et al., 1999). These data strongly suggest that PDZRN3 specifically regulates the surface levels of MuSK through the combination of its specific binding to MuSK and its E3 ubiquitin ligase activity.
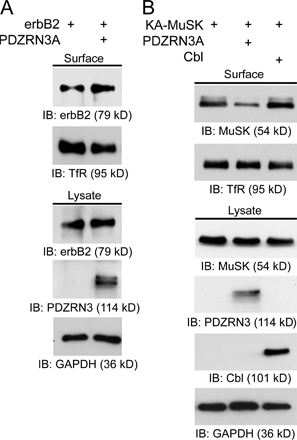
The ubiquitin system is best known for its function of adding polyubiquitin chains to target proteins, leading to the degradation of polyubiquitinated proteins through the 26S proteasome. However, a more recently discovered function of the ubiquitin system is to regulate membrane protein endocytosis through mono- or poly-ubiquitination (Hicke, 2001; Barriere et al., 2006; Hawryluk et al., 2006). To test whether PDZRN3 regulates MuSK activity through endocytosis, we compared the relative endocytosis of MuSK in the absence or presence of PDZRN3. To measure the endocytosis of MuSK, surface proteins from two identical sets of cells were biotinylated at 4°C. One set of biotinylated cells was kept on ice and used to measure initial surface levels of MuSK. The other set of biotinylated cells was moved to 37°C for 10 min to allow endocytosis to occur, and the remaining biotinylated surface proteins were then cleaved. Surface and endocytosed MuSK were detected by Western blot. The relative endocytosis was calculated as the ratio of endocytosed versus initial surface MuSK. As expected, expression of PDZRN3A reduced the surface level of MuSK (Fig. 4 E). This reduction of surface expression of MuSK was accompanied by increased endocytosis of MuSK (Fig. 4 E). In the presence of PDZRN3A, the ratio of endocytosed MuSK to initial surface MuSK is twice that of MuSK alone (Fig. 4, E and F).
Ubiquitination of membrane proteins generally leads to endocytosis and subsequent degradation through the lysosome (Hicke, 2001; Marmor and Yarden, 2004). Consistent with our observation that PDZRN3 down-regulates the surface level of MuSK by increasing its endocytosis, we found that the addition of chloroquine (CLQ), a lysosome inhibitor that blocks lysosomal degradation and membrane recycling (Tietze et al., 1980; Levkowitz et al., 1999; Rocca et al., 2001; Vecchione et al., 2003), largely abolished the effect of PDZRN3 on MuSK surface expression. In contrast, blocking 26S proteasome function with MG-132 did not affect the PDZRN3-mediated down-regulation of MuSK surface expression (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200610060/DC1).
PDZRN3 regulates agrin-induced AChR clustering in myotubes
To investigate the functional significance of PDZRN3 in regulating MuSK signaling, we examined MuSK-dependent AChR clustering in cultured myotubes. We used cultured C2C12 myoblasts, which differentiate into myotubes upon serum deprivation. The clustering of AChRs on these myotubes can be efficiently induced by the addition of exogenous agrin (Pun et al., 1997), thus providing a system to dissect agrin–MuSK signaling without the interference of other factors from nerve terminals. Agrin stimulation of wild-type myotubes leads to the formation of numerous large clusters of AChRs (Fig. 5, A and G). However, agrin-induced AChR clustering is significantly attenuated in myotubes overexpressing either form of PDZRN3 (Fig. 5; A–A′′, B–B′′, G). Importantly, the deletion of the RING domain largely abolishes this effect (Fig. 5; C–C′′, D–D′′, G). Furthermore, coexpression of MuSK with PDZRN3 in myotubes blocks the effect of PDZRN3 on agrin-induced AChR clustering (Fig. 5; E–E′′, F–F′′, G), suggesting that overexpression of MuSK can antagonize PDZRN3. These data are consistent with our observation that PDZRN3 down- regulates the surface levels of MuSK when coexpressed in COS-7 cells and point to a potential role for PDZRN3 as an E3 ubiquitin ligase regulating postsynaptic development at the NMJ.
Figure 5.

Overexpression of PDZRN3 in myotubes disrupts agrin-induced clustering of AChRs. (A and B) Transfected myotubes were stimulated with agrin and stained with α-BTX (green) and PDZRN3 (red). Overexpression of PDZRN3A (A) or PDZRN3B (B) attenuates agrin-induced clustering of AChRs in cultured myotubes. Arrows indicate transfected myotubes and arrowheads indicate untransfected myotubes. (C and D) Myotubes were transfected with PDZRN3 constructs lacking the RING domain (ΔRING). After agrin stimulation, myotubes were stained with α-BTX (green) and PDZRN3 (red). Deletion of the RING domain abolishes the effect of PDZRN3 on agrin-induced AChR clustering. (E and F) Myotubes were transfected with MuSK and PDZRN3 constructs. After agrin stimulation, myotubes were stained with α-BTX (green) and PDZRN3 (red). Coexpression of MuSK with PDZRN3 blocks the effect of PDZRN3 on agrin-induced AChR clustering. Bar, 50 μm. (G) Quantification of the number of AChR clusters in cultured myotubes. N (the number of myotubes) is shown inside bars (from three separate experiments). ***, P < 0.001.
Knockdown of PDZRN3 in myotubes leads to increased surface levels of MuSK
To further understand the role of PDZRN3 in regulating MuSK signaling, we performed a loss-of-function study using small interfering RNA (siRNA)–mediated knockdown of endogenous PDZRN3 in cultured myotubes. We designed four siRNAs targeting different regions of the PDZRN3 sequence, tested their ability to knockdown PDZRN3 expression in cotransfected COS-7 cells (unpublished data), and selected an effective one for use in myotubes.
Myotubes are multinucleated cells fused from individual myoblasts. To maximize the number of myoblasts expressing the siRNA constructs, we transfected myoblasts with siRNA constructs containing a Neo cassette in the vector. We then pooled all stably transfected myoblasts using G418 selection. These stably transfected myoblasts were cultured to fuse into myotubes, and they were found to fuse in a fashion indistinguishable from that of wild-type C2C12 cells (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200610060/DC1). Western blot analysis of endogenous PDZRN3 showed that myotubes expressing siRNA constructs have significantly reduced levels of endogenous PDZRN3 compared with myotubes expressing scrambled siRNA constructs (Fig. 6, A and B).
Figure 6.

Knockdown of PDZRN3 by siRNA increases the surface expression of MuSK and enhances the sensitivity of myotubes to agrin stimulation. (A and B) pSuper vector based siRNA constructs for PDZRN3 or scrambled sequence (SCR) were stably transfected in C2C12 myoblasts. PDZRN3 proteins in myotubes fused from these cells are reduced to ∼25% of the controls (mean ± SEM, n = 6). **, P < 0.01. (C and D) Surface levels of MuSK in these stably siRNA transfected C2C12 myotubes were determined by biotinylation assays. Knockdown of PDZRN3 in myotubes significantly increased the surface levels of MuSK (mean ± SEM, n = 6). **, P < 0.01. (E and F) Myotubes were treated with various concentrations of agrin and stained with α-BTX for AChR clusters. The number of agrin-induced AChR clusters at the lowest concentration of agrin (0.01 nM) is significantly increased in myotubes fused from C2C12 cells stably transfected with the PDZRN3 RNAi construct. In E, triangles (Δ) represent siRNA-PDZRN3 and squares (□) represent siRNA-scrambled (SCR). Data are shown as mean ± SEM, n = 20 from three separate experiments. *, P = 0.01. Images in F show representative views of the myotubes stimulated with 0.01 nM agrin. Bar (F), 50 μm. (G) Knockdown of PDZRN3 in cultured myotubes diminishes agrin-induced ubiquitination of MuSK. Lysates were prepared from agrin-treated (1 nM for 2 h) and nontreated C2C12 myotubes expressing siRNA for PDZRN3 (siRNA) or a scrambled sequence (SCR). MuSK was immunoprecipitated with an anti-MuSK antibody and ubiquitination of MuSK was detected with an anti-ubiquitin antibody. As in the wild-type myotubes, ubiquitination of MuSK is increased upon agrin stimulation in myotubes expressing a scrambled sequence. This agrin-induced increase of MuSK ubiquitination is diminished in myotubes expressing siRNA against PDZRN3.
To determine the effect of knockdown of PDZRN3 on MuSK signaling, we first measured the surface level of MuSK using biotinylation assays. As shown in Fig. 6 (C and D), the surface level of MuSK in myotubes expressing the siRNA construct is significantly increased, indicating that endogenous PDZRN3 plays an important role in regulating the surface level of MuSK on myotubes. We next tested whether the increased surface level of MuSK in PDZRN3 knockdown myotubes also leads to an enhanced response to agrin stimulation. When stimulated with a saturating concentration of agrin (1 nM), myotubes expressing the PDZRN3 siRNA construct and the scrambled siRNA construct showed a similar response as measured by the number of AChR clusters (last data point in Fig. 6 E), suggesting that myotubes stably transfected with siRNA constructs are capable of forming AChR clusters when stimulated with agrin. We then determined the dose–response curve of agrin stimulation. We found that at low concentrations of agrin, myotubes expressing the siRNA construct showed an enhanced response as indicated by the increased number of AChR clusters (Fig. 6, E and F). Furthermore, increased MuSK ubiquitination induced by agrin stimulation was not detected in myotubes expressing the PDZRN3 siRNA construct (Fig. 6 G). Together with the results from the gain-of-function studies, these data suggest a mechanism by which the activity of MuSK is regulated by the synapse-associated E3 ubiquitin ligase PDZRN3 to regulate postsynaptic development.
Overexpression of PDZRN3 in skeletal muscle perturbs the growth and maturation of the NMJ in transgenic mice
To test whether modulation of PDZRN3 levels affect the development of the NMJ in vivo, we generated transgenic mice which overexpress PDZRN3 in skeletal muscle. We used the well-characterized promoter/enhancer elements from the myosin light chain (MLC) 1f/3f gene to drive HA-tagged PDZRN3A expression (Fig. 7 A). MLC 1f/3f promoter/enhancer elements have been shown to drive high levels of transgene expression in skeletal muscle but not in nonmuscle cells (Rosenthal et al., 1989; Feng et al., 2000).
Figure 7.

Overexpression of PDZRN3 in skeletal muscle of transgenic mice perturbs the development of the NMJ. (A) A diagram of the transgene, in which HA-tagged PDZRN3A was flanked by promoter and enhancer sequences from the myosin light chain (MLC) 1f/3f gene to drive muscle specific expression. (B) RT-PCR shows that the HA-PDZRN3A transgene is expressed in skeletal muscle, but not in the spinal cord, brain, heart, kidney, lung, or liver of a P21 transgenic mouse. mRNA of the ribosomal protein rig/S15 was amplified as a control (bottom panel). (C) mRNA from the HA-PDZRN3A transgene is detected by RT-PCR in the skeletal muscle from E15.5 but not E14.5 embryos. (D) Western blot reveals the presence of HA-PDZRN3A protein in skeletal muscle from a transgenic E15.5 embryo. Blot was reprobed for β-tubulin as a loading control. (E) Western blot of muscles from P21 transgenic mice (TG) and wild-type littermates (WT) probed with an anti-HA antibody and an anti-PDZRN3 antibody. (F) Cross sections of muscles from P21 transgenic and wild-type littermates stained with α-BTX and an anti-HA antibody. HA-PDZRN3A is localized at the NMJ of transgenic mice. Bar, 10 μm. (G) Cross-sections of muscles from P21 transgenic and wild-type littermates stained with α-BTX and an anti-MuSK antibody. Four representative NMJs are shown for each genotype. MuSK expression at the NMJ is much lower in transgenic mice than that in wildtype littermates. Bar, 10 μm. (H) Quantification of fluorescence intensity of MuSK staining at the NMJ in transgenic mice and wild-type littermates (mean ± SEM, n = 20), ***, P < 0.001. (I and J) Wholemount staining of gluteus muscles with α-BTX and antibodies against neurofilament (NF) and synaptophysin (SYP). NMJs from P0, P8, and P21 transgenic mice (J) are smaller than those from wild-type littermates (I). NMJs in P8 and P21 transgenic mice are much less complex than those from wild-type littermates. Bars, 10 μm. (K) Quantification of areas of NMJs from transgenic and wildtype littermates (mean ± SEM; n is shown inside the bar [from 2–3 mice]). ***, P < 0.001. (L) The complexity of NMJs is quantified by the number of perforations in each NMJ. NMJs from P21 transgenic mice have fewer perforations than those from wild-type littermates (n is 111 for wild-type and 103 for transgenic mice).
Four transgenic founder lines were established and the line with the highest HA-PDZRN3A expression (named MLC-PDZRN3A) was selected for further study. MLC-PDZRN3A transgenic mice were born at an expected rate and have normal body size up to 1 wk after birth. However, by the age of 3 wk, the transgenic mice were significantly smaller than wild-type littermates, with their bodyweight at 50–75% of the wild-type littermates. No other phenotypical abnormalities were observed and most of the transgenic mice survive to adulthood. Histological examination of muscles showed no sign of muscle degeneration/regeneration (unpublished data).
RT-PCR showed that HA-PDZRN3A transcripts are specifically expressed in the skeletal muscle (Fig. 7 B). The expression of mRNA and protein of HA-PDZRN3A could be detected as early as E15.5 (Fig. 7, C and D). Quantification of proteins from muscles of P21 mice indicates that there is a 1.8-fold increase of PDZRN3 proteins in transgenic mice compared with wild-type littermates (182 ± 6.9% of wildtype controls, n = 3; Fig. 7 E). Immunostaining showed that, like endogenous PDZRN3, HA-PDZRN3A is concentrated at the NMJ as revealed by co-staining with anti-HA antibodies and α-BTX (Fig. 7 F).
To determine the effects of overexpression of PDZRN3A on the development of the NMJ, we first examined MuSK expression at the NMJ in MLC-PDZRN3A transgenic mice by immunostaining with a polyclonal anti-MuSK antibody (Herbst et al., 2002). In muscles of wild-type mice, MuSK is highly concentrated at the NMJ and precisely colocalized with AChRs (Fig. 7 G). In the MLC-PDZRN3A transgenic mice, a similar pattern of MuSK staining was observed. However, the intensity of the immunostaining of MuSK in the MLC-PDZRN3A transgenic mice was only 40% of that in wild-type littermates (Fig. 7, G and H), indicating that overexpression of PDZRN3A leads to reduced MuSK expression at the NMJ.
We next examined the development of the NMJ using wholemount staining of gluteus muscle with anti-synaptophysin/anti-neurofilament antibodies and α-BTX. In wild-type mice, NMJs grow from “plaque-like” at P0 to “donut-like” by the end of the first postnatal week (Fig. 7 I). At 3 wk of age, NMJs in wild-type mice are well developed with complex patterns of multi-bifurcated presynaptic nerve terminals and “pretzel-like” perforated postsynaptic AChR clusters (Fig. 7 I). In the MLC-PDZRN3A transgenic mice, however, NMJs were much smaller in size at all three stages (P0, P8, and P21) examined (Fig. 7, J and K). Morphologically, NMJs in the transgenic mice remained plaque-like at P8. This is even more profound at P21; both presynaptic nerve terminals and postsynaptic AChR clusters showed much less complexity (Fig. 7 J) compared with the wild-type littermates (Fig. 7 I). Postsynaptic AChR clusters of many NMJs were still plaque-like with few perforations, a hallmark of immature NMJs. To measure the complexity of the NMJ quantitatively, we counted the perforations of each NMJ and used this number as a “complexity index”. As shown in Fig. 7 L, NMJs in MLC-PDZRN3A transgenic mice contained many fewer perforations. These data strongly suggest that overexpression of PDZRN3A in muscle severely perturbs the growth and maturation of the NMJ.
Discussion
Although the importance of ubiquitin-dependent pathways in vertebrate synaptic development and plasticity has been well recognized, the specific E3 ubiquitin ligases involved in the ubiquitination of synaptic proteins are largely unknown (Colledge et al., 2003; DiAntonio and Hicke, 2004). In this study, we identified PDZRN3, a synapse-associated RING-type E3 ubiquitin ligase, as an important regulator of synaptic growth and maturation at the NMJ. We show that PDZRN3 is highly expressed in muscle and specifically localized to the postsynaptic site of the NMJ. PDZRN3 directly interacts with MuSK, and this interaction is enhanced by agrin stimulation. Coexpression of PDZRN3 and MuSK in heterologous cells promotes the ubiquitination of MuSK, and leads to enhanced endocytosis and reduced surface expression of MuSK. These effects require the RING domain of PDZRN3, which contains the E3 ubiquitin ligase activity. Overexpression of PDZRN3 leads to attenuated MuSK-dependent AChR clustering in cultured myotubes. Knockdown of endogenous PDZRN3 by RNAi in myotubes results in increased surface levels of MuSK and enhanced AChR clustering in response to agrin stimulation. Furthermore, overexpresion of PDZRN3 in muscle of transgenic mice perturbs the growth and maturation of the NMJ. Together, these data provide strong evidence that PDZRN3 functions as a synapse-associated E3 ubiquitin ligase to regulate the postsynaptic development of the NMJ.
MuSK is essential for all aspects of postsynaptic differentiation of the NMJ (Sanes and Lichtman, 2001; Burden, 2002; Willmann and Fuhrer, 2002). Therefore, modulation of MuSK level/activity might serve as a central point of regulation in the formation and maintenance of the NMJ. In cultured myotubes, agrin-induced MuSK phosphorylation is transient, suggesting the existence of mechanisms rapidly regulating MuSK level and/or phosphorylation (Fuhrer et al., 1997; Mittaud et al., 2004). Transgenic expression of a constitutively active MuSK in mice leads to the formation of extrasynaptic clusters of AChRs (Jones et al., 1999). Furthermore, patients with a missense mutation of MuSK that leads to low levels of MuSK expression have reduced AChR clusters and show signs of myasthenic syndrome (Chevessier et al., 2004). This demonstrates the importance of precise control of MuSK level/activity in the development and function of the NMJ. Although several MuSK-interacting proteins have been identified that may serve as points of regulation (Strochlic et al., 2001, 2004; Luo et al., 2002, 2003; Finn et al., 2003; Bromann et al., 2004; Madhavan et al., 2005; Okada et al., 2006), our study presents a molecular mechanism of direct regulation of MuSK activity through the modulation of its surface levels.
The enrichment of PDZRN3 mRNA in the synaptic region of the muscle and the concentration of PDZRN3 protein at the NMJ are consistent with our findings of its role in regulating MuSK signaling. However, a recent study also showed that PDZRN3 is expressed in cultured myoblasts and that its expression is up-regulated during the differentiation of C2C12 myoblasts into myotubes (Ko et al., 2006). Knockdown of PDZRN3 in cultured C2C12 cells inhibits the formation of myotubes, though overexpression of PDZRN3 has no effect on the differentiation of myoblasts into myotubes (Ko et al., 2006). This is in contrast with results of our RNAi experiments, which show no effects on the formation of myotubes. In our study, C2C12 cells expressing the RNAi construct for PDZRN3 grow and fuse in a fashion indistinguishable from wild-type C2C12 myotubes. Furthermore, these myotubes have a similar response to high concentrations of agrin stimulation as control myotubes. Currently, the reason for this discrepancy is not clear. One obvious difference is the method of expressing RNAi constructs in C2C12 cells. Ko et al. (2006) used a transient expression approach by transfecting the same batch of C2C12 cells for three consecutive times at 24-h intervals to reach high transfection efficiency. We selected stably transfected C2C12 cells to enrich cells expressing the RNAi construct. In general, the expression level of exogenous gene is lower in stably transfected cells than in transiently transfected cells due to lower copy numbers. We saw ∼75% reduction of endogenous PDZRN3 proteins in myotubes expressing the RNAi construct for PDZRN3. Future gene knockout studies in mice will likely resolve this difference.
In mice, NMJs start to form around E14.5 (Lin et al., 2001). We have shown that PDZRN3 protein is first detected at the NMJ around E16.5, suggesting that the role of PDZRN3 at the NMJ is unlikely to be in the initial formation of the NMJ, but rather in the growth and/or maturation of the NMJ. This role is further supported by our study of transgenic mice. Overexpression of PDZRN3 in skeletal muscle leads to reduced MuSK expression at the NMJ and smaller and less mature NMJs. Because PDZRN3 contains multiple protein–protein interaction domains, we cannot exclude the possibility that overexpression of PDZRN3 may affect additional signaling pathways in muscles. However, our results are consistent with the proposed role of synaptic E3 ubiquitin ligases in regulating homeostatic growth of synapses in Drosophila and C. elegans, in which mutations of E3 ubiquitin ligases lead to aberrant growth of synapses (Schaefer et al., 2000; Wan et al., 2000; Zhen et al., 2000; DiAntonio et al., 2001).
Although ubiquitination-mediated protein degradation through the 26S proteasome is the best characterized function of the ubiquitin system, increasing evidence suggests that ubiquitination-mediated endocytosis and lysosomal degradation play an important role in regulating cell surface abundance of membrane proteins (Hicke, 2001). Initial studies from yeast indicate that monoubiquitination of membrane proteins is sufficient for triggering endocytosis (Hicke, 2001). However, recent studies suggest that polyubiquitination, rather than monoubiquitination, is required as a signal for endocytosis in mammalian cells (Barriere et al., 2006; Hawryluk et al., 2006). Our data strongly suggest that PDZRN3 functions as an E3 ubiquitin ligase to regulate surface levels of MuSK through ubiquitination-dependent endocytosis. Ubiquitination-mediated endocytosis has been shown to be a key mechanism of down-regulating receptor tyrosine kinase activity in many cellular processes (Marmor and Yarden, 2004). Recently, a putative Ariadne-like ubiquitin ligase (PAUL) has been found to be present at the NMJ and to interact with MuSK (Bromann et al., 2004). Together, these synaptically localized E3 ubiquitin ligases may play an important role in postsynaptic development at the NMJ through the regulation of surface levels of MuSK.
Materials and methods
Antibodies and constructs
Rabbit anti-PDZRN3 antibody (PDZRN3-10B) and guinea pig anti-PDZRN3 antibody (PDZRN3-3B) were raised against hexahistidine tagged amino acids 83–134 and 468–628 of PDZRN3A, respectively. Anti-MuSK C-19 (Santa Cruz Biotechnology, Inc.), anti-MuSK N-19 (Santa Cruz Biotechnology, Inc.), anti-MuSK (Abcam), anti-MuSK #83033 (a gift from Dr. Steve Burden, New York University, New York, NY), anti-ubiquitin (Sigma-Aldrich), anti-β-tubulin (Sigma-Aldrich), anti-FLAG M2 (Sigma-Aldrich), anti-β-actin (Sigma-Aldrich), anti-transferrin receptor (Zymed Laboratories), anti-GAPDH (Zymed Laboratories), and anti-synaptophysin antibodies (Synaptic Systems GmbH) were used for immunoprecipitation and Western blot. AlexaFluor 488–, rhodamine-, and AlexaFluor 647–conjugated α-bungarotoxin (Invitrogen) were used for staining of AChRs.
PDZRN3AΔRING, PDZRN3AΔRZ, and PDZRN3AΔPDZ were generated by deleting amino acids 1–51, 1–157, and 246–338 of PDZRN3A, respectively. Kinase-dead MuSK was generated by replacing Lys608 with Ala, and kinase-active MuSK was generated by deleting amino acids 99–492 from the ectodomain of MuSK (Zhou et al., 1999).
Coimmunoprecipitation
C2C12 myotubes or transfected COS-7 cells were harvested and lysed in lysis buffer containing PBS, 1% Triton X-100, and Complete protease inhibitors (Roche). Immunoprecipitation was performed by incubating samples with appropriate antibodies and 25 μl of protein A agarose beads (Invitrogen) for 2–4 h. Proteins were eluted by the addition of loading buffer and analyzed by Western blot.
Immunohistochemistry
Tibialis anterior muscle was denervated, sectioned, and stained as described previously (Young et al., 2005). Fluorescent images were taken using an Axioskop2 Plus microscope (Carl Zeiss MicroImaging, Inc.) fitted with an AxioCam CCD camera (Carl Zeiss MicroImaging, Inc.) through a 20× objective (NA = 0.75) with acquisition software AxioVision 3.1 (Carl Zeiss MicroImaging, Inc.). Images were then adjusted with Adobe Photoshop 6.0 (cropping and brightness/contrast adjustments). Final figures were mounted and labeled using Adobe Illustrator 9.0.
C2C12 myoblast transfection
C2C12 myoblasts were plated in 6-well plates and transfected with a total of 2 μg DNA and 3 μl FuGENE 6 (Roche) per well. 36–48 h after transfection, myoblasts were switched to fusion medium (DME with 2% horse serum) to allow myoblasts to fuse into myotubes for 4–5 d. After stimulation with 1 nM agrin (R&D Systems) for 12–16 h, myotubes were fixed and stained with appropriate antibodies.
Biotinylation and endocytosis assay
C2C12 myotubes or transfected COS-7 cells were washed with PBS and surface biotinylated as described previously (Ehlers, 2000). For the endocytosis assay, two sets of identically transfected COS-7 cells were prepared and surface biotinylated. One set of samples was moved to 37°C for 10 min to allow endocytosis to occur. The remaining surface biotin was then cleaved with glutathione cleavage buffer (50 mM glutathione, 75 mM NaCl, 10 mM EDTA, 1% BSA, and 0.075 N NaOH). The other set was kept on ice without subsequent cleavage and used to determine the initial surface levels of MuSK. The relative endocytosis of MuSK from the cell surface was then calculated by determining the ratio of biotinylated MuSK after cleavage to the initial surface-biotinylated MuSK.
Ubiquitination assays
The in vitro ubiquitination assay was performed by incubating GST-FLAG-RING (GST and FLAG tagged RING domain of PDZRN3A) with 5 nM yeast E1, 100 nM of the indicated E2, and ubiquitin (Boston Biochem) in ubiquitination buffer (50 mM Tris-HCl, pH 7.5, 4 mM MgCl2, 1 mM DTT, 1 mM ATP, 10 mM creatine phosphate, and 16 IU/ml creatine phosphokinase) to a final volume of 40 μl. Samples were incubated at 30°C for 2 h and subsequently analyzed by Western blot. To detect ubiquitination of MuSK, transfected COS-7 cells or C2C12 myotubes stimulated with 10 nM agrin for 2 h were harvested in lysis buffer containing 2% SDS. After incubating at 4°C for 1 h, samples were diluted 1:20 using lysis buffer without SDS, and MuSK was immunoprecipitated and analyzed by Western blot with an anti-ubiquitin antibody (Sigma-Aldrich).
GST pull-down assay
GST or GST-UbcH5B proteins were immobilized on glutathione-sepharose beads (Novagen). Whole cell lysate from PDZRN3A transfected COS-7 cells was added and incubated with these beads at 4°C for 1–2 h. Proteins were then eluted and analyzed by Western blot.
RNA interference
The full-length mouse PDZRN3A sequence was copied into the siRNA design center of DHARMACON (http://www.dharmacon.com/sidesign/default.aspx?source=0) and four oligo sequences against PDZRN3A were generated. These oligo sequences were inserted into pSuper vector and cotransfected with PDZRN3A into COS-7 cells. Western blot analysis showed that one oligo (sequence 5′-GTCGGTGACTACTGTATAA-3′) had the most dramatic effect in knocking down expression of PDZRN3A. C2C12 stable cell lines expressing either this oligo or another oligo with a scrambled sequence were then generated by transfection of C2C12 cells with subsequent G418 selection.
Transgenic mice
The MLC-PDZRN3A transgene was constructed by inserting HA-tagged mouse PDZRN3A cDNA in the vector containing the promoter and enhancer elements from the rat MLC 1f/3f gene (Feng et al., 2000). Transgenic mice were generated by injection of DNA into the fertilized oocyte using standard pronuclear injection techniques. Transgenic founders were subsequently backcrossed to C57BL6/J mice for two to four generations before analysis. Genotypes were determined by PCR, and wild-type littermates of transgenic mice were used as controls.
Online supplemental material
Fig. S1 A shows weak staining of extrasynaptic muscle membrane by anti-PDZRN3 antibody; B and C shows that MuSK staining persists after denervation. Fig. S2 shows how regulation of surface levels of MuSK by PDZRN3 is specific. Fig. S3 describes how down-regulation of surface levels of MuSK by PDZRN3 is blocked by the lysosome inhibitor chloroquine, but not the proteasome inhibitor MG-132. Fig. S4 shows C2C12 cells stably transfected with PDZRN3 siRNA fuse in a fashion indistinguishable from controls. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200610060/DC1.
Supplementary Material
Acknowledgments
We are grateful to Dr. Joshua Sanes for MuSK constructs and to Dr. Steve Burden for the anti-MuSK antibody. We thank Dr. Fan Wang and Dr. Wolfgang Liedtke for comments that improved the manuscript. We are grateful to the members of the Feng lab for support and critical reading of the manuscript, with special thanks to Bridget Kelly and Molly Heyer.
This work was supported by an NIH grant, an Alfred P. Sloan Research Fellowship, the Klingenstein Fellowship in the Neurosciences, and a Beckman Young Investigator Award to G. Feng.
Abbreviations used in this paper: AChR, nicotinic acetylcholine receptor; MLC, myosin light chain; MuSK, muscle-specific receptor tyrosine kinase; NMJ, neuromuscular junction; PDZRN3, PDZ domain containing RING finger 3; siRNA, small interfering RNA.
References
- Barriere, H., C. Nemes, D. Lechardeur, M. Khan-Mohammad, K. Fruh, and G.L. Lukacs. 2006. Molecular basis of oligoubiquitin-dependent internalization of membrane proteins in Mammalian cells. Traffic. 7:282–297. [DOI] [PubMed] [Google Scholar]
- Bezakova, G., and M.A. Ruegg. 2003. New insights into the roles of agrin. Nat. Rev. Mol. Cell Biol. 4:295–308. [DOI] [PubMed] [Google Scholar]
- Bowe, M.A., and J.R. Fallon. 1995. The role of agrin in synapse formation. Annu. Rev. Neurosci. 18:443–462. [DOI] [PubMed] [Google Scholar]
- Bowen, D.C., J.S. Park, S. Bodine, J.L. Stark, D.M. Valenzuela, T.N. Stitt, G.D. Yancopoulos, R.M. Lindsay, D.J. Glass, and P.S. DiStefano. 1998. Localization and regulation of MuSK at the neuromuscular junction. Dev. Biol. 199:309–319. [DOI] [PubMed] [Google Scholar]
- Bromann, P.A., J.A. Weiner, E.D. Apel, R.M. Lewis, and J.R. Sanes. 2004. A putative ariadne-like E3 ubiquitin ligase (PAUL) that interacts with the muscle-specific kinase (MuSK). Gene Expr. Patterns. 4:77–84. [DOI] [PubMed] [Google Scholar]
- Burden, S.J. 2002. Building the vertebrate neuromuscular synapse. J. Neurobiol. 53:501–511. [DOI] [PubMed] [Google Scholar]
- Burgess, R.W., Q.T. Nguyen, Y.-J. Son, J.W. Lichtman, and J.R. Sanes. 1999. Alternatively spliced isoforms of nerve- and muscle-derived agrin: their roles at the neuromuscular junction. Neuron. 23:33–44. [DOI] [PubMed] [Google Scholar]
- Chapman, A.P., S.C. Courtney, S.J. Smith, C.C. Rider, and P.W. Beesley. 1992. Ubiquitin immunoreactivity of multiple polypeptides in rat brain synaptic membranes. Biochem. Soc. Trans. 20:155S. [DOI] [PubMed] [Google Scholar]
- Chevessier, F., B. Faraut, A. Ravel-Chapuis, P. Richard, K. Gaudon, S. Bauche, C. Prioleau, R. Herbst, E. Goillot, C. Ioos, et al. 2004. MUSK, a new target for mutations causing congenital myasthenic syndrome. Hum. Mol. Genet. 13:3229–3240. [DOI] [PubMed] [Google Scholar]
- Colledge, M., and S.C. Froehner. 1998. Signals mediating ion channel clustering at the neuromuscular junction. Curr. Opin. Neurobiol. 8:357–363. [DOI] [PubMed] [Google Scholar]
- Colledge, M., E.M. Snyder, R.A. Crozier, J.A. Soderling, Y. Jin, L.K. Langeberg, H. Lu, M.F. Bear, and J.D. Scott. 2003. Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron. 40:595–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeChiara, T.M., D.C. Bowen, D.M. Valenzuela, M.V. Simmons, W.T. Poueymirou, S. Thomas, E. Kinetz, D.L. Compton, E. Rojas, J.S. Park, et al. 1996. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 85:501–512. [DOI] [PubMed] [Google Scholar]
- DiAntonio, A., and L. Hicke. 2004. Ubiquitin-dependent regulation of the synapse. Annu. Rev. Neurosci. 27:223–246. [DOI] [PubMed] [Google Scholar]
- DiAntonio, A., A.P. Haghighi, S.L. Portman, J.D. Lee, A.M. Amaranto, and C.S. Goodman. 2001. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature. 412:449–452. [DOI] [PubMed] [Google Scholar]
- Ehlers, M.D. 2000. Reinsertion or degradation of AMPA receptors determined by activity dependent endocytic sorting. Neuron. 28:511–525. [DOI] [PubMed] [Google Scholar]
- Feng, G., M.B. Laskowski, D.A. Feldheim, H. Wang, R. Lewis, J. Frisen, J.G. Flanagan, and J.R. Sanes. 2000. Roles for ephrins in positionally selective synaptogenesis between motor neurons and muscle fibers. Neuron. 25:295–306. [DOI] [PubMed] [Google Scholar]
- Finn, A.J., G. Feng, and A.M. Pendergast. 2003. Postsynaptic requirement for Abl kinases in assembly of the neuromuscular junction. Nat. Neurosci. 6:717–723. [DOI] [PubMed] [Google Scholar]
- Fuhrer, C., J.E. Sugiyama, R.G. Taylor, and Z.W. Hall. 1997. Association of muscle-specific kinase MuSK with the acetylcholine receptor in mammalian muscle. EMBO J. 16:4951–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam, M., P.G. Noakes, L. Moscoso, F. Rupp, R.H. Scheller, J.P. Merlie, and J.R. Sanes. 1996. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 85:525–535. [DOI] [PubMed] [Google Scholar]
- Hawryluk, M.J., P.A. Keyel, S.K. Mishra, S.C. Watkins, J.E. Heuser, and L.M. Traub. 2006. Epsin 1 is a polyubiquitin-selective clathrin-associated sorting protein. Traffic. 7:262–281. [DOI] [PubMed] [Google Scholar]
- Herbst, R., E. Avetisova, and S.J. Burden. 2002. Restoration of synapse formation in Musk mutant mice expressing a Musk/Trk chimeric receptor. Development. 129:5449–5460. [DOI] [PubMed] [Google Scholar]
- Hicke, L. 2001. Ubiquitin and proteasomes: protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2:195–201. [DOI] [PubMed] [Google Scholar]
- Jennings, C.G.B., S.M. Dyer, and S.J. Burden. 1993. Muscle-specific trk-related receptor with a kringle domain defines a distinct class of receptor tyrosine kinases. Proc. Natl. Acad. Sci. USA. 90:2895–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G., C. Moore, S. Hashemolhosseini, and H.R. Brenner. 1999. Constitutively active MuSK is clustered in the absence of agrin and induces ectopic postsynaptic-like membranes in skeletal muscle fibers. J. Neurosci. 19:3376–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo, P., and J.M. Kaplan. 2004. The anaphase-promoting complex regulates the abundance of GLR-1 glutamate receptors in the ventral nerve cord of C. elegans. Curr. Biol. 14:2057–2062. [DOI] [PubMed] [Google Scholar]
- Katoh, M., and M. Katoh. 2004. Identification and characterization of PDZRN3 and PDZRN4 genes in silico. Int. J. Mol. Med. 13:607–613. [PubMed] [Google Scholar]
- Kim, E., and M. Sheng. 2004. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 5:771–781. [DOI] [PubMed] [Google Scholar]
- Ko, J.A., Y. Kimura, K. Matsuura, H. Yamamoto, T. Gondo, and M. Inui. 2006. PDZRN3 (LNX3, SEMCAP3) is required for the differentiation of C2C12 myoblasts into myotubes. J. Cell Sci. 119:5106–5113. [DOI] [PubMed] [Google Scholar]
- Kumar, S., W.H. Kao, and P.M. Howley. 1997. Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J. Biol. Chem. 272:13548–13554. [DOI] [PubMed] [Google Scholar]
- Levkowitz, G., H. Waterman, S.A. Ettenberg, M. Katz, A.Y. Tsygankov, I. Alroy, S. Lavi, K. Iwai, Y. Reiss, A. Ciechanover, et al. 1999. Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol. Cell. 4:1029–1040. [DOI] [PubMed] [Google Scholar]
- Lin, W., R.W. Burgess, B. Dominguez, S.L. Pfaff, J.R. Sanes, and K.F. Lee. 2001. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature. 410:1057–1064. [DOI] [PubMed] [Google Scholar]
- Luo, Z.G., Q. Wang, J.Z. Zhou, J. Wang, Z. Luo, M. Liu, X. He, A. Wynshaw-Boris, W.C. Xiong, B. Lu, and L. Mei. 2002. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron. 35:489–505. [DOI] [PubMed] [Google Scholar]
- Luo, Z.G., H.S. Je, Q. Wang, F. Yang, G.C. Dobbins, Z.H. Yang, W.C. Xiong, B. Lu, and L. Mei. 2003. Implication of geranylgeranyltransferase I in synapse formation. Neuron. 40:703–717. [DOI] [PubMed] [Google Scholar]
- Marmor, M.D., and Y. Yarden. 2004. Role of protein ubiquitylation in regulating endocytosis of receptor tyrosine kinases. Oncogene. 23:2057–2070. [DOI] [PubMed] [Google Scholar]
- Madhavan, R., X.T. Zhao, M.A. Ruegg, and H.B. Peng. 2005. Tyrosine phosphatase regulation of MuSK-dependent acetylcholine receptor clustering. Mol. Cell. Neurosci. 28:403–416. [DOI] [PubMed] [Google Scholar]
- Mittaud, P., A.A. Camilleri, R. Willmann, S. Erb-Vogtli, S.J. Burden, and C. Fuhrer. 2004. A single pulse of agrin triggers a pathway that acts to cluster acetylcholine receptors. Mol. Cell. Biol. 24:7841–7854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake, S., K.P. Mullane-Robinson, N.P. Lill, P. Douillard, and H. Band. 1999. Cbl-mediated negative regulation of platelet-derived growth factor receptor-dependent cell proliferation. J. Biol. Chem. 274:16619–16628. [DOI] [PubMed] [Google Scholar]
- Nitkin, R.M., M.A. Smith, C. Magill, J.R. Fallon, Y.M. Yao, B.G. Wallace, and U.J. McMahan. 1987. Identification of agrin, a synaptic organizing protein from Torpedo electric organ. J. Cell Biol. 105:2471–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noakes, P.G., W.D. Phillips, T.A. Hanley, J.R. Sanes, and J.P. Merlie. 1993. 43K protein and acetylcholine receptors colocalize during the initial stages of neuromuscular synapse formation in vivo. Dev. Biol. 155:275–280. [DOI] [PubMed] [Google Scholar]
- Okada, K., A. Inoue, M. Okada, Y. Murata, S. Kakuta, T. Jigami, S. Kubo, H. Shiraishi, K. Eguchi, M. Motomura, et al. 2006. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 312:1802–1805. [DOI] [PubMed] [Google Scholar]
- Patrick, G.N., B. Bingol, H.A. Weld, and E.M. Schuman. 2003. Ubiquitin-mediated proteasome activity is required for agonist-induced endocytosis of GluRs. Curr. Biol. 13:2073–2081. [DOI] [PubMed] [Google Scholar]
- Pun, S., Y.P. Ng, J.F. Yang, N.Y. Ip, and K.W. Tsim. 1997. Agrin-deficient myotube retains its acetylcholine receptor aggregation ability when challenged with agrin. J. Neurochem. 69:2555–2563. [DOI] [PubMed] [Google Scholar]
- Rocca, A., C. Lamaze, A. Subtil, and A. Dautry-Varsat. 2001. Involvement of the ubiquitin/proteasome system in sorting of the interleukin 2 receptor beta chain to late endocytic compartments. Mol. Biol. Cell 12:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal, N., J.M. Kornhauser, M. Donoghue, K.M. Rosen, and J.P. Merlie. 1989. Myosin light chain enhancer activates muscle-specific, developmentally regulated gene expression in transgenic mice. Proc. Natl. Acad. Sci. USA. 86:7780–7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes, J.R., and J.W. Lichtman. 2001. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat. Rev. Neurosci. 2:791–805. [DOI] [PubMed] [Google Scholar]
- Schaefer, A.M., G.D. Hadwiger, and M.L. Nonet. 2000. rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron. 26:345–356. [DOI] [PubMed] [Google Scholar]
- Serdaroglu, P., V. Askanas, and W.K. Engel. 1992. Immunocytochemical localization of ubiquitin at human neuromuscular junctions. Neuropathol. Appl. Neurobiol. 18:232–236. [DOI] [PubMed] [Google Scholar]
- Song, Y., J.A. Panzer, R.M. Wyatt, and R.J. Balice-Gordon. 2006. Formation and plasticity of neuromuscular synaptic connections. Int. Anesthesiol. Clin. 44:145–178. [DOI] [PubMed] [Google Scholar]
- Strochlic, L., A. Cartaud, V. Labas, W. Hoch, J. Rossier, and J. Cartaud. 2001. MAGI-1c: a synaptic MAGUK interacting with MuSK at the vertebrate neuromuscular junction. J. Cell Biol. 153:1127–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strochlic, L., A. Cartaud, A. Mejat, R. Grailhe, L. Schaeffer, J.P. Changeux, and J. Cartaud. 2004. 14-3-3 gamma associates with muscle specific kinase and regulates synaptic gene transcription at vertebrate neuromuscular synapse. Proc. Natl. Acad. Sci. USA. 101:18189–18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tietze, C., P. Schlesinger, and P. Stahl. 1980. Chloroquine and ammonium ion inhibit receptor-mediated endocytosis of mannose-glycoconjugates by macrophages: apparent inhibition of receptor recycling. Biochem. Biophys. Res. Commun. 93:1–8. [DOI] [PubMed] [Google Scholar]
- Torres, R., B.L. Firestein, H. Dong, J. Staudinger, E.N. Olson, R.L. Huganir, D.S. Bredt, N.W. Gale, and G.D. Yancopoulos. 1998. PDZ proteins bind, cluster, and synaptically colocalize with Eph receptors and their ephrin ligands. Neuron. 21:1453–1463. [DOI] [PubMed] [Google Scholar]
- Valenzuela, D.M., T.N. Stitt, P.S. DiStefano, E. Rojas, K. Mattsson, D.L. Compton, L. Nunez, J.S. Park, J.L. Stark, D.R. Gies, et al. 1995. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 15:573–584. [DOI] [PubMed] [Google Scholar]
- van Roessel, P., D.A. Elliott, I.M. Robinson, A. Prokop, and A.H. Brand. 2004. Independent regulation of synaptic size and activity by the anaphase-promoting complex. Cell. 119:707–718. [DOI] [PubMed] [Google Scholar]
- Vecchione, A., A. Marchese, P. Henry, D. Rotin, and A. Morrione, 2003. The Grb10/Nedd4 complex regulates ligand-induced ubiquitination and stability of the insulin-like growth factor I receptor. Mol. Cell. Biol. 23:3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, H.I., A. DiAntonio, R.D. Fetter, K. Bergstrom, R. Strauss, and C.S. Goodman. 2000. Highwire regulates synaptic growth in Drosophila. Neuron. 26:313–329. [DOI] [PubMed] [Google Scholar]
- Willmann, R., and C. Fuhrer. 2002. Neuromuscular synaptogenesis: clustering of acetylcholine receptors revisited. Cell. Mol. Life Sci. 59:1296–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X., S. Arber, C. William, L. Li, Y. Tanabe, T.M. Jessell, C. Birchmeier, and S.J. Burden. 2001. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron. 30:399–410. [DOI] [PubMed] [Google Scholar]
- Young, P., J. Nie, X. Wang, C.J. McGlade, M.M. Rich, and G. Feng. 2005. Interaction of ErbB2 with LNX1 E3 ubiquitin ligase: potential role in the development of perisynaptic Schwann cells at the neuromuscular junction. Mol. Cell. Neurosci. 30:238–248. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., A.N. Hegde, and K.C. Martin. 2003. The ubiquitin proteasome system functions as an inhibitory constraint on synaptic strengthening. Curr. Biol. 13:887–898. [DOI] [PubMed] [Google Scholar]
- Zhen, M., X. Huang, B. Bamber, and Y. Jin. 2000. Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron. 26:331–343. [DOI] [PubMed] [Google Scholar]
- Zhou, H., D.J. Glass, G.D. Yancopoulos, and J.R. Sanes. 1999. Distinct domains of MuSK mediate its abilities to induce and to associate with postsynaptic specializations. J. Cell Biol. 146:1133–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}