

Abstract
We identify a mitochondrial E3 ubiquitin ligase, MARCH5, as a critical regulator of mitochondrial fission. MARCH5 RING mutants and MARCH5 RNA interference induce an abnormal elongation and interconnection of mitochondria indicative of an inhibition of mitochondrial division. The aberrant mitochondrial phenotypes in MARCH5 RING mutant–expressing cells are reversed by ectopic expression of Drp1, but not another mitochondrial fission protein Fis1. Moreover, as indicated by abnormal clustering and mitochondrial accumulation of Drp1, as well as decreased cellular mobility of YFP-Drp1 in cells expressing MARCH5 RING mutants, MARCH5 activity regulates the subcellular trafficking of Drp1, likely by impacting the correct assembly at scission sites or the disassembly step of fission complexes. Loss of this activity may account for the observed mitochondrial division defects. Finally, MARCH5 RING mutants and endogenous Drp1, but not wild-type MARCH5 or Fis1, co-assemble into abnormally enlarged clusters in a Drp1 GTPase-dependent manner, suggesting molecular interactions among these proteins. Collectively, our data suggest a model in which mitochondrial division is regulated by a MARCH5 ubiquitin-dependent switch.
Introduction
In cells mitochondria fuse and divide continuously with balanced rates, resulting in the formation of highly dynamic reticular networks (Nunnari et al., 1997). Dynamic rearrangements of the mitochondrial network contribute to diverse cellular processes (Youle and Karbowski, 2005; Chan, 2006; McBride et al., 2006). For example, the proper regulation of mitochondrial fission and fusion rates appears to be vital for Ca2+ buffering (Szabadkai et al., 2006) and maintenance of synaptic plasticity (Li et al., 2004). Mitochondrial fragmentation and fusion inhibition are also closely associated with outer mitochondrial membrane (OMM) permeabilization and subsequent release of certain proteins from the mitochondria to the cytosol, early during apoptosis (Lee et al., 2004; Youle and Karbowski, 2005; Estaquier and Arnoult, 2007). The contribution of a balanced mitochondrial network for cellular fitness was further illuminated by reports showing that mutations of fusion proteins Opa1 and Mfn2 are associated with the neurodegenerative diseases dominant optic atrophy and Charcot-Marie-Tooth disease, respectively (for review see Chan, 2006; McBride et al., 2006).
Mitofusins (Fzo1p in yeast), Opa1 (Mgm1p in yeast), and Drp1 (Dnm1p in yeast) are large dynamin-related GTPases essential for mitochondrial fusion and fission (Otsuga et al., 1998; Labrousse et al., 1999; Mozdy et al., 2000; Santel and Fuller, 2001; Smirnova et al., 2001; Shaw and Nunnari, 2002; Chen et al., 2003; Olichon et al., 2007). In addition to Dnm1p, mitochondrial fission in yeast depends on at least three additional factors. Fis1p is an approximate 18-kD, C-tail–anchored integral protein of the OMM (Mozdy et al., 2000) with a tetratricopeptide repeat domain serving as a receptor for Mdv1p (Tieu and Nunnari, 2000; Shaw and Nunnari, 2002; Tieu et al., 2002; Suzuki et al., 2003) or its structural homologue Caf4p (Griffin et al., 2005). On mitochondria Dnm1p (as well as Drp1) localizes to relatively large punctiform structures, particularly abundant at prospective scission sites as well as at mitochondrial tips (Bleazard et al., 1999; Labrousse et al., 1999; Smirnova et al., 2001). As detailed recently (Bhar et al., 2006; Naylor et al., 2006), mitochondrial Dnm1p complexes are dynamic structures formed and maintained as a result of transient interactions of different components of the fission machinery. The N-terminal extension and the C-terminal WD-40 repeat of Mdv1p or Caf4p are thought to act as adaptors linking Fis1p and Dnm1p into the mitochondrial division process (Tieu et al., 2002; Griffin et al., 2005). In contrast to yeast, mitochondrial division in other organisms, including mammalian cells, is less well understood. As in budding yeast, mammalian cells require Drp1 (Smirnova et al., 2001) and Fis1 (Yoon et al., 2003; Stojanovski et al., 2004) for mitochondrial fission, suggesting that mitochondrial division mechanisms are to some extent conserved. However, the mechanisms of mitochondrial translocation of Drp1 as well as scission foci assembly in mammalian cells remain elusive.
The conjugation of the 76-amino acid protein ubiquitin to substrate proteins is involved in the regulation of a variety of cellular processes (Hershko and Ciechanover, 1992; Welchman et al., 2005) ranging from selective protein degradation to DNA repair (Huang and D'Andrea, 2006) and membrane protein trafficking (Staub and Rotin, 2006). Recent findings indicate that ubiquitylation plays a direct role in mitochondrial membrane remodeling (Neutzner and Youle, 2005; Escobar-Henriques et al., 2006; Nakamura et al., 2006; Yonashiro et al., 2006). For example, it has been shown that treatment with the mating pheromone α-factor induces a 26S proteasome-dependent degradation of the mitofusin Fzo1p, resulting in the fragmentation of mitochondria (Neutzner and Youle, 2005; Escobar-Henriques et al., 2006).
In this study we demonstrate that the mitochondrial E3 ubiquitin ligase MARCH5 (MarchV) (Bartee et al., 2004; Nakamura et al., 2006), also known as MITOL (Yonashiro et al., 2006), participates in the regulation of mitochondrial division.
Results
Expression of RING domain mutants of MARCH5 changes the organization of the mitochondrial network
RING finger–containing proteins comprise a large family of potential ubiquitin ligases involved in regulation of 26S proteasome-dependent degradation of misfolded and superfluous proteins (Hershko and Ciechanover, 1992) and in signal transduction in various cellular locations (Joazeiro and Weissman, 2000). Using bioinformatics combined with molecular biology techniques, we cloned several uncharacterized RING finger proteins associated with mitochondria. One of the mitochondrial proteins identified in our screen was MARCH5 (RNF153/MarchV), which has been previously described to associate with the ER (Bartee et al., 2004), and more recently shown to localize to mitochondria (Nakamura et al., 2006; Yonashiro et al., 2006). MARCH5 consists of four predicted transmembrane domains and an N-terminal RINGv-type RING finger domain critical for ubiquitin transfer activity of several E3 ubiquitin ligases (Fig. 1 A). MARCH5 was recently shown to mediate ubiquitylation of some mitochondrial proteins, including mitochondrial morphogenesis regulators Fis1 and Drp1 (Nakamura et al., 2006; Yonashiro et al., 2006). However, the role of MARCH5 in the regulation of mitochondrial network dynamics remains to be clarified.
Figure 1.

Mutations of the MARCH5 RING domain induce abnormal morphology of mitochondria. MARCH5 structure and the sequence homology of RINGv domains of various E3 ubiquitin ligases. The conserved histidine 43 (red) and cysteines 65 and 68 (blue) that are predicted to affect MARCH5 RING finger domain activity were mutated resulting in H43W and C65S,C68S mutants (A). Mitochondrial morphology in HeLa cells expressing wild type MARCH5-YFP, (green; B), MARCH5H43W-YFP (green; C and D), and MARCH5C65S,C68S-YFP (green; E) was analyzed by confocal microscopy. Mitochondria (red) were revealed by staining for cytochrome c. Number of cells with control-like tubular mitochondria, abnormally connected mitochondria, and with mitochondria predominantly localizing in perinuclear regions were scored (F). Data represent the average ± SD of four transfections per condition with 100 cells counted each time.
When fused with YFP and expressed in HeLa cells, both YFP-MARCH5 (unpublished data) and MARCH5-YFP localized to mitochondria (Fig.1 B), consistent with two recent studies that showed mitochondrial localization of endogenous MARCH5 (Nakamura et al., 2006; Yonashiro et al., 2006). We failed to detect any association of MARCH5 with the ER (unpublished data). To analyze the function of MARCH5 we mutated a conserved Zn2+-binding histidine residue inside the RING domain to tryptophan (MARCH5H43W) (Fig. 1 A). Based on the available RING finger protein structure (Katoh et al., 2003; Bottomley et al., 2005), the substitution of His-43 with tryptophan is predicted to inhibit Zn2+ coordination and therefore the overall function of the RING domain. Mitochondrial morphology in cells transiently transfected with MARCH5, or MARCH5H43W expression vectors was accessed by confocal microscopy with immunostaining of cytochrome c to visualize mitochondria (Fig. 1, B–D). As described previously (Yonashiro et al., 2006), overexpression of wild-type MARCH5 did not affect the overall organization of the mitochondrial network in various cell types as determined by cytochrome c (Fig. 1, B and F; unpublished data) and Tom20 staining patterns (unpublished data). This indicates that MARCH5 is well tolerated even at high expression levels and appears not to be a rate-limiting factor in mitochondrial morphogenesis. However, contrary to previous observations reporting fragmentation of the mitochondrial network as a consequence of MARCH5 inhibition (Nakamura et al., 2006; Yonashiro et al., 2006), the majority of cells (52 ± 8.7%) expressing MARCH5H43W displayed highly interconnected mitochondria (Fig.1, C, D, and F). The aberrant mitochondria range in morphology from thin and elongated, often forming tubule nets (Fig. 1 C), to highly interconnected thickened tubular and blebbed structures (Fig. 1 D). In cells expressing higher amounts of MARCH5H43W a further increase in the diameter and rounding of mitochondria was associated with the perinuclear accumulation of these organelles (30.5 ± 10%; Fig.1 F; Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200611064/DC1). In 17.5 ± 10.4% of MARCH5H43W-expressing cells, a control-like organization of the mitochondrial network was observed (Fig. 1 F). We also constructed another MARCH5 RING mutant by replacing cysteines 65 and 68 with serines (MARCH5C65S,C68S), as this mutation has been already shown to inhibit MARCH5 activity (Yonashiro et al., 2006). Consistent with the effect of MARCH5H43W, expression of MARCH5C65S,C68S led to the formation of interconnected mitochondria in ∼63% of cells (Fig. 1, E and F). The remaining ∼31% of cells displayed a perinuclear accumulation of mitochondria (Fig. 1 F and Fig. S1 B), or normal mitochondrial networks (6 ± 2%; Fig. 1 F). The range of mitochondrial phenotypes also resembled those induced by MARCH5H43W-YFP. The expression of a MARCH5 construct missing the entire RING domain (MARCH5Δ1-71) also led to the formation of abnormally interconnected mitochondria, although in a smaller percentage of cells (29.7 ± 5.9% cells with abnormally elongated mitochondria and 16.6 ± 8.3% with perinuclear clustering of mitochondria; Fig. 1 F; Fig. S1, C and D). These data indicate that inactivation of the MARCH5 RING domain induces an abnormal interconnection of the mitochondrial network.
MARCH5 RNAi induces mitochondrial elongation
To further test the role of MARCH5 in the regulation of mitochondrial dynamics, we applied short hairpin RNA interference (shRNAi)–mediated down-regulation of MARCH5 protein expression. HeLa cells were transfected with control RNAi vector targeting GFP (control RNAi cells; Lee et al., 2004), or a construct targeting MARCH5 (MARCH5 RNAi cells). As shown by Western blot, the MARCH5 RNAi led to a substantial down-regulation of MARCH5 expression compared with control RNAi cells (Fig. 2 A). We also tested the expression levels of Mfn2, Drp1, and Fis1, mitochondrial dynamics proteins reported to interact with MARCH5 (Nakamura et al., 2006; Yonashiro et al., 2006). Whole cell lysates from control RNAi and MARCH5 RNAi cells analyzed by Western blot show that a reduction of MARCH5 expression was not associated with apparent changes in the protein levels of Mfn2, Drp1, and Fis1 (Fig. 2 A). To analyze mitochondrial morphology, control RNAi (Fig. 2 B) and MARCH5 RNAi (Fig. 2 C) cells were immunostained for Tom20. In ∼40% of MARCH5 RNAi cells, mitochondria formed elongated and interconnected tubules (Fig. 2, C and D). In contrast to the previous reports, we did not detect a significant fragmentation of mitochondria in MARCH5 RNAi cells (5.67 ± 2.31% of MARCH5 RNAi cells, compared with 6.67 ± 4.72% of control RNAi cells; Fig. 2 D).
Figure 2.

MARCH5 RNAi-induced abnormal mitochondrial interconnection. (A) GFP RNAi or MARCH5 RNAi cells harvested 9 d after transfection with GFP- and MARCH5-silencing vectors were analyzed for MARCH5 by Western blot. The expression levels of Mfn2, Drp1, and Fis1 proteins implicated in the regulation of mitochondrial dynamics were also tested. Anti-α-tubulin antibodies were used as a loading control. (B–D) Mitochondrial morphology, as revealed by anti-Tom20 mAbs, in GFP RNAi and MARCH5 RNAi cells was analyzed by confocal microscopy. Number of cells with control-like tubular mitochondria, abnormally connected, and fragmented mitochondria was scored in GFP RNAi and MARCH5 RNAi cells (D). Data represent the average ± SD of three transfections per condition with 150 cells counted each time.
Mitochondrial network volume and connectivity in cells expressing MARCH5 RING domain mutants
To further characterize the effect of MARCH5 activity, we analyzed mitochondrial network unit size and contiguity using a mitochondrial matrix–targeted photoactivable variant of GFP (mito-PAGFP) (Karbowski et al., 2004) in cells expressing different MARCH5 constructs. To this end in double-transfected cells, 4-μm wide regions of interests (ROIs) (Fig. 3, A and B; black squares) were briefly irradiated with a 413-nm laser followed by imaging with a 488-nm laser immediately after 413-nm photoactivation (Fig. 3, A and B; middle panels). This results in a photoactivation of mito-PAGFP within the ROIs. The 488-nm confocal imaging, performed immediately after ROI photoactivation, reveals the area of mitochondrial network units located within the ROIs and those with matrix compartments contiguous with the ROIs (Karbowski et al., 2004). We found that, under the conditions applied here, in control (unpublished data) and wild-type MARCH5-expressing cells (Fig. 3 A) the activated mitochondria were usually more restricted to the photoactivated ROIs, whereas in MARCH5H43W (Fig. 3 B) and MARCH5C65S,C68S (unpublished data) expressing cells the mito-PAGFP signal redistributes further from the ROIs. This indicates an increased size of mitochondrial network units upon inhibition of MARCH5 activity. To quantify the relative area covered by ROI-activated mitochondria we also photoactivated the whole imaging fields. This results in depiction of all the mitochondria within the same cells (Fig. 3, A and B; right panels). The quantification of the ratio of area covered by ROI-activated mitochondria (r) to that covered by the whole cell mitochondria (w) revealed a significant increase in mitochondrial unit size in cells expressing MARCH5H43W (r/w = 0.595 ± 0.179) and MARCH5C65S,C68S (r/w = 0.626 ± 0.199), compared with control (r/w = 0.243 ± 0.086) and wild-type MARCH5 (r/w = 0.240 ± 0.074) expressing cells (Fig. 3 C).
Figure 3.
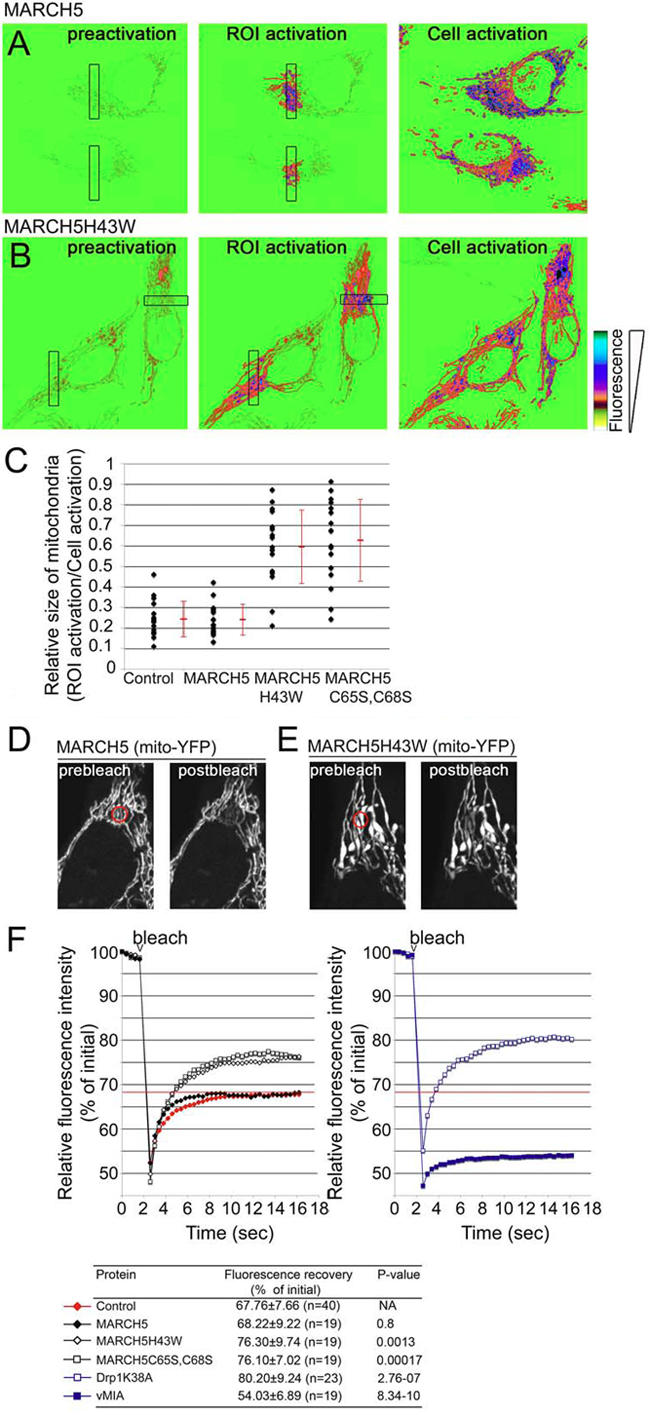
Quantification of mitochondrial volume and connectivity in cells expressing MARCH5 RING mutants. The mito-PAGFP diffusion-based assay (A–C) and FRAP (D–F) were used to investigate the relative size of mitochondrial network units in wild-type MARCH5 and MARCH5 RING mutant–expressing cells. For the mito-PAGFP based mitochondrial interconnectivity assay, cells were cotransfected with CFP-tagged WT MARCH5 (A and C), MARCH5H43W (B and C), MARCH5C65S,C68S (C) together with mito-PAGFP or transfected with mito-PAGFP alone (C). The pseudocolored images of typical cells expressing wild-type MARCH5 (A) and MARCH5H43W (B) are shown. The degree of mitochondrial interconnectivity was quantified using MetaMorph image analysis software (C). Red bars show averages ± SD of 17 measurements per experimental group, as indicated. Black diamonds represent values obtained from each single cell measurement (C). HeLa cells transiently cotransfected with CFP-tagged WT MARCH5, MARCH5H43W, MARCH5C65S,C68S-YFP together with mito-YFP or transfected with mito-YFP alone were assayed by FRAP for mitochondrial volume. The typical control, mito-YFP expressing cell (D), and cell expressing MARCH5H43W and mito-YFP (E) used in FRAP experiments are shown before and directly after photobleaching of regions of interest indicated with red rings. FRAP curves obtained after averaging of 19 single cell analysis per group, as well as the degree of FRAP in the indicated experimental groups are shown in F.
To analyze mitochondrial network complexity, we also applied FRAP, as described previously (Szabadkai et al., 2004; Karbowski et al., 2006), in cells expressing MARCH5-CFP, MARCH5H43W-CFP, or MARCH5C65S,C68S-CFP together with a mitochondrial matrix–targeted yellow fluorescent protein (mito-YFP). To measure FRAP, 3-μm ROIs (Fig. 3, D and E; red circles) in double-transfected cells were photobleached to ∼50% of the initial mito-YFP fluorescence by irradiating with a high power laser. This was followed by the analysis of the YFP fluorescence recovery every 400 msec for ∼14 s. Under conditions in which mito-YFP fluorescence in control cells recovered to 65.35 ± 7.6% of prebleach fluorescence, as accessed at the last time points of each measurement, fluorescence recoveries in MARCH5H43W and MARCH5C65S,C68S-expressing cells were significantly higher (Fig. 3 F; 76.3 ± 9.74 and 76.1 ± 7.02, respectively). Confirming the immunofluorescence results (Fig. 1, B and F), mito-PAGFP analysis (Fig. 3, A–C), and published data (Yonashiro et al., 2006), there was no statistically significant difference in FRAP between control and wild-type MARCH5-expressing cells (Fig. 3 F; 68.22 ± 9.22, P > 0.2). This demonstrates that ectopic expression of wild-type MARCH5 does not affect the overall volume of mitochondria, whereas the increased FRAP in the MARCH5 RING mutant–expressing cells reflects increased mitochondrial connectivity. We also performed control FRAP experiments in cells expressing Drp1K38A, a dominant-negative mutant of mitochondrial fission protein Drp1, or in cells transfected with the viral mitochondria-associated inhibitor of apoptosis (vMIA) (Fig. 3 F). It has been shown that Drp1K38A induces aberrant mitochondrial elongation (Smirnova et al., 2001), whereas vMIA causes mitochondrial fragmentation (McCormick et al., 2003; Karbowski et al., 2006). There was a significant increase of FRAP in Drp1K38A-expressing cells (Fig. 3 F; 80.2 ± 9.24), and a decrease in vMIA-expressing cells (Fig. 3 F; 54.03 ± 6.89). There was no significant difference between mitochondrial FRAP values in cells expressing MARCH5 RING mutants and Drp1K38A (MARCH5H43W/ Drp1K38A, P > 0.1; and MARCH5C65S,C68S/Drp1K38A, P > 0.1), leading to the conclusion that MARCH5 RING domain mutation-induced mitochondrial defects are qualitatively and quantitatively comparable to those induced by Drp1K38A, an established inhibitor of mitochondrial division (see also Supplemental text and Fig. S2 and Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200611064/DC1).
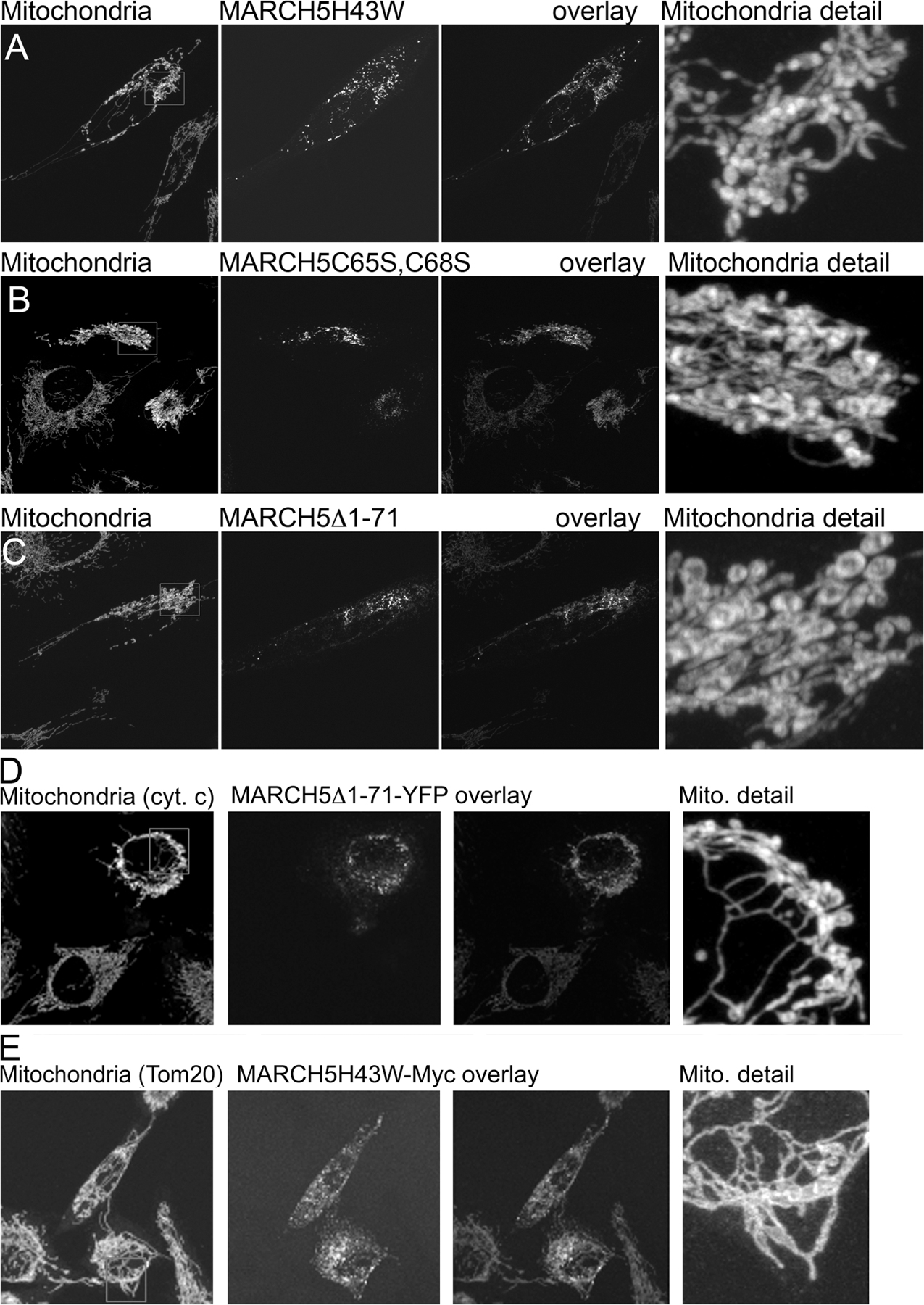
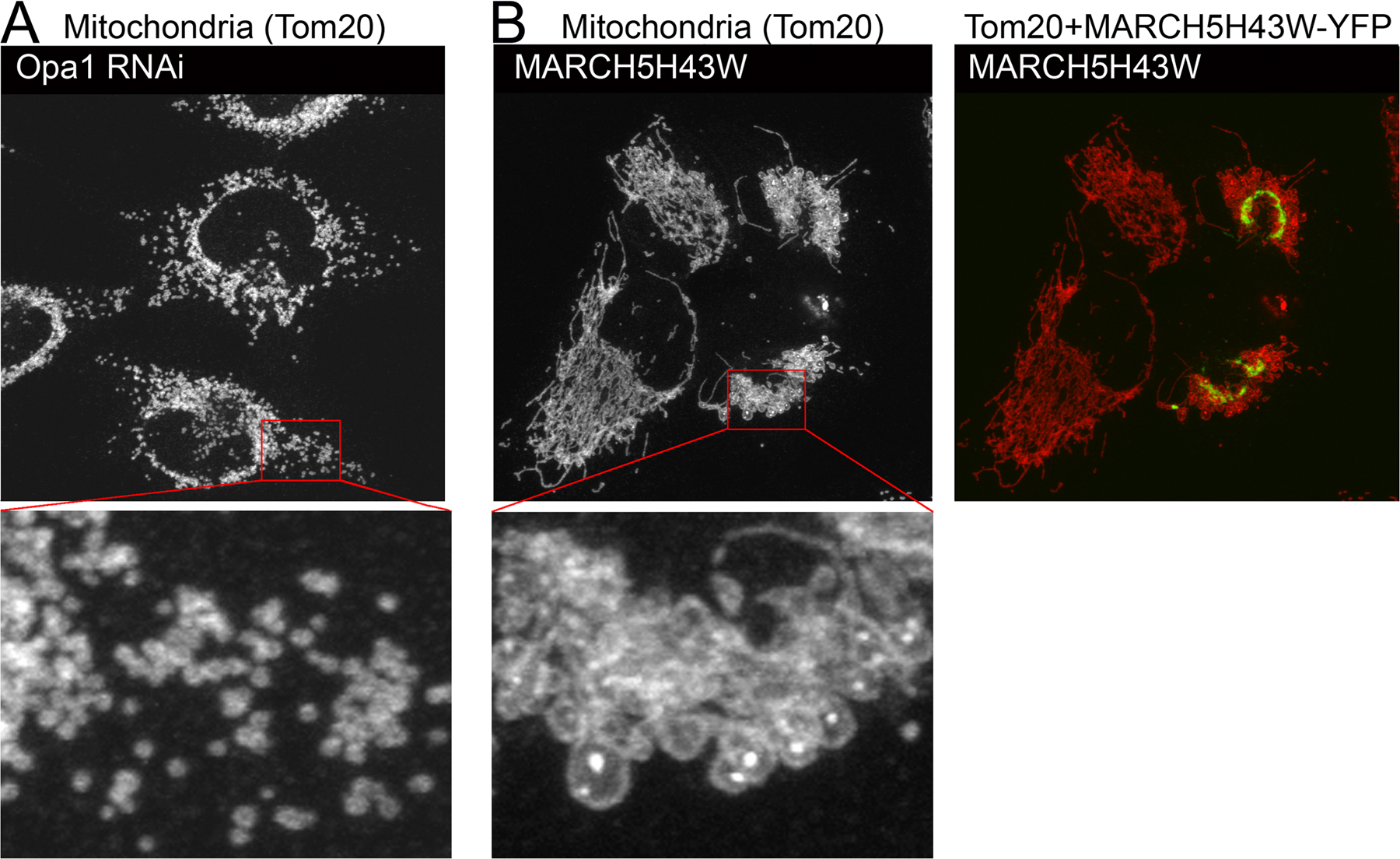
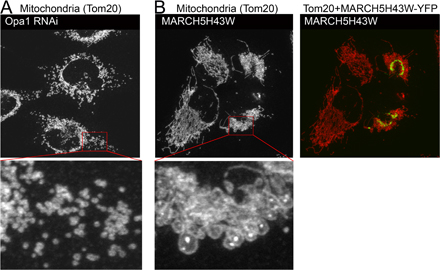
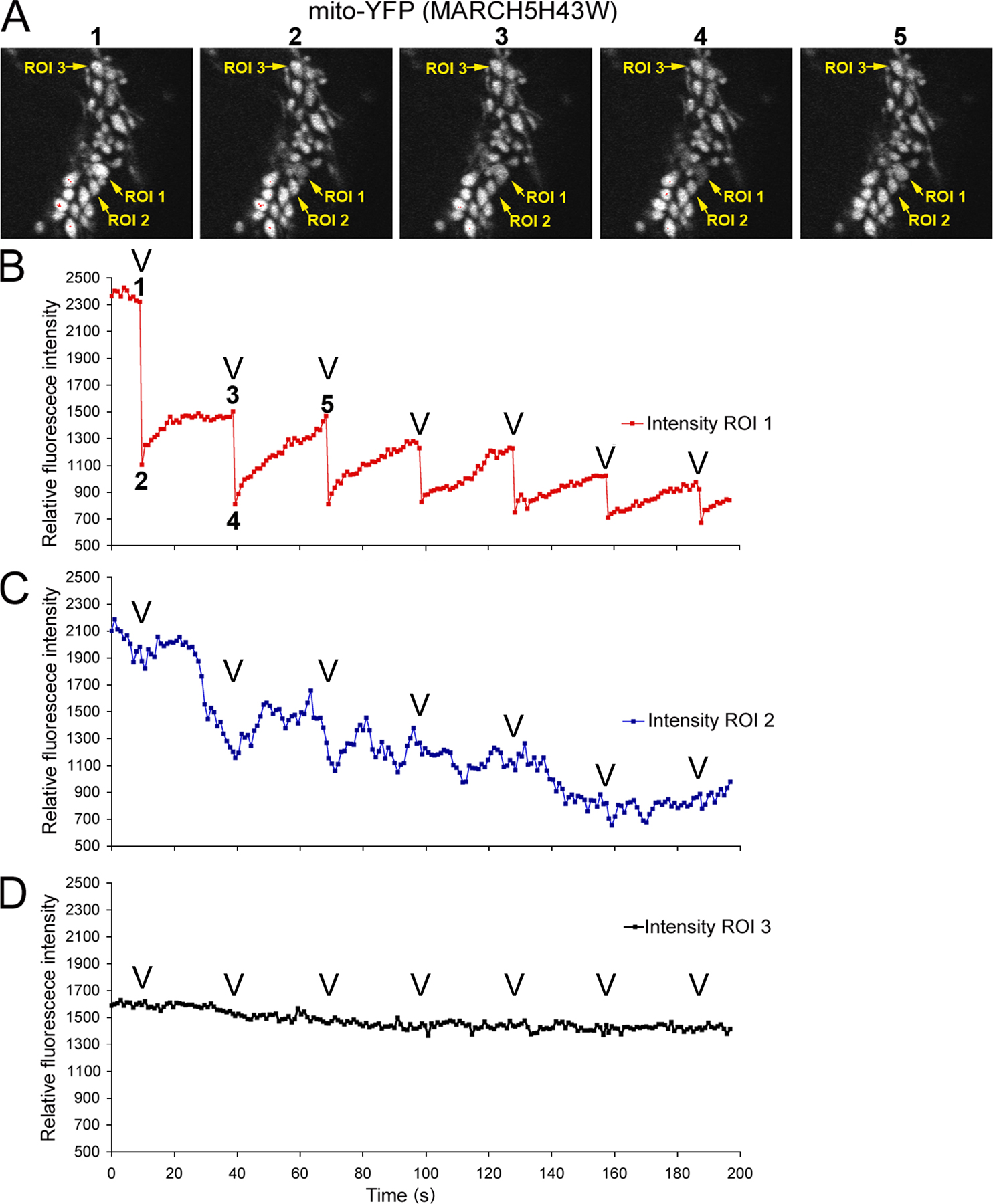
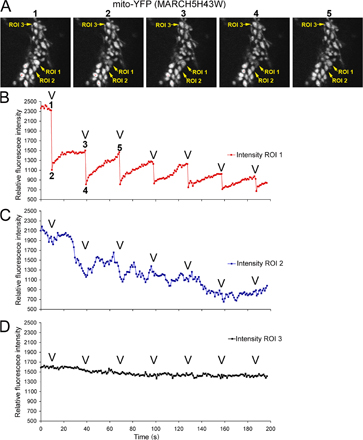
Our observations appear to contradict recently published interpretations of mitochondrial phenotypes in MARCH5 RING domain mutant–expressing cells (Nakamura et al., 2006; Yonashiro et al., 2006). It has been reported that mitochondrial fragmentation, but not elongation, is a predominant response to inhibition of MARCH5 activity. We found that in all cases analyzed here, after expression of: (1) N- and C-terminal YFP fusion of MARCH5H43W; and (2) C-terminal fusions of MARCH5C65S,C68S and MARCH5Δ1-71 (Fig. S2 A), as well as Myc-tagged MARCH5H43W (Fig. S2 B), normal morphology or an abnormal elongation and connection of mitochondria prevail in cells expressing low to moderate levels of the mutant proteins. One explanation is that the enlarged and rounded perinuclear mitochondria that are more apparent in cells producing high levels of MARCH5H43W, MARCH5C65S,C68S, and MARCH5Δ1-71 could be interpreted as “fragmented” at low magnification (Fig. S1, A–C). Yet, when compared with small vesicular mitochondria formed as a result of unbalanced division when mitochondrial fusion is impaired in Opa1 RNAi cells (Fig. S3 A) or Mfn1KO and Mfn2KO mouse embryonic fibroblasts (MEFs) (see Fig. 7), the round perinuclear organelles in MARCH5 mutant–expressing cells are much larger (Fig. S3 B). Furthermore, expression of the MARCH5 RING mutants does not cause an obvious increase in mitochondrial number (Fig. S3, A and B; unpublished data), expected upon activation of mitochondrial division, suggesting that mitochondrial fission does not cause the MARCH5 inhibition-induced mitochondrial aberrations. In fact, it has been demonstrated, both in mammalian cells (Smirnova et al., 2001) and in yeast (Griffin et al., 2005), that strong fission defects can lead to the perinuclear collapse and a rounding of mitochondria, whereas lesser inhibition of this process induces the formation of spread, net-like mitochondria. Taking into consideration that only relatively high levels of MARCH5 mutant expression can stimulate perinuclear collapse and rounding of mitochondria (unpublished data), one can picture that the degree of fission in MARCH5H43W or MARCH5C65S,C68S transfected cells is inversely proportional to the expression levels of the mutant proteins, as expected to occur in the case of dominant-negative mutants. As a consequence of this relation, in the most extreme cases mitochondrial alterations comparable to those induced by “strong” dominant-negative mutants of Drp1 may take place (Smirnova et al., 2001). We therefore attempted to test whether round collapsed mitochondria in MARCH5 mutant–expressing cells are separate units or whether they form aberrant but interconnected mitochondrial networks. To attain this we used another FRAP-based approach (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200611064/DC1). Multiple cycles of bleach (marked with “V” in Fig. S4, B–D) and fluorescence recovery were applied to seemingly separate round mitochondria (ROI 1 in Fig. S4 A; bleached area diameter ∼2 μm), while avoiding direct photobleaching of the adjacent mitochondria. Changes of fluorescence intensities of the photobleached ROI 1 (Fig. S4 B), a directly adjacent mitochondrion (ROI 2; Fig. S4 C), and a mitochondrion located relatively far from the bleached area (ROI 3; Fig. S4 D) were plotted as a function of time. We found that efficient fluorescence recoveries in ROI 1 occur after each bleach that were associated with a gradual depletion, but also slightly delayed fluorescence recovery in ROI 2 and little change in ROI 3. From these data we can conclude that an exchange of mito-YFP occurs between ROI 1 and ROI 2, and that the mitochondrion in ROI 3 is separate from ROIs 1 and 2. Moreover, because the mitochondrion located in ROI 2 also recovers mito-YFP fluorescence, it most likely is also connected with another mitochondria. Because mitochondrial matrix–targeted GFP can diffuse freely with the rate of 15–19 msec (Partikian et al., 1998) in normal tubular mitochondria in HeLa cells, the delayed response of ROI 2 indicates that it is likely connected with a mitochondrion located within ROI 1 by abnormally thin mitochondrial tubules. This is consistent with the observations that seemingly separate, round mitochondria visible in Dnm1p mutant Caenorhabditis elegans are able to exchange matrix-targeted GFP (Labrousse et al., 1999). In sum, based on the data discussed above, we conclude that in cells expressing MARCH5H43W and MARCH5C65S,C68S an abnormal interconnection of mitochondria is the main morphological manifestation and that inhibition of MARCH5 activity increases the volume and decreases the number of mitochondrial units within the network. This points to a possible inhibition of mitochondrial fission or activation of fusion by the loss of MARCH5 activity.
Figure 7.

Expression of MARCH5 RING mutants restores tubular mitochondria in Mfn1KO and Mfn2KO cells. (A) Expression levels of endogenous Mfn2 in wild-type MARCH5 and MARCH5H43W-expressing cells was analyzed by Western blot. The whole cell lysate (WCL) and mitochondria-enriched heavy membrane (HM) fractions were obtained from HeLa cells incubated in the presence and absence of MG132 and transfected with Myc-tagged MARCH5 and MARCH5H43W. Proteins were resolved using SDS-PAGE and then immunostained with anti-Mfn2 polyclonal antibodies. The loading was controlled by immunostaining with anti-Hsp60 antibodies. Mfn2KO cells transfected with MARCH5-YFP (B; yellow on overlay image) or MARCH5H43W-YFP (C and D; green on overlay images) were stained with anti-cytochrome c mAbs to reveal mitochondria (red on overlay images) and analyzed by confocal microscopy. In E, cells were scored for the morphology of mitochondria. Data represent averages ± SD of three transfections with 100 cells counted each time.
Submitochondrial coalescence of MARCH5 RING domain mutants
During the analysis of mitochondrial morphology in wild-type MARCH5-YFP, MARCH5H43W-YFP, and MARCH5C65S,C68S-YFP expressing cells we noticed that while wild-type MARCH5 decorated mitochondria uniformly (Fig. 1 B), MARCH5H43W (Fig. 1, C and D; Fig. 4 A) and MARCH5C65S,C68S (Fig. 1 E) localized mainly to submitochondrial aggregates with some diffusely circumscribing the OMM. To quantify the degree of MARCH5 association with the OMM, cells were transfected with wild-type MARCH5-YFP, MARCH5H43W-YFP, or MARCH5C65S,C68S-YFP followed by immunostaining for Tom20 and confocal image acquisition (Fig. 4 A). The quantification of the colocalization of green channel pixels (MARCH5 and MARCH5 RING mutants) with red channel pixels (Tom20) was performed using the “colocalization” function of the image analysis software Volocity. The association degree of different variants of MARCH5 with the OMM is depicted as a Pearson's Correlation coefficient (r) (Fig. 4 B) where the r-values between −0.3 ± 0.3 indicate little or no association, +0.3 ± 0.7 weak positive association, and +0.7 ± 1.0 strong positive association. Using this analysis we found that MARCH5-YFP, which localizes to the OMM in a diffuse manner with only occasional submitochondrial coalescence and extramitochondrial localization, shows a high degree of association with the OMM (r = 0.77 ± 0.03; Fig. 4 B). MARCH5H43W-YFP (r = 0.46 ± 0.11; Fig. 4, A and B) and MARCH5C65S,C68S-YFP (r = 0.36 ± 0.09; Fig. 4 B) display a distinctly lower degree of association with the OMM, indicating that mutations perturbing the activity of the MARCH5 RING domain lead to a redistribution of this protein in the OMM.
Figure 4.

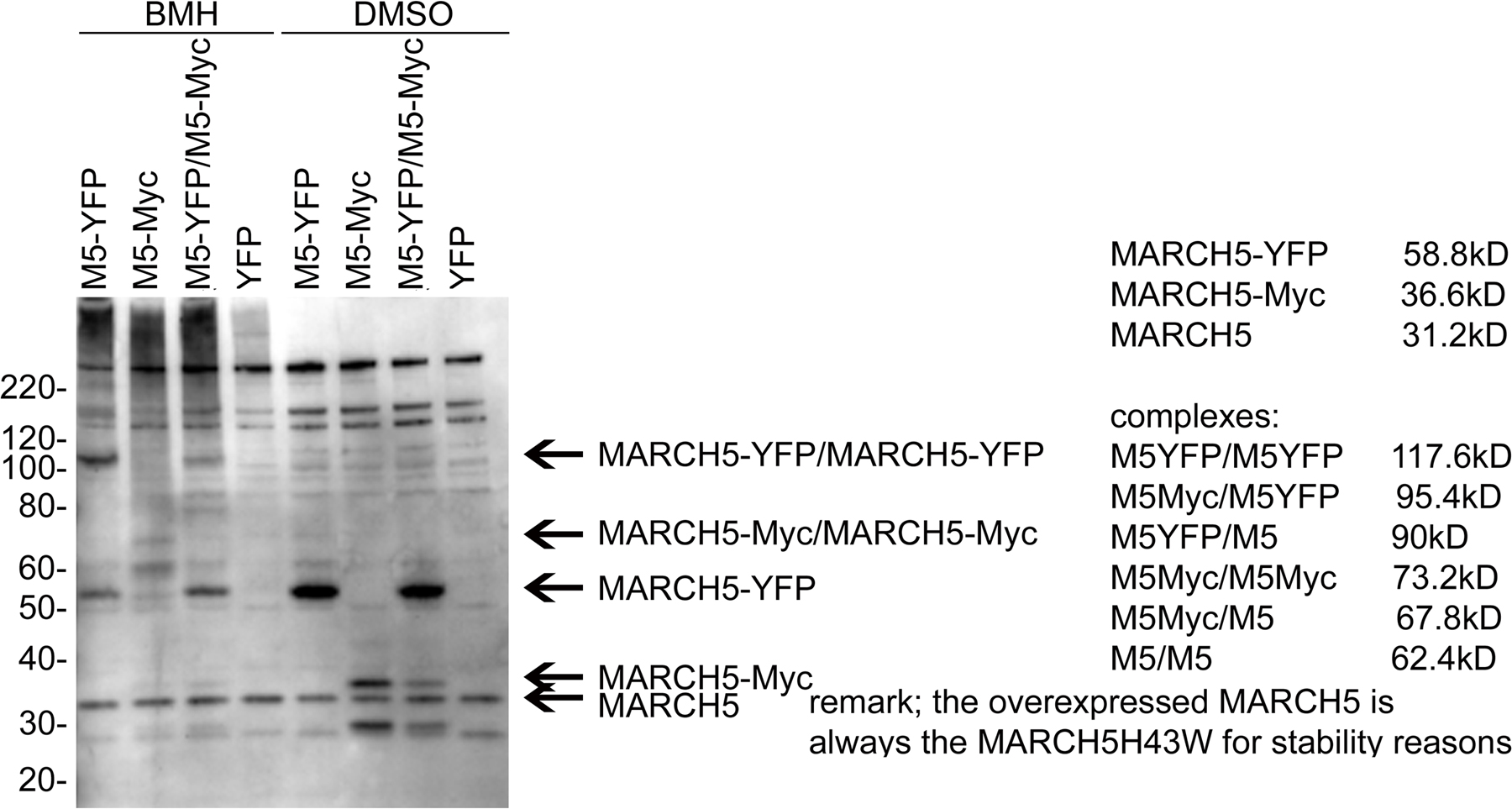
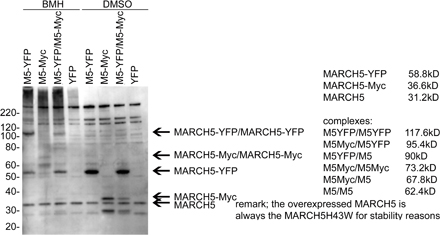
Submitochondrial coalescence of MARCH5 RING mutants. HeLa cells expressing wild-type MARCH5-YFP, MARCH5H43W-YFP, and MARCH5C65S,C68S-YFP were immunostained for Tom20, and analyzed by confocal microscopy. An example of a cell expressing MARCH5H43W-YFP (green) and immunostained for Tom20 (red) that was used for the analysis is shown in A. The colocalization degree of Tom20 with MARCH5 proteins was performed using the image analysis software Volocity (B). The values shown in B represent Pearson's Correlation units (r) that reveal the degree of association of pixels in different channels of the confocal image. (C) The oligomeric states of Myc-tagged wild-type MARCH5 and MARCH5H43W were analyzed by Western blot in cells treated with the chemical cross-linker BMH or with the solvent DMSO. Mitochondria- enriched heavy membrane fractions (HM) were subjected to SDS-PAGE and Western blot analysis. MARCH5 was detected with anti-Myc mAbs. The loading was controlled with anti Hsp60 mAbs. Asterisk represents non-specific band.
Next, we studied whether inhibition of the MARCH5 RING domain may affect oligomerization of this protein that may contribute to the focal appearance on mitochondria. To achieve this, mitochondria-enriched heavy membrane fractions (HM) were treated with the chemical cross-linker Bis- Maleimidohexane (BMH) and analyzed by Western blot for Myc-tagged MARCH5 (Fig. 4 C). We found, in contrast to wild-type MARCH5, which was mostly detected as a 33-kD monomer and an ∼65-kD form, that MARCH5H43W also formed higher molecular weight complexes. The ∼65-kD form of MARCH5 most likely represents homodimers of this protein (Fig. S4). The molecular components of the high molecular weight complexes containing the MARCH5 RING mutant are currently unknown. However, most likely these structures contribute to the formation of MARCH5 RING mutant submitochondrial foci. This difference in submitochondrial localization and oligomerization of the MARCH5 RING mutants compared with the wild-type protein suggests that protein complexes containing inactive MARCH5 and its substrates are stabilized and/or abnormally enlarged, likely due to perturbed ubiquitylation.
MARCH5 RING domain mutations lead to abnormal mitochondrial assembly of the fission protein Drp1
We investigated whether certain OMM-associated proteins, including those participating in mitochondrial morphogenesis, also accumulate in MARCH5H43W- and MARCH5C65S,C68S- enriched mitochondrial aggregates. Cells transfected with MARCH5-YFP and MARCH5H43W-YFP, in the presence or absence of cotransfection with mammalian expression vectors encoding Myc-tagged integral membrane proteins of the OMM, OMP25 and Fis1, were immunostained with anti-Myc antibodies or antibodies detecting endogenous Tom20, Tom22, endophilin B1, and Drp1. Although changes in the distribution of Tom20 (Fig. 4 A), Tom22, OMP25 and endophilin B1, as well as YFP-tagged mitochondrial fusion proteins Mfn1 and Mfn2 were not noticeable (unpublished data), an abnormal localization pattern of the fission protein Drp1 was apparent (Fig. 5, A–B′). In the most extreme cases, occurring in cells with a high degree of MARCH5H43W-YFP expression, formation of extremely enlarged perinuclear clusters of Drp1 was observed (Fig. S5 A, available at http://www.jcb.org/cgi/content/full/jcb.200611064/DC1). Consistent with MARCH5H43W, expression of MARCH5C65S,C68S-YFP also induced abnormal mitochondrial clustering of Drp1 (Fig. 5, C and C′). Furthermore, confocal analysis (Fig. 5, B–C′) and quantification of Pearson's Correlation values (Fig. 5 D) of cells expressing MARCH5H43W-YFP or MARCH5C65S,C68S-YFP and stained for Drp1 revealed clear colocalization of these proteins (Fig. 5 D), suggesting that MARCH5 participates in the regulation of Drp1. However, the fission protein Fis1 (Yoon et al., 2003; Stojanovski et al., 2004) did not localize to these MARCH5/Drp1-positive structures (Fig. S5 B). Because yeast Fis1p is required to recruit Dnm1p early during mitochondrial scission (Shaw and Nunnari, 2002), MARCH5 could be involved in mitochondrial scission downstream of human Fis1.
Figure 5.

Drp1 colocalizes with mitochondrial complexes of MARCH5 RING mutants. Cells transfected with MARCH5-YFP (A; green), MARCH5H43W-YFP (B; green) and MARCH5C65S,C68S-YFP (C; green) were immunostained for Drp1 (red) and analyzed by confocal microscopy. The inserts in A–C show magnified fragments (white squares) of the respective cells. Relative fluorescence intensities of Drp1 (red lines) and MARCH5 variants (green lines) located in cell areas indicated in the respective images were measured using Zeiss LSM510 software (A′–C′). Note the high degree of the MARCH5 and Drp1 linescan overlap in MARCH5 RING mutant–expressing cells. The overall degree of Drp1 colocalization with different variants of MARCH5 was analyzed using the colocalization function of Volocity software (D). Values shown represent Pearson's Correlation units (see text). Drp1 RNAi (E, F, and H) and control RNAi (G and H) cells obtained as described previously (Lee et al., 2004), were transfected with MARCH5-YFP (E and H) and MARCH5H43W-YFP (F–H), immunostained for Tom20 to reveal the OMM (red in overlay images) and analyzed by confocal microscopy. The bottom panels in E and F show magnified fragments of cells indicated by red squares. Arrows indicate peroxisomes, arrowheads mitochondria. The association of MARCH5 and MARCH5H43W with Tom20 in control RNAi and Drp1 RNAi cells was quantified (H). The degrees of association are shown as Pearson's Correlation units (r).
Membrane translocation of the Drp1 homologue, dynamin, and dynamin-mediated endocytic vesicle scission are regulated by binding and hydrolysis of GTP (Hinshaw, 2000; Praefcke and McMahon, 2004). Therefore, we tested whether Drp1 colocalization with MARCH5 depends on Drp1 GTPase activity. Cells were cotransfected with MARCH5H43W-YFP and GTPase- inactive Drp1K38A, immunostained for Drp1 and analyzed by confocal microscopy. As shown before (Yoon et al., 2001), unlike overexpressed wild-type Drp1 that correctly localizes to small submitochondrial foci (Smirnova et al., 2001; Karbowski et al., 2002), Drp1K38A forms enlarged aggregates that are only partly associated with mitochondria. We found that these Drp1K38A containing structures do not colocalize with MARCH5H43W-YFP (Fig. S5 C), whereas overexpressed wild-type Drp1 does (unpublished data). This suggests that the GTPase activity of Drp1 is required for its translocation to MARCH5H43W-YFP–enriched complexes on the OMM. Interestingly, there was no detectable effect of Drp1K38A on the submitochondrial localization of wild-type MARCH5-YFP and MARCH5H43W-YFP. In contrast, knockdown of Drp1 expression resulted in the redistribution of MARCH5H43W-YFP from foci (Fig. 5 G) into a more diffuse mitochondrial localization (Fig. 5, E and F) that colocalized with Tom20 to a significantly greater extent than when expressed in control RNAi cells (Fig. 5 H). This demonstrates that MARCH5H43W-YFP clusters are to some extent Drp1 dependent. However, MARCH5 RNAi did not induce abnormal distribution of Drp1, as revealed by immunofluorescence and Western blot (unpublished data). Collectively, these data suggest a functional interdependence between MARCH5 and Drp1.
MARCH5 RING domain mutants stabilize mitochondrial fission complexes
The data showing that expression of MARCH5H43W or MARCH5C65S,C68S has a strong impact on the localization of Drp1, and that Drp1 expression impacts MARCH5H43W distribution, suggest that MARCH5 is involved in the regulation of the mitochondrial fission complexes. The abnormal organization of Drp1 on mitochondria in MARCH5H43W and MARCH5C65S,C68S, but not wild-type MARCH5-expressing cells (Fig. 5, A–D), suggests that the transition between certain mitochondrial steps of the Drp1-dependent fission cycle is blocked by the inhibition of MARCH5 activity. In mammalian cells as well as in yeast, Drp1/Dnm1p cycles between the cytosol and mitochondria (Smirnova et al., 2001; Legesse-Miller et al., 2003). It is possible that the formation of abnormal fission complexes in response to the expression of MARCH5 mutants is caused by changes in the subcellular dynamics of Drp1. To test this hypothesis, the mobility of YFP-Drp1 in cells expressing MARCH5-CFP, MARCH5H43W-CFP, and MARCH5C65S,C68S-CFP (MARCH5/Drp1 plasmid ratio 5:1) or in control cells was analyzed by FRAP (Fig. 6 A). We confirmed that, like endogenous Drp1 (Fig. 5 A), the subcellular localization of YFP-Drp1 is altered by the expression of MARCH5 RING mutants and that the YFP tag does not influence colocalization of these proteins (unpublished data). To measure FRAP, a 4-μm strip through YFP-Drp1–expressing cells was photobleached by irradiation with a high power laser and subsequent fluorescence recovery was monitored over ∼20 s. The recovery curves were normalized, with the last time point values of the fastest recovering wild-type MARCH5 taken as 100%, and immediate postbleach values of each sample (1.06 s postbleach) as 0%. As shown in Fig. 6 A, a distinct decline in the mobility of assembled complexes of YFP-Drp1 in cells expressing MARCH5H43W or MARCH5C65S,C68S compared with YFP-Drp1, or Drp1 and wild-type MARCH5 cotransfected cells was evident.
Figure 6.

MARCH5 RING mutations decrease cellular mobility of Drp1. HeLa cells transiently transfected with YFP-Drp1 with or without cotransfection with MARCH5-CFP, MARCH5H43W-CFP, and MARCH5C65S,C68S-CFP were analyzed for cellular mobility of Drp1 with the FRAP assay (A). The averaged recovery curves and the FRAP values quantified at 1.73 s and 19.66 s after photobleach in the indicated experimental groups are shown at the bottom (A). The degree of Drp1 association with mitochondria was analyzed by Western blot (B). The whole cell lysate (WCL) and mitochondria- enriched heavy membrane (HM) fractions were obtained from HeLa cells transfected with Myc-tagged, wild-type MARCH5 and MARCH5H43W. Proteins were resolved using SDS-PAGE and then immunostained for Drp1. The loading was controlled with anti-Hsp60 antibodies. The HM fractions obtained from cells expressing Myc-tagged, wild-type MARCH5, and MARCH5H43W were also treated with a chemical cross-linker BMH, or with solvent DMSO. The samples were analyzed by Western blot for Drp1 (C).
We also tested the effects of MARCH5 activity on the expression level, mitochondrial association (Fig. 6 B), and the oligomerization (Fig. 6 C) of endogenous Drp1. Whole cell lysates (WCL) and heavy membrane (HM) fractions obtained from HeLa cells expressing Myc-tagged, wild-type MARCH5 or MARCH5H43W were analyzed by Western blot (Fig. 6 B). The data show that, while the total expression level of Drp1 is not influenced by MARCH5 RING domain activity, the quantity of Drp1 associating with mitochondria is slightly increased in MARCH5H43W-expressing cells and, to a lesser degree, in cells expressing wild-type MARCH5 (Fig. 6 B). The WCL and HM fractions used in these analyses were obtained. Thus, one can conclude that the MARCH5 RING mutants affect the cellular dynamics of Drp1, but not the degradation of this protein (see also Fig. 2 A). Next, to reveal the oligomeric state of mitochondria-associated Drp1, aliquots of the HM fractions were also treated with BMH (Fig. 6 C). Using this approach, we did not detect any clear alterations in the Drp1 oligomerization. These data suggest that the organization of the subunits forming the light microscopy-detectable Drp1 complexes is not influenced by MARCH5 activity.
In sum, we conclude that the cellular mobility of Drp1, but not proteasome-dependent degradation of this protein is regulated by MARCH5 activity.
MARCH5 RING mutants reverse mitochondrial defects in Mfn1 and Mfn2 knock-out cells
It has been reported that in addition to Drp1 and Fis1 (Nakamura et al., 2006; Yonashiro et al., 2006), proteins required for mitochondrial division, MARCH5 also coimmunoprecipitates with Mfn2 (Nakamura et al., 2006), a mitochondrial fusion protein. Therefore, we analyzed the effect of wild-type MARCH5 and MARCH5H43W expression on the proteasome-dependent degradation of Mfn2 (Fig. 7 A). To inhibit a proteasome-dependent degradation of Mfn2, cells were treated with the proteasome inhibitor MG132. Mitochondria-enriched HM fractions were obtained from MG132-treated or untreated HeLa cells expressing Myc-tagged, wild-type MARCH5 or MARCH5H43W. Although proteasomal activity is required for Mfn2 degradation, MARCH5 does not influence Mfn2 levels in either MG132-treated or untreated cells (Fig. 7 A).
The relative contribution of MARCH5 to mitochondrial division and fusion is not known. By analyzing whether wild-type MARCH5 and MARCH5 RING mutants can complement mitochondrial defects in Mfn1 (Mfn1KO) and Mfn2 (Mfn2KO) knock-out MEFs, we tested the degree to which MARCH5 activity influences mitochondrial fission versus fusion. It has been shown that the abnormally fragmented mitochondria in Mfn1KO and Mfn2KO MEFs are formed as a result of reduced fusion and unrestrained fission of these organelles (Chen et al., 2003). The ectopic expression of Drp1K38A, a strong inhibitor of mitochondrial division, reverses the mitochondrial defects in Mfn1KO and Mfn2KO cells (Chen et al., 2003). This indicates that the residual fusion of mitochondria in these cells is sufficient to rebuild tubular mitochondria. We found that, in contrast to wild-type MARCH5 (Fig. 7, B and E), when expressed in Mfn1KO or Mfn2KO cells, both MARCH5H43W (Fig. 7, C–E) and MARCH5C65S,C68S (Fig. 7 E) distinctly reversed mitochondrial fragmentation, inducing the formation of normal reticular mitochondria (Fig. 7, C and E) or, in the most extreme cases, induced abnormally interconnected organelles (Fig. 7, D and E). The MARCH5 RING mutant–induced reconstitution of reticular mitochondria in Mfn1KO and Mfn2KO cells indicates that the activity of the MARCH5 RING domain is a strong determinant of mitochondrial membrane dynamics.
Because MARCH5 coimmunoprecipitates with the mitochondrial fusion protein Mfn2 (Nakamura et al., 2006), it is possible that, due to a partial functional redundancy between Mfn1 and Mfn2 (Chen et al., 2003), MARCH5 RING mutants may stimulate the activity of Mfn2 in Mfn1KO cells, and Mfn1 activity in Mfn2KO cells. Therefore, we tested the relative contribution of mitochondrial fusion to the MARCH5 RING mutant– induced elongation of Mfn2KO cell mitochondria (Fig. 8). We measured mitochondrial fusion in control Mfn2KO cells (Fig. 8, A and D), and in Mfn2KO cells expressing MARCH5H43W (Fig. 8, C and D) using a mito-PAGFP-dilution–based assay (Karbowski et al., 2004). As a positive control we used Mfn2KO cells ectopically expressing Mfn2 (Fig. 8, B and D). The relative rates of mitochondrial fusion were quantified at different time points after ROI photoactivation, as indicated (Fig. 8). At 45 min after ROI photoactivation, mito-PAGFP fluorescence dilution in Mfn2KO MEFs was dramatically increased by ectopic expression of Mfn2 (Fig. 8, A, B, and D; Δmito-PAGFP fluorescence ∼28.5% in Mfn2KO MEFs, compared with Δmito-PAGFP fluorescence ∼76% in Mfn2KO MEFs expressing Mfn2; P = 1.176 × 10−18). These data indicate that the mitochondrial fusion in Mfn2KO cells was reconstituted by the reexpression of Mfn2. In contrast, mitochondrial fusion in Mfn2KO MEFs was only slightly improved by the expression of MARCH5H43W (Fig. 8, A, C, and D; at 45 min after photoactivation Δmito-PAGFP fluorescence ∼39.7% in Mfn2KO MEFs expressing MARCH5H43W; P = 0.026). These MARCH5H43W-induced improvements of mitochondrial fusion in Mfn2KO MEFs suggest that to some degree mitochondrial fusion could be regulated by MARCH5 activity. However, we found that there is no additional affect of MARCH5 RING mutants on mitochondrial morphology in Drp1 RNAi cells (Fig. 5 F), whereas overexpression of Mfn2 dramatically elongates mitochondria in Drp1 activity-deficient cells (Santel and Fuller, 2001). Thus, one can conclude that MARCH5 RING mutants reconstitute tubular mitochondria in Mfn1KO MEFs and Mfn2KO MEFs mainly through their inhibition of mitochondrial fission.
Figure 8.

Effect of MARCH5H43W on mitochondrial fusion rates in Mfn2KO cells. Mitochondrial fusion in Mfn2KO MEFs (A and D), Mfn2KO MEFs ectopically expressing Mfn2 (B and D), or MARCH5H43W-CFP (C and D) was analyzed using mito-PAGFP dilution based assay. Details of the cell areas containing photoactivated mito-PAGFP (red squares) are shown below the respective images. The images of cells before photoactivation were obtained with the higher detector gain. The dilution rates of mito-PAGFP over the time of the experiment were quantified (D). Data was normalized, with the mito-PAGFP fluorescence at 30 s after photoactivation used as 100%. Data represent averages ± SD of 62 (Mfn2KO cells), 53 (Mfn2KO cells expressing MARCH5H43W), and 32 (Mfn2KO cells ectopically expressing Mfn2) ROI measurements (D).
Ectopic expression of Drp1 compensates for MARCH5 RING domain mutation–induced mitochondrial defects
Altogether, the data presented above suggest that the abnormal mitochondrial interconnection in MARCH5H43W or MARCH5C65S,C68S expressing cells is due to an inhibition of Drp1-dependent mitochondrial fission. Therefore, we tested whether the ectopic expression of Drp1 could reverse this mitochondrial phenotype. Cells were cotransfected with MARCH5 (Fig. 9 C) or MARCH5 RING mutants (Fig. 9, A– C; green on overlay images) as well as Drp1 (MARCH5/Drp1 molar ratio 1:5), followed by immunostaining for Drp1 (red on overlay images) and Tom20 (blue on overlay images), to reveal mitochondrial morphology. Cells expressing elevated levels of Drp1 together with different variants of MARCH5 (Fig. 9, A and B, labeled with asterisk) were scored for mitochondrial morphology (Fig. 9 C). We found a substantial reversal of the aberrant mitochondrial phenotypes in cells cotransfected with MARCH5 mutants and Drp1. In 64.25% of the cells expressing MARCH5H43W and 65.5% of cells expressing MARCH5C65S,C68S together with elevated levels of Drp1, mitochondrial network organization resembled that of control cells. The overexpression of Drp1 alone only slightly affected mitochondrial structure (Fig. 9 C), inducing mitochondrial fragmentation in <10% of the cells. This indicates that ectopic expression of Drp1 specifically compensates for the MARCH5 RING mutant–induced alterations of mitochondria and suggests further that MARCH5 RING mutants may compete with Drp1 for certain factors involved in mitochondrial fission. Because expression of the mitochondrial fission protein Fis1 (Yoon et al., 2003; Stojanovski et al., 2004) does not reverse mitochondrial alterations induced by expression of MARCH5 RING mutants (unpublished data; Fig. 5 E) we conclude that MARCH5 regulates mitochondrial fission downstream of Fis1. This conclusion is also supported by the fact that expression of MARCH5 RING mutants had no detectable impact on the submitochondrial distribution of Fis1 (Fig. S5 B).
Figure 9.

Ectopic expression of Drp1 reverses MARCH5 mutation-induced mitochondrial morphology defects. HeLa cells were transiently transfected with MARCH5-YFP, MARCH5H43W-YFP (A; green), and MARCH5C65S,C68S-YFP (B; green) in the presence of cotransfection with Drp1 and then immunostained with anti-Tom20 polyclonal antibodies (A and B; blue) and with anti-Drp1 mAbs (A and B; red). Asterisk represents the transfected cell in the field. Cells were scored for the morphology of mitochondria. Data represent averages ± SD of four transfections with 100 cells counted each time (C).
Discussion
The data described in this study indicate that MARCH5, an integral ubiquitin E3 ligase of the OMM, is required for mitochondrial division in mammalian cells. This conclusion is based on the following evidence: (1) Expression of MARCH5 RING mutants induces abnormal interconnection of mitochondria, as well as an increase in size, and decrease in the number of mitochondrial network units. (2) MARCH5 RNAi induces mitochondrial elongation. (3) The subcellular distribution and mitochondrial association of Drp1, a protein essential for mitochondrial division, is significantly influenced by MARCH5 RING domain activity and the localization of MARCH5 RING mutants on mitochondria is altered by knockdown of Drp1 expression. Moreover, MARCH5 RING mutants colocalize with Drp1-enriched mitochondrial foci. (4) Ectopic expression of Drp1 reverses the MARCH5 RING mutant–induced mitochondrial abnormalities, suggesting strong functional interdependence of MARCH5 and Drp1. (5) MARCH5 RING mutants reverse mitochondria defects in Mfn1KO and Mfn2KO MEFs, most likely through the inhibition of mitochondrial division.
The data showing the formation of abnormally enlarged clusters of Drp1, mitochondrial accumulation of Drp1, as well as an inhibition of Drp1 mobility in MARCH5 RING mutant–expressing cells support the hypothesis that MARCH5 is a positive regulator of mitochondrial division. Since it has been reported before that MARCH5 can be coimmunoprecipitated only with the ubiquitylated form of Drp1 (Nakamura et al., 2006), the mitochondrial scission complexes are likely regulated by MARCH5-mediated conjugation of ubiquitin. Consistent with this, a reduction of MARCH5 ubiquitin ligase activity apparent after expression of MARCH5 RING mutants or by RNAi leads to major defects in mitochondrial division. The opposite phenotype, mitochondrial fragmentation, could be expected to occur if MARCH5 mediated degradation of Drp1, Fis1, or any positive regulator of mitochondrial division. The data showing no clear effect of the MARCH5 activity on the degradation of Drp1 or Mfn2, as well as the rescue of the fragmented mitochondria in Mfn1KO and Mfn2KO MEFs by MARCH5 RING mutants, support the model of a “molecular switch”. In this scenario, MARCH5 could be essential for the activity of mitochondrial fission complexes, for example by affecting Drp1 GTPase activity or influencing protein interactions within the mitochondrial fission machinery. Increased colocalization of MARCH5 RING mutants and Drp1, as well as abnormal mitochondrial association of Drp1 that could occur due to inhibition or delay of a specific step of the fission reaction, perhaps via perturbed ubiquitylation of an essential fission protein, further support this hypothesis. Yet, it cannot be excluded that MARCH5 regulates the stability of a currently unknown protein repressor of mitochondrial fission. The abnormal accumulation of such a factor in MARCH5 RING mutant–expressing cells could lead to the inhibition of fission complexes and elongation of mitochondria.
It has been proposed that conjugation of the ubiquitin-like molecule SUMO-1 stabilizes Drp1 in mammalian cells (Harder et al., 2004), especially in the context of apoptosis induction (Wasiak et al., 2007). This implies that conjugation of SUMO-1 could be the main determinant of Drp1trafficking onto or off mitochondria, however participation of a ubiquitin-dependent pathway, as well as yet-uncharacterized E3 ubiquitin ligases, cannot be excluded. Indeed, we have identified several novel RING finger proteins that localize to the mitochondria (unpublished data), some of which could mediate ubiquitin-dependent degradation of proteins from the OMM, including mitochondrial morphogenesis factors. However, as specific Lys residues of the target proteins may be modified by ubiquitin, as well as SUMO, resulting in different functional consequences, it is also possible that both sumoylation and ubiquitylation participate in regulation of Drp-1–dependent mitochondrial fission.
Materials and methods
Cell culture and transfection
HeLa cells (American Type Culture Collection) were cultured in complete Dulbecco's minimum essential medium supplemented with 10% heat- inactivated fetal calf serum, 100 U/ml penicillin, and 100 μg/ml streptomycin in 5% CO2 at 37°C. Wild-type, Mfn1KO, and Mfn2 KO cells were cultured as described previously (Karbowski et al., 2004, 2006). Transfections were performed using FuGENE 6 (Roche) according to the manufacturer's instructions.
Mammalian expression vectors, mutagenesis, and RNA interference
Human MARCH5 (GenBank/EMBL/DDBJ accession no. NM_017824) was amplified using the proof reading polymerase Pfx (Invitrogen) from IMAGE clone #3905766 (Invitrogen) with the following primers: 5′-ATCGCCTCGAGCCATGCCGGACCAAGCCCTACA-3′ and 5′-ACTAGGGATCCCGTGCTTCTTCTTGTTCTGGATAATT-3′ and cloned into the XhoI and BamHI site of YFP-N1 and YFP-C1 (BD Biosciences) to generate YFP-MARCH5 and MARCH5-YFP. MARCH5H43W-YFP and MARCH5C65S,C68S-YFP were generated using the mutagenic primers: 5′-GAGGATCTACAAAATGGGTTTGGCAAGCTTGTCTACAACGCTGGGTG-3′ and 5′-CACCCAGCGTTGTAGACAAGCTTGCCAAACCCATTTTGTAGATCCTC-3′ for MARCH5H43W-YFP and 5′-GTACAGCCAGAGTGGCATCTCCATGGTCCAATGCTGAATACCTAATAG-3′ and 5′-CTATTAGGTATTCAGCATTGGACCATGGAGATGCCACTCTGGCTGTAC-3′ for and MARCH5C65S,C68S-YFP and Pfu polymerase (Stratagene) with MARCH5-YFP as template. MARCH5Δ1-71 was obtained using the following primers: 5′-ATCGCCTCGAGCCATG GCTGAATACCTAATAGTTTTTCC-3′ and 5′-ACTAGGGATCCCGTGCTTCTTCTTGTTCTGGATAATT-3′ and cloned XhoI/BamHI into YFP-N1 to generate MARCH5Δ1-71-YFP. All constructs were verified by sequencing. The mouse Mfn2 expression vector (Chen et al., 2003) was provided by D. Chan (Caltech, Pasadena, CA).
The short hairpin RNAi was performed as described previously (Lee et al., 2004). The oligonucleotides used for MARCH5 shRNAi generation were: 5′-CTGGAATTCGAGGAACAGGACAACCTATGAAGCTTGATAGGTTGTCTTGTTCTTCGAATTCTAGTTGTTTTTTG-3′, and 5′-GATCCAAAAAACAACTAGAATTCGAAGAACAAGACAACCTATCAAGCTTCATAGGTTGTCCTGTTCCTCGAATTCCAGCG-3′. Drp1 shRNAi- and control GFP shRNAi-generating vectors were used as described previously (Lee et al., 2004).
Confocal microscopy analysis
For confocal microscopy analyses cells were cultured in 2-well chambered Borosilicate coverglasses (Nunc). Images were captured using an LSM510 Meta imaging station (Carl Zeiss MicroImaging, Inc.). The images of immunostained, fixed cells were acquired with 100×/1.4 color corrected Plan-Apochromat (Carl Zeiss MicroImaging, Inc.) objective lens. For live cell imaging, cells were maintained at 35°C for no longer than 30 min and imaged using 100×/1.45 α-Plan-FLUAR objective lens (Carl Zeiss MicroImaging, Inc.). FRAP analysis of mitochondrial volume was performed as described previously (Szabadkai et al., 2004; Karbowski et al., 2006), unless stated otherwise. Subcellular mobility of Drp1 was determined using FRAP analysis of YFP-Drp1. Cells were cotransfected with YFP-Drp1 and CFP fused MARCH5 constructs in the molar ratio 1:5. The 4-μm strip was photobleached in YFP-Drp1 expressing cells using a 30.0-mV argon laser set to 514 nm with 50% of the laser power output and 100% of transmission. This was followed by the time-lapse 12-bit image acquisition of YFP-Drp1 fluorescence recovery with the same laser set to 50% of the laser power output and 1% of transmission. Time-lapse imaging was performed for 19.66 s after photobleaching with the interval of 400 ms. The detector gain was maintained at the levels below saturation of the brightest structures, resulting in FRAP of mostly focal Drp1. The FRAP curves were obtained using LSM510 software (Carl Zeiss MicroImaging, Inc.). The photoactivation studies of mitochondrial matrix targeted photoactivable GFP (mito-PAGFP) were performed as described previously (Karbowski et al., 2004). For the mito-PAGFP and FRAP analysis in cotransfected cells, cells expressing WT MARCH5 and MARCH5 RING mutants were identified by CFP fluorescence.
Immunofluorescence and colocalization analysis
Cells were fixed with 4% paraformaldehyde in PBS for 30 min, permeabilized with 0.15% Triton X-100 for 20 min, and then blocked with 10% BSA for 45 min at room temperature. The cells were then immunostained for 90 min at RT, or overnight at 4°C with the following primary antibodies: anti-cytochrome c mAbs (BD Biosciences; 1:250), anti-Tom20 mAbs (BD Biosciences; 1:250), anti-Tom20 polyclonal antibodies (Santa Cruz Biotechnology, Inc.; 1:500), anti-Dlp1 mAbs (Drp1; clone 8; BD Biosciences; 1:100), and anti-Myc mAbs (Santa Cruz Biotechnology, Inc., 1:1,000). Cells were washed and stained with AlexaFluor (488-, 546- and 633)- conjugated secondary antibodies (BD Biosciences; 1:250) for 1 h at RT. For the colocalization assay the projections of 12-bit confocal z-sections covering the entire depth of the cell, obtained with an interval of 0.25 μm, were used. Images in all experimental groups were obtained with the same settings of the confocal microscope, except for detector gain adjustments in the green channel that were performed to normalize saturations levels. Images were analyzed using the colocalization menu of the Volocity software (Improvision). The ROIs covering the entire area of the cell were created, followed then by the adjustment of the red-channel lower threshold used in order to exclude background staining from further analysis. The colocalization statistics were determined from the points included within ROIs and presented as Pearson's Correlation values that can vary between −1 and 1. Pearson's Correlation reflects the linear relationship between two variables. When applied to image analysis it can be used to establish the statistical significance of the association of pixels from different channels of confocal images, and therefore to determine the colocalization degree of different proteins in the cell. A correlation of zero means that there is no linear relationship between the variables whereas a correlation of 1 indicates perfect positive linear relationship.
MARCH5 antibody generation
For antibody production, full-length MARCH5 was amplified using 5′-AACTAGCATATGCCGGACCAAGCCCTACA-3′ and 5′-ACTAGGGATCCTTATGCTTCTTCTTGTTCTGGATA-3′ and cloned into pET15b (CLONTECH Laboratories, Inc.) using NdeI/BamHI to obtain pET15b-MARCH5. Escherichia coli were transformed with pET15b-MARCH5. The inclusion bodies containing full-length HIS6MARCH5 were isolated and solubilized in denaturizing bind buffer (20 mM Tris, pH 7.4, 500 mM NaCl, 40 mM imidazole, and 6M Guanindium HCl) and purified using HISTrap HP (GE Healthcare) according to the manufacturer's suggestions. HIS6MARCH5 was refolded by dialysis against refolding buffer (20 mM Tris, pH 8.0, 500 mM NaCl, 500 mM Arginin/HCl, and 0.2% Triton X-100). MARCH5-specific antibodies were raised using recombinant full-length HIS6MARCH5 injected into a New Zealand White rabbit. The antibodies were affinity purified from rabbit serum using HIS6MARCH5 bound to HiTrap NHS-activated HP (GE Healthcare) according to the manufacturer's recommendations. Anti-Mfn2 polyclonal antibodies were obtained using the same protocol, with the 1–405 Mfn2 fragment used to immunize rabbits.
Cell fractionation, chemical cross-linking, and Western blot
For Western blot analysis HeLa cells were transfected with Myc3MARCH5, Myc3MARCH5H43W, or control vector (YFP-N1; CLONTECH Laboratories, Inc.) using Effectene (QIAGEN) according to the manufacturer's recommendation. Cells were harvested 18 h after transfection and then suspended in cell lysis buffer (250 mM Sucrose, 20 mM Hepes/KOH, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, and complete protease inhibitor [Roche]). Cells were disrupted by 15 passages through a 25-gauge needle (with 1-ml syringe). The heavy membrane fraction (HM) was obtained by centrifugation at 10,000 g for 10 min. For cross-linking experiments the HM fraction was resuspended in lysis buffer and incubated on ice for 30 min either with 1 mM BMH (Pierce Chemical Co.) or with DMSO as control. The reaction was stopped by addition of 100 mM glycine, and samples were boiled in SDS PAGE sample buffer and analyzed by Western blot. The commercial antibodies used in Western blot analysis were: anti-Drp1 (Dlp1) mAbs (clone 8; BD Biosciences), anti-Hsp60 mAbs (StressGen Biotechnologies), anti-α-tubulin mAbs (Sigma-Aldrich), and anti-Fis1 polyclonal antibodies (Biovision).
Online supplemental material
Supplemental text describes additional analyses of mitochondrial phenotypes in MARCH5-expressing cells. Figs. S1 and S2 show additional examples of mitochondrial phenotypes in MARCH5 RING mutant transfected cells. Fig. S3 shows FRAP analysis in cells expressing high amounts of MARCH5H43W. Fig. S4 shows analyses of the oligomeric state of MARCH5. Fig. S5 demonstrates the degree of colocalization of Drp1, Drp1K38A, and Fis1 with MARCH5H43W. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200611064/DC1.
Supplementary Material
Acknowledgments
We thank Dr. David Chan for the gift of Mfn1 and Mfn2 knockout cells, as well as for mouse Mfn2 construct, Dr. Zheng Li for critical reading of the manuscript, Kristi Norris and Dr. DerFen Suen for Mfn2 antibodies, Dr. Pascaline Clerc for help with quantification of colocalization data, Dr. Carolyn Smith for help with confocal microscopy, the NINDS DNA sequencing facility, and Susan Smith for technical assistance.
This work was supported by the Intramural Research Program of the National Institutes of Health, NINDS, and the institutional support of the University of Maryland Biotechnology Institute (to M. Karbowski).
M. Karbowski and A. Neutzner contributed equally to this paper.
Abbreviations used in this paper: HM, heavy membrane; MEF, mouse embryonic fibroblast; OMM, outer mitochondrial membrane; ROI, region of interest.
References
- Bartee, E., M. Mansouri, B.T. Hovey Nerenberg, K. Gouveia, and K. Fruh. 2004. Downregulation of major histocompatibility complex class I by human ubiquitin ligases related to viral immune evasion proteins. J. Virol. 78:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhar, D., M.A. Karren, M. Babst, and J.M. Shaw. 2006. Dimeric Dnm1-G385D interacts with Mdv1 on mitochondria and can be stimulated to assemble into fission complexes containing Mdv1 and Fis1. J. Biol. Chem. 281:17312–17320. [DOI] [PubMed] [Google Scholar]
- Bleazard, W., J.M. McCaffery, E.J. King, S. Bale, A. Mozdy, Q. Tieu, J. Nunnari, and J.M. Shaw. 1999. The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast. Nat. Cell Biol. 1:298–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottomley, M.J., G. Stier, D. Pennacchini, G. Legube, B. Simon, A. Akhtar, M. Sattler, and G. Musco. 2005. NMR structure of the first PHD finger of autoimmune regulator protein (AIRE1). Insights into autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) disease. J. Biol. Chem. 280:11505–11512. [DOI] [PubMed] [Google Scholar]
- Chan, D.C. 2006. Mitochondria: dynamic organelles in disease, aging, and development. Cell. 125:1241–1252. [DOI] [PubMed] [Google Scholar]
- Chen, H., S.A. Detmer, A.J. Ewald, E.E. Griffin, S.E. Fraser, and D.C. Chan. 2003. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 160:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Henriques, M., B. Westermann, and T. Langer. 2006. Regulation of mitochondrial fusion by the F-box protein Mdm30 involves proteasome-independent turnover of Fzo1. J. Cell Biol. 173:645–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estaquier, J., and D. Arnoult. 2007. Inhibiting Drp1-mediated mitochondrial fission selectively prevents the release of cytochrome c during apoptosis. Cell Death Differ. 14:1086–1094. [DOI] [PubMed] [Google Scholar]
- Griffin, E.E., J. Graumann, and D.C. Chan. 2005. The WD40 protein Caf4p is a component of the mitochondrial fission machinery and recruits Dnm1p to mitochondria. J. Cell Biol. 170:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder, Z., R. Zunino, and H. McBride. 2004. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr. Biol. 14:340–345. [DOI] [PubMed] [Google Scholar]
- Hershko, A., and A. Ciechanover. 1992. The ubiquitin system for protein degradation. Annu. Rev. Biochem. 61:761–807. [DOI] [PubMed] [Google Scholar]
- Hinshaw, J.E. 2000. Dynamin and its role in membrane fission. Annu. Rev. Cell Dev. Biol. 16:483–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, T.T., and A.D. D'Andrea. 2006. Regulation of DNA repair by ubiquitylation. Nat. Rev. Mol. Cell Biol. 7:323–334. [DOI] [PubMed] [Google Scholar]
- Joazeiro, C.A., and A.M. Weissman. 2000. RING finger proteins: mediators of ubiquitin ligase activity. Cell. 102:549–552. [DOI] [PubMed] [Google Scholar]
- Karbowski, M., Y.J. Lee, B. Gaume, S.Y. Jeong, S. Frank, A. Nechushtan, A. Santel, M. Fuller, C.L. Smith, and R.J. Youle. 2002. Spatial and temporal association of Bax with mitochondrial fission sites, Drp1, and Mfn2 during apoptosis. J. Cell Biol. 159:931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski, M., D. Arnoult, H. Chen, D.C. Chan, C.L. Smith, and R.J. Youle. 2004. Quantitation of mitochondrial dynamics by photolabeling of individual organelles shows that mitochondrial fusion is blocked during the Bax activation phase of apoptosis. J. Cell Biol. 164:493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski, M., K.L. Norris, M.M. Cleland, S.Y. Jeong, and R.J. Youle. 2006. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 443:658–662. [DOI] [PubMed] [Google Scholar]
- Katoh, S., C. Hong, Y. Tsunoda, K. Murata, R. Takai, E. Minami, T. Yamazaki, and E. Katoh. 2003. High precision NMR structure and function of the RING-H2 finger domain of EL5, a rice protein whose expression is increased upon exposure to pathogen-derived oligosaccharides. J. Biol. Chem. 278:15341–15348. [DOI] [PubMed] [Google Scholar]
- Labrousse, A.M., M.D. Zappaterra, D.A. Rube, and A.M. van der Bliek. 1999. C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol. Cell. 4:815–826. [DOI] [PubMed] [Google Scholar]
- Lee, Y.J., S.Y. Jeong, M. Karbowski, C.L. Smith, and R.J. Youle. 2004. Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol. Biol. Cell. 15:5001–5011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legesse-Miller, A., R.H. Massol, and T. Kirchhausen. 2003. Constriction and Dnm1p recruitment are distinct processes in mitochondrial fission. Mol. Biol. Cell. 14:1953–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Z., K. Okamoto, Y. Hayashi, and M. Sheng. 2004. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 119:873–887. [DOI] [PubMed] [Google Scholar]
- McBride, H.M., M. Neuspiel, and S. Wasiak. 2006. Mitochondria: more than just a powerhouse. Curr. Biol. 16:R551–R560. [DOI] [PubMed] [Google Scholar]
- McCormick, A.L., V.L. Smith, D. Chow, and E.S. Mocarski. 2003. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 77:631–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozdy, A.D., J.M. McCaffery, and J.M. Shaw. 2000. Dnm1p GTPase-mediated mitochondrial fission is a multi-step process requiring the novel integral membrane component Fis1p. J. Cell Biol. 151:367–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, N., Y. Kimura, M. Tokuda, S. Honda, and S. Hirose. 2006. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep. 7:1019–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor, K., E. Ingerman, V. Okreglak, M. Marino, J.E. Hinshaw, and J. Nunnari. 2006. Mdv1 interacts with assembled dnm1 to promote mitochondrial division. J. Biol. Chem. 281:2177–2183. [DOI] [PubMed] [Google Scholar]
- Neutzner, A., and R.J. Youle. 2005. Instability of the mitofusin Fzo1 regulates mitochondrial morphology during the mating response of the yeast Saccharomyces cerevisiae. J. Biol. Chem. 280:18598–18603. [DOI] [PubMed] [Google Scholar]
- Nunnari, J., W.F. Marshall, A. Straight, A. Murray, J.W. Sedat, and P. Walter. 1997. Mitochondrial transmission during mating in Saccharomyces cerevisiae is determined by mitochondrial fusion and fission and the intramitochondrial segregation of mitochondrial DNA. Mol. Biol. Cell. 8:1233–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olichon, A., G. Elachouri, L. Baricault, C. Delettre, P. Belenguer, and G. Lenaers. 2007. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 14:682–692. [DOI] [PubMed] [Google Scholar]
- Otsuga, D., B.R. Keegan, E. Brisch, J.W. Thatcher, G.J. Hermann, W. Bleazard, and J.M. Shaw. 1998. The dynamin-related GTPase, Dnm1p, controls mitochondrial morphology in yeast. J. Cell Biol. 143:333–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partikian, A., B. Olveczky, R. Swaminathan, Y. Li, and A.S. Verkman. 1998. Rapid diffusion of green fluorescent protein in the mitochondrial matrix. J. Cell Biol. 140:821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praefcke, G.J., and H.T. McMahon. 2004. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat. Rev. Mol. Cell Biol. 5:133–147. [DOI] [PubMed] [Google Scholar]
- Santel, A., and M.T. Fuller. 2001. Control of mitochondrial morphology by a human mitofusin. J. Cell Sci. 114:867–874. [DOI] [PubMed] [Google Scholar]
- Shaw, J.M., and J. Nunnari. 2002. Mitochondrial dynamics and division in budding yeast. Trends Cell Biol. 12:178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova, E., L. Griparic, D.L. Shurland, and A.M. van der Bliek. 2001. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol. Biol. Cell. 12:2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub, O., and D. Rotin. 2006. Role of ubiquitylation in cellular membrane transport. Physiol. Rev. 86:669–707. [DOI] [PubMed] [Google Scholar]
- Stojanovski, D., O.S. Koutsopoulos, K. Okamoto, and M.T. Ryan. 2004. Levels of human Fis1 at the mitochondrial outer membrane regulate mitochondrial morphology. J. Cell Sci. 117:1201–1210. [DOI] [PubMed] [Google Scholar]
- Suzuki, M., S.Y. Jeong, M. Karbowski, R.J. Youle, and N. Tjandra. 2003. The solution structure of human mitochondria fission protein Fis1 reveals a novel TPR-like helix bundle. J. Mol. Biol. 334:445–458. [DOI] [PubMed] [Google Scholar]
- Szabadkai, G., A.M. Simoni, M. Chami, M.R. Wieckowski, R.J. Youle, and R. Rizzuto. 2004. Drp-1-dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+-mediated apoptosis. Mol. Cell. 16:59–68. [DOI] [PubMed] [Google Scholar]
- Szabadkai, G., A.M. Simoni, K. Bianchi, D. De Stefani, S. Leo, M.R. Wieckowski, and R. Rizzuto. 2006. Mitochondrial dynamics and Ca2+ signaling. Biochim. Biophys. Acta. 1763:442–449. [DOI] [PubMed] [Google Scholar]
- Tieu, Q., and J. Nunnari. 2000. Mdv1p is a WD repeat protein that interacts with the dynamin-related GTPase, Dnm1p, to trigger mitochondrial division. J. Cell Biol. 151:353–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tieu, Q., V. Okreglak, K. Naylor, and J. Nunnari. 2002. The WD repeat protein, Mdv1p, functions as a molecular adaptor by interacting with Dnm1p and Fis1p during mitochondrial fission. J. Cell Biol. 158:445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasiak, S., R. Zunino, and H.M. McBride. 2007. Bax/Bak promote sumoylation of DRP1 and its stable association with mitochondria during apoptotic cell death. J. Cell Biol. 177:439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welchman, R.L., C. Gordon, and R.J. Mayer. 2005. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 6:599–609. [DOI] [PubMed] [Google Scholar]
- Yonashiro, R., S. Ishido, S. Kyo, T. Fukuda, E. Goto, Y. Matsuki, M. Ohmura-Hoshino, K. Sada, H. Hotta, H. Yamamura, et al. 2006. A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J. 25:3618–3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, Y., K.R. Pitts, and M.A. McNiven. 2001. Mammalian dynamin-like protein DLP1 tubulates membranes. Mol. Biol. Cell. 12:2894–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon, Y., E.W. Krueger, B.J. Oswald, and M.A. McNiven. 2003. The mitochondrial protein hFis1 regulates mitochondrial fission in mammalian cells through an interaction with the dynamin-like protein DLP1. Mol. Cell. Biol. 23:5409–5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle, R.J., and M. Karbowski. 2005. Mitochondrial fission in apoptosis. Nat. Rev. Mol. Cell Biol. 6:657–663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}