

Abstract
Double-strand break (DSB) damage in yeast and mammalian cells induces the rapid ATM (ataxia telangiectasia mutated)/ATR (ataxia telangiectasia and Rad3 related)-dependent phosphorylation of histone H2AX (γ-H2AX). In budding yeast, a single endonuclease-induced DSB triggers γ-H2AX modification of 50 kb on either side of the DSB. The extent of γ-H2AX spreading does not depend on the chromosomal sequences. DNA resection after DSB formation causes the slow, progressive loss of γ-H2AX from single-stranded DNA and, after several hours, the Mec1 (ATR)-dependent spreading of γ-H2AX to more distant regions. Heterochromatic sequences are only weakly modified upon insertion of a 3-kb silent HMR locus into a γ-H2AX–covered region. The presence of heterochromatin does not stop the phosphorylation of chromatin more distant from the DSB. In mouse embryo fibroblasts, γ-H2AX distribution shows that γ-H2AX foci increase in size as chromatin becomes more accessible. In yeast, we see a high level of constitutive γ-H2AX in telomere regions in the absence of any exogenous DNA damage, suggesting that yeast chromosome ends are transiently detected as DSBs.
Introduction
DNA damage such as double-strand breaks (DSBs) causes rapid alterations of chromatin structure, including the posttranslational modification of histones through the activated PI3K-like kinases ATM (ataxia telangiectasia mutated) and ATR (ataxia telangiectasia and Rad3 related; Shiloh, 2003; McGowan and Russell, 2004; Kitagawa and Kastan, 2005; Harrison and Haber, 2006). The best studied of these modifications is the phosphorylation of histone H2AX, an isotype of histone H2A. Phosphorylated H2AX (γ-H2AX) is detected in mammalian cells within several minutes after ionizing radiation; this modification spreads along a megabase of chromatin surrounding a DSB (Rogakou et al., 1998, 1999). The precise roles of this modification are still under investigation, but γ-H2AX formation appears to be required for and/or maintain the association of proteins involved in DNA repair and damage signaling, including Nbs1, Mdc1, and 53BP1 (Celeste et al., 2003; Fernandez-Capetillo et al., 2003; Furuta et al., 2003). The absence of γ-H2AX impairs DNA repair, most notably the repair of sister chromatids (Bassing et al., 2002; Celeste et al., 2002; Xie et al., 2004).
In Saccharomyces cerevisiae, the major H2A histone contains a phosphorylatable SQE site at its C terminus, and, for simplicity, we will refer to the yeast's histone H2A as H2AX. In budding yeast, a single HO nuclease-induced DSB also promotes the modification of chromatin around the break over a domain of ∼100 kb (Shroff et al., 2004). The presence of γ-H2AX in yeast leads to the recruitment of both cohesins (Strom et al., 2004; Unal et al., 2004) and the Smc5/6 complex (De Piccoli et al., 2006; Lindroos et al., 2006) and, consequently, promotes sister chromatid repair of ionizing radiation. Moreover, there is a γ-H2AX–dependent association of chromatin remodeling complexes such as Ino80, SWR1, and NuA4 at the damage site (Downs et al., 2004; Morrison et al., 2004; van Attikum et al., 2004). The alteration of chromatin through γ-H2AX has been suggested to provide the platform to recruit or maintain activities needed for the efficient repair of DSB damage (Shiloh, 2003; McGowan and Russell, 2004; Kitagawa and Kastan, 2005; Harrison and Haber, 2006). In addition, the presence of γ-H2AX acts as a signal to retard cells from reentering the cell cycle after DNA is repaired (Keogh et al., 2006); this signaling may be important to maintain genomic integrity.
Although there have been extensive studies to understand the role of γ-H2AX in the DSB-responsive pathway, the mechanism by which γ-H2AX spreads along a global chromatin in response to a DSB is still not clear. Whether there are boundaries to γ-H2AX spreading is not known. We have investigated the factors responsible for the extent of spreading of γ-H2AX around a site-specific DSB. To understand the alteration of chromatin structure after DSB formation and its involvement in the DSB-responsive pathway, we decided to study the extent and the regulation of γ-H2AX spreading in budding yeast. Our finding that γ-H2AX cannot form in regions of silenced heterochromatin led us to explore similar questions in mammalian cells. In this study, we report that γ-H2AX is largely excluded from regions of heterochromatin both in yeast and in mammalian cells but that heterochromatin is not a barrier to spreading γ-H2AX beyond that region. We report the surprising finding that in yeast, subtelomeric regions in budding yeast are constitutively modified by γ-H2AX, suggesting that telomeres are at least transiently recognized as a form of DSB damage.
Results
Regulation of γ-H2AX spreading from a single DSB
To examine how far γ-H2AX spreads along the chromosome in response to a DSB, we performed chromatin immunoprecipitation (ChIP) with an antibody specific to budding yeast γ-H2AX (Shroff et al., 2004). A DSB was generated at MAT by expressing a galactose-inducible HO endonuclease. Because the homologous sequences HML and HMR were deleted in this strain, the DSB at MAT could not be repaired by homologous recombination (Moore and Haber, 1996), and, thus, both of the DSB-responsive kinases Mec1 and Tel1 could be activated by the persistence of DSB. 1 h after HO induction, a DSB had formed in > 90% of the cells. The extent of γ-H2AX surrounding the DSB was examined by quantitative PCR using primer pairs for sites on either side of the DSB (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1) compared with results before HO cleavage. Consistent with the previous studies (Shroff et al., 2004; Unal et al., 2004; De Piccoli et al., 2006), γ-H2AX was enriched over a ∼50-kb region on either side of the DSB except for the regions very close to the DSB (Fig. 1 A). The highest increase of γ-H2AX was seen 10–30 kb from the either side of the DSB, but increased signal could be seen as far as 40–50 kb from the DSB. Previous studies have shown that either Tel1 or Mec1 could carry out γ-H2AX modification in asynchronous cells and that in G1-arrested cells, in which Mec1 is inactive and there is very little resection of DSB ends (Ira et al., 2004), Tel1 was necessary and sufficient to modify the entire region (Shroff et al., 2004). Now, we report that in G2-arrested mec1Δ cells, in which ends are continually resected, Tel1 itself can nevertheless fully modify the region in G2-arrested cells (Fig. 1 B).
Figure 1.

γ-H2AX spreads ∼50 kb on either side of a DSB. (A) Distribution of γ-H2AX in response to a DSB either at MAT in chromosome III (black squares) or at ∼97 kb from the left end of chromosome VI (white squares). γ-H2AX ChIP values on either side of the DSB were examined by quantitative PCR using primer pairs at the points indicated. The signal at each locus was normalized to the signal of the control locus, CEN8, and the increase seen 1 h after DSB induction was calculated by normalizing with the results before HO induction. (B) Distribution of γ-H2AX in response to a DSB at MAT in chromosome III either in the wild-type (wt; black squares) or mec1Δ cells (gray squares). γ-H2AX ChIP values on either side of the DSB were examined by quantifying PCR fragments on the agarose gel with Quantity One. (C) Changes in mRNA levels in genes near an HO-induced DSB at MAT. In yeast cells arrested in G2 in nocodazole, HO endonuclease was expressed, and changes in mRNA levels were analyzed by microarray. The location of HO cleavage is marked with an arrow. No change in gene expression is seen for genes lying within 50 kb of the DSB in the first hour, although as the DNA in this region becomes single stranded by 5′ to 3′ resection at roughly 4 kb/h, the levels of mRNA decrease (green) progressively as a function of distance from the DSB. Complete data can be found at http://db-dev.yeastgenome.org/cgi-bin/expression/expressionConnection.pl. A version of this figure was previously published by Lee et al. (2000).
One possible reason for modifying histones over such a large domain would be to eliminate transcription that might compete with DNA repair proteins in binding to DNA. We report that the modification of histones over this domain does not appear to affect the state of transcription in this domain. Cells were arrested in nocodazole so that we would not monitor changes in cell cycle–regulated genes when the HO-induced population arrested at G2/M. About 150 genes were either turned off or turned on in response to DNA damage (Lee et al., 2000); the complete data can be found at http://db-dev.yeastgenome.org/cgi-bin/expression/expressionConnection.pl. Here, we focus on genes surrounding the DSB (the genes between MATα1 and TSM1 are indicated; Fig. 1 C). 1 h after HO-induced creation of an unrepaired break at MAT, there were no notable changes in gene expression even though γ-H2AX modification can be seen in 15 min (Shroff et al. 2004). Over time, gene expression progressively stopped for genes further from the DSB; these changes correlate with the time it takes for these sequences to be rendered single stranded by 5′ to 3′ resection, moving at 4 kb/h (Lee et al., 2000).
The lack of additional spreading beyond 50 kb at 1 h after HO induction could be caused by the presence of barrier sequences. To test whether the spreading of γ-H2AX was limited by a barrier sequence to the right of the DSB, we deleted the normal HO cleavage site and inserted a site 17 kb to the right. In this circumstance, the entire region of γ-H2AX was displaced to the right (Fig. 2 A). Because γ-H2AX spread further to the right and did not extend as far as it had to the left, the spreading of γ-H2AX does not seem to be limited by boundaries. A similar result was found when we deleted the original HO cleavage site and inserted an HO cut site ∼600 bp from the centromere of chromosome III, CEN3 (Fig. 2 B). Again, spreading of γ-H2AX covered ∼50 kb on both sides of the DSB. These results also demonstrated that the budding yeast's small centromere, which lacks pericentric heterochromatin (Bloom and Carbon, 1982), is not a barrier to γ-H2AX spreading.
Figure 2.

The second DSB does not affect the extent of γ-H2AX spreading. (A) Lack of a barrier to γ-H2AX spreading to the right of MAT. A 117-bp HO cut site was inserted 17 kb to the right of the normal cleavage site, which was deleted (triangles) in the cells lacking the normal HO recognition site. There are general shifts of γ-H2AX modification with the displaced cut sites relative to cleavage at MAT (circles). The position of each DSB is pointed by arrowheads. (B) Lack of a barrier to γ-H2AX spreading to the right of CEN3. A 117-bp HO cut site was inserted 600 bp to the left of CEN3 in the cells lacking the normal HO recognition site (squares). γ-H2AX ChIP values on either side of the DSB were measured by quantitative PCR. The extent of γ-H2AX modification with the displaced cut site is similar to that with the normal HO recognition site at MAT (circles). The position of each DSB is pointed to by arrowheads, and the position of CEN3 is denoted with a square. (C) Simultaneous DSBs at MAT (black squares) and chromosome VI (white squares) did not change the extent of γ-H2AX spreading, as measured either on chromosome III or chromosome VI. (D) Simultaneous DSBs on chromosome III, at MAT, and ∼600 bp to the left of CEN3 did not significantly increase the extent of γ-H2AX spreading. The position of each DSB is marked by arrowheads, and the position of CEN3 is denoted with a square.
As a further test, we introduced an HO recognition site into the left arm of chromosome VI in the strain lacking the HO recognition sequence at MAT. γ-H2AX spreading on chromosome VI was nearly identical to that on chromosome III (Fig. 1 A). By creating a strain with DSBs on both chromosomes III and VI, we showed that the extent of γ-H2AX spreading was largely not affected by the presence of a second DSB in a different chromosome, although there seems to be a slight increase of the signal at 50 kb distal from the DSB on chromosome III (Fig. 2 C). We also examined γ-H2AX spreading when two DSBs were created in the same chromosome, chromosome III, with one DSB at MAT and the other 600 bp to the left of CEN3. The distance between the two DSBs is ∼86 kb. With two DSBs, the overall extent was similar to the results expected if one added the results of strains with only a DSB at MAT or at CEN3 (Fig. 2 D; also see B). γ-H2AX covered the 86 kb of chromatin between the two breaks and spread ∼50 kb to the left of the DSB near CEN3, as it did with the single DSB next to CEN3. There does seem to be a modest increase in γ-H2AX spreading around the DSB at MAT. Thus, the presence of a second DSB in the same chromosome did not significantly affect the extent of γ-H2AX spreading at each DSB.
γ-H2AX formed after DSB formation does not seem to be turned over or replaced around the DSB before loss by resection. The Mec1 and Tel1 kinases, which are responsible for creating γ-H2AX, can be inactivated by the PI3KK inhibitor caffeine (Fig. 3 A; Vaze et al., 2002). If γ-H2AX was rapidly turned over and replaced by unphosphorylated H2AX and/or by the alternative histone H2A.Z (Papamichos-Chronakis et al., 2006), we would expect to lose the γ-H2AX ChIP signal after inhibiting the Mec1 and Tel1 kinase activities by caffeine. Caffeine treatment mimics the deletion of both Mec1 and Tel1 in that it prevents γ-H2AX formation after methylmethanesulfonate (MMS) treatment (Fig. 3 A). 30 min after HO induction, we treated cells with 10 mg/ml of caffeine and examined γ-H2AX on the chromatin located 20 kb from the DSB 30 min and 1 h later, long before 5′ to 3′ resection of the DNA end would reach this region. The amount of γ-H2AX remaining at this site was not affected by caffeine treatment (Fig. 3 B), showing that γ-H2AX is not removed from DNA by rapid turnover.
Figure 3.

γ-H2AX is not removed from DNA by rapid turnover. (A) Cells were grown to log phase and treated with 0.1% MMS for 1 h. 10 or 20 mg/ml of caffeine was added at the same time when MMS was added to the culture. The activity of Mec1p and Tel1p kinases was examined by their ability to generate γ-H2AX, as shown by Western blot analysis. The CPY (carboxy peptidase Y) protein was used as a loading control. (B) A DSB was generated at MAT by HO induction. After 30 min, 10 mg/ml of caffeine was added into the cell culture to inhibit continued Mec1 and Tel1 kinase activity. γ-H2AX ChIP signals before HO induction as well as 0.5, 1, and 2 h after HO induction were examined.
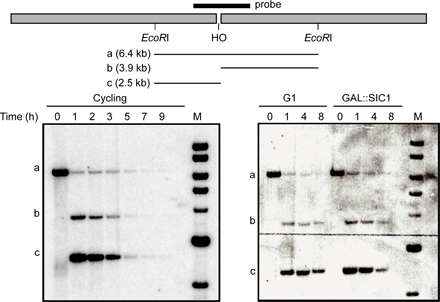
Our previous study showed that γ-H2AX is lost when a region becomes single stranded by DNA end resection, presumably because histones, or at least H2A–H2B dimers, are lost (Keogh et al. 2006). Here, we confirm and extend this conclusion, showing that there is a progressive loss of γ-H2AX from the region beginning near the DSB (Fig. 4 A, top). To establish that the loss of γ-H2AX depends on DNA resection, we looked at γ-H2AX when DNA end resection was inactive either in G1-arrested cells or in the cells overexpressing the CDK1/Clb inhibitor Sic1 (Ira et al. 2004). The formation of HO-induced γ-H2AX was not impaired by arrest. In both conditions, a substantial amount of γ-H2AX modification persisted as long as 8 h after HO induction, when resection was severely impaired (Fig. 4 A, middle and bottom). In fact, there was a slight increase in the level of γ-H2AX 10 kb away from the DSB 4 h after HO induction when resection was blocked. These results show that the loss of γ-H2AX does not occur without DNA resection. The decrease of γ-H2AX ChIP signals 2 or 10 kb from the DBS at 8 h after HO induction is likely caused by residual DNA resection (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1).
Figure 4.

Dynamics of γ-H2AX as DNA end resection proceeds. (A) γ-H2AX ChIP values either at 2 kb or at 10 kb to the right of the DSB at MAT were measured 1, 4, and 8 h after HO induction and were normalized with the value before HO induction (0 h). Three different cultures were examined: asynchronous cells (top), G1-arrested cells (middle), and SIC1-overexpressing cells (bottom). Quantitative PCR analysis was performed to measure γ-H2AX ChIP signals. (B) γ-H2AX ChIP values at 70 kb to the right of the DSB at MAT in three different cultures: asynchronous cells (white bars), G1-arrested cells (gray bars), and SIC1-overexpressing cells (black bars). (C) Cellular level of γ-H2AX after HO induction. A DSB was generated at MAT in derivatives of JKM179 deleted for HML and HMR: wild type (wt), mec1Δ, and tel1Δ. Cells were collected at 1, 4, and 8 h after HO induction as well as before HO induction, and protein extracts were subjected to Western blot analysis against either γ-H2AX or CPY. (D) γ-H2AX ChIP values at 70 kb to the right of the DSB at MAT were examined by quantitative PCR in the wild-type (white bars), mec1Δ (gray bars), and tel1Δ cells (black bars).
The results we have shown so far demonstrate that once γ-H2AX is formed in the first 30–60 min, there is little additional spreading of the modification to adjacent regions in the next few hours. But, if one looks at later times, when γ-H2AX is lost from single-stranded DNA at sites as far as 10 kb from the DSB, there is a large increase of γ-H2AX in the intact chromatin 70 kb away from the DSB (Fig. 4 B). We also note that there was a slight general increase (approximately two- to threefold) of γ-H2AX at sites on other chromosomes that did not suffer a DSB 8 h after DSB formation (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1). Similarly, Western blot analysis reveals a striking increase in γ-H2AX 4 and 8 h after HO induction compared with the level 1 h after HO induction (Fig. 4 C). However, when resection was blocked, the amount of γ-H2AX 70 kb away from the DSB did not significantly increase (Fig. 4 B). We suggest that single-strand DNA generated by resection results in the displacement of γ-H2AX but, at the same time, mediates the repositioning of kinases responsible for γ-H2AX so that more distant regions can now be modified.
To examine whether Mec1 and Tel1 kinase are responsible for the late spreading of γ-H2AX, we looked at γ-H2AX by the Western blot analysis either in mec1Δ or tel1Δ cells. As we showed previously (Keogh et al. 2006), there is a substantial increase in total γ-H2AX 4–8 h after HO induction; however, this increase is absent in mec1Δ cells but clearly seen in tel1Δ cells (Fig. 4 C). In addition, γ-H2AX ChIP signal in the chromatin 70 kb away from the DSB did not increase in mec1Δ cells 4 and 8 h as resection proceeded compared with that 1 h after HO induction (Fig. 4 D). The Mec1-interacting ATRIP protein Ddc2 is known to associate with replication protein A–coated single-strand DNA (Zou and Elledge, 2003). Therefore, we suggest that Mec1 is recruited to the newly generated single-strand DNA by DNA end resection and then can phosphorylate γ-H2AX in adjacent chromatin domains that were not initially modified when Mec1 and Tel1 were bound near the DSB end.
Previously, we showed that the histone phosphatase complex containing Pph3 is responsible for dephosphorylating γ-H2AX, but only after it had been released from chromatin (Keogh et al. 2006). However, the cellular pool of γ-H2AX continues to increase despite the loss of γ-H2AX from single-stranded DNA (Fig. 4, A [top] and C). One source of the increase can be the late spreading of γ-H2AX to more distant regions, but there may also be a decrease in the rate of dephosphorylation of released γ-H2AX. Thus, in checkpoint-arrested cells, with an unrepaired DSB, γ-H2AX may not be dephosphorylated immediately after it is released from chromosomes. Alternatively but not exclusively, some of the released γ-H2AX may be reincorporated randomly into the chromosomes and/or H2AX in the random chromosomes may be moderately phosphorylated in a Mec1-dependent manner.
The heterochromatic silent HML and HMR loci are refractory to γ-H2AX formation
The distribution of γ-H2AX across CEN3 from a nearby DSB both in strains with a second DSB at MAT (Fig. 2 C) and with a single DSB near CEN3 (Fig. 2 A) showed that the centromere-specific chromatin structure did not inhibit γ-H2AX spreading. However, in contrast to the centromeres of fission yeast and of most other organisms, centromeres of budding yeast are very small and not especially heterochromatic (Bloom and Carbon, 1982). To examine whether the presence of heterochromatin affects γ-H2AX modification, we examined silent heterochromatic HML and HMR loci, which have highly positioned, largely deacetylated nucleosomes (Weiss and Simpson, 1998; Ravindra et al., 1999). An HO cut site was introduced 7 kb to the right of HML. After DSB formation, γ-H2AX spread ∼50 kb from the right side of the DSB, as seen with DSBs at other sites; however, spreading of γ-H2AX in the left side of the DSB stopped at HML (Fig. 5 A, left). The lack of γ-H2AX increase at HML relative to a site on another chromosome is also seen in the 10 kb between HML and the telomere; however, the small fold increase in this region after HO-induced damage turns out to be attributable to an unexpectedly high level of γ-H2AX in subtelomeric regions in the absence of DNA damage (Fig. 5 A, right), as we discuss in detail below.
Figure 5.

Effect of heterochromatin in HML and HMR on γ-H2AX spreading. (A) A DSB (arrowhead) was generated at ∼7 kb from HML (gray box). The γ-H2AX ChIP signal was examined by quantitative PCR. The increase in γ-H2AX after HO induction (left) was calculated by normalizing the γ-H2AX ChIP signal at 1 h after HO induction (gray squares) with that before HO induction (black squares; right). The position of the telomere is marked with a black box. (B) The HMR sequences, including its own silencers (gray box), was introduced ∼41 kb from the left end of chromosome III, in which HML was replaced by LEU2 (white box). A DSB (arrowhead) led to increased γ-H2AX ∼50 kb to the right in the absence (gray squares) or presence (black squares) of the ectopic HMR sequence, but there is much less modification over HMR. (C) A DSB was generated ∼7 kb to the right of HMLα-inc (gray box). The level of γ-H2AX in a SIR3 (black diamonds) or sir3Δ (gray diamonds) strain are compared. Error bars represent one SEM.
To separate the possible overlapping effects of HML and the telomere on γ-H2AX spreading, we inserted the heterochromatic HMR locus ∼27 kb to the right of the HO cut site and ∼40 kb from the telomere in a strain in which the normal HML sequence was replaced by LEU2 marker (Fig. 5 B). After DSB induction, γ-H2AX spread ∼50 kb to the right of the DSB, but there was much less enrichment of γ-H2AX within the HMR chromatin (Fig. 5 B). Although γ-H2AX increased 19-fold to the right of the inserted HMR sequences (at ∼38 kb from the DSB), there was only a fourfold increase within HMR chromatin, which was 10 kb more proximal to the DSB. It is possible that the estimate of modification within HMR is too high because it is possible that there is some ChIP of γ-H2AX that is actually in the immediately adjacent regions, as not all sonicated fragments will be of the average 500-bp size. However, in any case, it is clear that HMR sequences are refractory to modification compared with sequences on either side. In the absence of the ectopic HMR domain, there was 15-fold γ-H2AX induction ∼30 kb away from the DSB, which is approximately the same distance from the DSB as the inserted HMR. These results suggest that the heterochromatic HML and HMR loci do not act as barriers to γ-H2AX spreading but are themselves refractory to γ-H2AX formation. These observations suggest that the spreading of γ-H2AX from a DSB does not occur by a processive mechanism in which the kinase modification of one histone provides a platform for the modification of an adjacent or nearby histone octamer, or at least the kinase must be able to reach over a 3-kb segment of heterochromatin.
As a further demonstration that heterochromatin and not simply chromatin covering specific sequences are unable to be modified, we desilenced the normal HML locus by deletion of the SIR3 gene. Here, the HMLα locus carries a single base pair mutation that prevents HO cleavage. After DSB induction, γ-H2AX was enriched over the unsilenced HML sequence in sir3Δ cells four or nine times more compared with modification in SIR3 cells (Fig. 5 C). We conclude that the inhibition of γ-H2AX formation depends on the silent chromatin status of HML. However, we note that the γ-H2AX epitope could be occluded in silent chromatin, preventing the detection of γ-H2AX by ChIP.
Phosphorylated H2AX occurs preferentially in open chromatin in mammalian cells
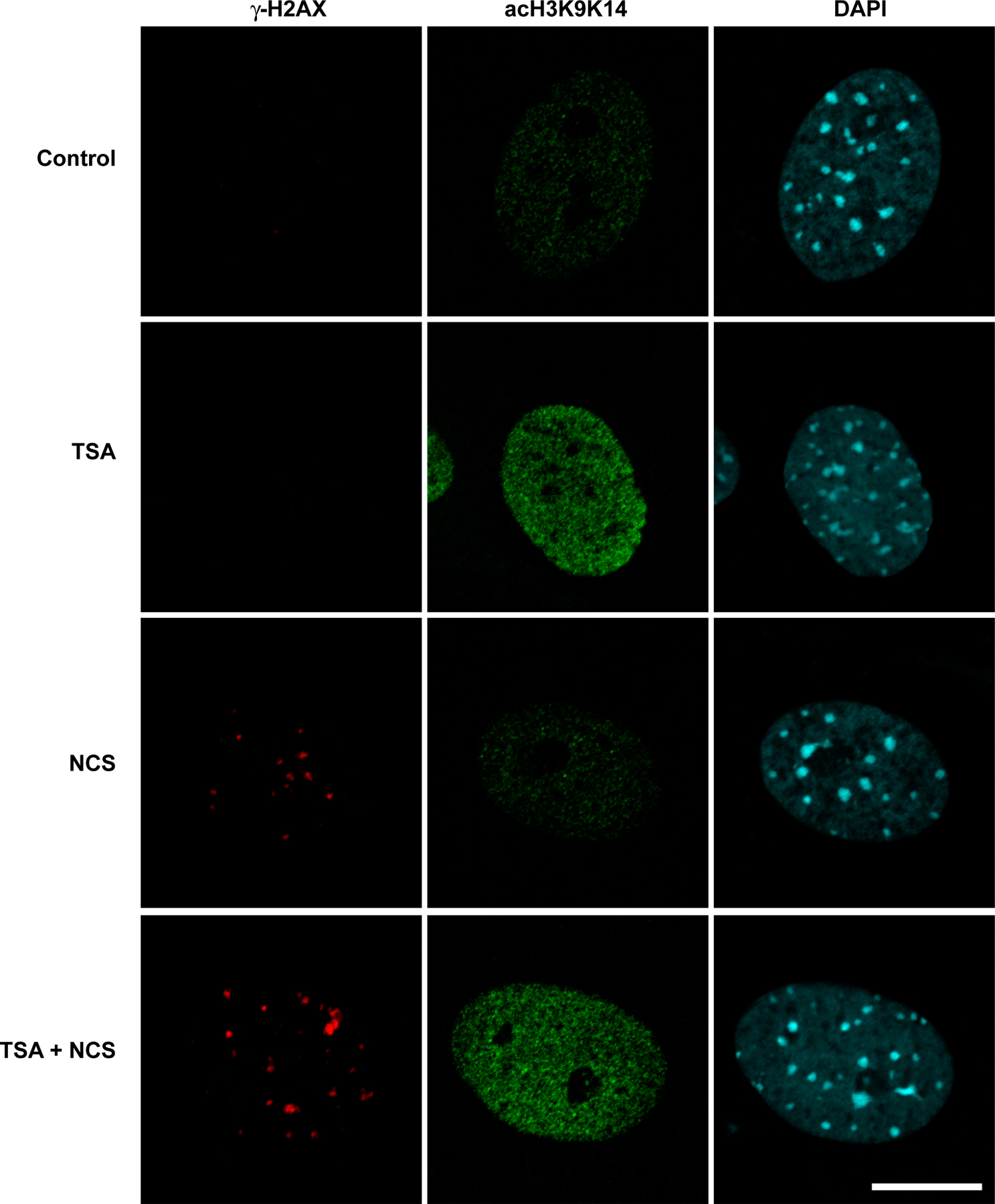
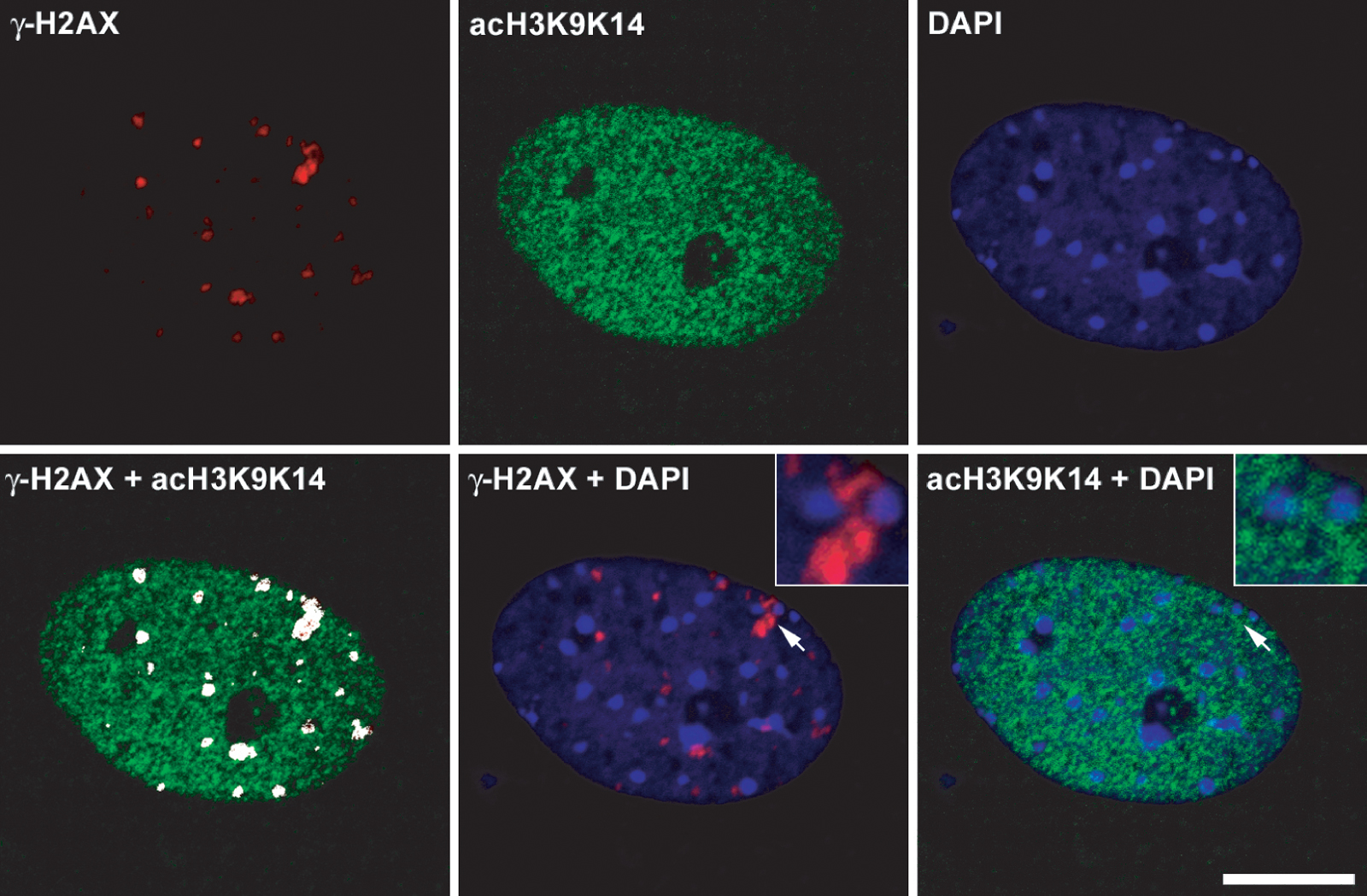
We next examined γ-H2AX distribution in mammalian cells. The introduction of DNA breaks is known to induce chromatin remodeling to a more open state, including when the DNA breaks occur within heterochromatin (Kruhlak et al., 2006). We hypothesized that if neighboring condensed regions of chromatin not containing DSBs are refractory to the spreading of γ-H2AX, treatment with a histone deacetylase inhibitor that has the potential to open chromatin would result in the increased spreading of phosphorylated H2AX in response to DNA damage. Therefore, we examined the distribution, mean fluorescence intensity, and size (volume) of γ-H2AX containing foci in primary mouse embryo fibroblasts treated with 1 μM trichostatin A (TSA) for 8 h before exposure with 10 ng/ml of the radiomimetic neocarzinostatin (NCS) for 1 h. We found that the mean number of γ-H2AX foci per nucleus increased slightly from 17.4 ± 5.9 in NCS-treated cells to 28.4 ± 7.1 in TSA plus NCS–treated cells (Fig. 6). TSA treatment led to a greater accumulation of acetylated histones but, in the absence of NCS treatment, did not influence the formation of γ-H2AX foci (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1). More strikingly, a subset of foci per nucleus (on average 5/28) had a volume >1.0 μm3 compared with ∼1/18 in non-TSA–treated cells. Altogether, there were 256 large foci out of 1,305 foci (n = 46) in TSA-pretreated cells compared with only 35/784 (n = 45) in the untreated cells (P < 0.0001).
Figure 6.

Distribution of phosphorylated H2AX in mouse embryo fibroblasts containing hyperacetylated histones. γ-H2AX foci in wild-type cells treated with either NCS or TSA and NCS. Confocal image stacks through the depth of the cell nucleus (z axis) were collected with an optical slice thickness of 800 nm. (A) Representative single optical slice of γ-H2AX foci in NCS-treated (top) and TSA + NCS–treated (bottom) cells. Images were collected with identical imaging parameters and contrast adjustments (histogram stretching). Arrows denote two separate γ-H2AX foci adjacent to pericentric heterochromatin that are shown at higher magnification in the top left and right insets. (B) The same cell nucleus as in A but shown as a 3D volume reconstruction, with the surface rendering of individual foci shown in red superimposed on the DAPI image, which is shown in blue. Images were background subtracted, and volume reconstructions were generated using Imaris software, from which the volumes of individual foci were measured (see Materials and methods). Bar, 5 μm.
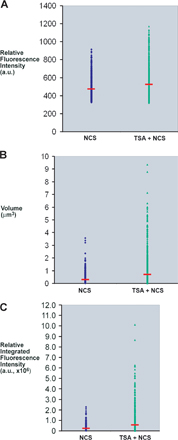
Interestingly, several of the larger γ-H2AX foci localized adjacent to pericentric heterochromatin (Fig. 6 A), but the γ-H2AX signal was restricted to the periphery of the heterochromatin and did not extend into the interior of the heterochromatin. Although the mean fluorescence intensity measured for the individual foci did not change dramatically in the NCS-treated cells compared with the TSA plus NCS–treated cells, the volume of the foci did increase, leading to a shift in the foci distribution to a greater total or integrated fluorescence intensity in the TSA plus NCS–treated cells (Fig. S4). This indicates that more total H2AX is phosphorylated in the larger foci compared with the smaller foci. Thus, chromatin remodeling to a more open state is accompanied by an increase in the total amount of H2AX phosphorylated within the damaged region.
We also examined the degree of colocalization of γ-H2AX with histone H3 acetylated at lysines 9 and 14 (acH3K9K14), a marker for highly acetylated histone H3 and a more open accessible chromatin state. The amount of acH3K9K14 increased in the TSA-treated cells (Fig. S5, available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1), and there was an increase in the colocalization coefficient of γ-H2AX colocalized with acH3K9K14 from 0.67 ± 0.13 (n = 20) in NCS-treated cells to 0.91 ± 0.09 (n = 20) in the TSA plus NCS–treated cells. Acetylated H3 (acH3K9K14) colocalized with γ-H2AX foci in cells not treated with TSA, but blocking histone deacetylase activity with TSA increased the amount of acetylated H3 and bolstered the association of γ-H2AX with acH3K9K14. Although more γ-H2AX colocalized with acH3K9K14 in the TSA plus NCS–treated cell, γ-H2AX and acH3K9K14 were found at the periphery of pericentric heterochromatin but not throughout the entire heterochromatin domain (Fig. S5). The accessibility of heterochromatin to immunostaining was substantiated by the localized labeling of methylated histone H3-K9, a marker for heterochromatin (unpublished data). Similar pretreatment with 5 mM sodium butyrate increased the volume of γ-H2AX foci in NCS-treated fibroblasts (unpublished data). Thus, global increases in histone acetylation leads to the further spreading of γ-H2AX in a subset of foci, and those foci are absent from heterochromatin.
Telomere-adjacent chromatin contains constitutive γ-H2AX throughout the cell cycle
In budding yeast, we find that γ-H2AX is constitutively located at telomeres (Fig. 5 A, right); consequently, when one examines the fold increase after creating DSB damage, there is only a small fold increase over the already high base line (Fig. 5 A, left). The high level of γ-H2AX near telomeres is seen when one compares γ-H2AX ChIP signals at telomeres with those at other locations, such as CEN8 (Fig. 5 A, right). We confirmed the high level of telomere-adjacent γ-H2AX at three chromosome ends by probing unique sequences near the left and right ends of chromosome III (Fig. 7 A) as well as near the left telomere of chromosome V (not depicted). The extent of γ-H2AX distribution at telomere-adjacent regions is ∼10 kb, which was smaller than that in response to an HO-induced DSB. As expected, there was no γ-H2AX signal at telomeres or elsewhere in cells in which the normal H2A gene is replaced by H2A-S129A (unpublished data). These results suggest that γ-H2AX is constitutively distributed on the telomere-adjacent chromatin, leading to a small increase fold of γ-H2AX after a DSB formation.
Figure 7.

Distribution of γ-H2AX near the telomere. (A) γ-H2AX ChIP at subtelomeric regions of chromosome III (left and right) in the absence of exogenous DNA damage normalized by the γ-H2AX ChIP signal at CEN8. The black boxes denote the positions of telomere sequences. The location of HML is indicated by the gray box. (B) cdc13-1 mutant cells were grown at 25°C in log phase and shifted to 37°C at the same time when HO was induced by adding galactose into the culture. γ-H2AX near the telomere, TEL03L, as well as ∼20 kb to the right of the HO-induced DSB at MAT (gray symbols) were examined by ChIP and normalized to the signal at CEN8. Samples were collected before temperature shift and HO induction (diamonds) and at 1 (squares), 2 (triangles), and 4 (circles) h. The arrowhead indicates the location of HO-induced DSB. The black box denotes the position of TEL03L. The location of HML is indicated by the gray box. (C) The constitutive level of γH2AX near TEL03L was examined in the sir4Δ strain (triangles) and in the yku80Δ strain (squares) by ChIP. The black box indicates TEL03L. (D) The constitutive level of γH2AX near TEL03L and TEL03R was examined in G1-arrested cells (diamonds) after α-factor arrest and in G2-arrested cells (squares) after nocodazole treatment. γ-H2AX was also examined in the G1-arrested tel1Δ strain (triangles). The black boxes indicate TEL03L and TEL03R. Error bars represent SEM.
Telomeric 3′ single-stranded DNA is associated with various proteins, including Cdc13, that prevent telomeres from activating DSB repair or cell cycle arrest (Garvik et al., 1995; Grandin et al., 2001). We asked whether more γ-H2AX would be generated in telomere-adjacent chromatin in response to decapping the telomere. In a cdc13-1 mutant strain at the nonpermissive temperature, the telomeric DNA is uncapped and degraded by 5′ to 3′ end resection, leading to the cell cycle arrest in G2 (Garvik et al., 1995; Grandin et al., 2001). We grew the cdc13-1 cells at 25°C and shifted the temperature to 37°C. At the same tine, HO endonuclease was induced to make a DBS at MAT. In response to the HO-induced DSB, we detected an ∼20-fold increase of γ-H2AX 20 kb away from the HO-induced DSB. Because of the constitutive presence of the modification near telomeres, there was only a twofold increase of γ-H2AX at the telomere-adjacent chromatin after the temperature shift (Fig. 7 B). Nonetheless, the absolute amounts of γ-H2AX normalized to the signal at CEN8 were almost the same at both locations (Fig. 7 B). Moreover, γ-H2AX spread further in response to uncapping telomeres, past HML, in which little γ-H2AX was induced, for example. Indeed, γ-H2AX near the telomeres was lost at 4 h after temperature shift, coinciding with the degradation of telomeric DNA (Booth et al., 2001). Thus, when telomeres are deprotected, they elicit the same modification as an HO-induced DSB.
Telomeres are localized at the nuclear periphery by either the yeast's Ku proteins or by Esc1, both of which require Sir4 for the telomere tethering. We asked whether the displacement of telomeres from the periphery would influence the level of constitutive γ-H2AX near the telomere by examining cells lacking either Yku80 or Sir4 (Taddei et al., 2004), but almost the same amount of γ-H2AX was detected near the telomere in both mutants (Fig. 7 C). In yku80Δ or sir4Δ mutants, gene silencing by telomere position effect (TPE) is defective; the fact that the level of γ-H2AX is the same in wild-type or TPE-defective mutants suggests that checkpoint-mediated chromatin modification is independent of TPE. To be replicated, telomeres likely transiently dissociate from their capping proteins; indeed, the cell cycle checkpoint kinases Mec1p and Tel1p are recruited to telomere ends, and both kinases influence telomere length (Chan et al., 2001; Pennaneach and Kolodner, 2004).
We hypothesized that γ-H2AX near the telomere could be generated when the telomere was associated with the kinases during DNA replication and, therefore, might be higher in G2 cells than in G1. We examined this possibility by comparing γ-H2AX at the telomere-proximal chromatin in G1-arrested cells with that in G2-arrested cells. Interestingly, a similar amount of γ-H2AX was detected in G2-arrested cells as well as in G1-arrested cells, in which the telomeres were fully capped and protected (Fig. 7 D). Indeed, γ-H2AX appeared even when tel1Δ cells were arrested in G1. This result was surprising because Tel1 has been shown to be the sole kinase responsible for γ-H2AX when G1-arrested cells suffer a DSB. These results lead us to suggest that γ-H2AX formed near the telomere in S and G2 persists on the chromatin until the next cell cycle. Consistent with this idea, we show in the previous sections that there is little turnover of γ-H2AX when kinases are inactivated; consequently, γ-H2AX at telomeres may persist for some time. On the other hand, Takata et al. (2004) showed that Mec1 associated with the short telomeres in G1-arrested tel1Δ cells. Therefore, it is also possible that H2AX at telomeres in G1 cells can be phosphorylated by Mec1 when Tel1 is not functional.
Discussion
We have shown that the extent of γ-H2AX spreading is not dependent on the location of DSB. There is a rather constant 50 kb of rapid modification (within 1 h) on either side of a DSB. This constraint is not apparently imposed by barrier sequences but may reflect a fundamental aspect of chromosome architecture that confines kinases associating with DSB ends from modifying more distant regions.
A second important finding is that histone H2AX in heterochromatin is not efficiently modified in response to DSBs either in budding yeast or in mammalian cells. This does not imply that the DNA damage response is necessarily less efficient if the lesion occurs within heterochromatin. When DSBs are generated within heterochromatin itself, the chromatin surrounding the lesion remodels to a more decondensed configuration that appears to permit the recruitment of repair factors and γ-H2AX formation (Kruhlak et al. 2006). When a DSB is generated outside of heterochromatin, it is possible that the hypoacetylated state of yeast histones at lysine residues in their N termini precludes the spreading of the C-terminal phosphorylation to heterochromatin; thus, eliminating histone deacetylation in yeast by deleting SIR3 restores γ-H2AX modification. A very important conclusion from our study is that the presence of a heterochromatic region does not preclude the modification of histones further away from the DSB. This finding argues that the process of modification is not strictly a processive hand-off process in which a newly phosphorylated γ-H2AX serves as a platform for Mec1 or Tel1 kinase to modify an adjacent histone. It is possible that the ends of the heterochromatic silent region form a loop that would allow a processive modification process to step over the silent region instead of traversing it nucleosome by nucleosome (Bystricky, K., personal communication).
In addition, we have documented that γ-H2AX is lost when DNA is rendered single stranded by resection. The loss of γ-H2AX is progressive, beginning from the DSB. On the contrary, there is no significant loss of γ-H2AX when DNA end resection is inhibited either in G1-arrested cells or in the cells overexpressing the CDK1/Clb inhibitor SIC1. An important new result is that chromatin that is far distal from the DSB and, thus, not initially modified eventually does become modified as resection proceeds. This late, distant modification is dependent on MEC1 but not on TEL1. Mec1 and its interacting partner Ddc2 are known to associate with replication protein A–coated single-strand DNA (Zou and Elledge, 2003). As resection proceeds, Mec1–Ddc2 can associate with newly generated single-stranded DNA and create γ-H2AX on still more distant regions. Thus, although 5′ to 3′ resection displaces the initially modified γ-H2AX/H2B dimer, it also provides new recognition sites for Mec1, leading to an extension of the modified chromatin domain.
Finally, we have found that chromatin near telomeres have constitutively high levels of γ-H2AX. Previously, the DSB-responsive kinases Mec1 and Tel1 were shown to be recruited to the telomeres at specific times in the cell cycle, playing roles in telomerase recruitment (Takata et al., 2004; Goudsouzian et al., 2006). Our current observation suggests that telomeres are at least transiently recognized as DSBs, although without triggering cell cycle arrest. Unlike what occurs with a single DSB, in which γ-H2AX is lost either because the locus is repaired or by extensive 5′ to 3′ resection (Keogh et al., 2006), γ-H2AX at undamaged telomeres seems to be persistent. Telomeres do not normally undergo extensive resection that would remove γ-H2AX nor do telomeres engage frequently in recombinational repair, so the modification is not removed. It is not possible to determine what proportion of any given telomere is modified by γ-H2AX in a population of cells; it is quite possible that there is a changing subset of ends that are detected as DSBs.
Materials and methods
Yeast strains
Strain JKM179, in which a galactose-induced DSB at MAT created by HO endonuclease cannot be repaired by homologous recombination, has been previously described (Moore and Haber, 1996). Strains lacking the HO cleavage site were selected as rare, imperfect, nonhomologous end-joining events from cells in which the HO endonuclease is continually expressed. The ectopic HO cut site cassette was constructed by inserting a 117-bp cleavage site derived from MATa adjacent to a HPH-MX marker, and the consequent 1.8-kb HO cassette containing the HPH marker was introduced at 217,940 bp on chromosome III to create the HO cut site 17 kb to the right of MAT (YJK1) by lithium acetate–mediated transformation. The HO cassette was also integrated on chromosome III either at 113,500 bp on chromosome III, 600 bp to the left of CEN3 (YJK2), or at 18,719 bp, which is 7 kb to the right of HML (YJK21). The HO cut site on chromosome VI was generated by integrating the HO cassette at 195,680 bp on chromosome VI (YFD032). To construct the strains containing two HO-inducible break sites, either JKM179 or YFD032 was transformed with the HO cassette for YJK9 (the second break near CEN3) or YJK15 (the second break on chromosome VI), respectively. To delete the endogenous HML domain (YJK52), YJK21 was transformed with BamHI-digested pJH455 (hmlΔ∷LEU2). The strain containing the ectopic HMRα at 41 kb away from the left end of chromosome III (YJK79) was constructed by crosses between a derivative of YJK52 containing pMATa and a derivative of XW426 (MATα hmlΔ∷LEU2 41kb∷LEU2∷HMRα hmrΔ∷ADE1). Deletions of YKU80 (YJK53) and SIR4 (YJK75) were created by PCR amplification of KAN-MX–marked gene deletions from a collection of yeast deletion mutants (Research Genetics) transformed into YJK52. To delete SIR3, a derivative of YJK21 containing HMLα-inc (YJK27) was transformed with sir3Δ∷KAN-MX, which was PCR amplified from a Research Genetics yeast deletion mutant collection. To overexpress the CDK1 inhibitor SIC1, the cells containing the gal promoter–fused SIC1 allele was used (Ira et al., 2004). YSL187 (MATa bar1Δ∷ADE3), a derivative of JKM179, was used for the α-factor arrest experiment. The cdc13-1 allele (YMV021) was introduced into JKM179 by pop-in/pop-out of a URA3-containing plasmid, pVL451, provided by V. Lundblad (Salk Institute, La Jolla, CA).
γ-H2AX analysis by ChIP
ChIP of γ-H2AX was performed as previously described (Shroff et al., 2004) using an antibody against yeast γ-H2AX, which was provided by C. Redon and W. Bonner (National Institutes of Health [NIH], Bethesda, MD). Cells were grown to a density between 5 ×106 cells and 1 × 107 cells/ml in yeast extract/lactate medium, and HO endonuclease was induced by adding 2% (wt/vol final concentration) galactose. DNA and proteins in the cells were cross-linked by the addition of 1.4% formaldehyde to 45 ml of cultures for 10 min. Cells were lysed with glass beads, and the extracts were sonicated to shear DNA to an average size of 0.5 kb. Immunoprecipitation samples were incubated with 50 ng/ml anti–γ-H2AX serum for 1 h at 4°C and bound to protein G–agarose beads for 1 h at 4°C. After a series of washing, samples were eluted from the beads followed by the reversal of cross-linking for 6 h at 65°C. Finally, proteins were removed from the sample by proteinase K and phenol extraction, and DNA was ethanol precipitated. The γ-H2AX ChIP signal was quantified by quantitative PCR with multiple primer pairs specific to chromatin regions surrounding the HO-induced DSB (Table S1). PCR was performed with a real-time PCR machine (Chromo 4; MJ Research) except for Fig. 1 B. With each primer pair, the number of amplification cycles that were required for the sample's response curve to reach a particular threshold fluorescence signal level was measured. The amount of chromatin immunoprecipitated template DNA for the reaction was then estimated from a standard curve based on serial dilution of a standard PCR product (Kubista et al. 2006). The ChIP signals in Fig. 1 B were measured by quantifying the band intensities on 1.5% agarose gels with Quantity One (Bio-Rad Laboratories). The band intensities were also converted to the initial amount of DNA template by being estimated from a standard curve. The ChIP signal at each locus was normalized to that at CEN8 in chromosome VIII, in which DSB was not induced. The fold increase of γ-H2AX was calculated by dividing the ChIP signal at 1 h after HO induction (T1) by that without HO induction (T0).
Western blot analysis
Cell extracts were prepared by trichloroacetic acid and were subjected to Western blot analysis. γ-H2AX was detected by polyclonal anti-yeast γ-H2AX antibody (1:10,000; a gift from C. Redon and W. Bonner). As a loading control, carboxy peptidase Y (CPY) was visualized with the monoclonal anti-CPY antibody (1:10,000; Invitrogen).
Immunofluorescence labeling and microscopy
Wild-type primary mouse embryo fibroblasts were grown on glass coverslips and treated with 10 ng/ml NCS for 1 h or with 1 μM TSA for 8 h and were treated with 10 ng/ml NCS for an additional hour before being fixed in 2% freshly prepared PFA in PBS for 5 min at room temperature. Cells were immunolabeled using the following antibodies, either individually or in combination: monoclonal antiphospho-H2AX antibody (clone JBW301; 1:1,000; Upstate Biotechnology), rabbit polyclonal antiacetylated histone H3 (K9 and K14; 1:1,000; Upstate Biotechnology), rabbit polyclonal lysine-methylated histone H3 (H3meK9; 1:200; Upstate Biotechnology), and goat anti–rabbit AlexaFluor488 (1:500) or goat anti–mouse AlexaFluor546 secondary antibody (Invitrogen). The cells were labeled for 30 min in PBS containing 100 nM DAPI, rinsed with PBS, and mounted on glass microscope slides in glycerol-based mounting media containing N-propyl gallate antifade (Sigma-Aldrich).
Confocal z-stack image series were collected using a fluorescence microscope (LSM510 META; Carl Zeiss MicroImaging, Inc.) equipped with a plan-Apochromat 63× NA 1.4 oil immersion objective lens (Carl Zeiss MicroImaging, Inc.) using 0.07 × 0.07-μm pixel sampling, optical slice thickness of 0.80 μm, and a step size of 0.2 μm. Individual optical slices were background subtracted and contrast adjusted only by stretching the histogram in a linear manner consistently for the different fluorescence channels. The adjusted images were organized into figures using Photoshop version 8.0 (Adobe).
For volume measurements, image stacks were background subtracted and imported into Imaris version 4.2 (Bitplane) image processing and analysis software using the Surpass volume rendering module. Maximum intensity projections were calculated for all fluorescence channels. Isosurface volume renderings of the γ-H2AX fluorescence channel were calculated without resampling or smoothing the dataset. The consistent threshold value, which is representative of signal above background from the negative control, was used in the isosurface calculation. Based on the visual inspection of all image stacks collected, the individual isosurface renderings were split into individual objects within a group using a value of 100 as the maximum number of objects per group. Statistics generated from the objects within the group were exported to a spreadsheet program, and the mean volume (micrometers3) was calculated. A snapshot of the γ-H2AX isosurface rendering superimposed onto the DAPI maximum intensity projection was saved and organized into figures using Photoshop version 8.0 (Adobe). The mean volume of 1.0 μm3 was empirically chosen as a cut-off value for significantly large γ-H2AX foci based on the fact that in the NCS-only treated cells, the vast majority (95%) of foci fell below 1.0 μm3 in size. Colocalization analysis was performed on 3D reconstructions from image stacks that were background subtracted and set at a consistent threshold value (the same γ-H2AX threshold value used above for individual foci volume measurements). The colocalization module in Imaris software (Bitplane) was used, and coefficients were measured for γ-H2AX colocalizing with acH3K9K14 within a range of 0 to 1.0.
Online supplemental material
Fig. S1 documents the inhibition of end resection at a DSB in Cdk1-inhbitied or G1-arrested cells. Fig. S2 shows the extent of γ-H2AX modification when a DSB was created on chromosome VI. Fig. S3 presents γ-H2AX foci and acetylated histone H3 in NCS-treated or TSA plus NCS–treated wild-type mouse embryonic fibroblasts. Fig. S4 summarizes the distributions of γ-H2AX foci in NCS- and TSA plus NCS–treated mouse embryonic fibroblasts in terms of mean fluorescence intensity, foci volume, or integrated fluorescence intensity. Fig. S5 presents the colocalization of γ-H2AX and acetylated histone H3 in mouse embryo fibroblasts containing hyperacetylated histones. Table S1 lists all primer pairs used in ChIP experiments. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200612031/DC1.
Supplementary Material
Acknowledgments
We thank Christophe Redon and William Bonner for γ-H2AX antibody, Victoria Lundblad for the cdc13-1 plasmid, and Hua-Tang Chen for technical help. Audrey Gasch, David Botstein, and Pat Brown provided invaluable microarray analysis. Moreshwar Vaze constructed the cdc13-1 strain. We also thank Michael Lichten for his comments on the manuscript.
This research was supported by NIH grants GM61799 and GM20056 to J.E. Haber as well as an unrestricted gift from Merck & Co. A. Nussenzweig and M. Kruhlak were supported by the Intramural Research Program of the NIH National Cancer Institute Center for Cancer Research. A. Nussenzweig is also supported by a grant from the A-T Children's Project.
Abbreviations used in this paper: ChIP, chromatin immunoprecipitation; CPY, carboxy peptidase Y; DSB, double-strand break; MMS, methylmethanesulfonate; NCS, neocarzinostatin; TPE, telomere position effect; TSA, trichostatin A.
References
- Bassing, C.H., K.F. Chua, J. Sekiguchi, H. Suh, S.R. Whitlow, J.C. Fleming, B.C. Monroe, D.N. Ciccone, C. Yan, K. Vlasakova, et al. 2002. Increased ionizing radiation sensitivity and genomic instability in the absence of histone H2AX. Proc. Natl. Acad. Sci. USA. 99:8173–8178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom, K.S., and J. Carbon. 1982. Yeast centromere DNA is in a unique and highly ordered structure in chromosomes and small circular minichromosomes. Cell. 29:305–317. [DOI] [PubMed] [Google Scholar]
- Booth, C., E. Griffith, G. Brady, and D. Lydall. 2001. Quantitative amplification of single-stranded DNA (QAOS) demonstrates that cdc13-1 mutants generate ssDNA in a telomere to centromere direction. Nucleic Acids Res. 29:4414–4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste, A., S. Petersen, P.J. Romanienko, O. Fernandez-Capetillo, H.T. Chen, O.A. Sedelnikova, B. Reina-San-Martin, V. Coppola, E. Meffre, M.J. Difilippantonio, et al. 2002. Genomic instability in mice lacking histone H2AX. Science. 296:922–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celeste, A., O. Fernandez-Capetillo, M.J. Kruhlak, D.R. Pilch, D.W. Staudt, A. Lee, R.F. Bonner, W.M. Bonner, and A. Nussenzweig. 2003. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat. Cell Biol. 5:675–679. [DOI] [PubMed] [Google Scholar]
- Chan, S.W., J. Chang, J. Prescott, and E.H. Blackburn. 2001. Altering telomere structure allows telomerase to act in yeast lacking ATM kinases. Curr. Biol. 11:1240–1250. [DOI] [PubMed] [Google Scholar]
- De Piccoli, G., F. Cortes-Ledesma, G. Ira, J. Torres-Rosell, S. Uhle, S. Farmer, J.Y. Hwang, F. Machin, A. Ceschia, A. McAleenan, et al. 2006. Smc5-Smc6 mediate DNA double-strand-break repair by promoting sister-chromatid recombination. Nat. Cell Biol. 8:1032–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs, J.A., S. Allard, O. Jobin-Robitaille, A. Javaheri, A. Auger, N. Bouchard, S.J. Kron, S.P. Jackson, and J. Cote. 2004. Binding of chromatin-modifying activities to phosphorylated histone H2A at DNA damage sites. Mol. Cell. 16:979–990. [DOI] [PubMed] [Google Scholar]
- Fernandez-Capetillo, O., A. Celeste, and A. Nussenzweig. 2003. Focusing on foci: H2AX and the recruitment of DNA-damage response factors. Cell Cycle. 2:426–427. [PubMed] [Google Scholar]
- Furuta, T., H. Takemura, Z.Y. Liao, G.J. Aune, C. Redon, O.A. Sedelnikova, D.R. Pilch, E.P. Rogakou, A. Celeste, H.T. Chen, et al. 2003. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 278:20303–20312. [DOI] [PubMed] [Google Scholar]
- Garvik, B., M. Carson, and L. Hartwell. 1995. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 15:6128–6138. (published erratum appears in Mol. Cell. Biol. 1996. 16:457). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goudsouzian, L.K., C.T. Tuzon, and V.A. Zakian. 2006. S. cerevisiae Tel1p and Mre11p are required for normal levels of Est1p and Est2p telomere association. Mol. Cell. 24:603–610. [DOI] [PubMed] [Google Scholar]
- Grandin, N., C. Damon, and M. Charbonneau. 2001. Cdc13 prevents telomere uncapping and Rad50-dependent homologous recombination. EMBO J. 20:6127–6139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, J.C., and J.E. Haber. 2006. Surviving the breakup: the DNA damage checkpoint. Annu. Rev. Genet. 40:209–235. [DOI] [PubMed] [Google Scholar]
- Ira, G., A. Pellicioli, A. Balijja, X. Wang, S. Fiorani, W. Carotenuto, G. Liberi, D. Bressan, L. Wan, N.M. Hollingsworth, et al. 2004. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 431:1011–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keogh, M.C., J.A. Kim, M. Downey, J. Fillingham, D. Chowdhury, J.C. Harrison, M. Onishi, N. Datta, S. Galicia, A. Emili, et al. 2006. A phosphatase complex that dephosphorylates gammaH2AX regulates DNA damage checkpoint recovery. Nature. 439:497–501. [DOI] [PubMed] [Google Scholar]
- Kitagawa, R., and M.B. Kastan. 2005. The ATM-dependent DNA damage signaling pathway. Cold Spring Harb. Symp. Quant. Biol. 70:99–109. [DOI] [PubMed] [Google Scholar]
- Kruhlak, M.J., A. Celeste, G. Dellaire, O. Fernandez-Capetillo, W.G. Muller, J.G. McNally, D.P. Bazett-Jones, and A. Nussenzweig. 2006. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 172:823–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubista, M., J.M. Andrade, M. Bengtsson, A. Forootan, J. Jonak, K. Lind, R. Sindelka, R. Sjoback, B. Sjogreen, L. Strombom, et al. 2006. The real-time polymerase chain reaction. Mol. Aspects Med. 27:95–125. [DOI] [PubMed] [Google Scholar]
- Lee, S., A. Pellicioli, J. Demeter, M. Vaze, A.P. Gasch, A. Malkova, P. Brown, T. Stearns, M. Foiani, and J.E. Haber. 2000. Arrest, adaptation and recovery following a chromosome double-strand break in Saccharomyces cerevisiae. Cold Spring Harb. Symp. Quant. Biol. 65:303–314. [DOI] [PubMed] [Google Scholar]
- Lindroos, H.B., L. Strom, T. Itoh, Y. Katou, K. Shirahige, and C. Sjogren. 2006. Chromosomal association of the Smc5/6 complex reveals that it functions in differently regulated pathways. Mol. Cell. 22:755–767. [DOI] [PubMed] [Google Scholar]
- McGowan, C.H., and P. Russell. 2004. The DNA damage response: sensing and signaling. Curr. Opin. Cell Biol. 16:629–633. [DOI] [PubMed] [Google Scholar]
- Moore, J.K., and J.E. Haber. 1996. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol. Cell. Biol. 16:2164–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison, A.J., J. Highland, N.J. Krogan, A. Arbel-Eden, J.F. Greenblatt, J.E. Haber, and X. Shen. 2004. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell. 119:767–775. [DOI] [PubMed] [Google Scholar]
- Papamichos-Chronakis, M., J.E. Krebs, and C.L. Peterson. 2006. Interplay between Ino80 and Swr1 chromatin remodeling enzymes regulates cell cycle checkpoint adaptation in response to DNA damage. Genes Dev. 20:2437–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennaneach, V., and R.D. Kolodner. 2004. Recombination and the Tel1 and Mec1 checkpoints differentially effect genome rearrangements driven by telomere dysfunction in yeast. Nat. Genet. 36:612–617. [DOI] [PubMed] [Google Scholar]
- Ravindra, A., K. Weiss, and R.T. Simpson. 1999. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating-type locus HMRa. Mol. Cell. Biol. 19:7944–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogakou, E.P., D.R. Pilch, A.H. Orr, V.S. Ivanova, and W.M. Bonner. 1998. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 273:5858–5868. [DOI] [PubMed] [Google Scholar]
- Rogakou, E.P., C. Boon, C. Redon, and W.M. Bonner. 1999. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 146:905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh, Y. 2003. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer. 3:155–168. [DOI] [PubMed] [Google Scholar]
- Shroff, R., A. Arbel-Eden, D. Pilch, G. Ira, W.M. Bonner, J.H. Petrini, J.E. Haber, and M. Lichten. 2004. Distribution and dynamics of chromatin modification induced by a defined DNA double-strand break. Curr. Biol. 14:1703–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom, L., H.B. Lindroos, K. Shirahige, and C. Sjogren. 2004. Postreplicative recruitment of cohesin to double-strand breaks is required for DNA repair. Mol. Cell. 16:1003–1015. [DOI] [PubMed] [Google Scholar]
- Taddei, A., F. Hediger, F.R. Neumann, C. Bauer, and S.M. Gasser. 2004. Separation of silencing from perinuclear anchoring functions in yeast Ku80, Sir4 and Esc1 proteins. EMBO J. 23:1301–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata, H., Y. Kanoh, N. Gunge, K. Shirahige, and A. Matsuura. 2004. Reciprocal association of the budding yeast ATM-related proteins Tel1 and Mec1 with telomeres in vivo. Mol. Cell. 14:515–522. [DOI] [PubMed] [Google Scholar]
- Unal, E., A. Arbel-Eden, U. Sattler, R. Shroff, M. Lichten, J.E. Haber, and D. Koshland. 2004. DNA damage response pathway uses histone modification to assemble a double-strand break-specific cohesin domain. Mol. Cell. 16:991–1002. [DOI] [PubMed] [Google Scholar]
- van Attikum, H., O. Fritsch, B. Hohn, and S.M. Gasser. 2004. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell. 119:777–788. [DOI] [PubMed] [Google Scholar]
- Vaze, M.B., A. Pellicioli, S.E. Lee, G. Ira, G. Liberi, A. Arbel-Eden, M. Foiani, and J.E. Haber. 2002. Recovery from checkpoint-mediated arrest after repair of a double-strand break requires srs2 helicase. Mol. Cell. 10:373–385. [DOI] [PubMed] [Google Scholar]
- Weiss, K., and R.T. Simpson. 1998. High-resolution structural analysis of chromatin at specific loci: Saccharomyces cerevisiae silent mating type locus HMLα. Mol. Cell. Biol. 18:5392–5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, A., N. Puget, I. Shim, S. Odate, I. Jarzyna, C.H. Bassing, F.W. Alt, and R. Scully. 2004. Control of sister chromatid recombination by histone H2AX. Mol. Cell. 16:1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou, L., and S.J. Elledge. 2003. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 300:1542–1548. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}