

Abstract
We describe a self-amplifying feedback loop that autoinduces Skp2 during G1 phase progression. This loop, which contains Skp2 itself, p27kip1 (p27), cyclin E–cyclin dependent kinase 2, and the retinoblastoma protein, is closed through a newly identified, conserved E2F site in the Skp2 promoter. Interference with the loop, by knockin of a Skp2-resistant p27 mutant (p27T187A), delays passage through the restriction point but does not interfere with S phase entry under continuous serum stimulation. Skp2 knock down inhibits S phase entry in nontransformed mouse embryonic fibroblasts but not in human papilloma virus–E7 expressing fibroblasts. We propose that the essential role for Skp2-dependent degradation of p27 is in the formation of an autoinduction loop that selectively controls the transition to mitogen-independence, and that Skp2-dependent proteolysis may be dispensable when pocket proteins are constitutively inactivated.
Introduction
Skp2 is the limiting component of the E3 ligase controlling the proteosomal degradation of p27kip1 (p27) in late G1/S phase (Carrano et al., 1999; Tsvetkov et al., 1999). Skp2 can also stimulate p27 degradation and S phase entry in serum-deprived cells (Sutterluty et al., 1999). Although Skp2 has several other substrates (Reed, 2003), the importance of p27 as a Skp2 substrate is emphasized by the finding that Skp2-p27 double-null mice lose most of the Skp2 knock out phenotype (Nakayama and Nakayama, 2005). It was therefore surprising that knockin mouse embryo fibroblasts (MEFs) expressing p27T187A, a Skp2-resistant p27 mutant, did not show a pronounced defect in mitogen-stimulated S phase entry (Malek et al., 2001).
Skp2 levels are inhibited posttranscriptionally by retinoblastoma protein (Rb) through its effects on anaphase-promoting complex/cyclosome and its activator Cdh1 (APC/CCdh1)–mediated Skp2 degradation (Hsu et al., 2002; Bashir et al., 2004; Ji et al., 2004; Wei et al., 2004; Binne et al., 2007). In this report, we describe a parallel regulation of Skp2 by Rb that results in the formation of a transcriptionally based Skp2 autoinduction loop. Interference with this loop selectively affects the transition to mitogen-independent cell cycle progression, also called the restriction point.
Results and discussion
A conserved E2F site links Skp2 regulation to Rb activation/inactivation
Transcript profiling indicates that E2F controls Skp2 gene expression (Markey et al., 2002; Vernell et al., 2003), and we found, in agreement with this data, that ectopic expression of human papilloma virus–E7 (E7), which inactivates pocket proteins and releases E2Fs, rescued Skp2 mRNA and protein expression in serum-starved MEFs (Fig. 1 A). We used zPicture (available at http://zpicture.dcode.org/) to identify conserved domains in the mouse versus the human, chimp, and dog Skp2 promoters, and rVista (available at http://rvista.dcode.org/) to look for putative E2F binding sites within the conserved domains. This analysis (Fig. 1 B) revealed an evolutionarily conserved E2F binding site in human, chimp, and dog Skp2 promoters that matches the E2F consensus (SCGSSAAA; Tao et al., 1997). The homologous mouse sequence (GCGCTAAA) differs from the consensus by one base (Fig. 1 B), but this same sequence acts as a functional E2F site in the E2F1 promoter (Neuman et al., 1994). The mouse sequence begins at position +114 relative to the transcription start site, between the transcription and translation start sites. It is the only E2F site we could identify in the mouse Skp2 promoter. Imaki et al. (2003) have reported that the Skp2 promoter contains a binding site for the transcription factor GABP. Interestingly, GABP can cooperate with E2F1 to regulate the transcription of target genes (Izumi et al., 2000).
Figure 1.

A conserved E2F site in the Skp2 promoter. (A) Vector- (V) or E7- (E7) transfected MEFs were incubated in 10% FCS or serum starved. Skp2 mRNA levels were determined by QPCR and plotted relative to the levels observed in the vector-transfected cells incubated with 10% FCS. A duplicate experiment was Western blotted for Skp2 and Cdk4 (loading control). (B) The conserved E2F site on the mouse, human, chimp, and dog Skp2 promoters. The numbers shown are relative to the known or putative transcription start sites. (C) MEFs transfected with the wild-type Skp2 promoter– (Swt) or E2F-mutated Skp2 promoter– (SEm) luciferase constructs were analyzed by ChIP using anti-E2F1 (E) or preimmune IgG (I). (D) Skp2 promoter activity in serum-stimulated MEFs expressing Swt or SEm promoter–luciferase constructs and either empty vector or E7. After 24 h in 10% FCS, Skp2 promoter–luciferase activity was plotted relative to the activity of the wild-type Skp2 promoter transfected with empty vector. (E–H) Serum-starved MEFs were stimulated with 10% FCS. (E) The levels of Skp2 and cyclin E1 mRNAs were determined by QPCR and plotted relative to the level of cyclin E mRNA in the starved cells. (F) Total cell lysates were Western blotted for Skp2 and Cdk4 (loading control). (G) Total cell lysates were Western blotted for Rb; the hyper- and hypophosphorylated forms are shown by the top and bottom arrows, respectively. (H) ChiP was performed using anti-E2F1 or control IgG, and the results were quantified by QPCR. The level of immunoprecipitated Skp2 promoter is plotted relative to the input (×10−3). Error bars show mean ± SD.
We generated luciferase constructs with the wild-type mouse Skp2 promoter, including one with a mutation in the conserved E2F site (Fig. 1 B), and transiently transfected them into MEFs. Chromatin immunoprecipitation (ChIP) performed with amplicons specific for the transfected genes showed that E2F1 bound to the transfected wild-type mouse Skp2 promoter but not to the transfected promoter with the E2F site mutation (Fig. 1 C). Moreover, mutation of the single E2F site completely blocked the activity of a Skp2 promoter–luciferase construct in serum-stimulated or E7-expressing MEFs (Fig. 1 D).
Skp2 mRNA levels fluctuate during cell cycle progression (Zhang et al., 1995). We found that Skp2 mRNA and protein expression are low in G0, gradually increase in early G1 phase, and further increase ∼15 h after mitogenic stimulation (Fig. 1, E and F, respectively). This second, late G1/S phase induction of Skp2 coincided with the hyperphosphorylation of Rb (Fig. 1 G). Moreover, this late G1/S phase induction of Skp2 mRNA closely matched the time-dependent increase in cyclin E1 mRNA (12–18 h; Fig. 1 E), a prototypic E2F1-regulated gene (DeGregori et al., 1995). ChIP was then used to examine the time-dependent binding of endogenous E2F1 to the mouse Skp2 promoter (Fig. 1 H). Indeed, the binding of endogenous E2F1 to the conserved site on the endogenous Skp2 promoter increased in late G1/S phase. Thus, the mid-to-late G1 phase induction of Skp2 (12–18 h after mitogen stimulation) is regulated by E2F activity.
Others (Zhang and Wang, 2006) have reported that the human Skp2 promoter contains three E2F-like sequences, one of which is the human homologue of the mouse E2F site reported in this paper (Fig. 1 B). However, these investigators concluded that a distinct, nonconserved E2F site (TTGCGCGCG) accounted for E2F-stimulated luciferase activity of the human Skp2 promoter. Although we cannot exclude the possibility that the human promoter relies on this nonconserved E2F site, it is curious that a consensus E2F site (CGCGCAAA) did not contribute to E2F-stimulated luciferase activity or interact with E2F in electrophoretic mobility shift assays in Zhang and Wang (2006). We also note that the amplicon used to show binding of E2F1 to this nonconserved site in the human promoter includes the conserved E2F site described in this report.
A Skp2 autoinduction loop in the G1/S transition
Our identification of a conserved E2F site in the mouse Skp2 gene allowed us to assemble Rb-E2F, Skp2, p27, and cyclin E–Cdk2 into a self-amplifying loop (Fig. 2 A). In this loop, the stimulatory effect of E2F on Skp2 gene expression would feed back to sustain Rb inactivation, E2F release, and further induction of the Skp2 gene in late G1 phase. Thus, this model predicts that Skp2 should induce itself and that this autoinduction should be detected as increased mRNA. Moreover, the autoinduction of Skp2 should occur in serum-deprived cells, because the loop is self-amplifying and does not require the presence of mitogens.
Figure 2.

A feedback loop for Skp2 autoinduction. (A) A positive feedback loop that perpetuates Skp2 induction and Rb inactivation in the absence of exogenous mitogens. (B) RNA from serum-starved MEFs infected with Ad-LacZ or Ad-hSkp2 was analyzed by QPCR; the levels of Skp2, cyclin E1, and cyclin A mRNAs are plotted relative to their levels in the Ad-LacZ–infected cells. (C) Skp2 promoter activity in cells transfected with an irrelevant control siRNA or Skp2 siRNA, infected with Ad-LacZ or Ad-E7, and stimulated with 10% FCS for 24 h. The Western blot shows representative Skp2 and actin levels in MEFs transfected with control (Co) siRNA or Skp2 (S2) siRNA, serum starved, and stimulated with 10% FCS for 24 h. Error bars show mean ± SD.
To test this model, we infected serum-free cultures of MEFs with an adenovirus encoding human Skp2 (Ad-hSkp2) and used a species-specific Skp2 primer probe set with quantitative real-time RT-PCR (QPCR) to detect the induction of mouse Skp2 mRNA. Ectopic expression of human Skp2 induced endogenous mouse Skp2 mRNA, as well as other known E2F target genes (cyclins E1 and A; Fig. 2 B and Fig. S1, A and B, available at http://www.jcb.org/cgi/content/full/jcb.200703034/DC1). Induction of mouse Skp2 mRNA and down-regulation of p27 by Ad-hSkp2 were seen when the serum-starved MEF were expressing ectopic Skp2 at near normal levels (compare FCS to 60 MOI Ad-hSkp2; Fig. S2) and when the infection was performed during or after the serum-starvation period (Fig. S3). Conversely, inhibition of the loop by RNAi-mediated knock down of Skp2 reduced serum-stimulated Skp2 promoter activation (Fig. 2 C). As expected, this Skp2 requirement was lost after pocket protein inactivation with E7 (Fig. 2 C).
The loop shown in Fig. 2 A predicts that the autoinduction of Skp2 requires Cdk activity, and we indeed found that the induction of endogenous Skp2 mRNA seen in response to ectopic Ad-hSkp2 in serum-starved MEFs was blocked by the Cdk inhibitor roscovitine (Fig. 3 A). Conversely, expression of cyclin E induced Skp2 mRNA and protein in serum-deprived MEFs (Fig. 3 B and Fig. S1 C). The autoinduction of Skp2 mRNA was efficiently inhibited in serum-deprived MEFs when transit through the loop was precluded by knockin of a Skp2-resistant p27 mutant, p27T187A (Fig. 3 C).
Figure 3.

Characteristics of the Skp2 autoinduction loop. (A) Serum-starved MEFs infected with Ad-LacZ or Ad-hSkp2 were pretreated for 3 h with DMSO or 20 μM roscovitine and then incubated for 24 h with BrdU. Skp2 mRNA was quantified by QPCR and plotted relative to its level in the Ad-LacZ–infected cells. BrdU incorporation analysis showed that roscovitine inhibited the effect of Ad-hSkp2 on S phase entry by >90%. (B) Serum-starved MEFs were infected with Ad-LacZ or Ad–hCyclin E. Skp2 mRNA was quantified by QPCR and plotted relative to its level in the Ad-LacZ–infected cells. (C) Skp2 mRNA in serum-starved wild-type (WT) and p27T187A (T187A) MEFs infected with Ad-LacZ or Ad-hSkp2 was determined by QPCR and plotted relative to its level in Ad-LacZ–infected wild-type cells. (D) Serum-starved MEFs were infected with increasing (3.7–100) MOI Ad-hSkp2 or 100 MOI Ad-LacZ. Lysates were examined for cyclin E1 mRNA (top), BrdU incorporation (middle), or protein expression (bottom) of Skp2, E2F1, and actin (loading control). Cyclin E1 mRNA levels were determined by QPCR and plotted relative to its level in the Ad-LacZ–infected cells. The Western blot includes a lysate from serum-starved MEFs treated for 24 h with 10% FCS to show the level of endogenous Skp2. The Western blots were derived from the same experiment. Error bars show mean ± SD.
A characteristic of positive feedback loops is that they yield “all-or-nothing” responses. Indeed, we observed stepwise increases in cyclin E1 gene induction (Fig. 3 D, top) and S phase entry (Fig. 3 D, middle) upon infection of serum- starved MEFs with increasing MOIs of Ad-hSkp2. The maximal responses occurred with near-normal levels of Skp2 protein and were clearly distinguishable from the gradual increase in Skp2 expression (Fig. 3 D, bottom). The induction of endogenous Skp2 mRNA and degradation of p27 also occurred in a stepwise fashion (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200703034/DC1). Marti et al. (1999) have reported that Skp2 degrades E2F1 in G2/M phase cells. However, Ad-hSkp2 did not decrease E2F1 levels in serum-deprived MEFs (Fig. 3 D, bottom), which is consistent with the fact that Skp2 stimulates S phase entry under these conditions. Collectively with Marti et al. (1999), this result suggests that Skp2 may switch from a positive to a negative regulator of E2F activity as cells progress from G1/S to G2/M. Ad-hCyclin E also had no effect on E2F1 levels (unpublished data).
Skp2 autoinduction and restriction point control
The Skp2 autoinduction loop has the potential to regulate S phase entry because it can perpetuate the down-regulation of p27 and, thereby, the activation of cyclin E–Cdk2, phosphorylation of Rb and release of E2Fs. However, Malek et al. (2001), using knockin of p27T187A, reported that Skp2-mediated p27 degradation is not required for S phase entry in serum-stimulated MEFs. Because the Skp2 autoinduction loop can function in mitogen-deprived cells (Fig. 2), we reasoned that these results could be reconciled if Skp2-mediated p27 degradation was essential only for the transition to mitogen independence, also called the restriction point (Blagosklonny and Pardee, 2002). We therefore used p27T187A MEFs to interrupt the Skp2 autoinduction loop and look for consequences on the restriction point.
To measure passage through the restriction point, serum-starved p27T187A or wild-type MEFs were stimulated with 10% FCS for selected times. The serum was removed, and the cells were incubated with serum-free medium and BrdU. S phase entry in the wild-type MEFs required mitogens for the first 10 h after serum stimulation and then quickly became mitogen independent (Fig. 4 A, WT). This rapid transition to mitogen independence was defective in primary p27T187A MEFs; these cells did not become mitogen independent until 16 h (Fig. 4 A, T187A). Importantly, this defect in restriction-point control was not caused by a general decrease in the rate of cycling because, as previously reported (Malek et al., 2001), the kinetics of S phase entry were nearly identical when wild-type and p27T187A MEFs were continuously exposed to mitogens (Fig. 4 B). Thus, Skp2-dependent degradation of p27, and probably the Skp2 autoinduction loop, regulates progression through the restriction point. A positive feedback loop should accelerate the transition to mitogen independence, and we indeed find that the rate of progression through the restriction point is decreased when transit through the loop is blocked in p27T187A MEFs (Fig. 4 A).
Figure 4.
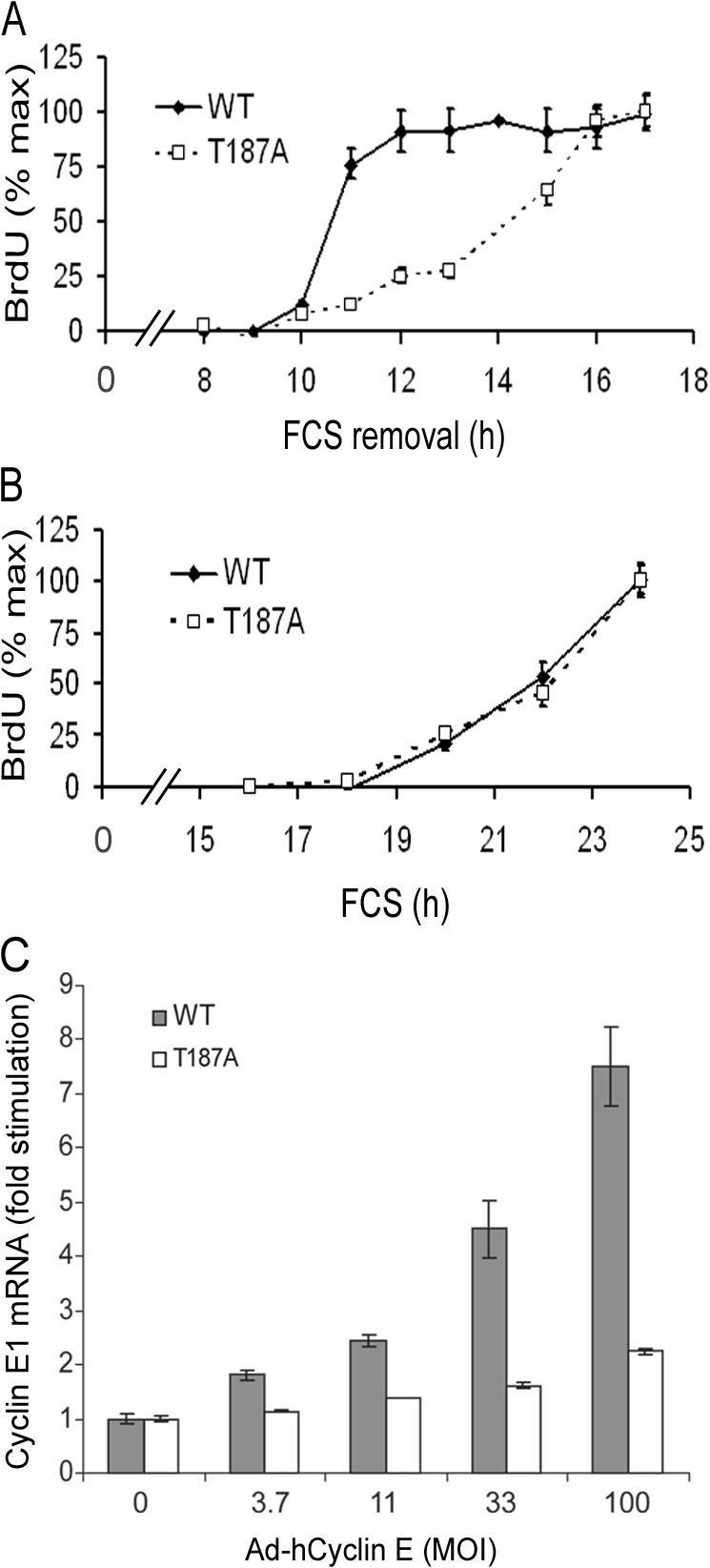
The Skp2 autoinduction loop controls the restriction point. (A) Restriction point analysis in wild-type (WT) and p27T187A (T187A) MEFs. Results are plotted relative to the BrdU incorporation observed at the last time point (34 and 39% for wild type and p27T187A MEFs, respectively). (B) S phase entry in wild-type and p27T187A MEFs continuously incubated with 10% FCS and BrdU. Results are plotted relative to the BrdU incorporation observed at the last time point (44 and 48% for wild-type and p27T187A MEFs, respectively). (C) Serum-starved wild-type (WT) and p27T187A (T187A) MEFs were infected with increasing (3.7–100) MOI of Ad–hCyclin E or 100 MOI Ad-LacZ (shown as 0 MOI Ad–hCyclin E). The level of mouse cyclin E1 mRNA was determined by QPCR (using a primer-probe set that recognizes mouse, but not human, cyclin E1 mRNA) and plotted relative to its level in the Ad-LacZ–infected cells. Error bars show mean ± SD.
Previous papers have proposed that passage through the restriction point is regulated by a positive feedback loop comprised of cyclin E and Rb-E2F (Dou et al., 1993; Blagosklonny and Pardee, 2002). In this model, cyclin E activation of Cdk2 would stimulate Rb phosphorylation and E2F-dependent transcription of the cyclin E1 gene, thereby furthering Rb inactivation and cyclin E1 induction. However, we reasoned that the repeated autoinduction of cyclin E might also require Skp2-mediated p27 degradation to allow for the activation of cyclin E–Cdk2. Based on this reasoning, the cyclin E autoinduction loop would be within, rather than separate from, the Skp2 autoinduction loop. To test this notion, we used a species-specific QPCR primer-probe set to mouse cyclin E1 mRNA to compare the efficiency of cyclin E autoinduction in wild-type and p27T187A MEFs infected with an adenovirus encoding human cyclin E. Indeed, we found that the autoinduction of cyclin E1 mRNA was barely detected in p27T187A MEFs (Fig. 4 C). This result strongly suggests that the major effects of cyclin E and Rb/E2F in restriction point control depend on their inclusion in the Skp2 autoinduction loop.
Others have reported that APC/CCdh1 stimulates Skp2 protein degradation (Bashir et al., 2004; Wei et al., 2004), and that APC/CCdh1 activity is inhibited by the E2F-dependent induction of Emi1 (Hsu et al., 2002). We therefore envision that the inactivation of pocket proteins and release of E2F controls the restriction point through the coordinated effects of the transcriptionally based Skp2 autoinduction loop described in this report and the posttranscriptionally based APC/CCdh1 pathway. Both of these effects would converge to increase the expression of Skp2 and degradation of p27. We note that p27T187A MEFs are not completely restrictionless, indicating that other Skp2 targets may also contribute to restriction point control. Alternatively, an independent positive feedback loop, perhaps in which Rb-E2F induces cyclin D1 (Ohtani et al., 1995), may cooperate with the loop described in this report.
In addition to its effect on cell cycle progression, the Skp2 autoinduction loop may contribute to cell cycle exit associated with pocket protein activation. Others have reported that Rb and p107 regulate p27 levels posttranscriptionally by acting as a scaffold for Skp2 and Cdh1 and thereby facilitating APC/CCdh1-dependent Skp2 proteolysis (Ji et al., 2004; Rodier et al., 2005; Binne et al., 2007). Interestingly, this rapid posttranscription down-regulation of Skp2 should inhibit the Skp2 autoinduction loop, which would in turn prevent Skp2 gene transcription and thereby enforce the quiescent state. Thus, coordinated transcriptional and posttranscriptional pocket protein effects on Skp2 levels may contribute to both the transition to mitogen independence and the G1 phase arrest that follows mitogen withdrawal.
Skp2 knock down or p27 overexpression inhibit S phase entry in serum-stimulated cells (Polyak et al., 1994; Toyoshima and Hunter, 1994; Zhang et al., 1995), whereas S phase entry is nearly normal in p27T187A MEFs (Malek et al., 2001; Fig. 4 B) cultured under similar conditions. These results imply that p27T187A MEFs (which have gone through mouse development in the absence of wild-type p27) may have acquired a compensatory mechanism that bypasses the need for Skp2-mediated p27 degradation in mitogen-bathed cells. In contrast, the restriction point defect is clearly seen in p27T187A MEFs, emphasizing that the role of Skp2-mediated p27 degradation in the transition to mitogen independence is essential.
Skp2 is dispensable in E7-expressing cells: implications for Skp2 in cellular transformation
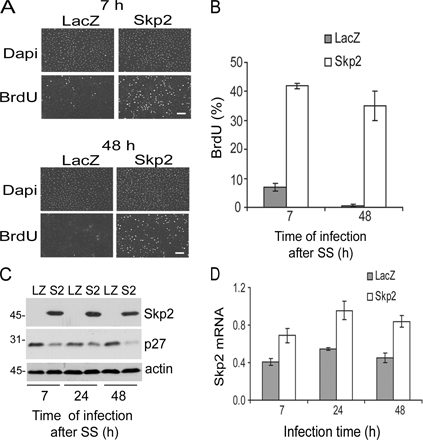
The results in Fig. 4 show that autoinduction of Skp2 is linked to efficient progression through the restriction point. However, this loop might become dispensable if Rb is inactivated by oncogenes during cellular transformation. To explore this possibility, we inactivated pocket proteins by ectopic expression of E7, knocked down Skp2 with siRNA, and determined the consequence of reduced Skp2 expression on S phase entry in serum-deprived MEFs (conditions where Skp2- mediated p27 degradation is required for the feedback loop and S phase entry; Figs. 2–4 ). Interestingly, S phase entry was not inhibited by knock down of Skp2 (Fig. 5, A and B), and a distinct Skp2 siRNA gave similar results (not depicted). We also found that Skp2 siRNA inhibited S phase entry in serum-stimulated MEFs, but even this Skp2 requirement was lost upon expression of E7 (Fig. 5, C and D).
Figure 5.

Effect of Skp2 siRNA in E7-expressing cells. (A) Serum-starved MEFs infected with either Ad-E7 (E7) or Ad-LacZ (LZ) were transfected with an irrelevant control (Co) siRNA or Skp2 (S2) siRNA and incubated with BrdU for 24 h in serum-free medium to assess S phase entry. (B) The experiment in A was repeated except that the collected cells were analyzed by Western blotting for Skp2 and actin (loading control). (C and D) The experiments in A and B were repeated except that the 24-h incubation with BrdU was performed in the presence of 10% FCS. Error bars show mean ± SD.
Elevated Skp2 expression is observed in cancers and has been considered a causative factor due, in part, to its effects on p27 (Gstaiger et al., 2001; Kamata et al., 2005; Zheng et al., 2005). However, our data indicate that high Skp2 expression may simply be a consequence of aberrant Rb inactivation and E2F release. Because many cancers constitutively activate upstream mitogenic signaling pathways that sustain Rb inactivation, the ability of Skp2 to sustain Rb inactivation through the feedback loop shown in Fig. 2 A may not be needed in cancer cells. Indeed, our results indicate that S phase entry induced by E7 is independent of Skp2. In fact, the notion that Skp2-mediated degradation of p27 may not be required for tumor development is supported by the finding that the p27T187A mutation does not delay tumorigenesis in mouse models of Ras-dependent lung and colon cancer (Timmerbeul et al., 2006).
Materials and methods
Cell culture
Spontaneously immortalized MEFs were maintained in DME (Invitrogen) with 5% FBS. For experimentation, near confluent cells were serum starved in DME with 1 mg/ml heat-inactivated, fatty acid–free BSA (DME-BSA; Sigma-Aldrich) for 48 h, trypsinized, reseeded at subconfluence, and stimulated largely as described previously (Welsh et al., 2001). Stimulated cells were washed once with cold PBS, scraped, collected by centrifugation, quick frozen, and stored at −80°C before analysis. S phase entry was determined by incorporation of BrdU (Kothapalli et al., 2003). Primary MEFs from wild-type and p27T187A knockin mice were maintained in 10% FCS and used at passages 2–5. To study progression through the restriction point, serum-starved wild-type and p27T187A MEFs were replated at subconfluence in 35-mm dishes and stimulated with 10% FCS for selected times. The FCS-containing medium was removed, and the cells were washed once with a 50-mM glycine, 150-mM NaCl, pH 2.8, acid wash buffer and twice with cold DME and were then incubated in DME-BSA for a total incubation of 17 h. BrdU was added to all of the plates, and the incubation continued for an additional 12 h before determining BrdU incorporation (relative to total DAPI-stained nuclei) by immunofluorescence microscopy. BrdU results show mean ± SD from multiple fields of view.
Skp2 promoter–luciferase constructs and assays
A 1.9-kb fragment of the mouse Skp2 promoter (bases −1,253 to 263) was cloned by PCR from ICR Swiss mouse DNA (Promega), ligated into the KpnI and BglII sites of pGL3 basic (Promega), and confirmed by DNA sequencing. Analysis of a 5′−Skp2 promoter–luciferase deletion series revealed that the minimal active promoter is from −362 to 263 (unpublished data); this construct was used for the luciferase assays shown. Mutagenesis of the E2F site at +114–121 was performed using the QuikChange multi site-directed mutagenesis kit (Stratagene) with the forward oligo (5′-GGGGATCACTCTAAGCCGAACTTTCAGACAGGAGTCTGGAAGGCAG-3′) and the reverse oligo (5′-CTGCCTTCCAGACTCCTGTCTGAAAGTTCGGCTTAGAGTGATCCCC-3′).
MEFs (∼60% confluent) in a six-well plate were transiently cotransfected as described previously (Bottazzi et al., 1999), but using 1 μg Skp2-pGL3, 0.1 ng cytomegalovirus–Renilla luciferase, 4 μl Lipofectamine (Invitrogen), and 6 μl Plus reagent (Invitrogen) per well. For experiments using E7, the cells were cotransfected with 0.5 μg of the firefly luciferase vector driven by wild-type (Swt) or E2F-mutated (SEm) Skp2 promoter, 0.1 ng CMV–Renilla luciferase, and either 0.5 μg of E7 plasmid or empty vector. After a 24-h incubation in 10% FCS, the cells were collected in passive lysis buffer, and luciferase activity was determined using the dual-luciferase reporter assay system (Promega). Measurements were performed in duplicate and recorded as mean ± SD. Skp2 luciferase activity was normalized to Renilla luciferase activity.
Transfections, infections, and RNAi
Unless noted otherwise in the figure legends, confluent MEFs were infected with adenoviruses after a 12-h incubation in serum-free DME-BSA. The cells were infected overnight at 100 MOI using adenoviruses encoding GFP, Ad-LacZ, Ad-E7 (provided by J. Meinkoth, University of Pennsylvania, Philadelphia, PA), human cyclin E1 (Ad–hCyclin E; provided by J. Albrecht, University of Minnesota, Minneapolis, MN, and S. Reed, University of California, San Diego, San Diego, CA), or Ad-hSkp2 (provided by K. Nakayama, Kyusu University, Fukuoka City, Fukuoka, Japan), and then incubated in fresh serum-free medium to obtain a total serum-free medium incubation time of 48 h. For plasmid transfections, MEFs were transiently transfected as described previously (Welsh et al., 2001) using 5 μg pCDNA3.1 (vector control), pCDNA3.1-based E7, or pcDNA3.1-based human E2F1. Transfected cells were incubated overnight in DME containing 10% FCS before use or serum starvation for 24–36 h. Transfections of siRNAs were performed as described previously (Walker et al., 2006), except that 100 nM irrelevant (human E cadherin; GAGUGAAUUUUGAAGAUUGtt) or mouse Skp2 (UUUGUCACUCCCUUUGCCCtt) siRNAs were used.
When adenoviral infection was combined with siRNA, near confluent MEFs were serum starved for 12 h, infected with either Ad-E7 or Ad-LacZ, and incubated for 24 h in DME-BSA. The medium was removed, and the infected cells were transfected with the irrelevant control siRNA or Skp2 siRNA. After an additional 24 h, the siRNA-containing medium was replaced with DME-BSA or 10% FCS DME with BrdU, and the incubation was continued for another 24 h. After a total of 84 h in serum-free medium, coverslips were collected for analysis of BrdU incorporation. In some experiments, the siRNA transfection also contained 0.05 μg of the wild-type Skp2 promoter–luciferase and 0.05 ng of Renilla luciferase vectors.
QPCR
Collected cell pellets were lysed in 0.5–1 ml of TRIzol (Invitrogen) to extract total RNA. Real-time PCR for mouse Skp2 and Cdk4 were performed as previously described (Stewart et al., 2004). Controls (unpublished data) demonstrated that the mouse Skp2 primer probe set did not detect human Skp2 mRNA. Mouse cyclin E1 mRNA, mouse cyclin A mRNA, and 18S rRNA levels were determined using assay-on-demand primer probe sets Mm00432367_ml, Mm00438064_ml, and Hs99999901_s1 (Applied Biosystems), respectively. Skp2 and cyclin E1 mRNAs were normalized to Cdk4 mRNA or 18S rRNA, neither of which varied reproducibly in response to any of the treatments used. Duplicate PCR reactions were run for each sample, and results are plotted as mean ± SD. Results shown in the figures are typically representative of three independent experiments.
ChIP
ChIPs were performed as described previously (Klein et al., 2007) using 106 MEFs per sample and 5 μg of either anti-E2F1 (C-20X; Santa Cruz Biotechnology, Inc.) or preimmune antibody control. One tenth of the final immunoprecipitated DNA (5 μl) was analyzed by QPCR with SYBR green to quantify the amount of immunoprecipitated Skp2 promoter. Primer sequences for mouse Skp2 were 5′-TGGTGATGGAACGTTGCTAGT-3′ (forward) and 5′-GGTGTCCACTGATTCAGGA-3′ ( reverse). ChIPs on MEFs transiently transfected with Skp2 promoter–luciferase constructs were performed as previously described (Klein et al., 2007) and analyzed by PCR using 5′-TGGTGATGGAACGTTGCTAGT-3′ (forward) and 5′-CTTTATGTTTTTGGCGTCTTCCA-3′ (reverse; encoding plasmid backbone sequence within the promoter–luciferase construct). The amplified PCR product (300 bp) was detected on a 1.5% agarose gel.
Western blotting
Western blotting was performed as described previously (Welsh et al., 2001) using 30–40 μg of total cellular protein and the following antibodies: Skp2 (SKP2-2B12; Invitrogen), Cdk4 (C-22 [Santa Cruz Biotechnology, Inc.] or DCS-31 [Invitrogen]), p27 (clone 57; BD Biosciences), Rb (Mab1; Invitrogen), E2F1 (C-20; Santa Cruz Biotechnology, Inc.) cyclin E (M-20; Santa Cruz Biotechnology, Inc.), and actin (1616R and C-2; Santa Cruz Biotechnology, Inc.). The resolved proteins were detected using ECL (GE Healthcare). Autoradiographs were digitized by scanning, and figures were assembled using Photoshop (Adobe).
Online supplemental material
Fig. S1 complements the mRNA analysis in Figs. 2 B and 3 B to show that infection with Ad-hSkp2 or Ad–hCyclin E leads to protein expression of the E2F targets, cyclin A, cyclin E, and Skp2. Fig. S2 shows that near endogenous levels of Skp2 can initiate the Skp2 autoinduction loop. Fig. S3 shows that Skp2 expression can initiate the Skp2 autoinduction loop even when cells are fully quiescent. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200703034/DC1.
Supplementary Material
Acknowledgments
We thank Jeffrey Albrecht, Keiichi Nakayama, Steve Reed, and Judy Meinkoth for reagents.
This work was supported by National Institutes of Health (NIH) grant HL083367 to R.K. Assoian. J. Walker was supported by NIH training grants F32-GM065031 and R25-CA101871. Y. Yung was supported by American Heart Association grant 0425489U.
Abbreviations used in this paper: Ad-hSkp2, adenovirus encoding human Skp2; APC/Ccdh1, anaphase-promoting complex/cyclosome and its activator Cdh1; ChIP, chromatin immunoprecipitation; E7, human papilloma virus–E7; MEF, mouse embryo fibroblast; p27, p27kip1; QPCR, quantitative real-time RT-PCR; Rb, retinoblastoma protein.
References
- Bashir, T., N.V. Dorrello, V. Amador, D. Guardavaccaro, and M. Pagano. 2004. Control of the SCF(Skp2-Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature. 428:190–193. [DOI] [PubMed] [Google Scholar]
- Binne, U.K., M.K. Classon, F.A. Dick, W. Wei, M. Rape, W.G. Kaelin Jr., A.M. Naar, and N.J. Dyson. 2007. Retinoblastoma protein and anaphase-promoting complex physically interact and functionally cooperate during cell-cycle exit. Nat. Cell Biol. 9:225–232. [DOI] [PubMed] [Google Scholar]
- Blagosklonny, M.V., and A.B. Pardee. 2002. The restriction point of the cell cycle. Cell Cycle. 1:103–110. [PubMed] [Google Scholar]
- Bottazzi, M.E., X. Zhu, R.M. Bohmer, and R.K. Assoian. 1999. Regulation of p21(cip1) expression by growth factors and the extracellular matrix reveals a role for transient ERK activity in G1 phase. J. Cell Biol. 146:1255–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano, A.C., E. Eytan, A. Hershko, and M. Pagano. 1999. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1:193–199. [DOI] [PubMed] [Google Scholar]
- DeGregori, J., T. Kowalik, and J.R. Nevins. 1995. Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol. Cell. Biol. 15:4215–4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou, Q.P., A.H. Levin, S. Zhao, and A.B. Pardee. 1993. Cyclin E and cyclin A as candidates for the restriction point protein. Cancer Res. 53:1493–1497. [PubMed] [Google Scholar]
- Gstaiger, M., R. Jordan, M. Lim, C. Catzavelos, J. Mestan, J. Slingerland, and W. Krek. 2001. Skp2 is oncogenic and overexpressed in human cancers. Proc. Natl. Acad. Sci. USA. 98:5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, J.Y., J.D. Reimann, C.S. Sorensen, J. Lukas, and P.K. Jackson. 2002. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 4:358–366. [DOI] [PubMed] [Google Scholar]
- Imaki, H., K. Nakayama, S. Delehouzee, H. Handa, M. Kitagawa, T. Kamura, and K.I. Nakayama. 2003. Cell cycle-dependent regulation of the Skp2 promoter by GA-binding protein. Cancer Res. 63:4607–4613. [PubMed] [Google Scholar]
- Izumi, M., M. Yokoi, N.S. Nishikawa, H. Miyazawa, A. Sugino, M. Yamagishi, M. Yamaguchi, A. Matsukage, F. Yatagai, and F. Hanaoka. 2000. Transcription of the catalytic 180-kDa subunit gene of mouse DNA polymerase alpha is controlled by E2F, an Ets-related transcription factor, and Sp1. Biochim. Biophys. Acta. 1492:341–352. [DOI] [PubMed] [Google Scholar]
- Ji, P., H. Jiang, K. Rekhtman, J. Bloom, M. Ichetovkin, M. Pagano, and L. Zhu. 2004. An Rb-Skp2-p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol. Cell. 16:47–58. [DOI] [PubMed] [Google Scholar]
- Kamata, Y., J. Watanabe, Y. Nishimura, T. Arai, M. Kawaguchi, M. Hattori, A. Obokata, and H. Kuramoto. 2005. High expression of skp2 correlates with poor prognosis in endometrial endometrioid adenocarcinoma. J. Cancer Res. Clin. Oncol. 131:591–596. [DOI] [PubMed] [Google Scholar]
- Klein, E.A., Y. Yung, P. Castagnino, D. Kothapalli, and R.K. Assoian. 2007. Cell adhesion, cellular tension and cell cycle control. Methods Enzymol. In press. [DOI] [PubMed]
- Kothapalli, D., S.A. Stewart, E.M. Smyth, I. Azonobi, E. Pure, and R.K. Assoian. 2003. Prostacylin receptor activation inhibits proliferation of aortic smooth muscle cells by regulating cAMP response element-binding protein- and pocket protein-dependent cyclin a gene expression. Mol. Pharmacol. 64:249–258. [DOI] [PubMed] [Google Scholar]
- Malek, N.P., H. Sundberg, S. McGrew, K. Nakayama, T.R. Kyriakides, and J.M. Roberts. 2001. A mouse knockin model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature. 413:323–327. [DOI] [PubMed] [Google Scholar]
- Markey, M.P., S.P. Angus, M.W. Strobeck, S.L. Williams, R.W. Gunawardena, B.J. Aronow, and E.S. Knudsen. 2002. Unbiased analysis of RB-mediated transcriptional repression identifies novel targets and distinctions from E2F action. Cancer Res. 62:6587–6597. [PubMed] [Google Scholar]
- Marti, A., C. Wirbelauer, M. Scheffner, and W. Krek. 1999. Interaction between ubiquitin-protein ligase SCFSKP2 and E2F-1 underlies the regulation of E2F-1 degradation. Nat. Cell Biol. 1:14–19. [DOI] [PubMed] [Google Scholar]
- Nakayama, K.I., and K. Nakayama. 2005. Regulation of the cell cycle by SCF-type ubiquitin ligases. Semin. Cell Dev. Biol. 16:323–333. [DOI] [PubMed] [Google Scholar]
- Neuman, E., E.K. Flemington, W.R. Sellers, and W.G. Kaelin Jr. 1994. Transcription of the E2F-1 gene is rendered cell cycle dependent by E2F DNA-binding sites within its promoter. Mol. Cell. Biol. 14:6607–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtani, K., J. DeGregori, and J. Nevin. 1995. Regulation of the cyclin E gene by transcription factor E2F1. Proc. Natl. Acad. Sci. USA. 92:12146–12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polyak, K., M.H. Lee, H. Erdjument-Bromage, A. Koff, J.M. Roberts, P. Tempst, and J. Massague. 1994. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 78:59–66. [DOI] [PubMed] [Google Scholar]
- Reed, S.I. 2003. Ratchets and clocks: the cell cycle, ubiquitylation and protein turnover. Nat. Rev. Mol. Cell Biol. 4:855–864. [DOI] [PubMed] [Google Scholar]
- Rodier, G., C. Makris, P. Coulombe, A. Scime, K. Nakayama, K.I. Nakayama, and S. Meloche. 2005. p107 inhibits G1 to S phase progression by down-regulating expression of the F-box protein Skp2. J. Cell Biol. 168:55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, S.A., D. Kothapalli, Y. Yung, and R.K. Assoian. 2004. Antimitogenesis linked to regulation of Skp2 gene expression. J. Biol. Chem. 279:29109–29113. [DOI] [PubMed] [Google Scholar]
- Sutterluty, H., E. Chatelain, A. Marti, C. Wirbelauer, M. Senften, U. Muller, and W. Krek. 1999. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1:207–214. [DOI] [PubMed] [Google Scholar]
- Tao, Y., R.F. Kassatly, W.D. Cress, and J.M. Horowitz. 1997. Subunit composition determines E2F DNA-binding site specificity. Mol. Cell. Biol. 17:6994–7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerbeul, I., C.M. Garrett-Engele, U. Kossatz, X. Chen, E. Firpo, V. Grunwald, K. Kamino, L. Wilkens, U. Lehmann, J. Buer, et al. 2006. Testing the importance of p27 degradation by the SCFskp2 pathway in murine models of lung and colon cancer. Proc. Natl. Acad. Sci. USA. 103:14009–14014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima, H., and T. Hunter. 1994. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 78:67–74. [DOI] [PubMed] [Google Scholar]
- Tsvetkov, L.M., K.H. Yeh, S.J. Lee, H. Sun, and H. Zhang. 1999. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr. Biol. 9:661–664. [DOI] [PubMed] [Google Scholar]
- Vernell, R., K. Helin, and H. Muller. 2003. Identification of target genes of the p16INK4A-pRB-E2F pathway. J. Biol. Chem. 278:46124–46137. [DOI] [PubMed] [Google Scholar]
- Walker, J.L., P. Castagnino, B.M. Chung, M.G. Kazanietz, and R.K. Assoian. 2006. Post-transcriptional destabilization of p21cip1 by protein kinase C in fibroblasts. J. Biol. Chem. 50:38127–38132. [DOI] [PubMed] [Google Scholar]
- Wei, W., N.G. Ayad, Y. Wan, G.J. Zhang, M.W. Kirschner, and W.G. Kaelin Jr. 2004. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature. 428:194–198. [DOI] [PubMed] [Google Scholar]
- Welsh, C.F., K. Roovers, J. Villanueva, Y. Liu, M.A. Schwartz, and R.K. Assoian. 2001. Timing of cyclin D1 expression within G1 phase is controlled by Rho. Nat. Cell Biol. 3:950–957. [DOI] [PubMed] [Google Scholar]
- Zhang, H., R. Kobayashi, K. Galaktionov, and D. Beach. 1995. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell. 82:915–925. [DOI] [PubMed] [Google Scholar]
- Zhang, L., and C. Wang. 2006. F-box protein Skp2: a novel transcriptional target of E2F. Oncogene. 25:2615–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, W.Q., J.M. Zheng, R. Ma, F.F. Meng, and C.R. Ni. 2005. Relationship between levels of Skp2 and P27 in breast carcinomas and possible role of Skp2 as targeted therapy. Steroids. 70:770–774. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}