

Abstract
The idea that conversion of glucose to ATP is an attractive target for cancer therapy has been supported in part by the observation that glucose deprivation induces apoptosis in rodent cells transduced with the proto-oncogene MYC, but not in the parental line. Here, we found that depletion of glucose killed normal human cells irrespective of induced MYC activity and by a mechanism different from apoptosis. However, depletion of glutamine, another major nutrient consumed by cancer cells, induced apoptosis depending on MYC activity. This apoptosis was preceded by depletion of the Krebs cycle intermediates, was prevented by two Krebs cycle substrates, but was unrelated to ATP synthesis or several other reported consequences of glutamine starvation. Our results suggest that the fate of normal human cells should be considered in evaluating nutrient deprivation as a strategy for cancer therapy, and that understanding how glutamine metabolism is linked to cell viability might provide new approaches for treatment of cancer.
Introduction
The differences between cancer and normal cells in consumption and metabolism of basic nutrients have been considered a promising cancer therapy target for several decades (for review see Thompson et al., 2005; Shaw and Cantley, 2006). The metabolism of glucose and glutamine has attracted a particular interest, as these molecules are the main nutrients consumed by cancer cells. Glucose metabolism has drawn much of the attention (for review see Garber, 2006) from the time when Otto Warburg found that some cancer cells consume more glucose than normal cells and proposed that a failure to make ATP by oxidative phosphorylation is the cause of cancer (Warburg, 1956). Many cancer cells indeed accumulate glucose derivatives faster than normal tissues, the ability that has been used successfully for cancer diagnosis and monitoring (for review see Rohren et al., 2004).
The more recent findings that glucose metabolism is linked to signaling pathways of cell growth and survival, and apoptosis in particular (for review see Hammerman et al., 2004; Fulda and Debatin, 2007) only reinforced the view of glucose metabolism as the Achilles heel of cancer cells. This view was also supported by the observation that depletion of glucose induced apoptosis in rodent cells that were transduced with MYC, an oncogene that is abnormally expressed in many human cancers (Nesbit et al., 1999), but not in the parental cell line (Shim et al., 1998). This finding was remarkable as it suggested that the very processes that make cells cancerous can make cells dependent on glucose.
However, the idea that restricting supply of glucose or imitating this restriction can kill cancer cells selectively by depleting ATP is not without its critics (for review see Zu and Guppy, 2004). Indeed, in contrast to Warburg's proposal, both normal and cancer cells can produce ATP by oxidative phosphorylation, and the contribution of glycolysis to ATP production varies greatly among both normal and cancer cells. Some cancer cells can even survive in vitro without glucose at all if supplied with glutamine and nucleosides (Reitzer et al., 1979; Wice et al., 1981; Linker et al., 1985).
Metabolism of glutamine, the major nutrient consumed by cancer cells besides glucose, has also been considered as a target of cancer therapy, even though with a much lower intensity than metabolism of glucose. The rate of glutamine transport is higher in cancerous than in normal cells (Bode et al., 2002), the activity of glutaminase, the first enzyme in glutamine metabolism, correlates with the growth rates of tumors (Knox et al., 1969; Linder-Horowitz et al., 1969; Perez-Gomez et al., 2005), and some cancer cell lines require glutamine or its metabolites for survival (for review see Medina, 2001; Fuchs and Bode, 2006). The ability to deplete glutamine is a therapeutically relevant component of asparaginase that has been used to treat acute lymphoblastic leukemia (Panosyan et al., 2004). Antibiotics DON (6-diazo-5-oxo-l-norleucine) and acivicin, which are glutamine analogues, were very effective in animal models of cancer, perhaps due to the ability to inhibit nucleotide synthesis, but proved to be unacceptably toxic in clinical trials (for review see Ahluwalia et al., 1990).
Surprisingly, why glutamine is required for cell viability, or, in other words, why depletion of glutamine would kill cells, is not entirely clear. On the one hand, glutamine is the most abundant amino acid in the body and is used by the cells in a variety of ways, including oxidation by the Krebs cycle to produce ATP (for review see Baggetto, 1992), providing nitrogen required for nucleotide synthesis, and being a precursor of glutathione, the major nonenzymatic cellular anti-oxidant (for review see Curi et al., 2005). However, glutamine is made by cells, which makes the requirement for exogenous glutamine appear unnecessary. Moreover, only a few percent of the consumed glutamine is used for macromolecular biosynthesis, while the rest is metabolized into molecules, such as lactate, that are released by the cells (for review see Newsholme et al., 1985). These observations raise the possibility that metabolism of glutamine not merely supports accelerated proliferation or an increased requirement for ATP, but is used for yet unrecognized pathways that are required for survival of cells and cancer cells in particular.
This study was designed to find nutrients that are required for cells that express abnormally high amounts of MYC but not for their normal counterparts. Considering that metabolism can vary among species, and that human rather than animal tumors are of primary interest, we used human cells. To our surprise, we found that in contrast to rodent cells (Shim et al., 1998) glucose depletion killed normal human cells irrespective of the activity of ectopic MYC and did so not by inducing apoptosis. On the other hand, depletion of glutamine induced apoptosis that depended on activity of ectopic MYC, which led us to investigate the mechanisms of this death. Overall, our study raises again the concern that the requirement of normal cells for glucose and intraspecies differences in glucose metabolism need to be considered in evaluating depletion of this nutrient for therapeutic needs. This study also suggests that learning why cells require glutamine to maintain their viability may provide new approaches for killing cancer cells selectively.
Results
To test how an abnormally high expression of MYC affects the requirement of human cells for nutrients, we used a well-characterized system in which MYC activity can be induced acutely. This system uses a fusion of MYC with a mutated hormone- binding domain of the estrogen receptor, ERTM-MYC (Eilers et al., 1989; Littlewood et al., 1995). ERTM-MYC is excluded from the nucleus, which makes the MYC moiety inactive transcriptionally, but translocates there upon binding a synthetic estrogen analogue 4-hydroxytamoxifen (OHT). We will refer to ERTM-MYC activated by OHT as active ERTM-MYC, and indicate active ERTM-MYC in the figures as MYCON and inactive as MYCOFF. This system allows studying immediate effects of MYC by minimizing the artifacts associated with clonal selection of cells that because of additional genetic changes avoided apoptosis induced by ectopic expression of MYC.
To further decrease the chances of clonal selection, we used retroviral transduction rather than transient transfection to generate populations of normal human fibroblasts IMR90 that expressed ERTM-MYC (IER-MYC) or ERTM (IER); the latter served to exclude effects of OHT unrelated to MYC activity. Adding OHT to the tissue culture medium caused translocation of ERTM-MYC to the nucleus (Fig. S1 A, available at http://www.jcb.org/cgi/content/full/jcb.200703099/DC1) and increased sensitivity of the cells to DNA damage (Fig. S1 B), which confirmed that the system functioned as expected (Eilers et al., 1989; Littlewood et al., 1995).
Glucose deprivation kills normal human fibroblasts not by apoptosis and largely independently of the MYC activity
To test the system, we analyzed how induction of ERTM-MYC activity affects the sensitivity of the cells to depletion of glucose. To our surprise, we found that in contrast to a previous study (Shim et al., 1998), which used rat cells transduced with MYC, depletion of glucose killed IER-MYC cells irrespective of ERTM-MYC activity (Fig. 1, A and B). The medium with 0.5 mM glucose, which is 2% of the normal concentration, killed the cells slower than a complete deficiency of the nutrient, but the rate was also independent of ER-Myc activity (unpublished data). Another difference with the report was that cells in our system died by a process other than apoptosis, as indicated by the absence of characteristic chromatin condensation (Fig. 1 C) and the failure of Bcl-2, an inhibitor of apoptosis, to prevent cell death (Fig. 1 D). These observations confirmed a previously raised concern that glucose depletion may be toxic not only to cancer but also to normal cells (Zu and Guppy, 2004), and suggested that interspecies differences in metabolism should be considered in extrapolating results obtained in nonhuman cells.
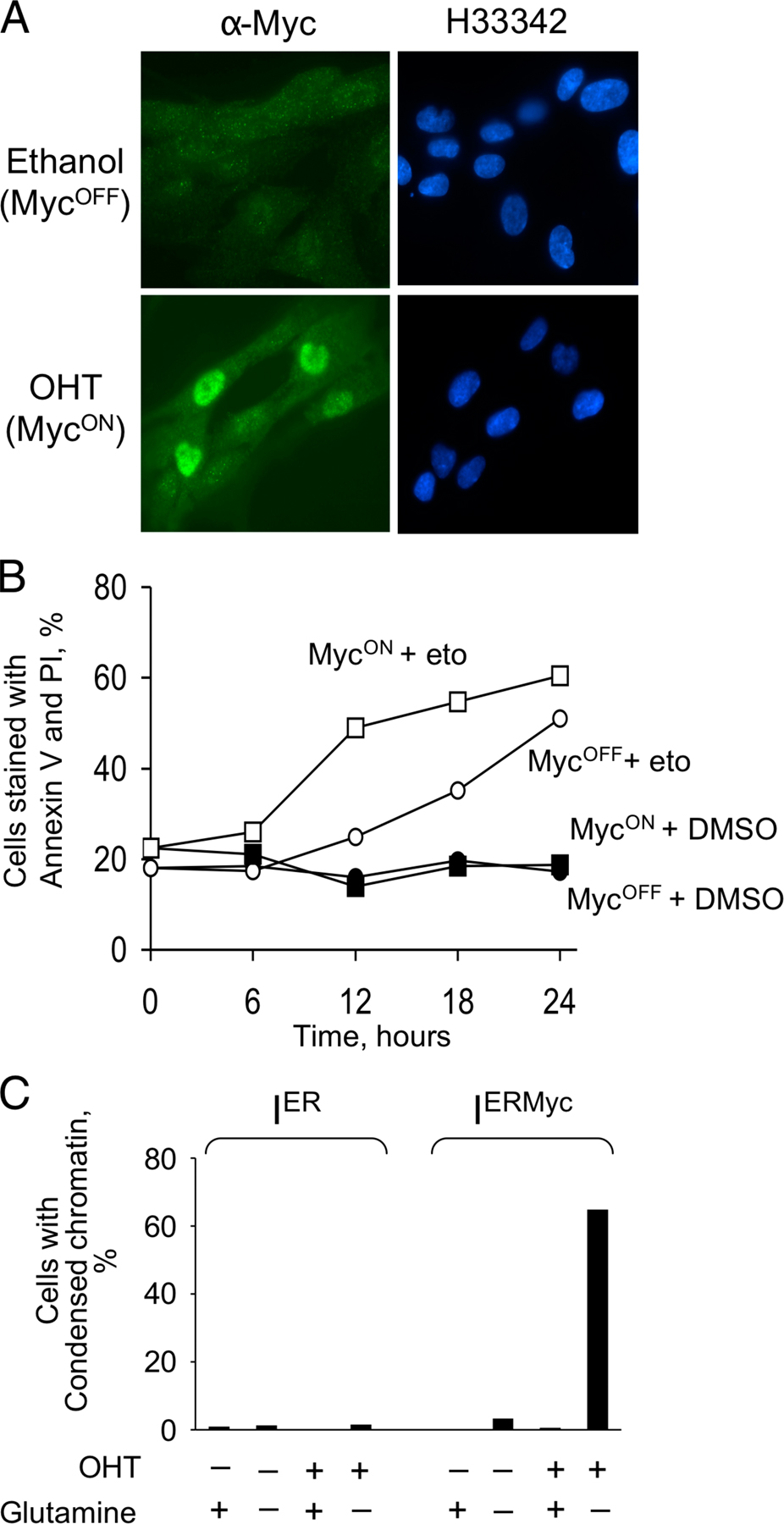
Figure 1.

Glucose deprivation kills human fibroblasts by a process that is different from apoptosis and is largely unaffected by the activity of MYC. (A) IER-MYC cells were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h. Media was then changed to glucose-free or to media containing 10 mM glucose and the cells were collected at the indicated time to measure cell viability. (B) The experiment was conducted as in A except cells were collected at 12 h (open bars) or 18 h (closed bars) after depletion. (C) IER-MYC cells were incubated as in A for 18 h, fixed, stained with Hoechst 33342, and analyzed by fluorescence microscopy. (D) IER-MYC fibroblasts were transduced with Bcl-2 or vector alone and incubated as in C and analyzed for cell viability by staining with Annexin V-FITC and propidium iodide (PI). Expression of Bcl-2 was confirmed by immunoblotting (not depicted). The averages of three independent experiments are presented in B and D with the error bars representing SDs.
Glutamine deprivation induces apoptosis selectively in cells with acutely activated ER-MYC
In contrast to deprivation of glucose, depletion of glutamine killed IER-MYC cells faster if ERTM-MYC was active (Fig. 2 A). Death was associated with the activity of MYC rather with that of OHT or ER because IER cells remained viable in the absence of glutamine irrespective of the OHT treatment (Fig. S1 C). The IER-MYC cells died by apoptosis, as indicated by the characteristic nuclear morphology of the dead cells (Fig. 2, B and C). Apoptosis was induced through the intrinsic pathway because Bcl-2 and a caspase-9 dominant-negative mutant (C9DN) prevented death (Fig. 3, A and B), whereas crmA, a viral inhibitor of the extrinsic pathway (Enari et al., 1995), had no effect (Fig. 3 D). Considering the difference in sensitivity to glutamine depletion, we asked how a deficiency in this amino acid can kill cells and why this effect would depend on expression of MYC.
Figure 2.

Glutamine deprivation induces MYC-dependent apoptosis. (A) IER-MYC cells were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h. Media was then changed to glutamine-free or to media containing 2 mM glutamine, and cells were collected at the indicated times to analyze cell viability. (B and C) IER-MYC were incubated as in A, except without glutamine for 18 h, fixed, stained with Hoechst 33342, visualized by fluorescence microscopy, and cells with condensed chromatin (indicated by white arrows in B) were counted as apoptotic. 200 to 300 nuclei were counted for each sample. The averages of three independent experiments are presented in A and C with errors bars indicating SD. Only MycON cells are shown in B.
Figure 3.

Glutamine deprivation induces apoptosis through the intrinsic pathway. (A) IER-MYC cells transduced with Bcl-2 or vector alone were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h, and then for 16 h with or without glutamine before analyzing for cell viability. (B) IER-MYC transduced with C9DN (IER-MYC–C9DN) or with the vector were analyzed as in A. (C) IER-MYC transduced with CrmA or vector alone were treated as in A except anti-Fas (α-CD95) antibody was added as indicated. Each graph represents averages of three independent experiments. Error bars indicate SD.
In principle, glutamine depletion could affect the apoptotic machinery directly, for example by modifying molecules that execute apoptosis, or indirectly by causing various types of stress, such as DNA damage, which in turn induces activation of apoptosis through various pathways. The second possibility seemed more likely because abnormal expression of MYC sensitizes cells to various agents that induce apoptosis (for review see Pelengaris and Khan, 2003) and because previous reports suggested that depletion of glutamine kills because of stress caused by deficiency in proteins, ATP, glutathione, or nucleotides (for review see Ahluwalia et al., 1990; Fuchs and Bode, 2006). These explanations appeared plausible as glutamine is indeed used for synthesis of these molecules (Fig. 4 A) and because glutamine analogues DON and acivicin are thought to kill cells by inducing nucleotide deficiency (Ahluwalia et al., 1990). Therefore we first tested whether any of these explanations can account for MYC-dependent apoptosis in our system.
Figure 4.

Preventing protein synthesis fails to induce apoptosis. (A) Pathways of glutamine metabolism. (B) IER-MYC cells were incubated in the presence of OHT (MYCON) for 24 h. The media was then changed to that deficient in the indicated amino acids and the cells were incubated for 19 h before analyzing their viability. An average of two experiments is shown. (C) IER-MYC were incubated as in B except rapamycin was added as indicated 18 h before analyzing the rate of apoptosis (top panel) and phosphorylation of S6 (bottom panels). One of two independent experiments is shown.
A deficiency in protein synthesis or inhibition of mTOR did not cause apoptosis induced by glutamine depletion
A deficiency in protein synthesis could explain apoptosis after glutamine depletion because glutamine is an amino acid, and MYC affects genes involved in protein synthesis (O'Connell et al., 2003), which regulates cell viability (Holcik and Sonenberg, 2005). However, omitting any of the essential amino acids from the tissue culture media failed to kill cells, at least over the time sufficient to kill by omitting glutamine (Fig. 4 B). Therefore, protein synthesis was not the cause of apoptosis induced by glutamine depletion.
The availability of nitrogen sources, and glutamine in particular, regulates the activity of mTOR, a kinase that controls protein synthesis, cell proliferation, and other processes that require nitrogen (for review see Sabatini, 2006). However, this kinase remained active in the absence of exogenous glutamine, as indicated by persistent phosphorylation of the mTOR target S6 (Fig. 4 C), while the inhibitor of mTOR rapamycin caused S6 dephosphorylation but had no effect on cell viability over the time sufficient to kill cells by glutamine depletion (Fig. 4 C). Hence, glutamine depletion caused apoptosis by affecting pathways that did not require mTOR activity.
A cell cycle arrest is not the cause of apoptosis
One of the models that explains the ability of MYC to facilitate apoptosis argues that cells in which expression of MYC is abnormally high die because they are unable to arrest in cell cycle when faced with a nutrient or growth factor deficiency (for review see Pelengaris and Khan, 2003). We noticed that IER-MYC cells ceased to proliferate irrespective of ERTM-MYC activity if either any of the essential amino acids or glutamine were absent (unpublished data). Therefore, deregulation of cell cycle arrest by itself could not explain cell death caused by glutamine depletion.
Apoptosis induced by glutamine depletion is not caused by ATP deficiency
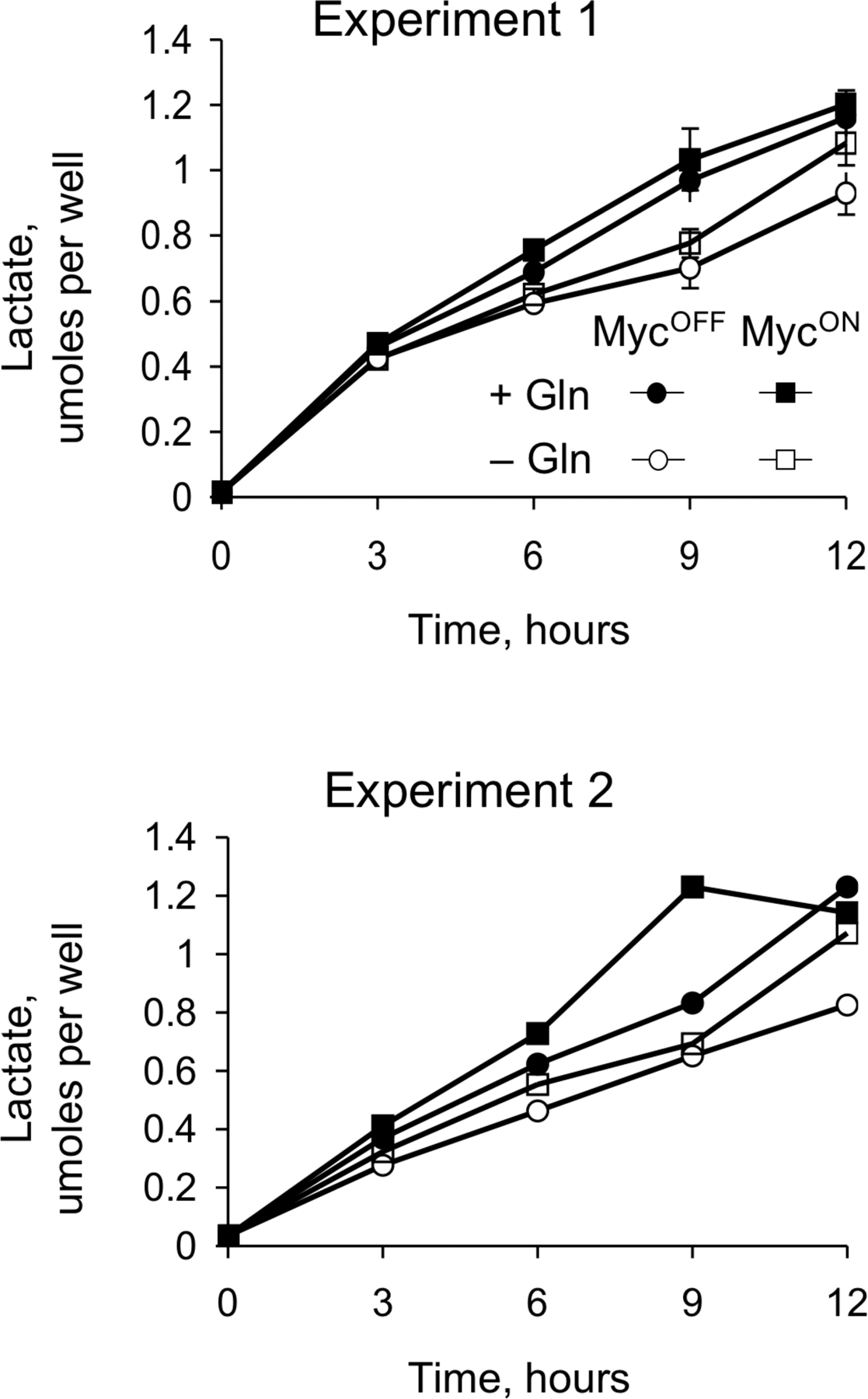
Several observations suggested that glutamine depletion could cause apoptosis by depleting ATP. Indeed, glutamine is a major source of ATP in rapidly dividing cells (for review see Baggetto, 1992; Curi et al., 2005), abnormal expression of MYC accelerates cell proliferation and changes metabolism of mitochondria (Morrish et al., 2003; Li et al., 2005), and even a transient decrease in ATP concentration can cause apoptosis (Izyumov et al., 2004; Shaw et al., 2004). To test the effects of glutamine depletion on ATP concentration we used IER-MYC that expressed C9DN (IER-MYC–C9DN), which prevents cell disassembly but not the preceding steps of apoptosis (Fearnhead et al., 1998), thus avoiding artifacts associated with cell decomposition. Depleting IER-MYC–C9DN cells of glutamine slightly decreased ATP concentration (Fig. 5 A). Yet, a much larger decrease induced by a combination of 2-deoxyglucose, an inhibitor of glycolysis, and antimycin A, an inhibitor of mitochondrial respiration, failed to kill IER-MYC cells as efficiently as did glutamine depletion (Fig. 5, A and B). Therefore, apoptosis caused by glutamine depletion could not be mediated by ATP deficiency alone. The observation that 2-deoxyglucose or antimycin had little effect on ATP concentration if used separately, but effectively decreased it if used together, indicated that either glycolysis or respiration alone could produce sufficient amounts of ATP in the cells we used.
Figure 5.

Glutamine depletion induces apoptosis not by causing ATP deficiency. IER-MYC–C9DN cells (A) or IER-MYC cells (B) were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h and then for 18 h in the indicated media before collecting them to measure concentration of ATP (A) or cell viability (B). An average of three independent experiments is presented in A with the error bars representing SD, and an average of two independent experiments in B.
Apoptosis is not caused by glutathione deficiency
Abnormally high activity of MYC could produce sufficient amounts of reactive oxygen species (ROS) to cause DNA damage, genomic instability, and apoptosis (Vafa et al., 2002), perhaps by affecting expression of mitochondrial proteins (Morrish et al., 2003; Li et al., 2005). MYC is also implicated in regulating concentration of glutathione (Benassi et al., 2006), the most abundant nonenzymatic antioxidant in the cell (Anderson et al., 1997). Because glutamine is a precursor of glutathione (Fig. 6 A), depleting this amino acid could trigger apoptosis by preventing inactivation of ROS. As expected, depletion of glutamine decreased glutathione concentration in IER-MYC–C9DN cells, as did l-buthionine sulfoximine (BSO), an inhibitor of l-glutamylcysteine synthase (Griffith and Meister, 1979; Anderson et al., 1997) (Fig. 6, A and B). However, BSO failed to induce apoptosis in IER-MYC cells (Fig. 6 C), suggesting that glutathione deficiency alone could not be the cause of apoptosis induced by glutamine depletion. This conclusion was consistent with our finding that depleting cysteine, which is required for glutathione synthesis (Fig. 6 A), also failed to induce apoptosis (Fig. 4 B), and a report that BSO failed to recapitulate apoptosis caused by glutamine deprivation in a hybridoma (Guerin et al., 2006).
Figure 6.

Depleting glutathione fails to induce apoptosis. (A) Biosynthesis of glutathione. (B) IER-MYC–C9DN were incubated with OHT (MYCON) or ethanol (MYCOFF) for 24 h, and then transferred to the medium depleted of glutamine or supplemented with 100 μM BSO. Cells were collected at the indicated time to measure reduced glutathione by flow cytometry (Materials and methods). (C) IER-MYC cells were treated as in A except the cells were collected at 24 h of incubation in the indicated media to determine the rate of apoptosis. Shown are the averages of two independent experiments.
Glutamine depletion does not cause DNA damage
Because the amido group of glutamine is used for nucleotide synthesis (Fig. 4 A), depletion of this amino acid could cause DNA damage, a well-known cause of apoptosis. Therefore, we tested whether glutamine depletion induces two reporters of DNA damage, an increase in concentration of p53 and phosphorylation of histone H2AX (Rogakou et al., 1998). As expected, p53 concentration was induced by DON, 6-methylmercaptopurine riboside (MMPR), an inhibitor of nucleotide synthesis (Stet et al., 1995), etoposide, which causes double-stranded DNA breaks, or by activation of ectopically expressed ERTM-MYC (Fig. 7). In contrast, glutamine depletion not only failed to increase the p53 concentration, but even reversed the increase induced by activation of ERTM-MYC (Fig. 7). Phosphorylation of H2AX, as detected by immunofluorescence, was induced by etoposide and by activation of ERTM-MYC, but not by glutamine depletion (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200703099/DC1). Hence, depletion of glutamine was unlikely to cause apoptosis by inducing DNA damage.
Figure 7.

Glutamine deprivation does not increase p53 concentration. IER-MYC cells were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h. The cells were then incubated either in the medium that was deficient in glutamine for 24 h, or for 36 h in the complete medium supplemented with DON (A), for 24 h supplemented with 1 mM MMPR (B), or for 18 h supplemented with 100 μM etoposide (C) before determining the rate of apoptosis (graphs) and the concentration of p53 by immunoblotting. The averages of two (A, B) or three (C) independent experiments are shown. Error bars in C are SDs.
Apoptosis caused by glutamine depletion is prevented by substrates of the Krebs cycle
Because glutamine can be converted via glutamate into 2-oxoglutarate, a Krebs cycle intermediate (Kovacevic, 1972; Baggetto, 1992), depletion of this amino acid could cause a deficiency of this cycle. Because the Krebs cycle is not only a source of NADH for oxidative phosphorylation, but also a “hub” of numerous metabolic pathways (http://www.tcd.ie/Biochemistry/IUBMB-Nicholson/minimaps.html), a deficiency in the Krebs cycle or its intermediates could have various consequences, including apoptosis, even if the normal ATP concentration is maintained by glycolysis (Albayrak et al., 2003; Zhou et al., 2003). Consistent with this possibility, oxaloacetate, which is a membrane-permeable Krebs cycle intermediate, or pyruvate, which can enter the cycle by converting into acetyl-CoA or oxaloacetate, completely prevented apoptosis in the absence of glutamine (Fig. 8, A and B). However, neither pyruvate nor oxaloacetate restored cell proliferation (Fig. 8 C; and unpublished data), which suggested that the rescue was not due to replenishing depleted glutamine but rather due to specific effect of these metabolites on cell viability.
Figure 8.

Oxaloacetate or pyruvate rescue glutamine-depleted cells from apoptosis but not from cell cycle arrest. (A and B) IER-MYC cells were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h and then in the indicated media for 18 h before measuring cell viability. An average of three (A) and two (B) independent experiments is presented. Error bars in A indicate SDs. (C) IER-MYC–C9DN cells were incubated with OHT for 24 h. Media was then changed to that containing 2 mM of the indicated supplements and incubated for 24 h in one experiment (filled bars) or 48 h in the second experiment (open bars) before counting the cells. The relative increase in cell number is shown. (D) IER-MYC–C9DN cells were treated either with ethanol or OHT and then incubated with or without glutamine for the indicated time before measuring the NADH to NAD+ ratio. Treatment with rotenon (Rot) was used as a positive control.
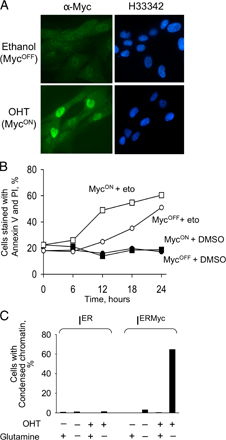
Pyruvate might affect cell viability by being a substrate of lactate dehydrogenase (LDH), which oxidizes NADH to NAD (Zielke et al., 1980). Because the NADH/NAD ratio determines rates of many processes, from glycolysis to histone modifications (for review see Ladurner, 2006), affecting this ratio could induce apoptosis. However, the NADH/NAD ratio remained unchanged (Fig. 8 D) and lactate was still produced (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200703099/DC1) after glutamine depletion, even though at a lower rate, indicating that pyruvate was unlikely to rescue from glutamine depletion by being a substrate of LDH.
The rescue by pyruvate and oxaloacetate could be explained if depletion of glutamine caused a deficiency in the Krebs cycle. To test this possibility we used capillary electrophoresis coupled to mass spectrometry (CE-MS), an approach that estimates concentration of multiple metabolites in one sample (Soga et al., 2003). The changes in ATP and glutathione concentrations detected by CE-MS reflected those obtained by biochemical methods (Fig. S4 and Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200703099/DC1), indicating that the approach was reliable in our hands in detecting relative changes in metabolite concentrations. As expected, glutamine deprivation caused rapid depletion of glutamate, 4-aminobutyrate, 5-oxoproline, and aspartate, which are products of glutamine metabolism (Fig. 9 and Table S1). All of the detected Krebs cycle intermediates, with the notable exception of citrate and isocitrate (Fig. 9), were depleted as well. These results indicated that glutamine depletion indeed inhibited the Krebs cycle progression. Therefore, the findings that the Krebs cycle is deficient and the substrates of the cycle rescue from glutamine depletion suggested that a deficiency in glutamine causes apoptosis by affecting the Krebs cycle.
Figure 9.

Glutamine deprivation depletes intermediates of the Krebs cycle. (A) IER-MYC–C9DN cells were incubated in the presence of OHT (MYCON) or ethanol (MYCOFF) for 24 h and then in the medium with or without glutamine for the indicated time. The cells were then collected at the indicated times to determine the concentration of intracellular metabolites by CE-MS (Materials and methods). Duplicate samples were prepared from independent plates. The result of one experiment is presented. The results of two more independent experiments are presented in Table S1. (B) A guide to the location of identified metabolites in the pathways of glutamine metabolism. The detected metabolites are indicated in red.
Modification of nutrient requirements by acute activation of ectopically expressed MYC vary with cell type
We found that depletion of glutamine but not glucose induces MYC-dependent apoptosis in human cells, whereas a previous report indicated that glucose depletion caused MYC-dependent apoptosis in a rat cell line (Shim et al., 1998). This difference could suggest that the requirement for nutrients varies with species and cell lines. To test this possibility, we compared several human cell lines that ectopically express MYC. Normal human foreskin fibroblasts (HFF) that express ERTM-MYC reacted to glutamine or glucose depletion similarly to IER-MYC (Fig. 10, A and B). However, human embryonic kidney epithelial cells immortalized by coexpression of hTERT and SV-40 large T antigen (HA1E) reacted to glutamine depletion as IER-MYC, but underwent apoptosis if depleted of glucose (Fig. 10, C and E). Human retinal pigmental cells (RPE) immortalized with hTERT (RPE-hTERT) remained alive in the absence of glutamine irrespective of the ectopic MYC expression, even though this expression sensitized the cells to etoposide (Fig. 10 F), and died without glucose irrespective of the ectopic MYC expression by a mechanism that was different from apoptosis (unpublished data). The observed diversity was consistent with the finding that metabolism of human cells is changed by oncogenes in various ways (Ramanathan et al., 2005) and indicated that how nutrient depletion affects cancers and normal cells may be difficult to conclude by extrapolating findings obtained in one or even a few experimental systems.
Figure 10.

The effect of MYC on requirements for glutamine and glucose depends on cell type. (A) Normal human foreskin fibroblasts (HFF) transduced with ERTM-MYC (HFFER-MYC) were incubated with either ethanol (MYCOFF) or OHT (MYCON) and then with or without glutamine for 48 h before scoring apoptotic nuclei. An average of two independent experiments is shown. (B) HFFER-MYC cells were incubated as in A, except the medium had no glucose and the viability determined by scoring cells stained with annexin V and PI. (C and D) Immortalized human kidney epithelial cells (HA1E) transduced with MYC-ERTM (HA1EMYC-ER) were incubated and scored for apoptosis as in A either at the indicated time points (C) or after 24 h of glutamine deprivation (D). In D an average of three independent experiments is presented. (E) HA1EMYC-ER were treated and scored for viability as in B. (F) RPE-MYC and RPE-Neo cells were incubated as indicated for 53 h before scoring for apoptosis. An average of three independent experiments is shown with the error bars indicating SDs.
Discussion
The first part of our study reiterated the previously raised concern (Zu and Guppy, 2004) that the knowledge about the relative requirement of normal and transformed cells for glucose is still insufficient to make a reliable judgment as to whether agents that mimic glucose deprivation can be effective in cancer therapy. In particular, our results demonstrated that dependence of cells on glucose and the effects of its depletion vary among cell types and can depend on the combination of oncogenes expressed in these cells.
Our finding that a deficiency in glutamine killed most of the human cells that we tested depending on the activity of MYC raised the possibility that the requirement for this nutrient can be explored for learning how to kill cancer cells in which MYC activity is abnormally high. Testing this possibility will require knowing why and how glutamine depletion kills, and how MYC activity synergizes with the depletion of glutamine to induce apoptosis.
How does glutamine depletion kill?
Several mechanisms were proposed previously to explain how glutamine depletion causes apoptosis, including a deficiency in ATP, protein synthesis, glutathione, or nucleotides, as well as a change in the cellular redox state (for review see Fuchs and Bode, 2006). We found no evidence that any of these mechanisms mediate apoptosis in our system, at least if considered alone. In principle, glutamine depletion could increase expression of ER-MYC, for example by stabilizing the protein, but we found that the concentration of this protein was unaffected by deficiency in either glutamine or glucose (unpublished data).
The clue to the cause of apoptosis may be provided by learning how pyruvate and oxaloacetate rescue from cell death. Glutamine provides cells with a carbon chain, which is required for various synthetic activities, and amino and amido groups, which are required for synthesis of nucleotides and nonessential amino acids. Because pyruvate and oxaloacetate contain no nitrogen but rescue from apoptosis induced by glutamine deficiency, the cause of apoptosis is likely to be associated with the fate of the glutamine carbon chain. This chain is the backbone of 2-oxoglutarate, an intermediate of the Krebs cycle. Indeed, our observations indicated that glutamine in our system is either the main source of Krebs cycle intermediates, as in some proliferating normal and cancer cells (Reitzer et al., 1979; Moreadith and Lehninger, 1984; Zielke et al., 1984; Kovacevic et al., 1991; Baggetto, 1992; Petch and Butler, 1994), or regulates the Krebs cycle activity.
How could a deficiency in the Krebs cycle cause apoptosis? This cycle has two main functions: to supply NADH and FADH for oxidative phosphorylation by reducing FAD+ and NAD+ and to link multiple metabolic pathways (Fig. 9 B). Although we found that a deficiency in oxidative phosphorylation does not cause apoptosis in our system, a change in the mitochondrial NADH/NAD+ and FADH/FAD+ ratios, which we did not measure, could affect other processes, including regulation of gene transcription (for review see Ladurner, 2006) and mitochondrial permeability (Jonas et al., 2004). Therefore, glutamine might support cell viability not by providing biosynthetic precursors, but by enabling the Krebs cycle to function as a catalyst of redox reactions (Kovacevic, 1972; Baggetto, 1992). This hypothesis is consistent with the observation that only a few percent of consumed glutamine is used by proliferating cells for biosynthesis (Newsholme et al., 1985).
Apoptosis could also be caused by a deficiency in numerous synthetic pathways that cross the Krebs cycle. For example, citrate is used for the synthesis of fatty acids whose deficiency might change permeability of mitochondrial membranes and affect their regulation by the Bcl-2 proteins (Green and Kroemer, 2004). Some of the Krebs cycle intermediates, such as fumarate, succinate, and 2-oxoglutarate, can function as messenger and cofactor molecules directly regulating activity of enzymes and thereby contributing to regulation of gene transcription and/or tumorigenesis (Selak et al., 2005; Chen et al., 2006). Alternatively, pyruvate, oxaloacetate, and other α-ketoacids could function as antioxidants by binding hydrogen peroxide (O'Donnell-Tormey et al., 1987; Nath et al., 1995). This mechanism would imply that glutamine depletion produced some ROS or weakened an antioxidant system of the cell. However, we detected no ROS accumulation after glutamine depletion and the antioxidants that we tested, tiron and N-acetyl-cysteine, failed to rescue the depleted cells from apoptosis (unpublished data). Finally, we also cannot exclude the possibility that the cells are killed by a combination of the mechanisms that we tested, or that glutamine metabolism has some poorly understood function whose effect we accidentally revealed.
How does ectopic activity of MYC synergize with glutamine depletion to induce apoptosis?
Ectopic expression of MYC makes various cells more sensitive to diverse stimuli that can induce apoptosis. Several hypotheses have been proposed to explain this sensitization (for review see (Adhikary and Eilers, 2005). One is that increased activity of MYC abrogates a DNA damage cell cycle checkpoint, thereby causing proliferation in the presence of unrepaired DNA breaks. The resulting massive DNA damage causes apoptosis. However, glutamine depletion caused cell cycle arrest irrespective of the ERTM-MYC activity (unpublished data), while in our hands depletion of essential amino acids caused cell cycle arrest but not apoptosis. Another hypothesis is that abnormally high expression of MYC induces the production of ROS, which have various deleterious effects, including DNA damage (Vafa et al., 2002). This effect could synergize with the inhibitory effect of glutamine depletion on the synthesis of reduced glutathione. However, we detected no increase in the concentration of ROS or DNA damage either after activation of ERTM-MYC or depleting glutathione. Moreover, glutamine depletion even prevented the increase in p53 concentration caused by activation of ERTM-MYC, which implied that in our experimental system glutamine metabolism links MYC activity and increase in p53 concentration. One explanation of this phenomenon is that increase in p53 concentration caused by abnormally high MYC activity is caused directly or indirectly by oxidative damage that is associated with a functioning respiratory chain that depends on the functioning Krebs cycle, but other explanations could also account for our observation and need to be explored.
Abnormally high expression of MYC might facilitate apoptosis by changing the expression of proteins that regulate apoptosis. For example, increasing the ratio of Bcl-2 proteins may ease permeabilization of the mitochondria after various kinds of stress (Maclean et al., 2003; Egle et al., 2004). Finally, MYC may affect multiple metabolic pathways (Osthus et al., 2000; O'Connell et al., 2003), including concentrations of some mitochondrial proteins (Morrish et al., 2003; Li et al., 2005), thus facilitating apoptosis. Overall, how abnormal expression of MYC facilitates apoptosis induced by glutamine remains to be determined.
Relevance of glutamine metabolism to cancer therapy
Treatments that induce glutamine deficiency or interfere with its metabolism as a substrate for nucleotide synthesis were highly successful in animals, but turned out to be unacceptably toxic in people and were largely abandoned (Ahluwalia et al., 1990). The finding that the effect of glutamine depletion on cell viability was different both quantitatively and qualitatively from DON, a glutamine mimetic that was highly effective in animal models but toxic in people when used in the clinic (Ahluwalia et al., 1990), suggests that the potential of glutamine metabolism as a target for cancer therapy may need revisiting. Indeed, we found that glutamine deprivation was much more potent than DON in inducing apoptosis and more selective for cells with ectopically activated MYC. In contrast to DON, glutamine depletion was not followed by an increase in p53 concentration, which indicated that these treatments activated different pathways and that glutamine deprivation may not need p53 to kill, which is an advantage as this protein is mutated in many cancers. Therefore, learning how depletion of glutamine kills cells may help to understand how the viability of cancer cells can be regulated.
Overall, our study is consistent with the view that metabolic changes associated with carcinogenesis, and the energy metabolism in particular, might be much more complex than Warburg postulated, as solving even apparently simple problems, such as how a deficiency in glutamine induces apoptosis, is likely to require to develop approaches that can routinely measure, analyze, and model the complex metabolic and signal transduction networks.
Materials and methods
Cell lines
Normal diploid human lung fibroblasts IMR-90 (American Type Culture Collection), normal diploid human foreskin fibroblasts (HFFs) (obtained from Donald Ganem, University of California at San Francisco, San Francisco, CA), and cell lines derived from them were maintained in DME (Invitrogen) without phenol red supplemented with 4.5 g/L d-glucose, 2 mM l-glutamine, 1 mM sodium pyruvate, 10% fetal bovine serum, and mixture of penicillin and streptomycin (100 g/ml each). Cells were used between passages 12 and 23. Human retinal epithelial RPE cells, immortalized with hTERT and transduced either with pMig-neo vector expressing c-MYC (RPE-MYC) or with the vector alone, were obtained from Dun Yang (University of California, San Francisco) and maintained in the DMEM media described above. Genetically defined immortalized kidney epithelial cells (HA1E) were obtained from Robert Weinberg (Whitehead Institute, Cambridge, MA). HA1E and HA1E-derived cells were grown in MEM-α medium (Invitrogen) supplemented with 10% fetal bovine serum and antibiotics as described above.
Retroviral transduction
Retroviral vectors pBABE-hygroER, expressing the estrogen receptor binding domain, and pBABE-hygroER-MYC expressing c-MYC fused to the estrogen receptor, were a gift from William Tansey (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY). pBABE-hygroER-MYC and pBABE-puro MYC-ER plasmids were described previously (Eilers et al., 1989).
cDNAs encoding caspase-9 Cys287 to Ser mutant (caspase 9 dominant negative, C9DN) (Fearnhead et al., 1998), crmA, Bcl-2, and human c-MYC were cloned into a MarX-IV-puro retroviral gene transfer vector (Hannon et al., 1999).
Retroviruses were produced and used to infect primary cells as described previously (Faleiro and Lazebnik, 2000). In the case when two vectors were transduced, cells were infected with supernatants containing two different viruses simultaneously. Cells were selected with appropriate selection agents and obtained cell clones were pooled.
Activation of estrogen receptor fusion protein
Cells were seeded at 1.2 × 105 cells per well into 6-well plates or 5 × 104 cells per well into 12-well plates and were allowed to attach for 7–12 h. ER, or the fusion of ER with MYC, were activated by adding 4-hydroxytamoxifen (OHT) (Sigma-Aldrich) to the media to a final concentration 200 nM. Ethanol was used as a vehicle. Cells were washed at 24 h once with PBS, supplemented with fresh medium, and used for the experiments. Unless indicated, all media used for the experiments contained ethanol or OHT.
Nutrient depletion and tissue culture
To deplete glutamine or glucose, cells were cultured in glutamine or glucose-free DMEM that contained neither phenol red nor pyruvate (Invitrogen) unless indicated. When these nutrients had to be present, glucose or glutamine was added into the media to the final concentrations, 10 mM and 2 mM, respectively. To prepare media deficient in one of the essential amino acids, Earle's balanced salt solution was supplemented with 4.5 g/L glucose (Sigma-Aldrich), MEM vitamin solution (Invitrogen), 0.37 mM sodium bicarbonate (Invitrogen), 24.8 μM ferric nitrate (Sigma-Aldrich), 2 mM glutamine, and all essential amino acids, except one, at the concentrations of complete DMEM. Media for all experiments was supplemented with 10% dialyzed FBS prepared by dialyzing 50 ml of FBS against 2 liters of PBS using a membrane with a molecular cut-off of 3,500 D. PBS was changed every 12 h for 72 h.
Cell death assays
Cells were harvested by combining floating cells in the media with adherent cells detached with 0.05% trypsin/0.53 mM EDTA solution (Invitrogen). Cells were washed with cold PBS, resuspended in 10 mM Hepes, 140 mM NaCl, and 2.5 mM CaCl2, pH 7.4, and incubated with 2 μg/ml of recombinant annexin V conjugated to FITC for 15 min and with 4 μg/ml of PI for 1 min and analyzed by Becton Dickinson LSR-II cell analyzer with FACSVantage DiVa software. For analyzing nuclear morphology, cells were resuspended in staining solution containing 4% parafomaldehyde, 50% glycerol, 0.1% Triton, and 2 μg/ml Hoechst 33342. To identify cells that failed to exclude Trypan Blue, the cells were collected and resuspended in 50 μl of PBS. The suspension was complemented with equal volume of 0.08% Trypan Blue solution and incubated on ice for 5 min. The Trypan Blue–containing cells were scored by light microscopy.
Cell cycle analysis
To analyze DNA content, 5 × 105 cells were collected by trypsinization, washed with PBS, fixed with 70% ethanol (−20°C) and kept at −20°C for at least 4 h. After washing with PBS, cells were resuspended in PBS containing 80 μg/ml RNase and 30 μg/ml PI. After incubating for 30 min at room temperature, samples were analyzed by LSR-II cell analyzer with ModFit software.
Immunofluorescence
Cells were grown on coverslips, fixed with 4% paraformadehyde, stained with appropriate primary antibody and a secondary antibody labeled with AlexaFluor 488, and mounted using Prolong antifade medium (Invitrogen). The images were taken with an Axiophot microscope (Carl Zeiss MicroImaging, Inc.) with Plan-Apochromat 63×/1.40 oil objective lens, using a CCD camera (DFC420C; Leica) and Firecam acquisition software (Leica). The images were formatted using Adobe Photoshop to fit the size of the figures. All changes in brightness or contrast, if any done, were applied to the entire image.
Measuring the intracellular concentration of ATP
Cells grown in 12-well plates were collected and washed with cold PBS. ATP was extracted by adding 1 ml of boiling water to the cell pellet as described previously (Yang et al., 2002). The extract was vortexed and cleared for 5 min at 12,000 g at 4°C. 700 μl of the cleared supernatant were taken to measure ATP by Luciferin-Luciferase kit (Sigma-Aldrich) following the manufacturer's instructions. The pellet was resuspended in the remaining 300 μl of the supernatant and used to determine protein concentration. The concentration of protein was also determined in the supernatant used to determine ATP concentration to correct for any protein left after centrifugation of the extract. The amount of protein in both fractions was combined to obtain the total protein amount in the extract.
Measuring reduced glutathione
The amount of reduced glutathione in cells was measured by flow cytometry as described previously (Sen et al., 1999). In brief, cells grown in 12-well plates were collected, resuspended in 0.5 ml of PBS containing 1% FBS, and incubated with 40 μM monobromobimane (Biochemika) for 10 min at room temperature. After incubation, cells were moved on ice and the fluorescence at 485 nm was measured by flow cytometry (DiVa; Becton Dickinson).
Measuring the concentrations of NADH and NAD
The intracellular concentrations of NADH and NAD were measured by enzymatic cycling assay as described previously (Lowry et al., 1961). In brief, cells grown in 6-well plates were collected and washed with cold PBS. NADH was extracted by adding 100 μl of 0.05 N NaOH and 0.5 mM cysteine and incubating cell suspension at 60°C for 10 min. NAD was extracted by adding 100 μl of 0.04 N HCl and 4 mM cysteine and incubating the suspension at room temperature for 15 min. NADH and NAD extracts were sonicated for 30 s and used in a cycling system consisting of lactic dehydrogenase and glutamic dehydrogenase to produce pyruvate in 2,000-fold yield in 30 min. Reaction was stopped by heating for 2 min at 100°C and the concentration of pyruvate was measured using lactic dehydrogenase reaction fluorimetrically. Protein was measured in the extracts by Bradford assay (Bio-Rad Laboratories).
Measuring concentration of lactate
Cells were grown in 12-well plates with 1 ml of media in each well. At the indicated time points the media was collected and pelleted at 2,000 g for 10 min at 4°C. The supernatant was snap-frozen in liquid nitrogen and stored at −80°C until the assay was performed. Lactate concentration was measured using lactate dehydrogenase reaction (Bergmeyer, 1974).
CE-MS analysis
Metabolites were extracted from adherent cells by a procedure adopted from Le Belle et al. (2002). In brief, cells were collected by trypsinization, pelleted, and resuspended in 1 ml of cold PBS. 50 μl of cell suspension was taken to measure protein concentration by BCA assay (Pierce Chemical Co.). The rest of the cells were pelleted and the pellets were frozen in liquid nitrogen. The mixture of methanol and chloroform (−20°C) in a 2:1 ratio (vol/vol; 360 μl/cell pellet) was added to the frozen cell pellet. The pellet–solvent mixture was allowed to thaw on ice and was then sonicated. The sonicated extracts were supplemented with internal standards, methionine sulfone and Pipes (pH 7.4), at 2 nmol each. To separate hydrophilic and lipophylic fractions, 120 μl of chloroform and 90 μl of water were added. Suspension was centrifuged at 14,000 g for 20 min at 4°C. Equal volumes of the upper aqueous fraction were dried in a SpeedVac concentrator. The extracted metabolites were resuspended in 50 μl of water and stored at −80°C.
Extracts were analyzed by capillary-electrophoresis mass spectrometry (CE-MS), according to the method devised by Soga et al. (2003) for the quantitation of anions in cellular extracts. Agilent CE system was interfaced to an Applied Biosystems QSTAR Pulsar I using Applied Biosystems' CE-MS interface. Electrophoresis was performed in positively coated 110 cm × 50 μm i.d. SMILE (+) capillary (Nacalay Tesque), using 50 mM ammonium acetate (pH 8.5) as electrolyte. Injection was done with 50 mbar overpressure for 30 s; separation was carried with a constant potential of −30 kV. The voltage of the electrosprayer was −4.5 kV (thus reducing the effective electrophoresis voltage to 25.5 kV), and sheat liquid 5 mM ammonium acetate in 50% methanol was delivered at a flow rate of 2 μl/min. The mass spectrometer was run in full MS mode, scanning m/z 50–600 at 3 Hz. Identification was based on retention time and exact mass measurements after alignment of both axes with reference compounds, and confirmed by spiked standards. For semi-quantitation, peak areas where normalized to that of the internal standard Pipes and to the amount of protein measured by the BCA assay. All processing steps were performed with custom routines written in the Matlab environment (The Mathworks).
Reagents and antibodies
Rabbit polyclonal antibodies to S6 and pS6 were obtained from Cell Signaling Technology and were used in 1:1,000 and 1:4,000 dilutions, respectively. Monoclonal antibody to p53, clone DO-1, was obtained from Calbiochem and used in 1:1,000 dilution. Anti-Fas activating antibody, clone CH11, was obtained from Upstate Biotechnology. Anti-pH2AX (Ser139) antibodies were from Upstate Biotechnology (1:1,000 dilution). The 9E10 antibody to Myc was obtained from the Cold Spring Harbor Laboratory Monoclonal Antibody Facility. Anti-actin antibodies were purchased from Sigma-Aldrich. Rapamycin was obtained from Biosource International. All other reagents were obtained from Sigma-Aldrich.
Online supplemental material
Fig. S1, A and B document that ER-MYC is activated as expected; Fig. S1 C demonstrates that the MYC moiety of ER-MYC makes cells sensitive to glutamine depletion. Fig. S2 visualizes localization of phosphorylated histone H2AX in cells deprived of glutamine or treated with etoposide. Fig. S3 documents that lactate production remains largely unchanged after depletion of glutamine and is independent of ER-MYC activity. Fig. S4 compares the concentrations of glutathione and ATP measured biochemically or by mass spectrometry. Table S1 shows how concentrations of the measured metabolites change after glutamine depletion. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703099/DC1.
Supplementary Material
Acknowledgments
We thank Michael J. Bishop and his laboratory (University of California at San Francisco), Jason Chesney, John Eaton, Andrew Lane, Teresa Fan (University of Louisville, Louisville, KY), and Arthur Cooper (Burke Institute, White Plains, NY) for helpful discussions and suggestions, and Bill Tansey (CSHL) for providing plasmids expressing ER-MYC.
This study was made possible by a National Institutes of Health grant (CA-13106-31) to Y. Lazebnik and a grant from BayGene to P. Oefner.
Abbreviations used in this paper: OHT, 4-hydroxytamoxifen; ROS, reactive oxygen species.
References
- Adhikary, S., and M. Eilers. 2005. Transcriptional regulation and transformation by Myc proteins. Nat. Rev. Mol. Cell Biol. 6:635–645. [DOI] [PubMed] [Google Scholar]
- Ahluwalia, G.S., J.L. Grem, Z. Hao, and D.A. Cooney. 1990. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 46:243–271. [DOI] [PubMed] [Google Scholar]
- Albayrak, T., V. Scherhammer, N. Schoenfeld, E. Braziulis, T. Mund, M.K. Bauer, I.E. Scheffler, and S. Grimm. 2003. The tumor suppressor cybL, a component of the respiratory chain, mediates apoptosis induction. Mol. Biol. Cell. 14:3082–3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, C.P., J. Tsai, W. Chan, C.K. Park, L. Tian, R.M. Lui, H.J. Forman, and C.P. Reynolds. 1997. Buthionine sulphoximine alone and in combination with melphalan (L-PAM) is highly cytotoxic for human neuroblastoma cell lines. Eur. J. Cancer. 33:2016–2019. [DOI] [PubMed] [Google Scholar]
- Baggetto, L.G. 1992. Deviant energetic metabolism of glycolytic cancer cells. Biochimie. 74:959–974. [DOI] [PubMed] [Google Scholar]
- Benassi, B., M. Fanciulli, F. Fiorentino, A. Porrello, G. Chiorino, M. Loda, G. Zupi, and A. Biroccio. 2006. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol. Cell. 21:509–519. [DOI] [PubMed] [Google Scholar]
- Bergmeyer, H.U. 1974. Lactate dehydrogenase assay with pyruvate and NADH. In Methods of Enzymatic Analysis. Vol. 2. H.U. Bergmeyer, editor. Academic Press, New York. pp. 574–579.
- Bode, B.P., B.C. Fuchs, B.P. Hurley, J.L. Conroy, J.E. Suetterlin, K.K. Tanabe, D.B. Rhoads, S.F. Abcouwer, and W.W. Souba. 2002. Molecular and functional analysis of glutamine uptake in human hepatoma and liver-derived cells. Am. J. Physiol. Gastrointest. Liver Physiol. 283:G1062–G1073. [DOI] [PubMed] [Google Scholar]
- Chen, Z., J. Zang, J. Whetstine, X. Hong, F. Davrazou, T.G. Kutateladze, M. Simpson, Q. Mao, C.H. Pan, S. Dai, et al. 2006. Structural insights into histone demethylation by JMJD2 family members. Cell. 125:691–702. [DOI] [PubMed] [Google Scholar]
- Curi, R., C.J. Lagranha, S.Q. Doi, D.F. Sellitti, J. Procopio, T.C. Pithon-Curi, M. Corless, and P. Newsholme. 2005. Molecular mechanisms of glutamine action. J. Cell. Physiol. 204:392–401. [DOI] [PubMed] [Google Scholar]
- Egle, A., A.W. Harris, P. Bouillet, and S. Cory. 2004. Bim is a suppressor of Myc- induced mouse B cell leukemia. Proc. Natl. Acad. Sci. USA. 101:6164–6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers, M., D. Picard, K.R. Yamamoto, and J.M. Bishop. 1989. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 340:66–68. [DOI] [PubMed] [Google Scholar]
- Enari, M., H. Hug, and S. Nagata. 1995. Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature. 375:78–81. [DOI] [PubMed] [Google Scholar]
- Faleiro, L., and Y. Lazebnik. 2000. Caspases disrupt the nuclear-cytoplasmic barrier. J. Cell Biol. 151:951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearnhead, H.O., J. Rodriguez, E.E. Govek, W. Guo, R. Kobayashi, G. Hannon, and Y.A. Lazebnik. 1998. Oncogene-dependent apoptosis is mediated by caspase-9. Proc. Natl. Acad. Sci. USA. 95:13664–13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, B.C., and B.P. Bode. 2006. Stressing out over survival: glutamine as an apoptotic modulator. J. Surg. Res. 131:26–40. [DOI] [PubMed] [Google Scholar]
- Fulda, S., and K.M. Debatin. 2007. HIF-1-regulated glucose metabolism: a key to apoptosis resistance? Cell Cycle. 6:790–792. [DOI] [PubMed] [Google Scholar]
- Garber, K. 2006. Energy deregulation: licensing tumors to grow. Science. 312:1158–1159. [DOI] [PubMed] [Google Scholar]
- Green, D.R., and G. Kroemer. 2004. The pathophysiology of mitochondrial cell death. Science. 305:626–629. [DOI] [PubMed] [Google Scholar]
- Griffith, O.W., and A. Meister. 1979. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine). J. Biol. Chem. 254:7558–7560. [PubMed] [Google Scholar]
- Guerin, P.J., T. Furtak, K. Eng, and E.R. Gauthier. 2006. Oxidative stress is not required for the induction of apoptosis upon glutamine starvation of Sp2/0-Ag14 hybridoma cells. Eur. J. Cell Biol. 85:355–365. [DOI] [PubMed] [Google Scholar]
- Hammerman, P.S., C.J. Fox, and C.B. Thompson. 2004. Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem. Sci. 29:586–592. [DOI] [PubMed] [Google Scholar]
- Hannon, G.J., P. Sun, A. Carnero, L.Y. Xie, R. Maestro, D.S. Conklin, and D. Beach. 1999. MaRX: an approach to genetics in mammalian cells. Science. 283:1129–1130. [DOI] [PubMed] [Google Scholar]
- Holcik, M., and N. Sonenberg. 2005. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 6:318–327. [DOI] [PubMed] [Google Scholar]
- Izyumov, D.S., A.V. Avetisyan, O.Y. Pletjushkina, D.V. Sakharov, K.W. Wirtz, B.V. Chernyak, and V.P. Skulachev. 2004. “Wages of fear”: transient threefold decrease in intracellular ATP level imposes apoptosis. Biochim. Biophys. Acta. 1658:141–147. [DOI] [PubMed] [Google Scholar]
- Jonas, E.A., J.A. Hickman, M. Chachar, B.M. Polster, T.A. Brandt, Y. Fannjiang, I. Ivanovska, G. Basanez, K.W. Kinnally, J. Zimmerberg, et al. 2004. Proapoptotic N-truncated BCL-xL protein activates endogenous mitochondrial channels in living synaptic terminals. Proc. Natl. Acad. Sci. USA. 101:13590–13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox, W.E., M.L. Horowitz, and G.H. Friedell. 1969. The proportionality of glutaminase content to growth rate and morphology of rat neoplasms. Cancer Res. 29:669–680. [PubMed] [Google Scholar]
- Kovacevic, Z. 1972. Possibility for the transfer of reducing equivalents from the cytosol to the mitochondrial compartment in Ehrlich ascites tumor cells by the malate-aspartate shuttle. Eur. J. Biochem. 25:372–378. [DOI] [PubMed] [Google Scholar]
- Kovacevic, Z., O. Brkljac, and K. Bajin. 1991. Control and function of the transamination pathways of glutamine oxidation in tumour cells. Biochem. J. 273:271–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladurner, A.G. 2006. Rheostat control of gene expression by metabolites. Mol. Cell. 24:1–11. [DOI] [PubMed] [Google Scholar]
- Le Belle, J.E., N.G. Harris, S.R. Williams, and K.K. Bhakoo. 2002. A comparison of cell and tissue extraction techniques using high-resolution 1H-NMR spectroscopy. NMR Biomed. 15:37–44. [DOI] [PubMed] [Google Scholar]
- Li, F., Y. Wang, K.I. Zeller, J.J. Potter, D.R. Wonsey, K.A. O'Donnell, J.W. Kim, J.T. Yustein, L.A. Lee, and C.V. Dang. 2005. Myc stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 25:6225–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder-Horowitz, M., W.E. Knox, and H.P. Morris. 1969. Glutaminase activities and growth rates of rat hepatomas. Cancer Res. 29:1195–1199. [PubMed] [Google Scholar]
- Linker, W., M. Loffler, and F. Schneider. 1985. Uridine, but not cytidine can sustain growth of Ehrlich ascites tumor cells in glucose-deprived medium with altered proliferation kinetics. Eur. J. Cell Biol. 36:176–181. [PubMed] [Google Scholar]
- Littlewood, T.D., D.C. Hancock, P.S. Danielian, M.G. Parker, and G.I. Evan. 1995. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 23:1686–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry, O.H., J.V. Passonneau, D.W. Schulz, and M.K. Rock. 1961. The measurement of pyridine nucleotides by enzymatic cycling. J. Biol. Chem. 236:2746–2755. [PubMed] [Google Scholar]
- Maclean, K.H., U.B. Keller, C. Rodriguez-Galindo, J.A. Nilsson, and J.L. Cleveland. 2003. c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol. Cell. Biol. 23:7256–7270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina, M.A. 2001. Glutamine and cancer. J. Nutr. 131:2539S–2542S (discussion 2550S-1S). [DOI] [PubMed] [Google Scholar]
- Moreadith, R.W., and A.L. Lehninger. 1984. The pathways of glutamate and glutamine oxidation by tumor cell mitochondria. Role of mitochondrial NAD(P)+-dependent malic enzyme. J. Biol. Chem. 259:6215–6221. [PubMed] [Google Scholar]
- Morrish, F., C. Giedt, and D. Hockenbery. 2003. c-MYC apoptotic function is mediated by NRF-1 target genes. Genes Dev. 17:240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath, K.A., E.O. Ngo, R.P. Hebbel, A.J. Croatt, B. Zhou, and L.M. Nutter. 1995. alpha-Ketoacids scavenge H2O2 in vitro and in vivo and reduce menadione-induced DNA injury and cytotoxicity. Am. J. Physiol. 268:C227–C236. [DOI] [PubMed] [Google Scholar]
- Nesbit, C.E., J.M. Tersak, and E.V. Prochownik. 1999. MYC oncogenes and human neoplastic disease. Oncogene. 18:3004–3016. [DOI] [PubMed] [Google Scholar]
- Newsholme, E.A., B. Crabtree, and M.S. Ardawi. 1985. The role of high rates of glycolysis and glutamine utilization in rapidly dividing cells. Biosci. Rep. 5:393–400. [DOI] [PubMed] [Google Scholar]
- O'Connell, B.C., A.F. Cheung, C.P. Simkevich, W. Tam, X. Ren, M.K. Mateyak, and J.M. Sedivy. 2003. A large scale genetic analysis of c-Myc-regulated gene expression patterns. J. Biol. Chem. 278:12563–12573. [DOI] [PubMed] [Google Scholar]
- O'Donnell-Tormey, J., C.F. Nathan, K. Lanks, C.J. DeBoer, and J. de la Harpe. 1987. Secretion of pyruvate. An antioxidant defense of mammalian cells. J. Exp. Med. 165:500–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osthus, R.C., H. Shim, S. Kim, Q. Li, R. Reddy, M. Mukherjee, Y. Xu, D. Wonsey, L.A. Lee, and C.V. Dang. 2000. Deregulation of glucose transporter 1 and glycolytic gene expression by c-Myc. J. Biol. Chem. 275:21797–21800. [DOI] [PubMed] [Google Scholar]
- Panosyan, E.H., R.S. Grigoryan, I.A. Avramis, N.L. Seibel, P.S. Gaynon, S.E. Siegel, H.J. Fingert, and V.I. Avramis. 2004. Deamination of glutamine is a prerequisite for optimal asparagine deamination by asparaginases in vivo (CCG-1961). Anticancer Res. 24:1121–1125. [PubMed] [Google Scholar]
- Pelengaris, S., and M. Khan. 2003. The many faces of c-MYC. Arch. Biochem. Biophys. 416:129–136. [DOI] [PubMed] [Google Scholar]
- Perez-Gomez, C., J.A. Campos-Sandoval, F.J. Alonso, J.A. Segura, E. Manzanares, P. Ruiz-Sanchez, M.E. Gonzalez, J. Marquez, and J.M. Mates. 2005. Co-expression of glutaminase K and L isoenzymes in human tumour cells. Biochem. J. 386:535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petch, D., and M. Butler. 1994. Profile of energy metabolism in a murine hybridoma: glucose and glutamine utilization. J. Cell. Physiol. 161:71–76. [DOI] [PubMed] [Google Scholar]
- Ramanathan, A., C. Wang, and S.L. Schreiber. 2005. Perturbational profiling of a cell-line model of tumorigenesis by using metabolic measurements. Proc. Natl. Acad. Sci. USA. 102:5992–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer, L.J., B.M. Wice, and D. Kennell. 1979. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 254:2669–2676. [PubMed] [Google Scholar]
- Rogakou, E.P., D.R. Pilch, A.H. Orr, V.S. Ivanova, and W.M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- Rohren, E.M., T.G. Turkington, and R.E. Coleman. 2004. Clinical applications of PET in oncology. Radiology. 231:305–332. [DOI] [PubMed] [Google Scholar]
- Sabatini, D.M. 2006. mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer. 6:729–734. [DOI] [PubMed] [Google Scholar]
- Selak, M.A., S.M. Armour, E.D. MacKenzie, H. Boulahbel, D.G. Watson, K.D. Mansfield, Y. Pan, M.C. Simon, C.B. Thompson, and E. Gottlieb. 2005. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 7:77–85. [DOI] [PubMed] [Google Scholar]
- Sen, C.K., S. Roy, and L. Packer. 1999. Flow cytometric determination of cellular thiols. Methods Enzymol. 299:247–258. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J., and L.C. Cantley. 2006. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 441:424–430. [DOI] [PubMed] [Google Scholar]
- Shaw, R.J., N. Bardeesy, B.D. Manning, L. Lopez, M. Kosmatka, R.A. DePinho, and L.C. Cantley. 2004. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 6:91–99. [DOI] [PubMed] [Google Scholar]
- Shim, H., Y.S. Chun, B.C. Lewis, and C.V. Dang. 1998. A unique glucose-dependent apoptotic pathway induced by c-Myc. Proc. Natl. Acad. Sci. USA. 95:1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soga, T., Y. Ohashi, Y. Ueno, H. Naraoka, M. Tomita, and T. Nishioka. 2003. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2:488–494. [DOI] [PubMed] [Google Scholar]
- Stet, E.H., R.A. De Abreu, J.P. Bokkerink, L.H. Lambooy, T.M. Vogels-Mentink, J.J. Keizer-Garritsen, and F.J. Trijbels. 1995. Reversal of methylmercaptopurine ribonucleoside cytotoxicity by purine ribonucleosides and adenine. Biochem. Pharmacol. 49:49–56. [DOI] [PubMed] [Google Scholar]
- Thompson, C.B., D.E. Bauer, J.J. Lum, G. Hatzivassiliou, W.X. Zong, F. Zhao, D. Ditsworth, M. Buzzai, and T. Lindsten. 2005. How do cancer cells acquire the fuel needed to support cell growth? Cold Spring Harb. Symp. Quant. Biol. 70:357–362. [DOI] [PubMed] [Google Scholar]
- Vafa, O., M. Wade, S. Kern, M. Beeche, T.K. Pandita, G.M. Hampton, and G.M. Wahl. 2002. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: a mechanism for oncogene-induced genetic instability. Mol. Cell. 9:1031–1044. [DOI] [PubMed] [Google Scholar]
- Warburg, O. 1956. On the origin of cancer cells. Science. 123:309–314. [DOI] [PubMed] [Google Scholar]
- Wice, B.M., L.J. Reitzer, and D. Kennell. 1981. The continuous growth of vertebrate cells in the absence of sugar. J. Biol. Chem. 256:7812–7819. [PubMed] [Google Scholar]
- Yang, N.C., W.M. Ho, Y.H. Chen, and M.L. Hu. 2002. A convenient one-step extraction of cellular ATP using boiling water for the luciferin-luciferase assay of ATP. Anal. Biochem. 306:323–327. [DOI] [PubMed] [Google Scholar]
- Zhou, W., P.J. Simpson, J.M. McFadden, C.A. Townsend, S.M. Medghalchi, A. Vadlamudi, M.L. Pinn, G.V. Ronnett, and F.P. Kuhajda. 2003. Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 63:7330–7337. [PubMed] [Google Scholar]
- Zielke, H.R., C.M. Sumbilla, D.A. Sevdalian, R.L. Hawkins, and P.T. Ozand. 1980. Lactate: a major product of glutamine metabolism by human diploid fibroblasts. J. Cell. Physiol. 104:433–441. [DOI] [PubMed] [Google Scholar]
- Zielke, H.R., C.L. Zielke, and P.T. Ozand. 1984. Glutamine: a major energy source for cultured mammalian cells. Fed. Proc. 43:121–125. [PubMed] [Google Scholar]
- Zu, X.L., and M. Guppy. 2004. Cancer metabolism: facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 313:459–465. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}