

Abstract
We have analyzed the spontaneous symmetry breaking and initiation of actin-based motility in keratocytes (fish epithelial cells). In stationary keratocytes, the actin network flow was inwards and radially symmetric. Immediately before motility initiation, the actin network flow increased at the prospective cell rear and reoriented in the perinuclear region, aligning with the prospective axis of movement. Changes in actin network flow at the cell front were detectable only after cell polarization. Inhibition of myosin II or Rho kinase disrupted actin network organization and flow in the perinuclear region and decreased the motility initiation frequency, whereas increasing myosin II activity with calyculin A increased the motility initiation frequency. Local stimulation of myosin activity in stationary cells by the local application of calyculin A induced directed motility initiation away from the site of stimulation. Together, these results indicate that large-scale actin–myosin network reorganization and contractility at the cell rear initiate spontaneous symmetry breaking and polarized motility of keratocytes.
Introduction
The coordinated movement of crawling cells depends on the proper spatial and temporal regulation of the actin cytoskeleton. Actin polymerization is biased toward the cell front, enabling protrusion of the leading edge, and myosin contraction is biased toward the cell rear, enabling traction of the cell body toward the front. In addition to the polarization of actin network dynamics, crawling cells exhibit a morphological polarization, with the cell front and rear being easily distinguishable.
In neutrophils, which respond to external gradients of chemoattractants, front-back polarization is initiated by protrusion of the leading edge closest to the chemoattractant source. However, stationary neutrophils in a uniform concentration of chemoattractant can also polarize spontaneously, initiate motility, and move in a random direction (Zigmond et al., 1981; Coates et al., 1992). In both cases, polarization is accompanied by the recruitment of PIP3 to the cell membrane (Servant et al., 2000) and the formation of actin ruffles at the leading edge (Weiner et al., 1999). Other cell types have also been reported to spontaneously break symmetry in the absence of any external cues, including fibroblasts, which polarize after plating onto glass coverslips coated with polylysine and ConA (Symons and Mitchison, 1991), and Walker carcinosarcoma cells (Fedier et al., 1999). This suggests that polarization and motility initiation do not require an external directional cue. Where one is present, it only induces a preferred directionality, but there must be an intrinsic mechanism for cell polarization.
In contrast to neutrophils, where polarization is initiated by protrusion, stationary lamellipodial fragments from fish epidermal keratocytes, which are circular, can be pushed at the rear by a stream of media from a micropipette. This induces rear retraction followed by protrusion of the front and the initiation of persistent motility (Verkhovsky et al., 1999). Similarly, in already polarized chick heart fibroblasts, rear retraction precedes front protrusion (Chen, 1979; Dunn and Zicha, 1995). So, although neutrophil polarization in response to a chemoattractant is initiated by protrusion at the front, lamellipodial fragments can polarize when physically pushed at the rear. However, it has not been determined how polarization arises and propagates in the absence of an external stimulus and what the events throughout spontaneous symmetry breaking may be.
The goal of this study is to establish quantitatively, for the first time, the sequence of structural events during spontaneous cell polarization. We chose rapidly moving fish epidermal keratocytes as a model system for our analyses. These cells are not known to respond to chemotactic stimuli; thus, the underlying mechanism of spontaneous symmetry breaking can be studied without cross talk from chemotaxis. Once keratocytes are polarized, they migrate in a persistent manner (Euteneuer and Schliwa, 1984). Furthermore, keratocytes are flat and, thus, are particularly amenable to high resolution live cell microscopy of morphodynamic events.
Our data indicate that the first signs of polarization do not emerge at the cell front or periphery but at the cell rear and perinuclear region. Rearrangements of the actomyosin network near the cell periphery and cell front occurred later in the polarization process, indicating that they were a consequence rather than a cause of symmetry breaking. This symmetry breaking was driven by contraction and was dependent on Rho kinase–mediated reorganization of the actomyosin network. Thus, we have identified an alternative paradigm for spontaneous cell symmetry breaking and motility initiation.
Results
Stationary keratocytes are radially symmetric and can spontaneously initiate motility
Keratocytes isolated from cichlid scales migrated from the scale as large epidermal sheets that could be disaggregated by incubation in 2.5 mM EGTA/85% PBS to form a mixture of smaller islands, individual motile polarized keratocytes, and individual stationary nonpolarized keratocytes. Stationary keratocytes were circular and radially symmetric (Fig. 1, A and C), which is in contrast to motile keratocytes, which were crescent shaped and bilaterally symmetric (Fig. 1, B and E). The network of actin filaments (filamentous actin [F-actin]) of stationary keratocytes was denser around the cell body than at the periphery and often formed circular bands around the cell body (Fig. 1 A). Although stationary keratocytes did not exhibit net translocation, transient protrusion and retraction occurred around the cell edge.
Figure 1.

F-actin network movement in stationary and motile keratocytes. (A and B) Keratocytes were fixed and stained with phalloidin to visualize F-actin. (A) F-actin in stationary keratocytes was denser near the cell body than the periphery. (B) Motile keratocytes had a criss-cross pattern of F-actin staining in the lamellipodia and F-actin bundles at the cell rear. (C and E) AF546-phalloidin FSM images of F-actin networks in live stationary (C) and motile (E) keratocytes. (D and F) F-actin flow field relative to the substratum extracted by multiframe correlation tracking of speckle movement. Vectors indicate network displacements between consecutive frames. Color encodes flow speed. (D) The F-actin network in stationary keratocytes flowed centripetally inwards. (F) The F-actin network in motile keratocytes had a small retrograde flow in the lamellipodium and large inwards movement at the rear sides. (G) During symmetry breaking and motility initiation of keratocytes, the cell must transition from being stationary with radially symmetric centripetal actin flow to being polarized with decreased actin flow speed at the cell front. C and E are available as Videos 1 and 2 (available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1).
In the lamellipodia of motile cells, F-actin generally moves from the leading edge to the cell body. To image F-actin network movement in the lamellipodia of stationary keratocytes, we used fluorescent speckle microscopy (FSM) with low levels of labeled phalloidin (Schaefer et al., 2002; Zhang et al., 2003; Vallotton et al., 2005), which binds specifically to F-actin and yields a higher signal/noise level than G-actin probes (Fig. 1, C and E; Schaefer et al., 2002; Vallotton et al., 2005). We measured the rate of F-actin network movement by adaptive multiframe correlation tracking of fluorescent speckle motion (Fig. 1, D and F; Ji and Danuser, 2005). In stationary keratocytes, F-actin flowed centripetally from the cell edge to the cell body (Fig. 1 D and Video 1, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1) at a rate of ∼25–60 nm/s. The flux was fastest at the cell periphery, decreasing gradually toward the cell body. This radial inward flow was consistent with previous studies of centripetal actin flow in circular, stationary lamellipodial fragments (Verkhovsky et al., 1999) and circular sea urchin coelomocytes (Edds, 1993). In motile keratocytes, it has been established that the lamellipodial F-actin network moves slowly rearwards with respect to the substratum as the cell moves rapidly forward (Jurado et al., 2005; Vallotton et al., 2005). In our experiments, we measured retrograde F-actin flow of ∼10 nm/s relative to the substratum in the lamellipodia (Fig. 1 F and Video 2) and faster inward flow at the rear sides of the cell (Fig. 1 F and Video 2) as observed previously (Theriot and Mitchison, 1991; Lee et al., 1993; Vallotton et al., 2005), where myosin-dependent contraction gathers the F-actin network toward the cell body (Anderson et al., 1996; Svitkina et al., 1997).
Stationary keratocytes could spontaneously break symmetry, polarize, and initiate motility in the absence of external cues at a frequency of ∼15% per 30 min. Given that the F-actin flow pattern and speed were different in stationary compared with motile keratocytes (Fig. 1 G), mechanisms that lead to symmetry breaking and morphological polarity should also alter F-actin dynamics within the cell. These changes could occur at the prospective cell front, prospective cell rear, or simultaneously at the front and rear.
Initial morphological changes during spontaneous symmetry breaking and motility initiation occur at the cell rear
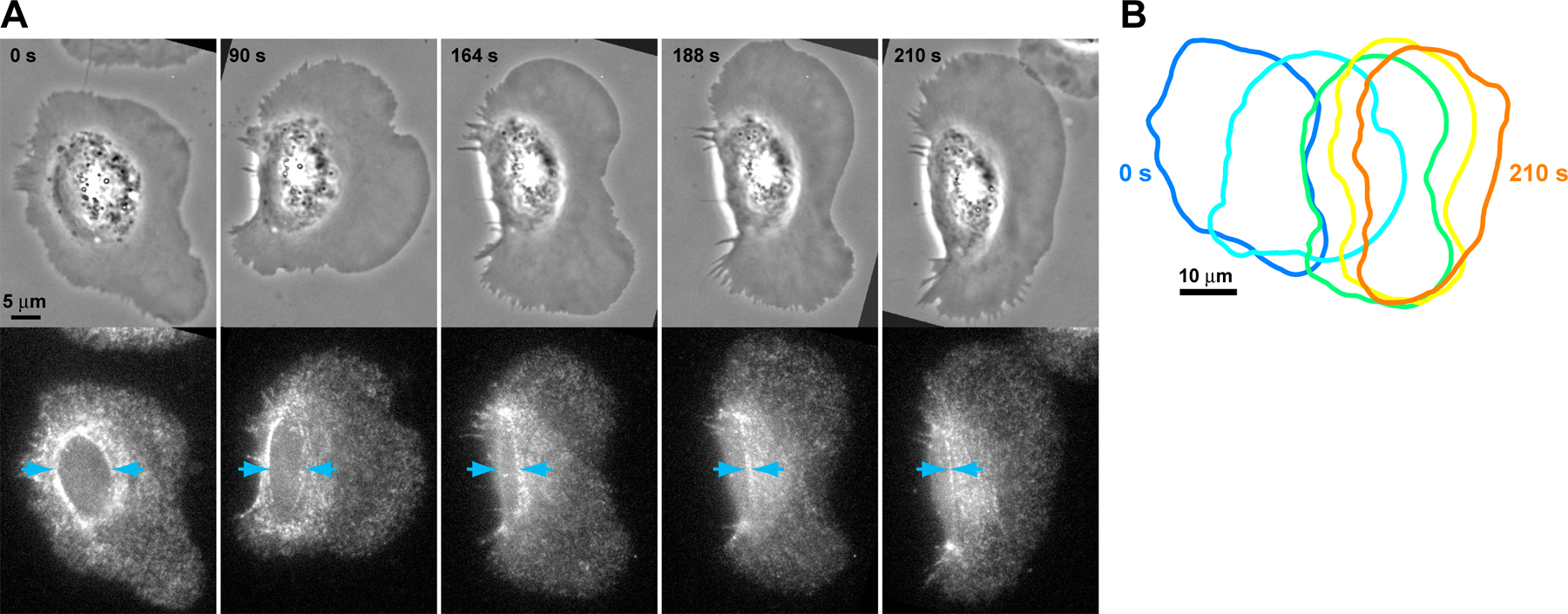
Not all stationary cells initiated motility, and stationary cells had variable protrusion and retraction at the cell edge. To determine the sequence of events leading to cell polarization and movement, we acquired time-lapse videos of keratocytes spontaneously initiating motility (Fig. 2 A and Videos 3 and 4, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1) and first examined the movement of the cell edge. We measured the position of the cell edge in each frame of a video using the gradient vector flow variation of the active contours fitting method (see Materials and methods and Fig. S1; Kass et al., 1988; Xu and Prince, 1998). Time sequences of cell edge outlines indicated that the prospective rear edge moved inwards before forward advancement of the front edge (Fig. 2 B). To quantify this, we used consecutive cell outlines to measure the extent of protrusion or retraction at points along the cell edge for each time point (Machacek and Danuser, 2006) and generated maps of cell edge movement over time (Fig. 2 C). A region of continuous retraction at the cell rear (Fig. 2 C, blue boxes) could reliably be identified in cells before the overt initiation of directed motility. There was also a region of continuous protrusion at the cell front (Fig. 2 C, red boxes), but this generally started after rear retraction was initiated (9/11 cells).
Figure 2.

The prospective rear edge moves before the front edge during motility initiation. (A) Phase-contrast image sequence of two representative keratocytes breaking symmetry and initiating motility. (B, left) Cell outline superimposed on the first and last image in the image sequence for the same cells as in A. (B, right) Time sequence of the cell outline during motility initiation, with color denoting time elapsed (in seconds). (C) Protrusion/retraction map of the cell edge (in polar coordinates) over time. Blue boxes are regions of continuous rear retraction. Red boxes are regions of continuous front protrusion. The rear of the cell exhibited three phases during motility initiation: I, slow rear retraction; II, fast rear retraction; and III, persistent movement. A is available as Videos 3 and 4 (available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1).
Although motility initiation was a continuous process, we typically observed three characteristic phases during motility initiation (Fig. 2 C). Phase I was a period of slow rear retraction (∼10– 40 nm/s) of highly variable duration, ranging from 20 to 430 s. In addition, by imaging cells with a fluorescent volume marker, we found that the lamellipodial thickness decreased at the prospective rear during phase I, whereas no changes in lamellipodial thickness were observed elsewhere (Fig. S2, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1). The types of transients observed in phase I were not unique to cells initiating motility but were also within the range of edge fluctuations observed in stationary cells. Phase II was a period of fast (∼50–180 nm/s) and sustained rear retraction in which the rear edge rapidly moved inwards toward the cell body for 40–170 s, leading to morphological polarization. In contrast to phase I, phase II seemed specific to those cells initiating motility. This was followed by phase III, in which the cell rear advanced persistently. Persistent protrusion of the cell front coordinated with movement of the cell rear usually began during phase III (8/11 cells).
Although the cell rear consistently retracted inwards during motility initiation, patterns of edge movement at the cell front were more variable and not predictive of cell polarization. For some cells, local transient movement of the cell front was observed during phases I and II, but these movements were comparable with those in stationary cells. Expansion of the cell sides also sometimes occurred during phases I and II. Because we observed cells in which there was no detectable movement of the front edge before persistent movement during phase III, we conclude that persistent movement of the front edge was not required for symmetry breaking and motility initiation. Together, these data suggested that symmetry breaking may be mediated by events at the prospective cell rear.
Initial events in symmetry breaking include an increase in actin network flow speed at the prospective cell rear and a reorientation of perinuclear actin network flow
Next, we examined transients in F-actin network flow during polarization and motility initiation using FSM and multiframe correlation tracking. We performed detailed tracking of F-actin network flow in five cells undergoing motility initiation. For each F-actin flow vector, we calculated the radial component of velocity (vr) and the centripetal deviation (φ), which was defined as the angular displacement between the radial component of velocity and the original velocity vector (Fig. 3 A). In stationary cells, the radial velocity generally decreased from the cell periphery to the center, and the centripetal deviation fluctuated around zero (Fig. 3 B and Video 5, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1). During phase I, slow edge retraction at the prospective cell rear was accompanied by a slight increase in radial velocity and an increase in the magnitude of centripetal deviation in the perinuclear region (Fig. 3 C, left; and Video 6), indicating that instead of flowing centripetally toward the cell body, F-actin flow was biased in the direction of prospective cell movement. Phase II was characterized by a large increase in the radial velocity at the cell rear and continued alignment of the F-actin flow in the perinuclear region toward the eventual direction of cell movement (Fig. 3 C, right; and Video 6).
Figure 3.

Initial events in symmetry breaking include an increase in F-actin network flow speed at the prospective cell rear and a reorientation of perinuclear F-actin network flow. (A) Representation of an F-actin flow field displacement vector by its radial (vr) and tangential (vθ) component. The centripetal deviation (φ) was positive for counter-clockwise rotation and negative for clockwise rotation of the original flow vector relative to vr. (B and C) F-actin flow field of a stationary cell (B) and a cell initiating motility (C) with the prospective direction of cell movement to the right. (left) F-actin network flow field; flow vectors are colored according to speed. (right) Scalar maps of radial velocity and centripetal deviation of F-actin flow vectors. White line is the delineation of the perinuclear and peripheral regions. (C, left) Phase I: the radial velocity increased slightly at the prospective cell rear. The magnitude of the centripetal deviation increased in the perinuclear region, corresponding to a reorientation of the F-actin flow along the prospective direction of motion. (right) Phase II: large increase in radial velocity at the prospective cell rear. The increased magnitude of centripetal deviation remained. (D and E, top) Time courses of radial velocity in the front (blue), rear (red), left (brown), and right (green) perinuclear (bold) and peripheral (standard) regions. Data in D originate from the cell in C and showed large transients before sustained polarization. (bottom) Time courses of centripetal deviation in the left (brown) and right (green) perinuclear (bold) and peripheral (standard) regions. Phases I and II are indicated by horizontal bars. B and C are available as Videos 5 and 6 (available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1).
To measure when and where changes in F-actin flow occurred during motility initiation, we distinguished F-actin dynamics in the perinuclear and peripheral regions of the lamellipodia based on the AF546-phalloidin fluorescence pattern. The F-actin network in the perinuclear region was denser and often formed circular bands, whereas the peripheral region had less dense labeling (Fig. 1, A and C). We also defined four sectors of equal angular size—front, back, and two sides—based on the eventual direction of cell movement. In the five cells tracked while initiating motility, the first sign of polarization was an increase in radial velocity (∼15–40%) at the rear perinuclear and peripheral regions and a reorientation of F-actin flow in the perinuclear region toward the prospective direction of movement, which was manifested in an increased magnitude of centripetal deviation (Fig. 3, D and E). These changes coincided with phase I. The changes in actin dynamics in the rear and side perinuclear regions were often accompanied by a decrease in the radial velocity in the front perinuclear region, suggesting a high coordination of F-actin dynamics in the perinuclear ring. However, no changes occurred in the front peripheral region during phase I. Notably, flow pattern variations similar to those observed in phase I were also observed in some cells while stationary (Fig. 3 D at 140–180s shows extreme variation before motility initiation). This suggests that flow transients of phase I are not specific to motility initiation and probably reflect inherent variation in F-actin flow in stationary cells. However, changes in F-actin flow characteristic of phase I always preceded phase II during motility initiation.
Approximately 5–20 s before the start of phase II, the rate of change of radial velocity increased in the rear perinuclear and peripheral regions, leading to the high radial velocity at the cell rear characteristic of phase II, approximately two- to threefold faster than basal speeds (Fig. 3, D and E; top). These changes were specific to the cell rear and were not observed at the prospective cell sides (Fig. 3 E, top). The increased magnitude of centripetal deviation in the perinuclear region was maintained during phase II and also occurred in the side peripheral regions (Fig. 3, D and E; bottom).
Together, these data suggest that symmetry breaking of the F-actin network in keratocytes is initiated in the perinuclear region and prospective cell rear. Phases of movement initiation defined by morphological analysis of the cell edge corresponded with specific changes in F-actin network flow, suggesting that variations in F-actin network movement trigger motility initiation.
Decrease in actin flow at the cell front occurs after initial polarization
In phases I and II, no changes in flow speed were detectable in the front peripheral region. However, the speed of F-actin retrograde flow with respect to the substratum in lamellipodia of fully polarized cells is lower than that of the centripetal flow in stationary cells (Fig. 1 G), implying that F-actin flow at the prospective cell front must decrease at some point. We followed cells that had initiated motility (Fig. 4 A), and, during phase III, in which the cell front and rear moved persistently in a coordinated fashion, we observed a period of maturation of the polarized form. The cell speed increased, accompanied by a decrease in F-actin retrograde flow speed relative to the substratum at the cell front (Fig. 4 B). Thus, flow changes at the front occurred only during the later stages of motility initiation and were a consequence, not a cause, of motility initiation. In addition, during maturation, the perinuclear F-actin bands collapsed toward the cell rear (Fig. S3 and Video 8, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1), where they formed an actin axle characteristic of motile cells (Fig. 1, B and E).
Figure 4.

Decrease in F-actin network flow at the cell front occurs during maturation of the polarized form. (A) F-actin network flow vectors relative to the substratum in the front central lamellipodium (boxed area) overlaid on an AF546-phalloidin FSM image. (B) Development of cell speed versus F-actin flow speed at the front relative to the substratum and parallel to the direction of movement over phase III (maturation) for five cells. During maturation, cells increased in speed, whereas the F-actin retrograde flow speed decreased in the lamellipodia.
Symmetry breaking and motility initiation require Rho kinase–dependent myosin activity
Microtubules and myosin II were heterogeneously distributed in the lamellipodium of stationary keratocytes (Fig. 5 A), and asymmetries in these distributions might influence cell polarization. Microtubules can regulate actin dynamics and are implicated in the establishment and maintenance of cell polarity during the migration of fibroblasts and epithelial cells (Waterman- Storer et al., 1999; Watanabe et al., 2005), whereas myosin has a polarized distribution in motile keratocytes (Svitkina et al., 1997). In addition, for neutrophils, polarization requires the recruitment of PIP3 to the cell membrane (Servant et al., 2000). To determine whether any of these molecules might contribute to motility initiation, we developed a quantitative assay for motility initiation based on the observation that the frequency of motility initiation was enhanced by increasing temperature (see Materials and methods Pharmacological treatments section).
Figure 5.

Motility initiation requires Rho kinase–dependent myosin activity. (A, top) Keratocyte fixed and stained for tubulin to visualize microtubules. (bottom) Live stationary keratocyte expressing YFP-myosin regulatory light chain. (B) Motility initiation frequency of stationary keratocytes within 30 min of a temperature shift from 20 to 30°C. Successful motility initiation was defined as persistent polarized morphology and cell movement over at least four cell lengths. Shape changers were cells that had irregular morphologies. Depolymerization of microtubules with 1 μg/ml nocodazole, inhibition of PI-3 kinase with 50 μM LY294002, and inhibition of MLCK with 10 or 25 μM ML-7 had no effect on the frequency of motility initiation (P = 0.7498, P = 0.0173, and P = 0.5562, respectively). Myosin II inhibition with 40 or 100 μM blebbistatin and Rho kinase inhibition with 10 or 25 μM Y-27632 decreased the frequency of motility initiation (P < 0.0001). In contrast, myosin phosphatase inhibition with 10 or 25 nM calyculin A increased the frequency of motility initiation (P < 0.0001). When two drug concentrations were used, there was no significant difference between the results (P > 0.2); results from the two concentrations were pooled. (C) Stationary keratocytes were treated with the indicated drugs for 10–20 min. F-actin was visualized by fixing the cells and staining with phalloidin. Treatment with ML-7 and calyculin A retained the circular bands of F-actin in the perinuclear region. Treatment with blebbistatin or Y-27632 reduced F-actin in the perinuclear region, and circular bands were no longer visible (yellow brackets). (D and E) Changes in perinuclear and peripheral F-actin radial velocity and directional coherence (see Materials and methods) before and after treatment with the indicated drugs. Error bars indicate the SD of the mean over time; gray lines indicate correspondence between data points representing the same cell before and after treatment. In some cases, the SD is smaller than the size of the data point. (D) Blebbistatin treatment decreased the perinuclear and peripheral radial velocity; conversely, calyculin A treatment increased the perinuclear and peripheral radial velocity. Y-27632 or ML-7 treatment had no effect. (E) Blebbistatin and Y-27632 treatment decreased the directional coherence of the actin flow in the perinuclear and peripheral zones. The effect was greater in the perinuclear zone than the peripheral zone. ML-7 and calyculin A had no effect.
Under control conditions, ∼45% of stationary cells spontaneously broke symmetry and initiated motility within 30 min of a temperature shift from 20 to 30°C (Fig. 5 B). Depolymerization of microtubules with nocodazole had no effect on the frequency of motility initiation, implying that microtubules were not required for motility initiation of keratocytes. Likewise, inhibition of PIP3 production by the addition of LY294002, a phosphatidylinositol phosphate 3-kinase (PI 3-kinase) inhibitor, had no effect (Fig. 5 B). To confirm that PIP3 was not required for motility initiation of keratocytes, we used a GFP-tagged pleckstrin homology (PH)–Akt construct (Kontos et al., 1998), which binds to the 3′ phosphorylated lipid products generated by PI 3-kinase, to visualize the PIP3 localization in keratocytes. GFP-PH-Akt was uniformly localized to the cell membrane. Notably, there was lack of enhancement of GFP-PH-Akt to the leading edge of motile keratocytes or to the periphery of stationary keratocytes. Furthermore, when circular stationary keratocytes were forced to polarize and initiate motility by pushing of the cell rear, no redistribution of GFP-PH-Akt was observed (unpublished data). Together, this suggests that PI 3-kinase and the lipid products of PI 3-kinase are not required for polarization and motility initiation of keratocytes.
However, the inhibition of myosin II with blebbistatin (Straight et al., 2003) decreased the fraction of cells initiating motility to 10% (Fig. 5 B), suggesting that myosin II was required for motility initiation. Nonmuscle myosin II is primarily regulated by Rho kinase, myosin light chain kinase (MLCK), and myosin light chain phosphatase. Used at concentrations effective in keratocytes, the Rho kinase inhibitor Y-27632 but not the MLCK inhibitor ML-7 (Jurado et al., 2005) mimicked the effect of myosin II inhibition by blebbistatin (Fig. 5 B). Rho kinase phosphorylates myosin light chain phosphatase, inhibiting its phosphatase activity and, thereby, increasing phosphorylation of the myosin II light chain. Calyculin A, an inhibitor of Ser/Thr protein phosphatases types I and IIA and a known inhibitor of myosin light chain phosphatase, inhibits dephosphorylation of the myosin II light chain, potentiating myosin II activity and leading to constriction of the central actin–myosin ring in sea urchin coelomocytes (Henson et al., 2003) and increased F-actin flow in newt lung epithelial cells (Vallotton et al., 2004). In motile keratocytes, calyculin A increases myosin-dependent inwards F-actin flow at the cell rear (unpublished data). Therefore, we hypothesized that calyculin A might facilitate symmetry breaking in keratocytes by increasing myosin II activity. Indeed, we found that 100% of cells initiated motility with calyculin A treatment (Fig. 5 B). Importantly, these drugs did not influence the morphology, polarity, or F-actin organization of motile keratocytes (Fig. S4, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1) and had little effect on the steady-state speed. Together, these data implicate myosin II activity regulated by Rho kinase as a critical factor in symmetry breaking and motility initiation of keratocytes.
Myosin-mediated actin network organization in the perinuclear region is required for symmetry breaking
To investigate the effect of myosin II or Rho kinase inhibition on F-actin organization, drug-treated cells were fixed and stained with fluorescent phalloidin to visualize the F-actin network. Cells treated with blebbistatin and Y-27632 were larger and flatter, and F-actin tended to accumulate around the cell periphery, whereas circular F-actin bands were absent from the perinuclear region (Fig. 5 C). In contrast, ML-7 had no detectable effect on the F-actin network (Fig. 5 C), which is consistent with its lack of effect on motility initiation. Myosin phosphatase inhibition by calyculin A at low concentrations also had no detectable effect (Fig. 5 C). In some cases, increased phalloidin staining in the perinuclear region was observed at higher concentrations or longer incubations with calyculin A. These results suggest that perturbation of F-actin network organization, particularly in the perinuclear region, inhibits motility initiation.
We also measured the effect of the various drugs on F-actin network flow. Cells were imaged by FSM before and after drug treatment, and F-actin movement was calculated by multiframe correlation tracking. We characterized the F-actin flows by radial velocity and directional coherence, a measure of how similar the orientations of the F-actin flow vectors in a local region were to each other. Perfectly coherent flow had a value of 1 (see Materials and methods section Analysis of displacement fields generated…). All drug treatments affecting symmetry breaking altered F-actin flow. Myosin II inhibition by blebbistatin decreased the radial velocity and coherence in the perinuclear and peripheral regions (Fig. 5, D and E), with a greater effect in the perinuclear region. The Rho kinase inhibitor Y-27632 decreased F-actin flow coherence, with a stronger effect in the perinuclear region than the peripheral region (Fig. 5 E). These changes in F-actin dynamics were consistent with the F-actin staining in cells showing perinuclear F-actin network disruption by myosin II or Rho kinase inhibition. In contrast, ML-7, which had no effect on the frequency of motility initiation or F-actin organization, also had no effect on F-actin flow (Fig. 5, D and E). Calyculin A, which increased the frequency of motility initiation, increased the radial velocity both in the perinuclear and peripheral regions (Fig. 5 D), supporting the notion that calyculin A promotes motility initiation by increasing actomyosin contractility.
Local increase in actin network flow stimulates symmetry breaking and motility initiation
Based on these results, we hypothesized that symmetry breaking could be induced by the local activation of myosin II. First, we tested whether calyculin A treatment of keratocytes promoted motility initiation via the same mechanism as in normal untreated cells. We imaged a cell initiating motility spontaneously in the presence of calyculin A. Like untreated cells undergoing spontaneous motility initiation, the action started by retraction of the prospective rear edge accompanied by an increase in F-actin flow at the prospective cell rear and reorientation of the perinuclear F-actin flow toward the prospective direction of movement (Fig. 6, A and B). Again, no change in F-actin flow was observed at the front peripheral region. Although the transients in F-actin network flow were faster, motility initiation by calyculin A stimulation appeared to operate through the same pathway as identified for untreated cells.
Figure 6.

Local application of calyculin A can stimulate motility initiation. (A and B) Changes in F-actin network radial velocity (A) and centripetal deviation (B) in phases I and II of spontaneous symmetry breaking in the presence of 20 nM calyculin A. (top) Scalar maps of the radial velocity and centripetal deviation. White line is the delineation of perinuclear and peripheral regions. (bottom) Time course of radial velocity and centripetal deviation. Phases I and II are indicated by horizontal bars. (A) The radial velocity at the cell rear increased during phases I and II. (B) The magnitude of the centripetal deviation increased during phases I and II, reflecting a reorientation of the F-actin flow along the prospective direction of motion. (C) Local application of calyculin A to one side of a stationary keratocyte. Red pseudocolor indicates the drug flow from the micropipette (positioned in the bottom left corner). The keratocyte polarized and initiated motility away from the source of calyculin A. C is available as Video 7 (available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1).
Next, we sought to stimulate asymmetric contraction with the expectation that we might direct symmetry breaking and motility initiation. We locally applied calyculin A to one side of a stationary cell and indeed found that the cell would break symmetry and move away from the source of calyculin A (Fig. 6 C and Video 7, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1). 39% (15/38 cells) initiated motility away from the calyculin A source within 5 min, with 29% (11/38 cells) polarizing without initiating motility, and only 26% (10/38 cells) remained stationary. In contrast, when cells were exposed to local perfusion with medium lacking drug, 13% (3/24 cells) initiated motility away from the pipette and 13% (3/24 cells) polarized without initiating motility, whereas most cells (67%; 16/24) remained stationary. We were unable to induce motility initiation by a local decrease in contractility at the presumptive front (via local application of blebbistatin or Y-27632); this may have been the result of unfavorable kinetics of drug entry or intracellular diffusion of the drug or drug–target complex. Thus, a local increase in actin–myosin contraction mediated by the local application of calyculin A could induce cell polarization from the cell rear.
Discussion
Symmetry breaking and motility initiation is driven by contraction and initiated at the prospective rear and perinuclear region
This study establishes the sequence of morphodynamic events and spatio-temporal reorganization of the F-actin network during spontaneous symmetry breaking of fish epidermal keratocytes. In contrast to polarization and motility initiation of neutrophils in response to chemoattractants, where initial morphological changes occur at the prospective cell front and periphery (Weiner et al., 1999; Servant et al., 2000), here, the first events were detected at the prospective cell rear and perinuclear region (Fig. 7 A). Changes at the cell front occurred later in motility initiation and appeared to be consequences rather than causes of cell polarization.
Figure 7.

Symmetry breaking and motility initiation are initiated at the cell rear and perinuclear region and propagate throughout the entire cell. (A) Model for symmetry breaking and motility initiation in keratocytes. (left) Schematic of F-actin network flow in the peripheral (gray) and perinuclear (purple) regions. (right) Cell cross section schematic. In a stationary keratocyte, the F-actin network flows centripetally inwards, driven by net polymerization at the periphery and myosin contraction and net depolymerization in the perinuclear region. In phase I, an increase in perinuclear contractility causes an increase in the F-actin network flow speed at the prospective cell rear and partial polarization of the perinuclear F-actin network flow in the direction of eventual cell movement. The rear edge retracts slightly and decreases in thickness. A further increase in F-actin network flow speed at the prospective cell rear, which is caused by a further increase in perinuclear contractility, leads to phase II. The rear edge retracts, and adhesive coupling to the substrate decreases at the rear. The cell body moves forward slowly. In phase III, perinuclear actin bands transform into an actin axle. F-actin flow speed at the cell front decreases relative to the substratum, whereas the adhesive coupling to the substrate and net F-actin polymerization increases. This leads to protrusion of the front edge, and the entire cell moves rapidly and persistently. (B, left) Symmetry breaking is driven by coordinated actin–myosin contraction and requires perinuclear F-actin bands. (right) Model of actin–myosin organization in stationary keratocytes. At the periphery of the lamellipodium, myosin bipolar filaments cross-link a dendritic F-actin network without contraction. Toward the cell body, the activity of large myosin II clusters contracts and reorganizes the dendritic network to form F-actin bands and bundles. This is dependent on Rho kinase.
In unpolarized stationary keratocytes, the F-actin network flows centripetally from the cell periphery to the cell body. Based on changes in cell morphology, we defined phases of motility initiation: phase I (slow rear retraction) and phase II (fast rear retraction). These phases were associated with characteristic changes in F-actin dynamics, suggesting that morphological changes are directly mediated by reorganization of the F-actin network (Fig. 7 A). During phase I, F-actin flow increased at the prospective cell rear and reoriented in the perinuclear region such that it was biased along the eventual direction of cell movement. The flow speed increase appeared to be tightly coupled to the change in flow orientation, as we were unable to distinguish whether one event occurred before the other. The radial velocity usually decreased in the front perinuclear region during phase I, but F-actin flow was never altered in the front peripheral region. The changes in F-actin flow observed during phase I is most likely a consequence of fluctuations in actin polymerization kinetics and increased perinuclear actomyosin contraction, similar to variations that can occur in stationary cells. In fact, cells exhibiting transient F-actin dynamics similar to those in phase I did not necessarily initiate motility and often remained stationary. Phase I appeared to be necessary but not sufficient for motility initiation.
For those cells that initiated motility, phase I was followed by phase II, which was characterized by a larger increase in F-actin flow speed at the cell rear. The increase in F-actin flow speed was similar to the increase in rear edge retraction speed, suggesting that increased actomyosin contractility during phase II drives inward movement of the cell edge at the rear accompanied by a loss of adhesive coupling to the substrate.
During phase III/maturation, the cell speed increases, and the F-actin retrograde flow speed relative to the substratum at the cell front decreases, probably reflecting an increased coupling of the actin–myosin network with the substrate. The increase in cell speed observed could not be accounted for solely by the decrease in F-actin retrograde flow speed, implying that increased polymerization at the cell front also occurs during phase III. Our data define a new paradigm for actomyosin-driven symmetry breaking and motility initiation in the absence of external cues, where symmetry breaking can be initiated by contraction and the initial events occur at the prospective cell rear and cell interior.
Symmetry breaking and motility initiation require Rho kinase–dependent actomyosin contractility
The requirement for Rho kinase and myosin II activity for symmetry breaking in our data is consistent with experiments identifying a requirement for Rho, Rho kinase, and myosin light chain phosphorylation in the spontaneous polarization and migration of Walker 256 carcinosarcoma cells (Wicki and Niggli, 2001; Gutjahr et al., 2005) and in the polarization of neutrophils in response to a chemoattractant (Xu et al., 2003). In stationary keratocytes, Rho kinase and myosin II were required for perinuclear F-actin band formation and normal F-actin flow. We suggest that the perinuclear F-actin bands in stationary keratocytes assemble by dynamic network contraction (Fig. 7 B), a model proposed for the behavior of the actomyosin network in lamellipodia of motile keratocytes (Svitkina et al., 1997). The dynamic network contraction model postulates that at the periphery of the lamellipodium, myosin bipolar filaments cross-link a dendritic F-actin network without contraction. Toward the rear of the lamellipodium, network contractility is enhanced as the size of the myosin clusters increases, and the myosin clusters are able to align F-actin into bundles. In stationary keratocytes, this bundling activity is dependent on Rho kinase. We propose that during symmetry breaking, asymmetric actomyosin contraction is transformed into the morphological events of cell polarization via the consequential asymmetry of F-actin flow. According to the network contraction model, this mechanical signal from the cell center to the cell periphery triggers an autocatalytic positive feedback loop in that the more asymmetric the flow, the more effective the contraction and bundle alignment leading to further flow asymmetry.
The implication of Rho kinase in perinuclear actin dynamics but not peripheral actin dynamics is consistent with Rho kinase activity in neuronal growth cones, which is responsible for the maintenance and movement of actin arcs in the center (C domain) of the growth cone lamellipodium but not for peripheral (P domain) F-actin retrograde flow (Zhang et al., 2003). Similarly, Rho kinase has been implicated in regulating myosin activity and stress fiber formation in the interior and rear of fibroblasts (Totsukawa et al., 2000), and, in neutrophils, Rho, Rho kinase, and activated myosin are restricted to and define the cell rear (Xu et al., 2003). However, unlike keratocytes, activation of backness signals alone (e.g., through the expression of a constitutively active myosin light chain mutant or the addition of fMLP (formyl-methionyl-leucine-phenylalanine) to pertussis toxin–treated cells) in neutrophils does not lead to correct polarization and cell migration. Instead, pertussis toxin–treated neutrophils exposed to a gradient of fMLP polarize in the opposite direction to normal, with their rear closest to the fMLP source, and do not initiate movement (Xu et al., 2003). This and other results indicate that frontness is normally dominant over backness in neutrophils and that chemotactic signals at the front are required for normal polarization. Likewise, neutrophil polarization depends on PIP3 localization to the cell front (Weiner et al., 2002), whereas spontaneous keratocyte polarization is independent of PIP3. Backness signals alone in keratocytes can trigger polarization possibly because, unlike neutrophils, keratocytes are not known to respond to chemoattractants and can polarize in the absence of external chemical cues.
It remains to be determined whether the propagation of organizational information from the cell rear to the cell front during polarization also involves chemical signals besides mechanical processes. It has been proposed for neutrophils and lamellipodial fragments that the functional incompatibility of the F-actin assemblies characteristic of the front and back causes them to segregate into separate domains (Verkhovsky et al., 1999; Xu et al., 2003). Furthermore, changes in actomyosin contractility around the cell body during motility initiation could generate hydrostatic pressure, which could affect protrusion at the leading edge (Charras et al., 2005). In simpler model systems of actin-based motility, such as polystyrene beads coated with the actin nucleating proteins ActA or N-WASP, spontaneous symmetry breaking and motility initiation depend on purely mechanical effects. For small beads in which actin turnover is rapid, symmetry breaking results from cooperative interaction among a large population of polymerizing actin filaments, amplifying small stochastic events to generate large-scale polarity (van Oudenaarden and Theriot, 1999). In large beads, symmetry breaking results from the accumulation of strain in the actin gel followed by release of elastic energy via breakage of cross-links or filaments (Sekimoto et al., 2004; van der Gucht et al., 2005).
In the case of symmetry breaking of entire cells, Rho kinase and myosin II activity might be required not only to regulate local contractility but also for the large-scale coordination and amplification of local contraction fluctuations throughout the cell. Motility initiation may be a purely stochastic event, resulting from intrinsic variation in actomyosin contractility. The increased frequency of symmetry breaking under calyculin A treatment supports this hypothesis. Calyculin A, which potentiates myosin II activity, may amplify intrinsic fluctuations in actomyosin contractility so that they more often exceed the threshold at which they initiate polarization. We could induce polarization by a local increase in contractility, but whether an absolute increase in contractility or merely an imbalance in contractility is required for symmetry breaking remains an open question.
Materials and methods
Keratocyte culture and labeling
Keratocytes were cultured as described previously (Lacayo et al., 2007). To obtain individual cells, sheets of keratocytes were disaggregated by incubating in 85% PBS and 2.5 mM EGTA, pH 7.4, for 4.5–5 min. YFP Xenopus laevis myosin regulatory light chain (a gift from A.F. Straight, Stanford University, Stanford, CA) and GFP-PH-Akt (a gift from T. Meyer, Stanford University, Stanford, CA) was transfected into keratocytes as described previously (Lacayo et al., 2007).
AlexaFluor546 phalloidin (AF546-phalloidin; Invitrogen) was used to visualize F-actin dynamics in live keratocytes using FSM (Zhang et al., 2003; Vallotton et al., 2005). AF546-phalloidin was introduced into keratocytes with a small volume electroporator for adherent cells (provided by M.N. Teruel and T. Meyer, Stanford University, Stanford, CA; Teruel and Meyer, 1997; Teruel et al., 1999). 2 μM AF546-phalloidin was premixed with 7.5 μM deoxy-ATP, 7.5 μM deoxy-GTP, and 5 μM deoxy-CTP in water for ∼15 min at room temperature to prevent phalloidin aggregation. Cells were electroporated with 20 μl of the phalloidin mixture with three pulses at 150 V and were allowed to recover for ∼10 min before viewing on an inverted microscope (Diaphot-300; Nikon). Phalloidin staining of F-actin and staining of microtubules were performed as described previously (Lacayo and Theriot, 2004; Yam and Theriot, 2004).
Microscopy and image acquisition
Phalloidin was conjugated to AlexaFluor546 or tetramethylrhodamine for live cell FSM or fixed cell labeling, respectively. Myosin regulatory light chain and PH-Akt were visualized in live cells by conjugation to YFP or GFP, respectively. FITC-conjugated secondary antibodies were used to detect the primary antibody in tubulin labeling of fixed cells. Live cell imaging was performed at room temperature or at 25°C by mounting the coverslips on a temperature-controlled chamber, with the exception of temperature shift experiments (see next section) in which the temperature was shifted from 20 to 30°C. Cells were imaged in culture media ( Leibovitz's L-15 medium without phenol red supplemented with 14.2 mM Hepes, pH 7.4, 10% FBS, and 1% antibiotic-antimycotic). Time-lapse phase contrast and epifluorescent images were acquired using an inverted microscope (Diaphot-300; Nikon) with a 40× NA 1.3 oil Fluor or 60× NA 1.4 oil plan-Apo objective (Nikon). For FSM of AF546-phalloidin, images were acquired every 2 or 3 s with the 60× oil objective. A 20× NA 0.4 air phase-contrast objective was also used for some image acquisition. Images of fixed cells were collected with a microscope (Axioplan 2; Carl Zeiss MicroImaging, Inc.) using a 63× NA 1.4 oil plan-Apochromat objective (Carl Zeiss MicroImaging, Inc.). All time-lapse and fixed images were collected with a cooled back-thinned CCD camera (MicroMax 512BFT; Princeton Instruments) with a 2× optovar attached using MetaMorph software version 6 (Molecular Devices). Adjustments to brightness and/or contrast were performed with MetaMorph or Photoshop (Adobe), and pseudocolor overlays were made with Photoshop.
Pharmacological treatments
Cells were treated with 1 μg/ml nocodazole (Sigma-Aldrich), 50 μM LY294002 (Calbiochem), 40 or 100 μM blebbistatin (a gift from A.F. Straight), 10 or 25 μM Y-27632 (Calbiochem), 10 or 25 μM ML-7 (Calbiochem), or 10 or 20 nM calyculin A (Upstate Biotechnology). For FSM of drug-treated cells, the same cell was imaged before and 3–10 min after addition of the drug. For pharmacological treatments in conjunction with temperature shifts, individual stationary cells were generated by treatment of the keratocyte culture with 2.5 mM EGTA and 85% PBS and were allowed to recover in full media at 20°C for 5–8 min. Stationary cells were imaged for 4 min. The drug was added, and, 4 min later, the temperature was increased to 30°C, and the cells were imaged for a further 30 min. The effect of various pharmacological treatments on the frequency of motility initiation assayed by temperature shift was analyzed using the Chi-square test with Prism version 3.03 for Windows (GraphPad).
Local perfusion of calyculin A was performed with a glass micropipette controlled by a micromanipulator. A fluorescent marker (AlexaFluor488; Invitrogen) was added to visualize the flow from the micropipette. An overall flow throughout the live cell chamber, which was generated by a peristaltic pump connected to two needles spaced ∼1 cm from each other ∼1 mm above the coverslip, made the calyculin A gradient steeper.
Automated cell outline determination and calculation of cell boundary movement
Cell outlines were calculated using a variation of the active contours algorithm (Kass et al., 1988) derived from the gradient vector flow method of Xu and Prince (1998). The method was custom written in MATLAB 7 (The MathWorks). First, a nonlinear sigmoidal scaling of pixel intensities was applied to the image to stretch the contrast of the cell margin while reducing the contrast of edge responses inside the cell, such as those around the cell body (Fig. S1). This was followed by bandpass filtering to reduce noise and fine detail. Then, the edge map was calculated as the squared magnitude of the image gradient. A vector field oriented toward the steepest edges and zero elsewhere was generated from the gradient of the edge map. This vector field was then diffused iteratively as described previously (Xu and Prince, 1998) and normalized. After diffusion, the vector field was oriented toward the edges of interest even in relatively smooth regions of the image. An active contour representation of the cell outline was initialized by the cell outline from the previous time point. For the first frame of a time-lapse sequence, the contour was initialized manually. Subsequently, the contour was deformed iteratively by minimizing an energy function that used the edge-oriented vector field to attract the contour to the position of image edges while controlling the internal tension and curvature of the contour (Kass et al., 1988). The contour minimizing this energy function was taken as the cell outline for that image (Fig. S1).
Using these cell outlines, the movement of the cell boundary was determined according to the mechanical model described by Machacek and Danuser (2006) using software provided by M. Machacek (The Scripps Research Institute, La Jolla, CA). The cell boundary positions were translated to polar coordinates, and the scalar map function (see Scalar map function section) was used to generate continuous space-time plots of protrusion and retraction.
Tracking of F-actin network movement
An adaptive multiframe correlation technique (Ji and Danuser, 2005) was used to measure the movement of fluorescent image features from the FSM image sequences and, thus, the velocity of F-actin network flow. We implemented modifications to the technique to remove stationary background features before correlation for tracking of F-actin flow in stationary cells. Signals from these background features were very weak but contributed to the integrated correlation score because of their intensity stability and consistency. In many cases, they created a false global maximum at zero displacement (Fig. S5 A, available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1). To eliminate signal contributions from stable background features, we calculated the time-averaged intensity of each pixel in the correlation window and subtracted the average map pixel by pixel from both the original and the shifted templates before correlation. This maintained the characteristic signal variation in a template for cross-correlation while removing stationary signals. Consequently, the global correlation maximum at zero displacement was suppressed, whereas the secondary local maximum associated with the true velocity was preserved (Fig. S5 B).
For stationary cells and cells initiating motility, flow fields were calculated for a sequence of overlapping 7 or 10 frame windows (for videos acquired at 3- or 2-s intervals, respectively), giving an upper bound to the temporal resolution of ∼10 s, which is sufficient to capture the events of motility initiation occurring on a time scale of 100–200 s. The tracking algorithm automatically adjusted the correlation template size between a length of 13 and 23 pixels (1 pixel = 0.1121 μm), depending on the local image contrast. This yielded an upper bound to the spatial resolution of 1.3 μm, which is more than an order of magnitude less than the size of the cell.
Once cells entered rapid motility, they moved too quickly to still assume stationary F-actin network flow over the 7–10 frames required for the application of multiframe correlation tracking. Therefore, the videos were transformed into a cell frame of reference using a rigid body approximation of whole cell motion (Wilson and Theriot, 2006). Multiframe correlation tracking was then performed on the transformed videos using sequences of overlapping five-frame windows and a template size adjusted between 13 and 23 pixels. The flow fields generated were transformed back into the original lab frame of reference for comparison with measurements from stationary cells.
To confirm the results obtained from adaptive multiframe correlation tracking, we analyzed a subset of our stationary cell FSM data with spatio-temporal image correlation spectroscopy (STICS). STICS was performed on three consecutive nonoverlapping 20-frame time windows with partially overlapping subregions of 16 × 16 pixels as previously described (Wiseman et al., 2004; Hebert et al., 2005). STICS averaged the actin network flow over longer time scales compared with multiframe correlation tracking, producing qualitatively similar results and trends with slightly lower absolute speeds.
Analysis of displacement fields generated by adaptive multiframe correlation tracking
The displacement fields (flow vectors) obtained by adaptive multiframe correlation tracking of F-actin network flow were filtered before subsequent analysis using custom code in MATLAB 7 (The MathWorks). First, measurements made within 7 pixels of the cell margin were discarded to avoid the risk that the algorithm was measuring movement of the cell edge rather than that of the F-actin network. Then, measurements that deviated substantially from measurements in the local neighborhood were removed. Because the search templates often overlapped, a certain degree of spatial coherence in the calculated displacements was a necessary consequence. Therefore, outliers in a local neighborhood must be the result of tracking error. For each vector, a dissimilarity score was calculated that compared the measured speed and direction with the mean speed and direction of measurements within 18 pixels. A threshold was fixed at 2.5 SDs above the mean dissimilarity of vectors in a displacement field. The measurement with the highest dissimilarity was removed, the dissimilarity scores were recalculated, and the process was repeated until no dissimilarity scores exceeded the initial threshold. Flow vectors in the perinuclear region were additionally filtered by removing vectors with a speed greater than 4× the mean speed in the perinuclear region.
For analysis of flow patterns in stationary cells before and during symmetry breaking, each flow vector was transformed into polar coordinates with the center of the polar coordinate system defined as the cell center. The radial component of velocity and the centripetal deviation (i.e., the angle between the radial velocity component and the original velocity vector) were calculated (Fig. 3 A). These data were presented as continuous scalar maps of the radial flow speed and centripetal deviation. The directional coherence of the flow vectors was defined as the similarity in orientation among vectors inside 1 of 12 radial sectors defined over the lamellipodium. For each sector, the flow vectors were normalized to unit speed and averaged. The magnitude of the mean vector was taken as the coherence score for the sector. The maximum coherence score was one (all vectors in the sector aligned along the same direction).
Scalar map function
For continuous space-time plots of protrusion and retraction and scalar field visualizations of radial velocity and centripetal deviation, it was necessary to resample data that were not measured regularly in the dimensions along which they were being plotted. A custom scalar map function implemented in MATLAB 7 (The MathWorks) was used to resample data on a regularly spaced grid. The value at each grid point was interpolated from measurements within a given radius and weighted using a Gaussian window centered at the grid point. Then, bicubic interpolation between grid points was performed to generate continuous images. Color maps were then applied to the continuous maps.
Online supplemental material
Fig. S1 illustrates the automated cell outline determination method. Fig. S2 shows that the prospective rear edge thickness decreases early during motility initiation. Fig. S3 illustrates the formation of an actin axle at the cell rear during maturation of the polarized form. Fig. S4 shows that perturbation of myosin II activity does not perturb the polarity of motile cells. Fig. S5 illustrates the removal of stationary background features in adaptive multiframe correlation tracking. Videos 1 and 2 show F-actin network movement in stationary and motile keratocytes, respectively, visualized with FSM. Videos 3 and 4 depict stationary keratocytes spontaneously breaking symmetry and initiating motility. Video 5 shows the radially symmetric F-actin flow field in a stationary keratocyte. Video 6 shows changes in the F-actin flow field in a keratocyte initiating motility. Video 7 shows a stationary keratocyte initiating motility in response to the local application of calyculin A. Video 8 illustrates the formation of an actin axle at the cell rear during the maturation of polarity. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200706012/DC1.
Supplementary Material
Acknowledgments
We are grateful to A.F. Straight for reagents. We thank M.N. Teruel and T. Meyer for the microporator and reagents and M. Machacek for providing software calculating cell protrusion. We appreciate comments on the manuscript from K. Keren and S. Yamada. We also thank S. Sivasankar for suggesting the use of nucleotides with AF546-phalloidin and D.J. Fleet and R. Szeliski for valuable feedback during development of the cell outline determination software.
P.T. Yam was supported by a Howard Hughes Medical Institute Predoctoral Fellowship, a Stanford Graduate Fellowship, and a Skye International Foundation Scholarship. C.A. Wilson and E.L. Barnhart were supported by the National Institute of General Medical Sciences. N.A. Dye was supported by a Stanford Graduate Fellowship. L. Ji and G. Danuser were funded by National Institutes of Health grants R01 GM67230 and U54 GM64346. B. Hebert was supported by a postgraduate scholarship fellowship from the Natural Sciences and Engineering Research Council of Canada. P.W. Wiseman acknowledges funding from the Natural Sciences and Engineering Research Council of Canada. J.A. Theriot was supported by the American Heart Association.
P.T. Yam's present address is Institut de recherches cliniques de Montreal, Montreal, Quebec H2W 1R7, Canada.
Abbreviations used in this paper: F-actin, filamentous actin; FSM, fluorescent speckle microscopy; MLCK, myosin light chain kinase; PH, pleckstrin homology; PI 3-kinase, phosphatidylinositol phosphate 3-kinase; STICS, spatio-temporal image correlation spectroscopy.
References
- Anderson, K.I., Y.L. Wang, and J.V. Small. 1996. Coordination of protrusion and translocation of the keratocyte involves rolling of the cell body. J. Cell Biol. 134:1209–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras, G., J. Yarrow, M. Horton, L. Mahadevan, and T. Mitchison. 2005. Non-equilibration of hydrostatic pressure in blebbing cells. Nature. 435:365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. 1979. Induction of spreading during fibroblast movement. J. Cell Biol. 81:684–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates, T.D., R.G. Watts, R. Hartman, and T.H. Howard. 1992. Relationship of F-actin distribution to development of polar shape in human polymorphonuclear neutrophils. J. Cell Biol. 117:765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, G., and D. Zicha. 1995. Dynamics of fibroblast spreading. J. Cell Sci. 108:1239–1249. [DOI] [PubMed] [Google Scholar]
- Edds, K.T. 1993. Effects of cytochalasin and colcemid on cortical flow in coelomocytes. Cell Motil. Cytoskeleton. 26:262–273. [DOI] [PubMed] [Google Scholar]
- Euteneuer, U., and M. Schliwa. 1984. Persistent, directional motility of cells and cytoplasmic fragments in the absence of microtubules. Nature. 310:58–61. [DOI] [PubMed] [Google Scholar]
- Fedier, A., P. Eggli, and H.U. Keller. 1999. Redistribution of surface-bound con A is quantitatively related to the movement of cells developing polarity. Cell Motil. Cytoskeleton. 44:44–57. [DOI] [PubMed] [Google Scholar]
- Gutjahr, M.C., J. Rossy, and V. Niggli. 2005. Role of Rho, Rac, and Rho-kinase in phosphorylation of myosin light chain, development of polarity, and spontaneous migration of Walker 256 carcinosarcoma cells. Exp. Cell Res. 308:422–438. [DOI] [PubMed] [Google Scholar]
- Hebert, B., S. Costantino, and P.W. Wiseman. 2005. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 88:3601–3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson, J.H., S.E. Kolnik, C.A. Fried, R. Nazarian, J. McGreevy, K.L. Schulberg, M. Detweiler, and V.A. Trabosh. 2003. Actin-based centripetal flow: phosphatase inhibition by calyculin-A alters flow pattern, actin organization, and actomyosin distribution. Cell Motil. Cytoskeleton. 56:252–266. [DOI] [PubMed] [Google Scholar]
- Ji, L., and G. Danuser. 2005. Tracking quasi-stationary flow of weak fluorescent signals by adaptive multi-frame correlation. J. Microsc. 220:150–167. [DOI] [PubMed] [Google Scholar]
- Jurado, C., J.R. Haserick, and J. Lee. 2005. Slipping or gripping? Fluorescent speckle microscopy in fish keratocytes reveals two different mechanisms for generating a retrograde flow of actin. Mol. Biol. Cell. 16:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, M., A. Witkin, and D. Terzopoulos. 1988. Snakes: active contour models. Int. J. Comp. Vision. 1:321–331. [Google Scholar]
- Kontos, C.D., T.P. Stauffer, W.P. Yang, J.D. York, L. Huang, M.A. Blanar, T. Meyer, and K.G. Peters. 1998. Tyrosine 1101 of Tie2 is the major site of association of p85 and is required for activation of phosphatidylinositol 3-kinase and Akt. Mol. Cell. Biol. 18:4131–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacayo, C.I., and J.A. Theriot. 2004. Listeria monocytogenes actin-based motility varies depending on subcellular location: a kinematic probe for cytoarchitecture. Mol. Biol. Cell. 15:2164–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacayo, C.I., Z. Pincus, M.M. Vanduijn, C.A. Wilson, D.A. Fletcher, F.B. Gertler, A. Mogilner, and J.A. Theriot. 2007. Emergence of large-scale cell morphology and movement from local actin filament growth dynamics. PLoS Biol. 10.1371/journal.pbio.0050233. [DOI] [PMC free article] [PubMed]
- Lee, J., A. Ishihara, J.A. Theriot, and K. Jacobson. 1993. Principles of locomotion for simple-shaped cells. Nature. 362:167–171. [DOI] [PubMed] [Google Scholar]
- Machacek, M., and G. Danuser. 2006. Morphodynamic profiling of protrusion phenotypes. Biophys. J. 90:1439–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer, A.W., N. Kabir, and P. Forscher. 2002. Filopodia and actin arcs guide the assembly and transport of two populations of microtubules with unique dynamic parameters in neuronal growth cones. J. Cell Biol. 158:139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekimoto, K., J. Prost, F. Julicher, H. Boukellal, and A. Bernheim-Grosswasser. 2004. Role of tensile stress in actin gels and a symmetry-breaking instability. Eur. Phys. J. E. Soft Matter. 13:247–259. [DOI] [PubMed] [Google Scholar]
- Servant, G., O.D. Weiner, P. Herzmark, T. Balla, J.W. Sedat, and H.R. Bourne. 2000. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 287:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight, A.F., A. Cheung, J. Limouze, I. Chen, N.J. Westwood, J.R. Sellers, and T.J. Mitchison. 2003. Dissecting temporal and spatial control of cytokinesis with a myosin II inhibitor. Science. 299:1743–1747. [DOI] [PubMed] [Google Scholar]
- Svitkina, T.M., A.B. Verkhovsky, K.M. McQuade, and G.G. Borisy. 1997. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J. Cell Biol. 139:397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons, M.H., and T.J. Mitchison. 1991. Control of actin polymerization in live and permeabilized fibroblasts. J. Cell Biol. 114:503–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruel, M.N., and T. Meyer. 1997. Electroporation-induced formation of individual calcium entry sites in the cell body and processes of adherent cells. Biophys. J. 73:1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruel, M.N., T.A. Blanpied, K. Shen, G.J. Augustine, and T. Meyer. 1999. A versatile microporation technique for the transfection of cultured CNS neurons. J. Neurosci. Methods. 93:37–48. [DOI] [PubMed] [Google Scholar]
- Theriot, J.A., and T.J. Mitchison. 1991. Actin microfilament dynamics in locomoting cells. Nature. 352:126–131. [DOI] [PubMed] [Google Scholar]
- Totsukawa, G., Y. Yamakita, S. Yamashiro, D.J. Hartshorne, Y. Sasaki, and F. Matsumura. 2000. Distinct roles of ROCK (Rho-kinase) and MLCK in spatial regulation of MLC phosphorylation for assembly of stress fibers and focal adhesions in 3T3 fibroblasts. J. Cell Biol. 150:797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallotton, P., S.L. Gupton, C.M. Waterman-Storer, and G. Danuser. 2004. Simultaneous mapping of filamentous actin flow and turnover in migrating cells by quantitative fluorescent speckle microscopy. Proc. Natl. Acad. Sci. USA. 101:9660–9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallotton, P., G. Danuser, S. Bohnet, J.-J. Meister, and A.B. Verkhovsky. 2005. Tracking retrograde flow in keratocytes: news from the front. Mol. Biol. Cell. 16:1223–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Gucht, J., E. Paluch, J. Plastino, and C. Sykes. 2005. Stress release drives symmetry breaking for actin-based movement. Proc. Natl. Acad. Sci. USA. 102:7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oudenaarden, A., and J.A. Theriot. 1999. Cooperative symmetry-breaking by actin polymerization in a model for cell motility. Nat. Cell Biol. 1:493–499. [DOI] [PubMed] [Google Scholar]
- Verkhovsky, A.B., T.M. Svitkina, and G.G. Borisy. 1999. Self-polarization and directional motility of cytoplasm. Curr. Biol. 9:11–20. [DOI] [PubMed] [Google Scholar]
- Watanabe, T., J. Noritake, and K. Kaibuchi. 2005. Regulation of microtubules in cell migration. Trends Cell Biol. 15:76–83. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer, C.M., R.A. Worthylake, B.P. Liu, K. Burridge, and E.D. Salmon. 1999. Microtubule growth activates Rac1 to promote lamellipodial protrusion in fibroblasts. Nat. Cell Biol. 1:45–50. [DOI] [PubMed] [Google Scholar]
- Weiner, O.D., G. Servant, M.D. Welch, T.J. Mitchison, J.W. Sedat, and H.R. Bourne. 1999. Spatial control of actin polymerization during neutrophil chemotaxis. Nat. Cell Biol. 1:75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, O.D., P.O. Neilsen, G.D. Prestwich, M.W. Kirschner, L.C. Cantley, and H.R. Bourne. 2002. A PtdInsP3- and Rho GTPase-mediated positive feedback loop regulates neutrophil polarity. Nat. Cell Biol. 4:509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki, A., and V. Niggli. 2001. The Rho/Rho-kinase and the phosphatidylinositol 3-kinase pathways are essential for spontaneous locomotion of Walker 256 carcinosarcoma cells. Int. J. Cancer. 91:763–771. [DOI] [PubMed] [Google Scholar]
- Wilson, C.A., and J.A. Theriot. 2006. A correlation-based approach to calculate rotation and translation of moving cells. IEEE Trans. Image Process. 15:1939–1951. [DOI] [PubMed] [Google Scholar]
- Wiseman, P.W., C.M. Brown, D.J. Webb, B. Hebert, N.L. Johnson, J.A. Squier, M.H. Ellisman, and A.F. Horwitz. 2004. Spatial mapping of integrin interactions and dynamics during cell migration by image correlation microscopy. J. Cell Sci. 117:5521–5534. [DOI] [PubMed] [Google Scholar]
- Xu, C., and J.L. Prince. 1998. Snakes, shapes, and gradient vector flow. IEEE Trans. Image Proc. 7:359–369. [DOI] [PubMed] [Google Scholar]
- Xu, J., F. Wang, A. Van Keymeulen, P. Herzmark, A. Straight, K. Kelly, Y. Takuwa, N. Sugimoto, T. Mitchison, and H.R. Bourne. 2003. Divergent signals and cytoskeletal assemblies regulate self-organizing polarity in neutrophils. Cell. 114:201–214. [DOI] [PubMed] [Google Scholar]
- Yam, P.T., and J.A. Theriot. 2004. Repeated cycles of rapid actin assembly and disassembly on epithelial cell phagosomes. Mol. Biol. Cell. 15:5647–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X.F., A.W. Schaefer, D.T. Burnette, V.T. Schoonderwoert, and P. Forscher. 2003. Rho-dependent contractile responses in the neuronal growth cone are independent of classical peripheral retrograde actin flow. Neuron. 40:931–944. [DOI] [PubMed] [Google Scholar]
- Zigmond, S.H., H.I. Levitsky, and B.J. Kreel. 1981. Cell polarity: an examination of its behavioral expression and its consequences for polymorphonuclear leukocyte chemotaxis. J. Cell Biol. 89:585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}