

Abstract
L-selectin requires a threshold shear to enable leukocytes to tether to and roll on vascular surfaces. Transport mechanisms govern flow-enhanced tethering, whereas force governs flow-enhanced rolling by prolonging the lifetimes of L-selectin–ligand complexes (catch bonds). Using selectin crystal structures, molecular dynamics simulations, site-directed mutagenesis, single-molecule force and kinetics experiments, Monte Carlo modeling, and flow chamber adhesion studies, we show that eliminating a hydrogen bond to increase the flexibility of an interdomain hinge in L-selectin reduced the shear threshold for adhesion via two mechanisms. One affects the on-rate by increasing tethering through greater rotational diffusion. The other affects the off-rate by strengthening rolling through augmented catch bonds with longer lifetimes at smaller forces. By forcing open the hinge angle, ligand may slide across its interface with L-selectin to promote rebinding, thereby providing a mechanism for catch bonds. Thus, allosteric changes remote from the ligand-binding interface regulate both bond formation and dissociation.
Introduction
Cell–cell interactions of selectins with glycosylated ligands mediate leukocyte tethering to and rolling on vascular surfaces during inflammation and immune responses (McEver, 2002). L-selectin is expressed on leukocytes, whereas P- and E-selectin are expressed on activated platelets and/or endothelial cells. The hydrodynamic environment of the circulation imposes kinetic and mechanical constraints on selectin–ligand interactions. For a flowing cell to tether, a selectin must encounter its ligand and interact with it before flow again separates the molecules. For a cell to roll, new interactions must form at the leading edge to replace those that dissociate at the trailing edge. Force applied to these interactions affects their dissociation rates and, hence, their lifetimes.
L-selectin requires a counterintuitive threshold shear to mediate both tethering and rolling (Finger et al., 1996; Alon et al., 1997). As flow increases to an optimal level, more cells tether and the cells roll more slowly. Above the flow optimum, fewer cells tether, and the cells roll more rapidly. Distinct physical mechanisms regulate flow-enhanced tethering and rolling. Transport augments tethering through the following three mechanisms: sliding of the cell bottom on the surface, Brownian motions of the cell, and rotational diffusion of L-selectin and its ligand (Yago et al., 2006). As flow increases, these mechanisms increase the collision frequency between L-selectin and its ligands, which favors productive interactions because the intrinsic docking rate is very rapid. Above the flow optimum, the tethering rate declines as the encounter times become shorter than the time scale for docking, and thus become limiting. It is not known whether, and if so how, changes in the structure of a selectin might affect its molecular diffusivity. Force augments rolling by decreasing the dissociation of L-selectin from its ligands. Normally, forces shorten the lifetimes of receptor–ligand interactions (slip bonds; Bell, 1978). However, at low levels force paradoxically prolongs the lifetimes of selectin–ligand interactions (catch bonds) before they convert to slip bonds at higher forces (Marshall et al., 2003; Sarangapani et al., 2004). Catch bonds are particularly evident for L-selectin (Sarangapani et al., 2004). As flow increases from the threshold to an optimal value, rolling becomes slower and more regular as force lengthens the lifetimes of L-selectin catch bonds (Yago et al., 2004). Above the flow optimum, rolling becomes faster and less regular as higher forces shorten the lifetimes of slip bonds. Several models explaining transitions from catch to slip bonds have been proposed (Zhu et al., 2005). Studies of bacterial variants support a model in which force applied to a linker region in the adhesin FimH allosterically regulates ligand binding (Thomas et al., 2002), perhaps by generating catch bonds (Thomas et al., 2006). However, the structural basis for catch bonds remains poorly understood.
Each selectin has an N-terminal C-type lectin domain, followed by an EGF-like module, a series of short consensus repeats, a transmembrane domain, and a cytoplasmic tail (McEver, 2002). Crystal structures of the lectin and EGF domains of P- and E-selectin have been published (Graves et al., 1994; Somers et al., 2000). The ligand-binding region is a broad shallow surface at the top of the lectin domain opposite to where the EGF domain is attached (Fig. 1 A; Somers et al., 2000). This region includes a Ca2+-coordination site that is shared with the fucose in sialyl Lewis x (sLex; NeuAcα2-3Galβ1-4[Fucα1-3]GlcNAcβ1-R), which is a capping structure on glycans of selectin ligands. The lectin domain forms other contacts with sialic acid and galactose, as well as with the sulfated components of some glycoproteins. P- and L-selectin bind to the N-terminal region of the leukocyte mucin P-selectin glycoprotein ligand-1 (PSGL-1) through cooperative interactions, with sLex capping a core 2 O-glycan, and with adjacent sulfated tyrosines and other amino acids (Leppänen et al., 2000, 2003; Somers et al., 2000). L-selectin also binds to the peripheral node addressin, which is a group of mucins on high endothelial venules of lymph nodes. The major binding determinant on the O-glycans of these mucins is 6-sulfo- sLex, a form of sLex with a sulfate ester attached to the C-6 position of GlcNAc (McEver, 2005).
Figure 1.

Selectin conformational changes regulated by an interdomain hinge. (A) X-ray structures of the lectin and EGF domains of P-selectin with a closed (left; PDB 1G1Q) and open angle (right; PDB 1G1S; Somers et al., 2000). The golden sphere at the top of the lectin domain represents a Ca2+ ion that is coordinated as part of the ligand-binding site. (B) X-ray (left) and MD-simulated (right) structures of the lectin and EGF domains of L-selectin with a closed (left) and open angle (right). The respective open- and closed-angle structures of P- and L-selectin align well. The boxed areas (left) highlight the putative hinge regions that are magnified in the insets. A hydrogen bond (dotted line) connects Tyr37 with Asn138, but not with Gly138. (C) RMSD between the corresponding backbone atoms from residues 121–156 (the EGF domain) of the simulated L-selectin structure and the crystal structures of closed-angle L-selectin (blue curve) or open-angle P-selectin (red curve) as a function of simulation time. The lectin domains were aligned by minimizing the RMSD between the backbone atoms from residues 1–120.
There are only a few noncovalent interactions between the lectin and EGF domains, most of which are conserved among the three selectins. Two P-selectin structures with different angles between the lectin and EGF domains have been described (Fig. 1 A; Somers et al., 2000). The structures suggest that the P-selectin lectin domain can pivot on a hinge over the EGF domain. We postulated that this conformational change is common to all three selectins and plays an important role in regulating the kinetic on/off-rate of selectin–ligand interactions. We show that eliminating a hydrogen bond to increase the flexibility of the interdomain hinge in L-selectin reduced the shear threshold for adhesion by increasing tethering through greater rotational diffusion and by strengthening rolling through augmented catch bonds with longer lifetimes at smaller forces. Thus, allosteric changes remote from the ligand-binding interface regulate both bond formation and dissociation.
Results
The interdomain hinge of L-selectin has dual conformations in dynamic equilibrium
At the putative hinge region of P-selectin, Tyr37 of the lectin domain is located close to Gly138 of the EGF domain (Fig. 1 A). As previously proposed (Somers et al., 2000), the lack of a side chain in Gly138 should favor flexibility between the domains. In contrast, E- and L-selectin have an Asn at residue 138, and published structures of E-selectin reveal a hydrogen bond between Tyr37 and the side chain of Asn138 in a closed-angle conformation (Graves et al., 1994; Somers et al., 2000). We have solved a crystal structure of the lectin and EGF domains of L-selectin (unpublished data), which also has a hydrogen bond between Tyr37 and Asn138 in a closed-angle conformation (Fig. 1 B, left). We used molecular dynamics (MD) simulations to examine whether L-selectin can assume an open-angle conformation (Videos 1 and 2, available at http://www.jcb.org/cgi/content/full/jcb.200606056/DC1). The closed-angle structure of L-selectin aligned well with the closed-angle structure of P-selectin (Somers et al., 2000), with only a 1.4-Å root mean square distance (RMSD) between the corresponding backbone atoms from residues 1–156. Five of seven L-selectin free-dynamics simulations (each for 6 ns) exhibited only small conformational fluctuations around the crystal structure, suggesting that the closed-angle conformation is stable (Video 1). However, transitions between the closed-angle and open-angle conformations were observed in two simulations (Video 2). The dynamics of conformational transitions were quantified by the time courses of RMSD between the corresponding backbone atoms of the simulated L-selectin structure and the closed-angle L-selectin crystal structure or the open-angle P-selectin crystal structure (Fig. 1 C). A RMSD value of ∼2 Å indicated good alignment, and a RMSD value of ∼15 Å indicated poor alignment. After 2.5 ns of free dynamics, the simulated L-selectin structure changed from the original closed-angle conformation to an open-angle conformation (Fig. 1 B, right), which lasted for 1.5 ns and aligned well with the open-angle crystal structure of P-selectin (Fig. 1 C). These results suggest that L-selectin primarily resides in the closed-angle conformation, but occasionally makes spontaneous transitions to the open-angle conformation. MD simulations also indicated that force applied to unbind PSGL-1 from P-selectin promoted transitions from the closed-angle to the open-angle conformation (unpublished data). Thus, the closed-angle and open-angle conformations are in dynamic equilibrium; force can shift this equilibrium to a higher fraction of time in the open-angle conformation and a lower fraction of time in the closed-angle conformation. The force required to set the same conformational equilibrium between the closed and open interdomain angle should be higher for L-selectin than for P-selectin, because it must disrupt the Tyr37–Asn138 hydrogen bond and overcome steric interference in the hinge to enable transition from the closed-angle to open-angle conformation.
Greater hinge flexibility of L-selectin increases tethering by enhanced rotational diffusion
How might the hinge flexibility of a selectin affect its interactions with ligand at an interface several nanometers from the hinge? A flexible hinge will increase the frequency of transitions between the closed- and open-angle conformations. This will facilitate rotational diffusion of the lectin domain, thereby contributing to the on-rate for ligand binding. Substituting Gly for Asn138 in L-selectin (L-selectinN138G) will eliminate the hydrogen bond between Try37 and Asn138. This might increase rotational diffusivity and enhance cellular on-rate. To test this hypothesis, we compared the tether rate, which is a metric of cellular on-rate, of microspheres bearing recombinant L-selectin or L-selectinN138G as they flowed over two distinct immobilized ligands. Microspheres with a 3-μm radius (r) bearing L-selectin or L-selectinN138G at a site density (m
r) of 750 μm−2 in media of 1 cP viscosity (μ) were flowed at different wall shear rates (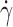 ) through a PSGL-1–coated chamber at a site density (m
l) of 120 μm−2 or coated with 6-sulfo-sLex at a density that supported rolling, but could not be precisely measured with IgM mAb. Tether rates plotted against the product
) through a PSGL-1–coated chamber at a site density (m
l) of 120 μm−2 or coated with 6-sulfo-sLex at a density that supported rolling, but could not be precisely measured with IgM mAb. Tether rates plotted against the product 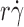 exhibited a biphasic shape characteristic of the shear threshold phenomenon, increasing initially, reaching a maximum, and then decreasing with further increase in
exhibited a biphasic shape characteristic of the shear threshold phenomenon, increasing initially, reaching a maximum, and then decreasing with further increase in 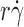 (Fig. 2, A and B). As predicted by our hypothesis, the tether rates were higher for L-selectinN138G than for L-selectin at all
(Fig. 2, A and B). As predicted by our hypothesis, the tether rates were higher for L-selectinN138G than for L-selectin at all 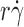 values tested for both ligands, whereas the
values tested for both ligands, whereas the 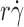 values where tether rates reached maximum were not significantly different.
values where tether rates reached maximum were not significantly different.
Figure 2.

Effects of interdomain hinge flexibility on flow-enhanced tethering. (A and B) Tether rates of microspheres of indicated radii bearing either L-selectin or L-selectinN138G perfused in media of indicated viscosities were plotted against the product 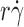 , where r is the microsphere radius and
, where r is the microsphere radius and  is the wall shear rate. The microspheres were coated with 750 molecules μm−2 of either L-selectin or L-selectinN138G, except in one case in B, where they were coated with 1,500 molecules μm−2 of L-selectinN138G (blue squares). The flow chamber floor in A was coated with PSGL-1 at either 120 (red triangles and green diamonds) or 240 (blue squares) molecules μm−2. The flow chamber floor in B was coated with a constant density of 6-sulfo-sLex. The data in A and B represent the mean ± the SD from three experiments. (C) Optimal
is the wall shear rate. The microspheres were coated with 750 molecules μm−2 of either L-selectin or L-selectinN138G, except in one case in B, where they were coated with 1,500 molecules μm−2 of L-selectinN138G (blue squares). The flow chamber floor in A was coated with PSGL-1 at either 120 (red triangles and green diamonds) or 240 (blue squares) molecules μm−2. The flow chamber floor in B was coated with a constant density of 6-sulfo-sLex. The data in A and B represent the mean ± the SD from three experiments. (C) Optimal 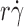 (peak locations of the p
ad/(m
r
m
l
r) vs.
(peak locations of the p
ad/(m
r
m
l
r) vs. 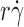 curves) versus microsphere diffusivity k
B
T/(6πμr) data for tethering to PSGL-1 (open circles; Yago et al., 2006) were replotted to provide calibration. The diffusivities of 3-μm radius microspheres bearing L-selectin (red triangle) or L-selectinN138G (green diamond) were plotted for comparison. (D) Maximum p
ad/(m
r
m
l
r) versus molecular diffusivity k
B
T/(6πμl) data for tethering to PSGL-1 (open circles; unpublished data) were replotted to provide calibration, which assumed the characteristic length for molecular diffusion as l = 100 nm. The same l value was used for the L-selectin datum (red triangle), which matched the calibration curve well. The l value for L-selectinN138G is predicted to be smaller. Using the measured maximum p
ad/(m
r
m
l
r) value, the L-selectinN138G datum point (green diamond) was located at the intercept of y = [p
ad/(m
r
m
l
r)] max (green dashed horizontal line) and the extrapolation of the calibration curve (red line). The increased molecular diffusivity for L-selectinN138G could be calculated from the x-axis value of this datum point (green dashed vertical line).
curves) versus microsphere diffusivity k
B
T/(6πμr) data for tethering to PSGL-1 (open circles; Yago et al., 2006) were replotted to provide calibration. The diffusivities of 3-μm radius microspheres bearing L-selectin (red triangle) or L-selectinN138G (green diamond) were plotted for comparison. (D) Maximum p
ad/(m
r
m
l
r) versus molecular diffusivity k
B
T/(6πμl) data for tethering to PSGL-1 (open circles; unpublished data) were replotted to provide calibration, which assumed the characteristic length for molecular diffusion as l = 100 nm. The same l value was used for the L-selectin datum (red triangle), which matched the calibration curve well. The l value for L-selectinN138G is predicted to be smaller. Using the measured maximum p
ad/(m
r
m
l
r) value, the L-selectinN138G datum point (green diamond) was located at the intercept of y = [p
ad/(m
r
m
l
r)] max (green dashed horizontal line) and the extrapolation of the calibration curve (red line). The increased molecular diffusivity for L-selectinN138G could be calculated from the x-axis value of this datum point (green dashed vertical line).
In another study, we demonstrated that tether rate is enhanced by the following three transport mechanisms: sliding of the cell or microsphere bottom on the chamber floor (proportional to 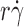 ), cell or microsphere Brownian motions (proportional to k
B
T/(6πμr), where k
B is the Boltzmann constant and T is the absolute temperature), and molecular diffusion (proportional to k
B
T/(6πμl), where l is a characteristic length in the molecular scale; Yago et al., 2006). The tether rate (TR) can be converted to the probability of adhesion per distance traveled by a flowing cell or microsphere, p
ad = −ln (1 − TR)/L ≈ TR/L, where L (= 224 μm) is the length of the microscopic field of view. It can then be normalized by dividing by the product m
r
m
l
r to remove the mass action effects caused by different numbers of interacting molecules in the contact area. For L-selectin–bearing microspheres tethering to PSGL-1, we showed that p
ad/(m
r
m
l
r) is determined by
), cell or microsphere Brownian motions (proportional to k
B
T/(6πμr), where k
B is the Boltzmann constant and T is the absolute temperature), and molecular diffusion (proportional to k
B
T/(6πμl), where l is a characteristic length in the molecular scale; Yago et al., 2006). The tether rate (TR) can be converted to the probability of adhesion per distance traveled by a flowing cell or microsphere, p
ad = −ln (1 − TR)/L ≈ TR/L, where L (= 224 μm) is the length of the microscopic field of view. It can then be normalized by dividing by the product m
r
m
l
r to remove the mass action effects caused by different numbers of interacting molecules in the contact area. For L-selectin–bearing microspheres tethering to PSGL-1, we showed that p
ad/(m
r
m
l
r) is determined by  , k
B
T/(6πμr), and k
B
T/(6πμl). In this study, k
B
T/(6πμr) and k
B
T/(6πμl) are used as respective metrics for microsphere and molecular diffusivity, based on the Stokes-Einstein relationship. In the previous study, we independently varied these values by using microspheres of different radii (r = 1, 2.25, and 3 μm) and media of different viscosities (μ = 1, 1.8, 2.6, and 4.2 cP). We found that p
ad/(m
r
m
l
r) increased with
, k
B
T/(6πμr), and k
B
T/(6πμl). In this study, k
B
T/(6πμr) and k
B
T/(6πμl) are used as respective metrics for microsphere and molecular diffusivity, based on the Stokes-Einstein relationship. In the previous study, we independently varied these values by using microspheres of different radii (r = 1, 2.25, and 3 μm) and media of different viscosities (μ = 1, 1.8, 2.6, and 4.2 cP). We found that p
ad/(m
r
m
l
r) increased with  until it reached maximum, when microsphere Brownian motions became the limiting transport mechanism. Increasing microsphere Brownian motions enabled sliding to further enhance tether rate (reaching maximum at higher
until it reached maximum, when microsphere Brownian motions became the limiting transport mechanism. Increasing microsphere Brownian motions enabled sliding to further enhance tether rate (reaching maximum at higher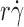 ). The
). The 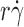 value where the p
ad/(m
r
m
l
r) curve reached maximum (optimal
value where the p
ad/(m
r
m
l
r) curve reached maximum (optimal 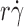 ) increased linearly with k
B
T/(6πμr) (Fig. 2 C). In addition, increasing molecular diffusion increased tether rate at all
) increased linearly with k
B
T/(6πμr) (Fig. 2 C). In addition, increasing molecular diffusion increased tether rate at all 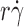 values, just as substitution of L-selectinN138G for L-selectin does (Fig. 2, A and B), and the maximum value of the p
ad/(m
r
m
l
r) versus
values, just as substitution of L-selectinN138G for L-selectin does (Fig. 2, A and B), and the maximum value of the p
ad/(m
r
m
l
r) versus 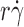 curve increased linearly with k
B
T/(6πμl) (Fig. 2 D). These previously defined linear relationships were used as calibrations to estimate the difference in molecular diffusivities of L-selectin and L-selectinN138G in the present study. As expected, microspheres bearing L-selectin or L-selectinN138G had comparable optimal
curve increased linearly with k
B
T/(6πμl) (Fig. 2 D). These previously defined linear relationships were used as calibrations to estimate the difference in molecular diffusivities of L-selectin and L-selectinN138G in the present study. As expected, microspheres bearing L-selectin or L-selectinN138G had comparable optimal 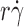 values that were similar to the previous data in the calibration curve because they had the same diffusivities calculated from the same radius (3 μm) and media viscosity (1 cP; Fig. 2 C). The characteristic length for molecular diffusion was taken as l = 100 nm in the data in Fig. 2 D, which serves as an order-of-magnitude estimate. A relative molecular diffusivity value of k
B
T/(6πμl) = 2.18 μm2/s could be calculated from the same l value for L-selectin in this study. Using this value as x-axis coordinate, and the measured maximum p
ad/(m
r
m
l
r) (= 1.56 × 10−9 μm2) as a y-axis coordinate, this L-selectin datum agreed with the previous data measured in media with 1-cP viscosity, regardless of the microsphere radius (Fig. 2 D). We hypothesized that the higher tether rate of microspheres bearing L-selectinN138G rather than L-selectin was caused by increased relative molecular diffusivity for L-selectinN138G. Using the measured value of maximum p
ad/(m
r
m
l
r) (= 3.08 × 10−9 μm2), a relative molecular diffusivity of 4.02 μm2/s for L-selectinN138G was extrapolated from the calibration curve, which was increased by 85% over that of L-selectin (Fig. 2 D).
values that were similar to the previous data in the calibration curve because they had the same diffusivities calculated from the same radius (3 μm) and media viscosity (1 cP; Fig. 2 C). The characteristic length for molecular diffusion was taken as l = 100 nm in the data in Fig. 2 D, which serves as an order-of-magnitude estimate. A relative molecular diffusivity value of k
B
T/(6πμl) = 2.18 μm2/s could be calculated from the same l value for L-selectin in this study. Using this value as x-axis coordinate, and the measured maximum p
ad/(m
r
m
l
r) (= 1.56 × 10−9 μm2) as a y-axis coordinate, this L-selectin datum agreed with the previous data measured in media with 1-cP viscosity, regardless of the microsphere radius (Fig. 2 D). We hypothesized that the higher tether rate of microspheres bearing L-selectinN138G rather than L-selectin was caused by increased relative molecular diffusivity for L-selectinN138G. Using the measured value of maximum p
ad/(m
r
m
l
r) (= 3.08 × 10−9 μm2), a relative molecular diffusivity of 4.02 μm2/s for L-selectinN138G was extrapolated from the calibration curve, which was increased by 85% over that of L-selectin (Fig. 2 D).
To further test our hypothesis, we designed a set of conditions to counter the predicted increase in the L-selectinN138G diffusivity. The medium viscosity was increased from 1 to 1.8 cP to reduce the diffusivity of L-selectinN138G by 80%. The radius of the microspheres was decreased from 3 to 1.5 μm to keep the product μr (and hence the microsphere diffusivity) approximately constant. The PSGL-1 site density was increased from 120 to 240 μm−2 to keep the normalizing factor m
r
m
l
r constant. Because the microsphere diffusivities, molecular diffusivities, and normalizing factors were matched, we predicted that the tether rate versus  curve for L-selectinN138G measured under the designed conditions would match the tether rate versus
curve for L-selectinN138G measured under the designed conditions would match the tether rate versus 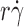 curve for L-selectin measured in 1-cP viscosity media with 3-μm radius microspheres. This was, indeed, the case (Fig. 2 A).
curve for L-selectin measured in 1-cP viscosity media with 3-μm radius microspheres. This was, indeed, the case (Fig. 2 A).
The tether rate curves for 6-sulfo-sLex (Fig. 2 B) were qualitatively similar to those for PSGL-1 (Fig. 2 A), but the former curves had smaller maximum tether rates that occurred at higher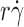 . Such quantitative differences reveal the impact of molecular docking rates specific to the ligands. These differences also place the optimal
. Such quantitative differences reveal the impact of molecular docking rates specific to the ligands. These differences also place the optimal 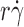 value from the L-selectin–6-sulfo-sLex tether rate curve significantly above the calibration curve in Fig. 2 C, and the maximum p
ad/(m
r
m
l
r) value significantly below the calibration curve in Fig. 2 D, because the calibration curves were based on interactions of L-selectin with PSGL-1 rather than 6-sulfo-sLex. This prevented estimating a relative change in the L-selectinN138G rotational diffusivity by comparing the measured maximum p
ad/(m
r
m
l
r) value from the L-selectin–6-sulfo-sLex tether rate curve to the calibration curve. Nevertheless, increasing rotational diffusivity of L-selectinN138G should augment tethering to molecularly distinct ligands because this mechanism does not require alterations of the ligand-binding surface. In the preceding paragraph, we estimated an 85% increase in diffusivity of L-selectinN138G over that of L-selectin by comparing the tether rate curves for interactions with PSGL-1 (Fig. 2 D). If this value represents the true difference in rotational diffusivities between the two selectins, instead of a fortuitous value that enabled curve fitting, it should account for the higher tether rate curve of L-selectinN138G over L-selectin for 6-sulfo-sLex (Fig. 2 B), as well as for PSGL-1 (Fig. 2 A). This hypothesis predicts that designing conditions to counter the increase in L-selectinN138G diffusivity would also align the designed L-selectinN138G curve with the original L-selectin curve for 6-sulfo-sLex. To test this prediction, we increased the medium viscosity from 1.0 to 1.8 cP and reduced the radius of the microspheres from 3.0 to 1.5 μm. Because the density of 6-sulfo-sLex could not be precisely measured, we increased the L-selectinN138G density from 750 to 1,500 μm−2. As predicted, the tether rate curve for these designed conditions aligned with the tether rate versus
value from the L-selectin–6-sulfo-sLex tether rate curve significantly above the calibration curve in Fig. 2 C, and the maximum p
ad/(m
r
m
l
r) value significantly below the calibration curve in Fig. 2 D, because the calibration curves were based on interactions of L-selectin with PSGL-1 rather than 6-sulfo-sLex. This prevented estimating a relative change in the L-selectinN138G rotational diffusivity by comparing the measured maximum p
ad/(m
r
m
l
r) value from the L-selectin–6-sulfo-sLex tether rate curve to the calibration curve. Nevertheless, increasing rotational diffusivity of L-selectinN138G should augment tethering to molecularly distinct ligands because this mechanism does not require alterations of the ligand-binding surface. In the preceding paragraph, we estimated an 85% increase in diffusivity of L-selectinN138G over that of L-selectin by comparing the tether rate curves for interactions with PSGL-1 (Fig. 2 D). If this value represents the true difference in rotational diffusivities between the two selectins, instead of a fortuitous value that enabled curve fitting, it should account for the higher tether rate curve of L-selectinN138G over L-selectin for 6-sulfo-sLex (Fig. 2 B), as well as for PSGL-1 (Fig. 2 A). This hypothesis predicts that designing conditions to counter the increase in L-selectinN138G diffusivity would also align the designed L-selectinN138G curve with the original L-selectin curve for 6-sulfo-sLex. To test this prediction, we increased the medium viscosity from 1.0 to 1.8 cP and reduced the radius of the microspheres from 3.0 to 1.5 μm. Because the density of 6-sulfo-sLex could not be precisely measured, we increased the L-selectinN138G density from 750 to 1,500 μm−2. As predicted, the tether rate curve for these designed conditions aligned with the tether rate versus 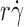 curve for L-selectin measured in 1-cP viscosity media with 3-μm radius microspheres (Fig. 2 B). Collectively, these data quantitatively demonstrate that eliminating the Tyr37–Asn138 hydrogen bond enhances cell tethering to different ligands by increasing the rotational diffusion of L-selectin.
curve for L-selectin measured in 1-cP viscosity media with 3-μm radius microspheres (Fig. 2 B). Collectively, these data quantitatively demonstrate that eliminating the Tyr37–Asn138 hydrogen bond enhances cell tethering to different ligands by increasing the rotational diffusion of L-selectin.
Greater hinge flexibility of L-selectin augments catch bonds by prolonging lifetimes with smaller forces
A flexible hinge might also affect force-dependent dissociation of L-selectin from its ligands. At low applied forces, the interdomain angle should remain mostly closed, as suggested by our MD simulations (Fig. 1 C; and Videos 1 and 2). Noncovalent interactions with the ligand may dissociate as it detaches from the lectin domain at the interface that is perpendicular to the direction of force (Fig. 3). As applied force increases, the equilibrium between the interdomain angles should shift toward a higher probability in the open conformation, which tilts the interface to align with the force direction, as suggested by MD simulations of the unbinding of P-selectin from PSGL-1 (unpublished data). Consequently, the ligand may slide across the lectin domain as preexisting interactions dissociate (Fig. 3). As observed in MD simulations of the unbinding of P-selectin from PSGL-1 (unpublished data), the sliding motion provides an opportunity for new interactions to replace those that are disrupted, or for the original interactions to reform before the ligand fully dissociates, thereby slowing dissociation. This force-dependent deceleration of dissociation is a hallmark of catch bonds (Marshall et al., 2003; Sarangapani et al., 2004). Thus, a flexible hinge between the lectin and EGF domains may allow force to allosterically elicit catch bonds with ligand by sliding and rebinding. Once the interdomain angle is fully open, further increases in force can no longer increase rebinding, resulting in the transition from catch bonds to slip bonds. Force-dependent sliding of ligand over a pivoting lectin domain might at least partially explain why P-selectin, whose interdomain hinge is predicted to be more flexible than that of L-selectin, forms augmented catch bonds with longer lifetimes that convert to slip bonds at lower force than L-selectin (Marshall et al., 2003; Sarangapani et al., 2004). We have formulated this sliding–rebinding mechanism into a mathematical model (see Materials and methods) whose solution exhibits catch–slip transitional bonds that fit the respective lifetime versus force relationships observed for P- and L-selectin interacting with PSGL-1 (Marshall et al., 2003; Sarangapani et al., 2004; unpublished data).
Figure 3.

Sliding–rebinding model for selectin–ligand interactions. The interdomain hinge is represented as a coiled spring. The yellow arrows indicate possible sequences of events. On the top left, a ligand binds to a selectin with a closed interdomain angle. A low applied force (f, short arrow) perpendicular to the binding interface favors ligand dissociation (bottom left). As applied force increases (f, long arrow), the equilibrium between the closed- and open-angle conformations shifts in favor of the open conformation (top right). This tilts the interface to align with the force direction, allowing the ligand to slide across the interface. Sliding allows new interactions to form or the original interactions to reform (rebinding). Eventually both old and new interactions break, and the ligand dissociates (bottom right).
The sliding–rebinding model also predicts that substituting Gly for Asn138 in L-selectin (L-selectinN138G) will reduce the force required to elicit catch bonds, prolong their lifetimes, and lower the force where catch bonds convert to slip bonds, even with molecularly distinct ligands. To test these predictions, we measured how force affected the lifetimes of interactions of L-selectin and L-selectinN138G with PSGL-1 and 6-sulfo-sLex. Both biomembrane force probe (BFP) and flow chamber experiments were used to obtain complementary data. For BFP experiments, interactions of L-selectin or L-selectinN138G coated on a target bead with PSGL-1 or 6-sulfo-sLex coated on a probe bead, were stressed through a red blood cell to allow lifetime measurements at various levels of constant force. For flow chamber experiments, microspheres displaying each selectin were perfused at various wall shear stresses over low densities of PSGL-1 or 6-sulfo-sLex. The lifetimes of transient tethers were measured by high-speed video microscopy. A large number of lifetime measurements were used to derive the mean lifetime (which equals the reciprocal off-rate 1/k off for first-order dissociation of single bonds) for each interaction at each tensile force (for BFP) or each wall shear stress (for the flow chamber). Both methods yielded similar results. As previously observed (Sarangapani et al., 2004; Yago et al., 2004), the lifetimes of L-selectin interactions with both ligands demonstrated a biphasic pattern characteristic of transitions from catch to slip bonds (Fig. 4). Initial increases in force prolonged mean lifetimes until an optimal value was reached; further increases in force shortened lifetimes. Although L-selectinN138G interactions with both ligands also exhibited transitions between catch and slip bonds (Fig. 4), the lifetimes in the catch bond regime were significantly longer and the transitions to slip bonds occurred at lower forces. In contrast, there was little difference in the lifetimes of L-selectin and L-selectinN138G interactions in the slip bond regime. The lifetime versus force curves were sensitive to the selectin and ligand used, although both selectins were captured by the same antibody and both biotinylated ligands were captured by streptavidin (see Materials and methods). The present L-selectin–PSGL-1 data also agree quantitatively with previous measurements using different capturing methods (Sarangapani et al., 2004) and using neutrophils expressing membrane-bound L-selectin (Yago et al., 2004). This strongly suggests that the data in Fig. 4 are dominated by lifetimes of selectin–ligand bonds rather than of antibody–antigen or biotin–streptavidin bonds. Because we demonstrated that L-selectin interacts as a monomer in the previous measurements (Sarangapani et al., 2004), the quantitative agreement with the current data suggests that they also represent monomeric interactions. However, the sliding–rebinding model of Fig. 3 can be translated to dimeric bonds (Zhu et al., 2005).
Figure 4.

Effects of interdomain hinge flexibility on catch bonds. (A and C) Interactions of L-selectin or L-selectinN138G with PSGL-1. (B and D) Interactions of L-selectin or L-selectinN138G with 6-sulfo-sLex. Bond lifetimes were measured by BFP (A and B) or by flow chamber (C and D) experiments (points), which were fitted by the sliding–rebinding model using Monte Carlo simulations (curves). The data in A and B represent the mean ± the SEM of ∼100 lifetime measurements. The data in C and D represent the mean ± the SD from five experiments. The fitting parameters areas follows: k
+1 = 30 s−1;  ; a = 0.2–0.4 Å, −f
1 = 5–40 pN; f
2 = 35–85 pN; and k
+2 = 350–5,400 s−1.
; a = 0.2–0.4 Å, −f
1 = 5–40 pN; f
2 = 35–85 pN; and k
+2 = 350–5,400 s−1.
Although the crystal structure of the P-selectin–PSGL-1 complex reveals many noncovalent interactions at the atomic level (Somers et al., 2000), they were simplified into two pseudoatomic interactions in this study to model the sliding–rebinding mechanism of forced-dissociation of a selectin–ligand complex (Fig. 3). It is assumed that each interaction binds at an association rate of k
+1 and unbinds at a dissociation rate that obeys the Bell equation (Bell, 1978), with a stress-free dissociation rate of 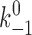 and an energy well of width a. After both preexisting interactions dissociate, the ligand is assumed to slide over a pivoting lectin domain to form a new interaction with a probability that increases with force in the range f
1 < f < f
2. It is further assumed that the system can return to the original state at a rebinding rate of k
+2 (see Materials and methods for details). Curve fitting by the model yielded excellent agreement with the data (Fig. 4). As expected, four (k
+1,
and an energy well of width a. After both preexisting interactions dissociate, the ligand is assumed to slide over a pivoting lectin domain to form a new interaction with a probability that increases with force in the range f
1 < f < f
2. It is further assumed that the system can return to the original state at a rebinding rate of k
+2 (see Materials and methods for details). Curve fitting by the model yielded excellent agreement with the data (Fig. 4). As expected, four (k
+1, 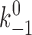 , a, and f
1) of the six fitting parameters could be the same for the curves of both selectins interacting with the same ligand. Compared with L-selectin, L-selectinN138G required less force (smaller f
2) to fully open the interdomain angle and exhibited a higher rebinding rate (larger k
+2). Thus, both experimental data and their theoretical fits strongly support the model for transitions from catch to slip bonds through force-dependent sliding of the ligand over a pivoting lectin domain that promotes rebinding. Although alterations in the hinge region might propagate conformational changes across the lectin domain to the ligand-binding interface, the model does not require such changes.
, a, and f
1) of the six fitting parameters could be the same for the curves of both selectins interacting with the same ligand. Compared with L-selectin, L-selectinN138G required less force (smaller f
2) to fully open the interdomain angle and exhibited a higher rebinding rate (larger k
+2). Thus, both experimental data and their theoretical fits strongly support the model for transitions from catch to slip bonds through force-dependent sliding of the ligand over a pivoting lectin domain that promotes rebinding. Although alterations in the hinge region might propagate conformational changes across the lectin domain to the ligand-binding interface, the model does not require such changes.
Greater hinge flexibility of L-selectin lowers the shear threshold for rolling
To determine whether the augmented catch bonds lowered the shear threshold for L-selectin-dependent rolling, we perfused microspheres bearing each selectin over higher densities of PSGL-1 or 6-sulfo-sLex. Rolling motions were visualized by high-speed video microscopy. As wall shear stress increased, the mean rolling velocities of microspheres displaying either selectin first decreased and then reached a minimum (Fig. 5, A and B), a characteristic of the shear threshold phenomenon that is mediated by catch bonds (Yago et al., 2004). At higher shear stresses, the rolling velocities again increased as catch bonds converted to slip bonds. Remarkably, the descending phases of the rolling velocity curves for L-selectinN138G shifted downward and to the left, with significantly slower mean velocities and with minimal velocities at much lower shear stresses than for L-selectin. Rolling motions at these suboptimal flow rates were more regular for L-selectinN138G than for L-selectin, with longer mean stop times (Fig. 5, C and D) and higher fractions of time in the stop phase (Fig. 5, E and F). In contrast, the ascending phases of the rolling velocity curves were similar for both selectins. These data confirm that substituting Gly for Asn138 lowers the shear threshold for L-selectin-dependent rolling.
Figure 5.

Effects of interdomain hinge flexibility on flow-enhanced rolling. Microspheres bearing matched densities of L-selectin or L-selectinN138G were perfused through a flow chamber containing immobilized PSGL-1 (A, C, and E) or 6-sulfo-sLex (B, D, and F). Rolling parameters are presented as mean rolling velocities (A and B), mean stop time (C and D), or fractional stop time (E and F). The data in A and B represent the mean ± the SD from five experiments. The data in C–F represent analyses of thousands of rolling steps collected from 10–15 microspheres rolling for 1 s at each wall shear-stress for each pair of selectin–ligand interactions.
Greater hinge flexibility of L-selectin causes flowing microspheres to aggregate with leukocytes
An unexplained property of circulating leukocytes is that they do not aggregate, even though they express both L-selectin and its ligand PSGL-1. We hypothesized that specific kinetic and mechanical properties of L-selectin–PSGL-1 interactions prevent leukocyte aggregation. Because of these properties, randomly colliding leukocytes might form bonds between L-selectin and PSGL-1, but the number of bonds would be too small and/or their lifetimes would be too short at small forces for stable adhesion. This hypothesis predicts that increasing interdomain flexibility will cause spontaneous aggregation of leukocytes because both bond formation and bond lifetime are increased. To test this hypothesis, we flowed neutrophils or mixtures of neutrophils and microspheres bearing L-selectin or L-selectinN138G in a shear field to promote collisions. Cells or microspheres were perfused in a flow chamber coated with the nonadherent protein human serum albumin (HSA) at a wall shear stress of 1 dyn/cm2. This shear stress level applies sufficient force to bonds at the trailing edge of a rolling cell, thereby promoting optimal L-selectin–dependent rolling on PSGL-1 when it is coated in the flow chamber (Fig. 5), but should exert much smaller forces on bonds bridging cells or microspheres in flowing aggregates (Long et al., 1999). High-speed video microscopy revealed that some free-flowing neutrophils formed doublets after they collided. However, doublet lifetimes were very brief, and the interacting cells dissociated within 0.02 to 0.03 s (Fig. 6 A and Video 3, available at http://www.jcb.org/cgi/content/full/jcb.200606056/DC1). Doublets of neutrophils and L-selectin microspheres were similarly short lived (Fig. 6 B and Video 4). In contrast, neutrophils formed stable doublets with L-selectinN138G microspheres that persisted until they flowed out of the field of view (Fig. 6 C and Video 5). To quantify this phenomenon, we perfused mixtures of neutrophils and microspheres that were labeled with different fluorescent dyes and fixed the mixtures after they exited the flow chamber. Flow cytometry revealed very few particles containing fluorescence markers for both neutrophils and L-selectin microspheres (Fig. 7 A), but many particles labeled for both neutrophils and L-selectinN138G microspheres (Fig. 7 B). Addition of the anti–L-selectin mAb DREG-56 blocked aggregate formation, confirming that aggregates developed through engagement of L-selectinN138G (Fig. 7 C). Similarly, fluorescence microscopy revealed few aggregates between neutrophils and L-selectin microspheres (Fig. 7 D), whereas neutrophils and L-selectinN138G microspheres formed many doublets and larger aggregates (Fig. 7 E). Such aggregates did not develop in the presence of DREG-56 (Fig. 7 F). These data suggest that a subtle change in the interdomain hinge of L-selectin is sufficient to cause flowing leukocytes to aggregate.
Figure 6.

Effects of interdomain hinge flexibility on lifetimes of doublets of neutrophils or microspheres in a flow field. Neutrophils or mixtures of neutrophils with microspheres bearing L-selectin or L-selectinN138G were perfused through a flow chamber coated with HSA. Representative images of randomly colliding cells and/or microspheres were collected at 250 frames/s. A doublet formed by collision of two neutrophils (A) or of a neutrophil and an L-selectin microsphere (B), each dissociated within 0.028 s. In contrast, a doublet formed by the collision of a neutrophil and an L-selectinN138G microsphere (C) persisted for at least 0.1 s, until it flowed out of the field of view.
Figure 7.

Quantification of neutrophil–microsphere aggregation in a flow field. Mixtures of neutrophils labeled with the red dye PKH26 and microspheres labeled with the green dye FITC were perfused through a flow chamber coated with HSA, as in Fig. 6. After exiting the flow chamber, the suspensions were fixed and analyzed by flow cytometry or by fluorescence microscopy. (A and D) Mixtures of neutrophils and L-selectin microspheres. (B and E) Mixtures of neutrophils and L-selectinN138G microspheres. (C and F) Mixtures of neutrophils and L-selectinN138G microspheres perfused in the presence of the anti–L-selectin mAb DREG-56. (A–C) Flow cytometry of ungated samples. The percentage of particles labeled with both dyes is listed in the top right quadrant. (D–F) Representative fluorescence micrographs. The data are representative of three independent experiments.
Discussion
Leukocyte adhesion to vascular surfaces requires that the kinetics of formation and dissociation of selectin–ligand bonds be regulated by the mechanics of blood flow. The shear threshold for tethering and rolling through L-selectin provides a striking illustration of this regulation. Transport mechanisms, including molecular diffusion, govern flow-enhanced tethering, whereas force governs flow-enhanced rolling through catch bonds. How an atomic-level structure dictates such mechanisms was not understood. In this study, we used crystal structures and MD simulations to formulate a model for these mechanisms. MD simulations revealed that the hinge between the lectin and EGF domains of L-selectin can assume dual conformations that are in dynamic equilibrium but allow rapid transitions. Applied force or changes in hinge flexibility can alter the stability of the conformations, tilt the dynamic equilibrium, and induce conformational transitions. Structural analysis identified a single residue that contributes to the rigidity of the interdomain hinge by forming a hydrogen bond. Eliminating this hydrogen bond demonstrated that hinge flexibility regulates both the kinetic and mechanical properties of L-selectin. Thus, the fine tuning of hinge flexibility modulates flow-enhanced cell adhesion and likely prevents inappropriate leukocyte aggregation.
Cells expressing a chimeric L-selectin, in which the native EGF domain was replaced with that of P-selectin, were shown to have a lowered shear threshold for adhesion because of an enhanced cellular on-rate that increased tethering to surface-bound ligand (Dwir et al., 2000, 2003). This property may have resulted, at least in part, from increased rotational diffusivity caused by substituting Gly for Asn138 in the chimeric molecule. Our results suggest that augmented catch bonds between the chimeric L-selectin and its ligands also may have contributed to the lowered shear threshold for adhesion.
That L-selectinN138G promotes aggregation of microspheres with free-flowing neutrophils suggests that the kinetic and mechanical properties of L-selectin–PSGL-1 interactions must be finely regulated to allow rolling on vascular surfaces, but not aggregation of flowing leukocytes. This regulation is essential because both L-selectin and PSGL-1 are constitutively expressed on the surfaces of leukocytes. In contrast, platelets mobilize P-selectin on their surfaces only after they are activated, which normally does not occur until they adhere to damaged vessel walls (McEver, 2002). Our model suggests that leukocytes will form aggregates with circulating activated platelets because the lifetimes of PSGL-1 bonds with P-selectin are longer than those with L-selectin at all force levels (Marshall et al., 2003; Sarangapani et al., 2004), perhaps in part because of the increased interdomain flexibility of P-selectin conferred by Gly138. Indeed, circulating platelet-leukocyte aggregates are observed in pathological disorders that increase platelet activation (Michelson et al., 2001).
Our findings exemplify how force-induced conformational changes allosterically regulate protein function, which may be applicable to other proteins. The sliding–rebinding model provides a structural explanation for catch bonds and may be applicable to interactions of other proteins with an interdomain hinge. For example, the adhesin FimH mediates shear stress-enhanced adhesion of bacteria to epithelial cells (Thomas et al., 2002), which may result from catch bonds between FimH and its mannosylated ligands (Thomas et al., 2006). MD simulations revealed that force applied at the ligand-binding site of the lectin domain extends a linker chain that connects the lectin domain to the adjacent pilin domain. Mutations in this segment decrease the shear threshold for bacterial adhesion (Thomas et al., 2002). Such mutations might make the linker region more flexible and reduce the force required to slide ligand across the binding interface. The increased flexibility could also favor bond formation through greater rotational diffusion. Interactions of glycoprotein Ibα with von Willebrand factor mediate flow-enhanced adhesion of platelets to damaged vascular surfaces (Savage et al., 1996; Doggett et al., 2003). Catch bonds might contribute to flow-dependent platelet adhesion, and a sliding–rebinding mechanism might explain why mutations in glycoprotein Ibα or von Willebrand factor that are remote from the interface can alter binding (Ajzenberg et al., 2000; Peng et al., 2005). Force-induced allosteric changes may also regulate catch–slip transitional bonds between actin and myosin (Guo and Guilford, 2006) and the conversion of integrins to their active conformations (Salas et al., 2002; Jin et al., 2004).
Materials and methods
Proteins and glycoconjugates
L-selectin–Ig containing the lectin domain, EGF domain, and both consensus repeats of human L-selectin fused to the Fc portion of human IgG1 was expressed as previously described (Yago et al., 2004). The cDNA encoding L-selectin was used as a template to alter the codon for Asn138 to Gly, using the QuikChange Mutagenesis kit (Stratagene). The mutations in the construct, termed L-selectinN138G, were confirmed by DNA sequencing. L-selectinN138G-Ig was expressed as described for L-selectin–Ig. The aminopropyl glycoside of 6-sulfo-sLex was biotinylated as previously described (Korchagina and Bovin, 1992). Soluble recombinant monomeric PSGL-1, anti–PSGL-1 mAb PL1, and anti–L-selectin mAb DREG-56 have been previously described (Yago et al., 2004). Anti–human IgG Fc polyclonal antibody was obtained from CHEMICON International, Inc.
MD simulations
A crystal structure of the lectin and EGF domains of human L-selectin served as the starting coordinate for MD simulations, except that the glycan attached to residue Asn66 was deleted to reduce the system size and to avoid the use of less reliable glycan force fields. Because this glycan extends out from Asn66 on the protein surface, its deletion is unlikely to affect the structure of the lectin and EGF domains. The molecule was solvated in a 90 × 60 × 60 Å3 TIP3 water box, together with 8 Ca2+ and 18 Cl− ions to neutralize the system, which included 29,969 atoms. MD simulations were performed using NAMD (Phillips et al., 2005) with a CHARMM22 all-atom force field (MacKerell et al., 1998). The system was first subjected to a two-step energy minimization. Each step consisted of 10,000 conjugate gradient iterations. In the first step, the heavy atoms of the protein were fixed and the rest of the atoms were allowed to move. In the second step, all atoms were allowed to move. After energy minimization, the system was heated gradually from 0 to 300 K in 100 ps. Then the system was equilibrated for 1 ns with pressure and temperature control. The temperature was held at 300 K using Langevin dynamics and the pressure was held at 1 atm by the Langevin piston method. Free dynamics were then simulated with the equilibrated system for 5 ns. A periodic boundary condition was adopted. Particle Mesh Ewald was used for electrostatic interactions, and a cutoff of 12 Å was used for van der Waals interactions. A total of seven independent simulations were performed, following the same procedure. The trajectory of each simulation was compared with the initial L-selectin crystal structure and with the P-selectin open-angle crystal structure (PDB 1G1S; Somers et al., 2000).
Sliding–rebinding model
The sliding–rebinding model can be formulated with increasing complexity, depending on the number of pseudoatomic interactions assumed to describe the noncovalent interactions distributed across the selectin–ligand interface. A minimal model was solved by Monte Carlo simulations. The simulation starts with two pseudoatomic interactions (Fig. 3) and advances in 1-μs time steps (Δt). To determine what would happen in each time step, a random number (uniformly distributed between 0 and 1) is compared with one of the probabilities given by equations 1, 2, 4, and 5, depending on the stage of the simulation. In the first stage, the dissociation from the initial bound state is simulated. The fate of each of the two pseudoatomic interactions in the current time step is determined using one of two probabilities, depending on whether that pseudoatomic interaction survived in the previous time step. If so, it would survive if the random number were smaller than the probability,
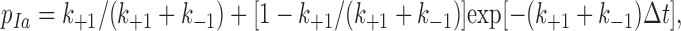 |
(1) |
or it would dissociate. If not, i.e., if in the previous time step the pseudoatomic interaction in question dissociated, but the other pseudoatomic interaction survived, it would associate if the random number were smaller than the probability,
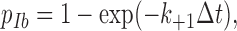 |
(2) |
or it would remain dissociated. In equation 1 and 2, k +1 is a constant association rate and k −1 is a force-dependent dissociation rate that obeys the Bell equation (Bell, 1978), as follows:
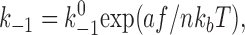 |
(3) |
where 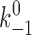 and a are model parameters, f is applied force, and n = 1 or 2, depending on whether one or both pseudoatomic interactions were intact in the previous time step. After both preexisting pseudoatomic interactions dissociate, the simulation advances to the second stage, which simulates sliding and the formation of a new interaction. In the next simulation step, a new pseudoatomic interaction would form if the random number were smaller than the probability, as follows:
and a are model parameters, f is applied force, and n = 1 or 2, depending on whether one or both pseudoatomic interactions were intact in the previous time step. After both preexisting pseudoatomic interactions dissociate, the simulation advances to the second stage, which simulates sliding and the formation of a new interaction. In the next simulation step, a new pseudoatomic interaction would form if the random number were smaller than the probability, as follows:
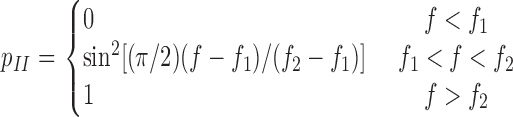 |
(4) |
or if the ligand would dissociate. Equation 4 relates the probability of forming a new interaction to force through the interdomain angle. If the applied force were below f 1, the interdomain angle would be confined to the closed conformation (as depicted in the top left portion of Fig. 3), so no new interaction would be allowed. Because spontaneous transition from the closed-angle to open-angle conformation was observed in MD simulations in the absence of applied force, a negative value is expected for f 1, i.e., compressive force. In the range f 1 < f < f 2, force shifts the equilibrium between the two hinge conformations toward what is more likely to be in the open-angle conformation, allows the binding interface to slide more readily, and increases the probability of forming a new pseudoatomic interaction (as depicted in the top right portion of Fig. 3). After the force exceeds f 2, the interdomain angle reaches maximum and p II can increase no further (i.e., p II = 1). Thus, a positive value is expected for f 2, i.e., tensile force. Formation of a new interaction advances the simulation to the third stage, where two tests are performed in the next time step. One compares a random number to the probability given by equation 1 to determine whether the new interaction would survive. The other test determines whether the original double-pseudoatomic interactions would reform (as depicted in Fig. 3). It would, if the random number were smaller than the rebinding probability,
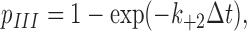 |
(5) |
where k +2 is a constant rebinding rate, which returns the simulation back to the first stage. The simulation continues until the ligand dissociates from the selectin when no new interaction forms after both old interactions dissociate or the new interaction dissociates before rebinding occurs. The accumulated time steps are the lifetime of the selectin–ligand complex in that simulation run, which is repeated 1,000,000 times for a given force to obtain an ensemble of exponentially distributed lifetimes and their average at that force.
The six model parameters (k
+1, 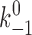 , a, f
1, f
2, k
+2) were adjusted to obtain a mean lifetime versus force curve that fit the experimental data for a given selectin–ligand pair. For each ligand interacting with two L-selectin molecules, the first three parameters (k
+1,
, a, f
1, f
2, k
+2) were adjusted to obtain a mean lifetime versus force curve that fit the experimental data for a given selectin–ligand pair. For each ligand interacting with two L-selectin molecules, the first three parameters (k
+1, 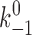 , and a) describing the rates of association and dissociation of the pseudoatomic interactions were kept the same for L-selectin and L-selectinN138G. Of the two parameters describing the probability (p
II) of forming a new pseudoatomic interaction after sliding, one (f
1) was also kept invariant. The other parameter (f
2) had to be smaller for L-selectinN138G than for L-selectin, to reflect the more flexible interdomain hinge of L-selectinN138G. For the same reason, the rebinding rate (k
+2) had to be larger for L-selectinN138G than for L-selectin. Fitting parameters are listed in the Fig. 4 legend, which are reasonably ranged except for k
+2. The unrealistically large k
+2 (compared with k
+1) results from the greatly reduced number of interactions assumed and the same kinetic rates assumed for the new interaction formed after sliding as the preexisting interactions. Monte Carlo simulations with a less simplified model consisting of three pseudoatomic interactions instead of two were able to fit the data with much smaller k
+2 values.
, and a) describing the rates of association and dissociation of the pseudoatomic interactions were kept the same for L-selectin and L-selectinN138G. Of the two parameters describing the probability (p
II) of forming a new pseudoatomic interaction after sliding, one (f
1) was also kept invariant. The other parameter (f
2) had to be smaller for L-selectinN138G than for L-selectin, to reflect the more flexible interdomain hinge of L-selectinN138G. For the same reason, the rebinding rate (k
+2) had to be larger for L-selectinN138G than for L-selectin. Fitting parameters are listed in the Fig. 4 legend, which are reasonably ranged except for k
+2. The unrealistically large k
+2 (compared with k
+1) results from the greatly reduced number of interactions assumed and the same kinetic rates assumed for the new interaction formed after sliding as the preexisting interactions. Monte Carlo simulations with a less simplified model consisting of three pseudoatomic interactions instead of two were able to fit the data with much smaller k
+2 values.
BFP
Our in-house–built BFP apparatus is a duplicate of that developed in the laboratory of E. Evans (University of British Columbia and Boston University; Evans et al., 2004). L-selectin-Ig or L-selectinN138G-Ig was captured by goat anti–human Fc antibody covalently precoupled to the target bead (3–4 μm in diameter), as previously described (Evans et al., 2004). The same protocol, but without the step of linking proteins to polyethyleneglycol polymers, was used to couple streptavidin-maleimide (Sigma-Aldrich) to the probe bead (2 μm in diameter), which captured either biotinylated PSGL-1 or 6-sulfo-sLex. The streptavidin also attached the probe bead to a biotinylated red blood cell, which, when pressurized by micropipette aspiration, served as an ultrasensitive force transducer. Low densities of selectins and ligands ensured infrequent adhesion (30%), which was specific, as EDTA and anti–L-selectin mAb DREG-56 abolished adhesion. The in-house online image analysis software tracked the red blood cell deflection with a 0.6-ms temporal resolution and 5-nm spatial resolution, which, for a spring constant of 0.3 pN/nm, translates to 1.5-pN force resolution. Driven by a computer-controlled piezoelectric translator, the force-clamp test cycle consisted of an approach of the target bead (1,500 nm/s) to touch the probe bead, a gentle (15 pN) and brief (0.1 s) contact period, a retraction of the target bead to load the selectin–ligand bond (if a bond was formed) at 1,000 pN/s to the desired level of force, and a waiting period during which the bond (if the bond survived ramping) was subject to a constant force until dissociation, which was then repeated thousands of times. The bond lifetime was measured from the moment when the bond force reached the desired level to the moment when the bond dissociated. A total of ∼800 lifetimes were measured at forces ranging from 3–90 pN for each interaction of L-selectin or L-selectinN138G with PSGL-1 or 6-sulfo-sLex, which were segregated into 7–9 force bins. For each bin, the natural log of the number of measurements with a lifetime greater than t was plotted versus t, which exhibited linear decay as predicted by first-order dissociation kinetics, except for longer lifetimes of probable multiple bonds that represented <8% of the total interactions, which were excluded as outliers. The reciprocal of the negative slope was found equal to the mean and SD of lifetime, as predicted by first-order dissociation kinetics. The mean ± the SEM of lifetimes in each bin were plotted against force for each interaction, as shown in Fig. 4, A and B.
Coupling of selectin-Ig to microspheres and selectin ligands to flow chambers
Each selectin-Ig was captured on polystyrene microspheres (6- or 3-μm diam; Polysciences, Inc.) coated with anti–human Fc polyclonal antibody (Yago et al., 2004). Matched densities of each selectin were confirmed by flow cytometry (Yago et al., 2004). Biotinylated PSGL-1 or 6-sulfo-sLex was captured on streptavidin (Pierce Chemical Co.) adsorbed to flow chamber floors (Yago et al., 2004).
Flow assays
Microspheres (2 × 106/ml in HBSS containing 0.5% HSA) were perfused in media without or with 3% (wt/vol) Ficoll (molecular weight 400,000; Sigma-Aldrich) at various flow rates over PSGL-1 or 6-sulfo-sLex in a parallel-plate flow chamber (Yago et al., 2004). The viscosities of the media without and with Ficoll were 1.0 and 1.8 cp, respectively, at room temperature as previously described (Yago et al., 2004). Images were captured with a digital video camera (Fastcam Super 10 K; Photron) at 250 frames/s. Tether rates were measured by a previously described method (Ramachandran et al., 1999; Yago et al., 2002). The tether rate, TR, was calculated by normalizing the number of observed tethering events in 1 min by the total number of microspheres flowing through the field of view in the same focal plane in the same period of time (Ramachandran et al., 1999). Mean rolling velocities were measured by tracking individual microspheres frame by frame. Rolling step analysis was performed with custom-designed macros prepared in Excel (Microsoft; Yago et al., 2004). In some experiments, microspheres were perfused in media containing 20 μg/ml DREG-56 or PL1, or 10 mM EDTA. All tethering and rolling events were specific because they were eliminated by inclusion of mAb or EDTA in the media.
Transient tether lifetimes were measured on low densities of PSGL-1 or 6-sulfo-sLex that did not support rolling or skipping (Yago et al., 2004). Images captured at 250 fps were replayed in slow motion, and durations of transient tethers were measured using frame-by-frame analysis. For each mean lifetime curve, five sets of lifetimes at each wall shear stress (∼100 tethering events in each set) were measured. At each wall shear stress, the exponentially distributed transient tether lifetimes in each set were averaged. The data are presented as mean ± SD of the five sets of average lifetimes.
Aggregation of flowing microspheres and neutrophils
Microspheres were labeled with FITC-conjugated anti–human Fc antibody (Sigma-Aldrich). In some experiments, microspheres directly conjugated with FITC (Polysciences) were coated with unlabeled anti-human Fc antibody. After blocking with HBSS containing 1% HSA, L-selectin-Ig, or L-selectinN138-Ig was captured on the labeled microspheres. Isolated human neutrophils (Yago et al., 2004) were labeled with the fluorochrome PKH26 (Sigma-Aldrich). Neutrophils were mixed with microspheres bearing L-selectin or L-selectinN138G (final concentration of 2 × 106/ml for both cells and microspheres) in HBSS without Ca2+ and Mg2+ containing 0.5% HSA. Immediately before perfusion, 1 M CaCl2 and 1 M MgCl2 were added to the suspensions to achieve final concentrations of 2 mM Ca2+ and 2 mM Mg2+. The cell/microsphere suspensions were perfused through a flow chamber coated with 1% HSA at a wall shear stress of 1 dyn/cm2. In some experiments, 20 μg/ml DREG-56 was added to the suspensions. After exiting the flow chamber, samples were collected and fixed with 1% paraformaldehyde. Some samples were analyzed by flow cytometry without gate selection on a FACScalibur instrument (Becton Dickinson). Other samples were visualized in a fluorescence microscope (ECLIPSE E800; Nikon) connected to a CCD digital camera (Dxm1200; Nikon). Digital images were stored with Nikon software. FITC and PKH26 images were merged using Photoshop (Adobe).
In some experiments, unlabeled neutrophils or mixtures of neutrophils with microspheres bearing L-selectin or L-selectinN138G were perfused at 1 dyn/cm2. Video microscopy images of interactions among flowing neutrophils or microspheres were captured at 250 frames/s.
Online supplemental information
The online material consists of five videos. Videos 1 and 2 are MD simulations of movements around the hinge between the lectin and EGF domains of L-selectin. Videos 3, 4, and 5 depict collisions of free-flowing neutrophils with other neutrophils or with microspheres bearing L-selectin or L-selectinN138G. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200606056/DC1.
Supplementary Material
Acknowledgments
We thank E.A. Evans, A. Leung, and K. Kinoshita for helping us install the BFP apparatus and for training in its operation. We thank the Interactive High Performance Computing Laboratory of the College of Computing and the High Performance Computing Center of the Office of Information Technology, Georgia Institute of Technology for computational resources.
This work was supported by grants from the National Institutes of Health.
J. Lou, T. Yago, and A.G. Klopocki contributed equally to this paper.
Abbreviations used in this paper: BFP, biomembrane force probe; HSA, human serum albumin; MD, molecular dynamics; PSGL, P-selectin glycoprotein ligand; RMSD, root mean square distance; sLex, sialyl Lewis x.
References
- Ajzenberg, N., A.S. Ribba, G. Rastegar-Lari, D. Meyer, and D. Baruch. 2000. Effect of recombinant von Willebrand factor reproducing type 2B or type 2M mutations on shear-induced platelet aggregation. Blood. 95:3796–3803. [PubMed] [Google Scholar]
- Alon, R., S.Q. Chen, K.D. Puri, E.B. Finger, and T.A. Springer. 1997. The kinetics of L-selectin tethers and the mechanics of selectin-mediated rolling. J. Cell Biol. 138:1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, G.I. 1978. Models for the specific adhesion of cells to cells: a theoretical framework for adhesion mediated by reversible bonds between cell surface molecules. Science. 200:618–627. [DOI] [PubMed] [Google Scholar]
- Doggett, T.A., G. Girdhar, A. Lawshe, J.L. Miller, I.J. Laurenzi, S.L. Diamond, and T.G. Diacovo. 2003. Alterations in the intrinsic properties of the GPIbα-VWF tether bond define the kinetics of the platelet-type von Willebrand disease mutation, Gly233Val. Blood. 102:152–160. [DOI] [PubMed] [Google Scholar]
- Dwir, O., G.S. Kansas, and R. Alon. 2000. An activated L-selectin mutant with conserved equilibrium binding properties but enhanced ligand recognition under shear flow. J. Biol. Chem. 275:18682–18691. [DOI] [PubMed] [Google Scholar]
- Dwir, O., A. Solomon, S. Mangan, G.S. Kansas, U.S. Schwarz, and R. Alon. 2003. Avidity enhancement of L-selectin bonds by flow: shear-promoted rotation of leukocytes turn labile bonds into functional tethers. J. Cell Biol. 163:649–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, E., A. Leung, V. Heinrich, and C. Zhu. 2004. Mechanical switching and coupling between two dissociation pathways in a P-selectin adhesion bond. Proc. Natl. Acad. Sci. USA. 101:11281–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, E.B., K.D. Puri, R. Alon, M.B. Lawrence, U.H. Von Andrian, and T.A. Springer. 1996. Adhesion through L-selectin requires a threshold hydrodynamic shear. Nature. 379:266–269. [DOI] [PubMed] [Google Scholar]
- Graves, B.J., R.L. Crowther, C. Chandran, J.M. Rumberger, S. Li, K.-S. Huang, D.H. Presky, P.C. Familletti, B.A. Wolitzky, and D.K. Burns. 1994. Insight into E-selectin/ligand interaction from the crystal structure and mutagenesis of the lec/EGF domains. Nature. 367:532–538. [DOI] [PubMed] [Google Scholar]
- Guo, B., and W.H. Guilford. 2006. Mechanics of actomyosin bonds in different nucleotide states are tuned to muscle contraction. Proc. Natl. Acad. Sci. USA. 103:9844–9849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, M., I. Andricioaei, and T.A. Springer. 2004. Conversion between three conformational states of integrin I domains with a C-terminal pull spring studied with molecular dynamics. Structure. 12:2137–2147. [DOI] [PubMed] [Google Scholar]
- Korchagina, E.Y., and N.V. Bovin. 1992. Synthesis of spacered trisaccharides with blood group A and B specificities and fragments and structural analogues of them. Sov. J. Bioorg. Chem. 18:153–165. [Google Scholar]
- Leppänen, A., S.P. White, J. Helin, R.P. McEver, and R.D. Cummings. 2000. Binding of glycosulfopeptides to P-selectin requires stereospecific contributions of individual tyrosine sulfate and sugar residues. J. Biol. Chem. 275:39569–39578. [DOI] [PubMed] [Google Scholar]
- Leppänen, A., T. Yago, V.I. Otto, R.P. McEver, and R.D. Cummings. 2003. Model glycosulfopeptides from P-selectin glycoprotein ligand-1 require tyrosine sulfation and a core 2-branched O-glycan to bind to L-selectin. J. Biol. Chem. 278:26391–26400. [DOI] [PubMed] [Google Scholar]
- Long, M., H.L. Goldsmith, D.F. Tees, and C. Zhu. 1999. Probabilistic modeling of shear-induced formation and breakage of doublets cross-linked by receptor-ligand bonds. Biophys. J. 76:1112–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell, A.D., Jr., D. Bashford, R.L. Bellott, R.L. Dunbrack Jr., J.D. Evanseck, M.J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, et al. 1998. All-atom empirical potential for molecular modeling and dynamics study of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- Marshall, B.T., M. Long, J.W. Piper, T. Yago, R.P. McEver, and C. Zhu. 2003. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 423:190–193. [DOI] [PubMed] [Google Scholar]
- McEver, R.P. 2002. Selectins: lectins that initiate cell adhesion under flow. Curr. Opin. Cell Biol. 14:581–586. [DOI] [PubMed] [Google Scholar]
- McEver, R.P. 2005. A sulfated address for lymphocyte homing. Nat. Immunol. 6:1067–1069. [DOI] [PubMed] [Google Scholar]
- Michelson, A.D., M.R. Barnard, L.A. Krueger, C.R. Valeri, and M.I. Furman. 2001. Circulating monocyte-platelet aggregates are a more sensitive marker of in vivo platelet activation than platelet surface P-selectin: studies in baboons, human coronary intervention, and human acute myocardial infarction. Circulation. 104:1533–1537. [DOI] [PubMed] [Google Scholar]
- Peng, Y., C.N. Shrimpton, J.F. Dong, and J.A. Lopez. 2005. Gain of von Willebrand factor-binding function by mutagenesis of a species-conserved residue within the leucine-rich repeat region of platelet glycoprotein Ibα. Blood. 106:1982–1987. [DOI] [PubMed] [Google Scholar]
- Phillips, J.C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R.D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran, V., M.U. Nollert, H. Qiu, W.-j. Liu, R.D. Cummings, C. Zhu, and R.P. McEver. 1999. Tyrosine replacement in P-selectin glycoprotein ligand-1 affects distinct kinetic and mechanical properties of bonds with P- and L-selectin. Proc. Natl. Acad. Sci. USA. 96:13771–13776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas, A., M. Shimaoka, S. Chen, C.V. Carman, and T. Springer. 2002. Transition from rolling to firm adhesion is regulated by the conformation of the I domain of the integrin lymphocyte function-associated antigen-1. J. Biol. Chem. 277:50255–50262. [DOI] [PubMed] [Google Scholar]
- Sarangapani, K.K., T. Yago, A.G. Klopocki, M.B. Lawrence, C.B. Fieger, S.D. Rosen, R.P. McEver, and C. Zhu. 2004. Low force decelerates L-selectin dissociation from P-selectin glycoprotein ligand-1 and endoglycan. J. Biol. Chem. 279:2291–2298. [DOI] [PubMed] [Google Scholar]
- Savage, B., E. Saldívar, and Z.M. Ruggeri. 1996. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. 84:289–297. [DOI] [PubMed] [Google Scholar]
- Somers, W.S., J. Tang, G.D. Shaw, and R.T. Camphausen. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. 103:467–479. [DOI] [PubMed] [Google Scholar]
- Thomas, W., M. Forero, O. Yakovenko, L. Nilsson, P. Vicini, E. Sokurenko, and V. Vogel. 2006. Catch-bond model derived from allostery explains force-activated bacterial adhesion. Biophys. J. 90:753–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, W.E., E. Trintchina, M. Forero, V. Vogel, and E.V. Sokurenko. 2002. Bacterial adhesion to target cells enhanced by shear force. Cell. 109:913–923. [DOI] [PubMed] [Google Scholar]
- Yago, T., A. Leppänen, H. Qiu, W.D. Marcus, M.U. Nollert, C. Zhu, R.D. Cummings, and R.P. McEver. 2002. Distinct molecular and cellular contributions to stabilizing selectin-mediated rolling under flow. J. Cell Biol. 158:787–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yago, T., J. Wu, C.D. Wey, A.G. Klopocki, C. Zhu, and R.P. McEver. 2004. Catch bonds govern adhesion through L-selectin at threshold shear. J. Cell Biol. 166:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yago, T., V.I. Zarnitsyna, A.G. Klopocki, R.P. McEver, and C. Zhu. 2006. Transport governs flow-enhanced cell tethering through L-selectin at threshold shear. Biophys. J. In press. [DOI] [PMC free article] [PubMed]
- Zhu, C., J. Lou, and R.P. McEver. 2005. Catch bonds: physical models, structural bases, biological function and rheological relevance. Biorheology. 42:443–462. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.