

Abstract
The voltage-dependent anion channel (VDAC) of the outer mitochondrial membrane mediates metabolic flow, Ca2+, and cell death signaling between the endoplasmic reticulum (ER) and mitochondrial networks. We demonstrate that VDAC1 is physically linked to the endoplasmic reticulum Ca2+-release channel inositol 1,4,5-trisphosphate receptor (IP3R) through the molecular chaperone glucose-regulated protein 75 (grp75). Functional interaction between the channels was shown by the recombinant expression of the ligand-binding domain of the IP3R on the ER or mitochondrial surface, which directly enhanced Ca2+ accumulation in mitochondria. Knockdown of grp75 abolished the stimulatory effect, highlighting chaperone-mediated conformational coupling between the IP3R and the mitochondrial Ca2+ uptake machinery. Because organelle Ca2+ homeostasis influences fundamentally cellular functions and death signaling, the central location of grp75 may represent an important control point of cell fate and pathogenesis.
Introduction
Mitochondria and ER of eukaryotic cells form two intertwined endomembrane networks, and their dynamic interaction controls metabolic flow, protein transport, intracellular Ca2+ signaling, and cell death (Ferri and Kroemer, 2001; Berridge et al., 2003; Szabadkai and Rizzuto, 2004; Yi et al., 2004; Brough et al., 2005; Levine and Rabouille, 2005). Mitochondrial Ca2+ uptake, via a yet to be identified Ca2+ channel of the inner mitochondrial membrane (the mitochondrial Ca2+ uniporter), regulates processes as diverse as aerobic metabolism (Hajnoczky et al., 1995), release of caspase cofactors (Pinton et al., 2001), and feedback control of neighboring ER or plasma membrane Ca2+ channels (Hajnoczky et al., 1999; Gilabert and Parekh, 2000). A corollary of the efficient mitochondrial Ca2+ uptake during IP3-induced Ca2+ release is the close apposition of ER and outer mitochondrial membranes (OMM; Mannella et al., 1998; Rizzuto et al., 1998b; Marsh et al., 2001; Frey et al., 2002). The molecular determinants of this crosstalk, however, are still largely unknown (Walter and Hajnoczky, 2005). Recently, PACS2, which is an ER-associated vesicular-sorting protein, was proposed to link the ER to mitochondria (Simmen et al., 2005). The knockdown of PACS2 led to stress-mediated uncoupling of the organelles, which was also reflected by the inhibition of Ca2+ signal transmission.
On the other side, the abundant OMM channel voltage-dependent anion channel 1 (VDAC1) was also suggested to participate in the interaction. It was shown to be present at ER–mitochondrial contacts and to mediate Ca2+ channeling to the intermembrane space from the high [Ca2+] microdomain generated by the opening of the inositol 1,4,5-trisphosphate receptor (IP3R; Gincel et al., 2001; Rapizzi et al., 2002). In addition, VDAC1 mediates metabolic flow through the OMM, forming an ATP microdomain close to the ER and sarcoplasmic reticulum Ca2+ ATPases (SERCAs; Ventura-Clapier et al., 2004; Vendelin et al., 2004), and both VDAC1 and VDAC2 take part in metabolic and apoptotic protein complexes (Cheng et al., 2003; Colombini, 2004; Lemasters and Holmuhamedov, 2006).
The transfer and assembly of components of cellular protein complexes were shown to be assisted by molecular chaperones, adding a novel function to their role in nascent protein folding (Young et al., 2003; Soti et al., 2005). Accordingly, Ca2+ binding–, heat shock–, and glucose-regulated chaperone family members are abundantly present along the Ca2+ transfer axis, linking the ER and mitochondrial networks. Well known examples are the Ca2+-binding chaperones of the ER lumen (Michalak et al., 2002), immunophilins interacting with ER Ca2+-release channels and the mitochondrial permeability transition pore (Bultynck et al., 2001; Forte and Bernardi, 2005), and several heat shock family members localized at the mitochondrial membranes, which are proposed to interact with the components of the mitochondrial permeability transition pore, such as VDAC (He and Lemasters, 2003; Gupta and Knowlton, 2005; Wadhwa et al., 2005). Still, their exact role at the ER–mitochondria interface is not well known, although recently, weak links between chaperones were proposed to stabilize signaling and organellar cellular networks (Csermely, 2004; Soti et al., 2005).
Considering the central position of VDAC at the ER–mitochondrial interface outlined in the previous paragraphs, we used VDAC1 as a starting point for protein biochemical studies, to explore molecular interactions between the ER and mitochondrial networks. We found that through the OMM-associated fraction of the glucose-regulated protein 75 (grp75) chaperone (Zahedi et al., 2006), VDAC1 interacts with the ER Ca2+-release channel IP3R. Organellar Ca2+ measurements, using targeted recombinant Ca2+ probes, confirmed functional interaction between the IP3R and the mitochondrial Ca2+ uptake machinery, which was abolished by grp75 knockdown.
Results
VDAC1, grp75, and IP3Rs are present in a macromolecular complex at the ER–mitochondria interface
We performed yeast two-hybrid screens of human liver and kidney LexA-AD–fused libraries, using rat VDAC1-LexA-DNA- BD fusion protein as bait. Among the putative interactors we found cytoskeletal elements, which were previously thought to participate in sorting of VDAC or in mitochondrial dynamics (Schwarzer et al., 2002; Varadi et al., 2004) and a group of chaperone proteins (Table I). To investigate whether the chaperones participate in mediating organelle interactions, we focused our attention on the human heat shock 70 kD protein 9B/grp75 (nt 1,456–2,089 from GenBank/EMBL/DDBJ under accession no. BC000478; aa 471–681). The yeast homologue of grp75 is part of the protein import motor associated with TIM23 in the mitochondrial matrix (Neupert and Brunner, 2002), but it was also found in the cytosol and in OMM-associated high molecular weight protein complexes (Ran et al., 2000; Danial et al., 2003; Zahedi et al., 2006). In addition, two further findings indicated that grp75 may be involved in ER–mitochondria Ca2+ transfer: first, its C-terminal domain reduced the voltage dependence and cation selectivity of VDAC1 (Schwarzer et al., 2002), and second, grp75 overexpression was shown to promote cell proliferation and protect against Ca2+-mediated cell death (Wadhwa et al., 2002a; Liu et al., 2005).
Table I.
VDAC1 interactors found by yeast two-hybrid screening
| Name | Accession number |
|---|---|
| DnaJ (Hsp40) homologue, subfamily A, member 1; DNAJA1 |
NM_001539 |
| filamin B, beta (actin-binding protein 278); FLNB | NM_001457 |
| heat shock 70-kd protein 5; HSPA5 | NM_005347 |
| heat shock 70-kd protein 9b; HSPA9B | BC024034 |
| protein phosphatase 1g (formerly 2c), magnesium-dependent, gamma isoform; PPM1G | NM_002707 |
| t complex–associated testis-expressed 1-like 1; TCTEL1 | D50663 |
| tetratricopeptide repeat domain 1; TCC1 | NM_0033114 |
| thioredoxin-like 1; TXNL1 | AF052659 |
| tubulin-specific chaperone c; TBCC | BC020170 |
| zinc finger–like protein 9; ZPR9 | AY046059 |
Yeast two-hybrid screening was carried using the pLexA system according to the protocol of Gyuris et al. (1993). For details see Supplementary materials and methods. Approximately 90% of the clones contained a sub-sequence of the ER-resident chaperone heat shock 70-kD protein 5 (HSPA5, grp-78), most probably reflecting the requirement of efficient folding of the VDAC1 protein in yeast. The results of sequencing of the remaining clones are shown in the table. One group of the putative interacting proteins were found to be cytoskeletal and signaling elements (shown in bold); another group (shown in normal) were found to be folding intermediates, presumably underlying the proper function of VDAC1.
Based on these findings, we used further biochemical approaches to investigate the role of grp75 at the ER–mitochondria contact sites. We took advantage of a previously developed method to purify a mitochondria-associated ER subfraction (mitochondria-associated membrane (MAM) fraction [Vance, 1990]). The MAM was previously shown to be enriched in lipid synthases and transferases (Vance, 2003), and it likely represents the membrane microdomain engaged in ER–mitochondrial Ca2+ transfer (Filippin et al., 2003; Szabadkai and Rizzuto, 2004; Yi et al., 2004). Indeed, immunoblot screening of the MAM fraction, purified from rat liver and HeLa cells, revealed the presence of grp75, as well as Ca2+ channels from both the OMM (VDAC1) and the ER (IP3R; Fig. 1 A). In liver cells, given the higher yield, the microsomal fraction and the different mitochondrial extracts (the crude mitochondrial pellet [Mito C], the low-density MAM, and the high-density mitochondrial fraction containing the matrix proteins [Mito P]) were separately analyzed. As expected, VDAC and grp75 are not enriched in the MAM, given that the former is highly expressed throughout the OM (because of its role in ion and metabolite diffusion) and the latter is mostly in the matrix (but the two pools show different macromolecular assembles; see following paragraph). Conversely, the IP3R, besides the microsomes, is present in the crude mitochondria and the MAM fractions and is absent in the purified mitochondria (Fig. 1 A).
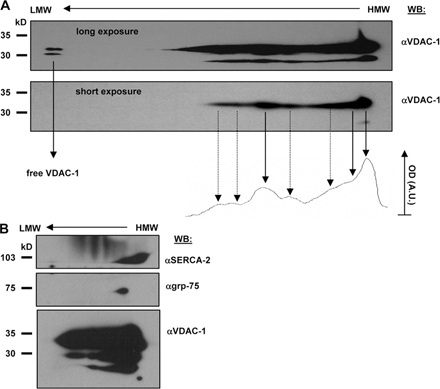
Figure 1.

IP3R, VDAC1, and grp75 colocalize on the MAM fraction. (A) Protein components of subcellular fractions prepared from rat liver and HeLa cells revealed by immunoblot analysis. Mito, mitochondria; MAM, light mitochondrial fraction; P, heavy mitochondrial fraction, enriched in matrix components; C, crude mitochondrial fraction before Percoll gradient separation. 10 μg of proteins were loaded on 10% SDS-polyacrylamide gels. The presence of IP3Rs was shown by using a non–isotype-specific monoclonal antibody. VDAC1 and grp75 were both present in the MAM, whereas it was free of contamination from inner membrane (Cox-II) and matrix (MnSOD; C) proteins. Different preparations are separated by the dotted line. Blots are representative of more than five experiments. (B) Blue-native and SDS-PAGE 2D separation of the MAM fraction (below BN) and Mito P proteins (above BN; for preparation of native subcellular fractions, see Materials and methods and A). The native fractions were solubilized and separated on an acrylamide gradient gel in the first dimension. The capillary gel was stacked over a 10% SDS-polyacrylamide gel and separated, and the proteins were immunoblotted against the IP3Rs, grp75, and VDAC1. A typical result of an immunoblot from three separate experiments is shown. (C) The MAM and Mito P fractions (50 μg of proteins) were subjected to proteinase K digestion (50 μg/ml) and the presence of grp75 and MnSOD was revealed by immunoblotting. Hyposmotic shock (50 mM mannitol, 5 mM Hepes, and 0.1 mM EGTA for 30 min at room temperature) was applied to the Mito P fraction to induce release of matrix proteins. (D–F) Coimmunoprecipitation of grp75 with IP3R and VDAC1. Total cellular proteins were used for immunoprecipitation with a polyclonal IP3R1 (D), a polyclonal VDAC (E), and a monoclonal grp75 (F) antibody, and the precipitated protein fractions were separated on 10% SDS-polyacrylamide gels and immunoblotted against IP3Rs, grp75, and VDAC1. The input homogenate fractions, the IgG controls, and the immunoprecipitates are shown.
We next investigated whether grp75 and the ER and mitochondrial Ca2+ channels are part of the macromolecular complex, and whether the grp75 pool involved is that present in the MAM fraction. For this purpose, we separately analyzed the MAM and the Mito P fractions by 2D Blue native (Fig. 1 B; first dimension, central part of the image) and SDS-PAGE protein separation. In the latter dimension, grp75, IP3R, and VDAC were revealed by immunoblotting. Although VDAC1 was present in different amounts in complexes of a wide molecular weight range, we found a specific complex characterized by the presence of VDAC, the IP3R, and grp75, suggesting their interaction in the native state. The specificity of the complex formation of VDAC, the IP3R, and grp75 was corroborated by the finding that SERCA2b showed different localization in the 2D separation (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1). In Mito P, grp75 was found in lower molecular weight complexes (<400 kD), which is similar to previous data in yeast (Dekker et al., 1997), confirming that grp75 is involved in high molecular weight protein–protein interactions only in the MAM. To confirm the different location of the two grp75 pools, we verified that whereas the matrix-localized grp75 was resistant to proteinase K digestion (similar to the matrix enzyme MnSOD; Fig. 1 C), grp75 of the MAM fraction was degraded by the enzyme, documenting its association with the cytosolic surface of mitochondria.
To further investigate the arrangement of the grp75–VDAC–IP3R complex, we used coimmunoprecipitation studies of the involved proteins. Immunoprecipitation of both IP3Rs and VDAC led to the coprecipitation of grp75 (Fig. 1, D and E, respectively), but no IP3R was found in the VDAC precipitate, and no VDAC was detectable in the IP3R precipitate. However, immunoprecipitation of grp75 led to the copurification of both VDAC and the IP3R (Fig. 1 F). These results strongly suggest that grp75 has a central role in setting up the protein complex with VDAC and the IP3R. Moreover, the interactions were detected both in the presence and absence of Mg2+-ATP (unpublished data), further suggesting the scaffolding, rather than chaperoning, function of grp75 in the complex.
Direct regulation of mitochondrial Ca2+ uptake by the IP3R ligand-binding domain
If the IP3R is in a macromolecular assembly with VDAC, we assumed that the mitochondrial Ca2+ uptake machinery might be regulated by the large cytoplasmic domain of the IP3R. This scheme was also supported by previous studies showing that the ligand-binding domain of the IP3R (aa 224–605; denoted as IP3R-LBD224-605), located on the surface of the cytoplasmic domain, participates in intramolecular interactions with other IP3R domains (Boehning and Joseph, 2000), as well as in linking the receptor with other protein partners (Bosanac et al., 2004). To assess a direct role of the IP3R in mitochondrial Ca2+ uptake, we coexpressed in HeLa cells mRFP1-tagged IP3R-LBD224-605 with cytosolic (cytAEQ) or mitochondrially targeted (mtAEQmut) aequorin-based Ca2+ probes, and evaluated global and organellar Ca2+ responses to agonist stimulation. After reconstitution with the aequorin cofactor coelenterazine, cells were challenged with histamine (in incremental doses from 1 to 100 μM), and luminescence was measured and converted to [Ca2+]. Recombinant expression of the IP3R-LBD224-605 caused a marked increase in mitochondrial Ca2+ uptake at each agonist concentration applied, in spite of reduced cytoplasmic Ca2+ response ([Ca2+]c), because of IP3 buffering and consequent reduction of IP3-induced Ca2+ release from the ER (see Fig. 2 [A and B] and Fig. S2 [available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1] for the lower agonist concentrations). The effect of the IP3R-LBD224-605 was presumably exerted on the OMM because targeting the IP3R-LBD224-605 to the OMM surface (by fusing to an N-terminal AKAP1 domain) augmented its stimulatory effect (see Fig. 3 A for intracellular localization of the mRFP1-tagged construct and Fig. 2 B for the effect on [Ca2+]m). Morphological imaging and mitochondrial loading with the potential-sensitive dye teramethylrhodamine methyl ester showed that the effect was not caused by changes in mitochondrial morphology (Fig. 3 A) or to the modification of mitochondrial membrane potential (not depicted).
Figure 2.

Effect of the IP3R ligand-binding domain on mitochondrial Ca2+ uptake. (A and C) HeLa cells were transfected with mitochondrially targeted (mtAEQmut; top) and cytosolic aequorin (bottom). Control traces are shown in black; traces from cells cotransfected with the IP3R-LBD224-605 (A) and the IP3R-LBD224-605 K508 mutant (C) are shown in gray. Traces are representative of >15 experiments from >5 preparations. (B) Effect of the cytosolic-, OMM-, and ER-targeted IP3R-LBD224-605 on peak mitochondrial and cytosolic Ca2+ responses (top and bottom, respectively). (D) Effect of the OMM-IP3R-LBD224-605 (K508A), the IP3-binding PH domain of the p130 PLC-like protein (OMM-p130-PH), and the OMM targeted N-terminal (1-604 aa) part of the IP3R (OMM-IP3R-LBD1-604), on mitochondrial (top) and cytoplasmic Ca2+ responses (bottom) after 100 μM histamine stimulation. Data in B and D were normalized to mean of the control group. Mean ± SEM of variation is shown as percentage. Cells were transfected, and [Ca2+] was measured as described in Materials and methods. Values are shown. *, P < 0.05; **, P < 0.01. For absolute values see Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1.
Figure 3.

Intracellular localization of OMM- and ER-targeted IP3R-LBD224-605. Cells were transfected with OMM-IP3R-LBD224-605-mRFP1 (A) or ER-IP3R-LBD224-605-mRFP1 (B) and loaded with the mitochondrial dye MitoTracker Green. Images on the left show mitochondrial structure, middle images show images of IP3R-LBD224-605-mRFP1 fluorescence, and images on the right show colocalization of the green and red signals. Insets show magnified images of the mitochondrial and ER networks. Bars: (A and B) 10 μm; (insets) 2 μm.
To confirm that activation of mitochondrial Ca2+ uptake can be exerted from the original site of the IP3R (i.e., from the ER membrane), we expressed IP3R-LBD224-605 fused to a C-terminal ER-targeting sequence derived from the yeast UBC6 protein (denoted as ER-IP3R-LBD224-605; Varnai et al., 2005). Expression of this construct reduced the steady-state ER [Ca2+] ([Ca2+]er) and IP3-induced Ca2+ release (Fig. S2 and Fig. 2 B, respectively), which were probably caused by direct activation of the IP3R, as previously reported for COS-7 cells (Varnai et al., 2005), although store depletion was incomplete in HeLa cells at the expression levels of this study. Still, most importantly, expression of the ER-targeted IP3R-LBD224-605 augmented mitochondrial Ca2+ accumulation after cellular stimulation by histamine, similar to what was observed upon expression of the OMM-targeted IP3R-LBD domain (Fig. 3 B shows the intracellular localization of ER-IP3R-LBD224-605; Fig. 2 B shows the stimulatory effect of ER-IP3R-LBD224-605 on [Ca2+]m). These results strongly suggested that the IP3R, acting from the ER surface, regulates mitochondrial Ca2+ uptake at an OMM site, independent of its Ca2+-channeling function.
Stimulation of mitochondrial Ca2+ uptake by the IP3R-LBD is the result of specific protein interactions at the ER–OMM interface
Based on our conclusions, we further investigated whether the effect of the N-terminal cytosolic domain of the IP3R reflects specific protein–protein interactions at the ER–mitochondrial contacts. We first verified that the effect of IP3R-LBD224-605 on mitochondrial Ca2+ uptake is independent of IP3 buffering. For this purpose, we used a point-mutated (K508A) IP3R-LBD224-605, which is unable to bind IP3. The K508 mutant increased the [Ca2+]m rise in a manner similar to the wild-type (although slightly less efficient), but, as expected, did not modify the [Ca2+]c response (Fig. 2, C and D). The capacity of an IP3-insensitive IP3R-LBD224-605 to enhance mitochondrial Ca2+ uptake was also confirmed in digitonin-permeabilized HeLa cells. In this case, mitochondrial Ca2+ uptake is exclusively dictated by the perfused [Ca2+], and it is totally independent of IP3R activity. In permeabilized cells, Ca2+ uptake was triggered by the perfusion of an intracellular buffer containing Ca2+ buffered at 1 μM. Under those conditions, in which protein interactions might have been affected by the application of digitonin, both the wild-type and the K508A OMM-IP3R-LBD224-605 increased the rate of mitochondrial Ca2+ uptake, although also in this case the wild-type was more efficient (14.71 ± 4.66% increase, n = 25, P < 0.01 vs. 6.58 ± 4.23% increase, n = 25, P > 0.05).
The notion that IP3 binding cannot account for the mitochondrial effect was further confirmed by the demonstration that a structurally unrelated IP3-binding protein domain, the PH domain of the PLC-like protein p130 (p130PH; Lin et al., 2005), targeted to the OMM, reduced both the [Ca2+]m and [Ca2+]c responses (Fig. 2 D). Interestingly, the reduction of the [Ca2+]m response was more pronounced for the OMM-targeted PH domain than for the untargeted cytosolic version of the IP3 buffer, although the two proteins were equally effective on [Ca2+]c. These data (Fig. S3, available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1) further stress the strict dependence of mitochondrial Ca2+ homeostasis on the ER– mitochondrial contacts, and thus on the Ca2+ release occurring in these microdomains.
The IP3R-LBD224-605 was also shown to play an important role in the regulation of IP3R channel activity by interacting with the N-terminal repressor domain (aa 1–223; Boehning and Joseph, 2000; Varnai et al., 2005). Still, expressing the entire N-terminal surface domain of the IP3R, targeted to the exterior of the OMM (OMM-IP3R1-604), augmented mitochondrial Ca2+ uptake (Fig. 2 D). These results exclude that the stimulatory effect of the IP3R-LBD224-605 was exerted through unmasking this intramolecular interaction in the endogenous IP3R; instead, they support a model in which the entire N-terminal IP3R exerts direct activation on the mitochondrial Ca2+ uptake machinery.
Finally, we investigated the regulatory activity on mitochondria of the IP3R-LBD224-605 when the [Ca2+]c rise is elicited in the cell by the opening of plasma membrane channels. Under those conditions, not only the [Ca2+]c rise is IP3R-independent, but the [Ca2+]c and ensuing [Ca2+]m increases are markedly slower and smaller than upon ER Ca2+ release. We thus measured [Ca2+]m after emptying the ER Ca2+ pool with the SERCA blocker tert-butyl-benzohydroquinone (tBHQ) in Ca2+-free medium and re-adding CaCl2. This protocol induces capacitative Ca2+ entry, causing a [Ca2+]c rise and subsequent mitochondrial Ca2+ uptake. As presented in Fig. 4, IP3R-LBD224-605–expressing cells showed an ∼60% increase in the influx-dependent [Ca2+]m response (top), even if the [Ca2+]c rise remained unaltered (bottom). This increase in [Ca2+]m was almost doubled, as compared with the effect after histamine-/IP3-induced Ca2+ release from the ER (Fig. 2 B). Thus, we concluded that local IP3 buffering masks the stimulatory effect of the IP3R-LBD224-605 upon ER Ca2+ release, and, indeed, the effect of the IP3R-LBD is established at the ER–mitochondrial contacts.
Figure 4.

The effect of IP3R-LBD224-605 on mitochondrial Ca2+ uptake after capacitative Ca2+ influx. [Ca2+]m and [Ca2+]c (top and bottom, respectively, in A and B) were measured in HeLa cells and transfected with mtAEQmut and cytAEQ, respectively. After ER depletion in Ca2+-free medium (100 μM KRB-EGTA; 4 min), Ca2+ influx was induced by the re-addition of 2 mM CaCl2 to the extracellular medium. (A) Representative traces of control (black traces) and OMM-IP3R-LBD224-605–cotransfected cells (gray traces) are shown. ([Ca2+]m peak in controls, 12.1 ± 2.11 μM; [Ca2+]m peak in OMM-IP3R-LBD224-605–expressing cells, 21.2 ± 4.00 μM; P = 0.05; [Ca2+]c peak in controls, 0.96 ± 0.04 μM; [Ca2+]c peak in OMM-IP3R-LBD224-605–expressing cells, 1.04 ± 0.03 μM). In B, data normalized to the mean ± the SEM of the control group are shown as percentages. For absolute values see Table S1. **, P < 0.01.
Down-regulation of grp75 abolishes the functional coupling between the IP3R and mitochondria
Because our proteomic studies suggested that the interaction of the VDAC and IP3R channels is mediated by grp75, we investigated whether the stimulatory effect of the OMM-targeted IP3R-LBD224-605 on mitochondrial Ca2+ uptake requires the presence of grp75. A first series of experiments showed that strong inhibition of grp75 expression (48 h after transfection) in itself strongly reduced mitochondrial Ca2+ uptake, most probably because of alterations of mitochondrial function through inhibition of protein import and Δψm loss (unpublished data). Thus, we opted for a lower silencing efficiency by conducting experiments 24 h after transfection (Fig. 5, inset). We expressed control and grp75 siRNAs in HeLa cells, cotransfecting them with the IP3R-LBD224-605 construct and mtAEQmut. Under those conditions, grp75 siRNA had no effect on the [Ca2+]m response to histamine stimulation (Fig. 5, A and B). However, the down-regulation of grp75 prevented the stimulatory effect on mitochondrial Ca2+ uptake of the IP3R-LBD224-605, which was expressed both on the OMM and the ER surface (Fig. 5 B). Thus, we concluded that grp75 is not only physically associated with the IP3R–VDAC1 complex, but is also necessary for functional coupling between these proteins. These results also show that although moderate knockdown of grp75 does not interfere with its function in the mitochondrial matrix, in accordance with previous results on mitochondrial protein import (Sanjuan Szklarz et al., 2005), the low amount of grp75 at the ER–mitochondrial contacts is a limiting factor for the stimulatory effect of the IP3R-LBD.
Figure 5.

Coupling of the ER and mitochondrial Ca2+ channels depends on the presence of grp75. Mitochondrial Ca2+ uptake was measured in control siRNA–transfected HeLa cells (control); after siRNA-driven down-regulation of grp75 (siRNA-grp75); control siRNA and OMM-IP3R-LBD224-605–transfected cells; and siRNA-grp75 and OMM-IP3R-LBD224-605 cotransfected cells. Cells were also cotransfected with the mtAEQmut probe and mitochondrial Ca2+ response to 100 μM histamine was measured as described in the Materials and methods. Inset shows the effect of grp75 siRNA on grp75 levels after 24 h of transfection. Controls transfected only with Lipofectamine showed no difference in respect to control siRNA (not depicted). (B) Silencing of grp75 reverts the stimulatory effect of IP3R-LBD224-605 targeted both to the OMM and ER surface. The percent increase of [Ca2+]m peaks normalized to the mean of controls are shown in cells cotransfected with mtAEQmut and control siRNA (siRNA-grp75) and OMM-IP3R-LBD224-605 or ER-IP3R-LBD224-605 after stimulation with 100 μM histamine. The stimulatory effect of both the OMM- and ER-targeted IP3R-LBD224-605 was inhibited after the cotransfection with siRNA-grp75 (+), whereas the control Ca2+ peaks remained unaffected. Data normalized to the mean ± the SEM of the control group are shown in percentages. For absolute values see Table S1. *, P < 0.05; **, P < 0.01.
In the final set of experiments, we further investigated the role of grp75 in mitochondrial Ca2+ uptake regulation by overexpressing the protein. Most likely caused by its differentially localized pools, grp75 appeared to modify mitochondrial Ca2+ uptake after IP3-induced Ca2+ release through diverse mechanisms. Indeed, as shown in Fig. 6 (A and B), overexpression of the wild-type protein led to reduced histamine-induced [Ca2+]m response. However, at the same time, it also significantly decreased the steady-state [Ca2+]er level (Fig. 6 B, right), thus, reducing the driving force for IP3-induced Ca2+ release, which in turn might be responsible for the dampened mitochondrial Ca2+ accumulation. This parallel reduction of [Ca2+]er and [Ca2+]m may reflect two different effects of grp75: (1) OMM-localized grp75, presumably through the interaction with the IP3R or other members of the ER Ca2+-handling machinery, may increase the Ca2+ leak from the ER through the IP3R, as previously shown for Bcl-2 (Pinton et al., 2000; Bassik et al., 2004); (2) matrix-localized grp75 may modify mitochondrial parameters (e.g., pH) or import of Ca2+-handling proteins, leading to altered mitochondrial Ca2+ uptake, as well as the ATP supply for ER Ca2+ accumulation through the SERCA pumps. To dissect these effects, we again used the approach of measuring IP3-independent mitochondrial Ca2+uptake after capacitative Ca2+ influx. In addition, to distinguish OMM-based effects from those in the mitochondrial matrix, we expressed a truncated grp75 lacking the N-terminal 51-aa mitochondrial-targeting sequence, and thus unable to enter the mitochondrial matrix. Ca2+ influx was induced by depleting the ER Ca2+ store with tBHQ in the absence of extracellular Ca2+, as described in the previous section (Fig. 4). This “cytosolic” form of grp75 (grp75cyt) did not change the bulk cytosolic [Ca2+] response to readdition of Ca2+ in the extracellular medium, but significantly increased mitochondrial Ca2+ accumulation (Fig. 6, C and D). Moreover, grp75cyt further potentiated the stimulatory effect of IP3R-LBD224-605 (Fig. 6, C and D), confirming the results obtained with siRNA grp75 and proving that the amount of grp75 present at the OMM in the VDAC–grp75–IP3R complex is a limiting factor of the positive effect of the IP3R-LBD on mitochondrial Ca2+ uptake. Lastly, by coexpressing grp75cyt and IP3R-LBD224-605, we achieved a very high stimulation of mitochondrial Ca2+ uptake rate during capacitative Ca2+influx (i.e., upon the increase of bulk [Ca2+]c to ∼1 μM). Thus, we concluded that the VDAC–grp75–IP3R complex renders mitochondria more sensitive at low extramitochondrial [Ca2+], as compared with higher local [Ca2+]c increases during IP3-induced Ca2+ release (compare the effect of IP3R-LBD224-605 on [Ca2+]m in Fig. 2 and Fig. 4 or Fig. 6). Indeed, by overexpression of grp75cyt we could not observe a significant increase in histamine-induced [Ca2+]m responses even if the steady-state [Ca2+]er remained unaltered (unpublished data).
Figure 6.

Effect of grp75 overexpression on mitochondrial Ca2+ responses and steady-state [Ca2+]er. (A and B) HeLa cells were cotransfected with mtAEQmut or erAEQmut probes (controls) and mouse grp75. [Ca2+]m was measured as described in Fig. 2, after stimulation with 100 μM histamine, as indicated in A. The percent variation (± SEM) of [Ca2+]m peaks normalized to the mean of controls are shown in B (left); the effect of grp75 on steady-state [Ca2+]er is shown on the right. Steady-state [Ca2+]er was measured after refilling of the ER in the presence of 1 mM CaCl2 in the extracellular medium (n = 10, from four separate experiments). Before measurements, erAEQmut-transfected cells were reconstituted with coelenterazine n, after ER Ca2+ depletion in a solution containing 0 [Ca2+], 600 μM EGTA, and 1 μM ionomycin, as previously described (Chiesa et al., 2001). For [Ca2+]m values see Table S1. [Ca2+]er in controls, 416 ± 19.3 μM; in grp75-overexpressing cells: 334 ± 13.6 μM; P < 0.05. *, P < 0.05. (C and D) [Ca2+]m (top) and [Ca2+]c (bottom) were measured in control and grp75cyt-expressing cells, after induction of capacitative Ca2+ influx, after ER depletion with tBHQ in Ca2+-free medium (100 μM KRB-EGTA; 4 min) and readdition of 2 mM CaCl2. Representative traces of controls, cells cotransfected with OMM-IP3R-LBD224-605, grp75cyt, or both are shown. The percent increase (± SEM) of [Ca2+]m peaks normalized to the mean of controls are shown in D. [Ca2+]m peak in controls, 9.7 ± 1.2 μM; in OMM-IP3R-LBD224-605–expressing cells, 13.0 ± 1.8 μM; grp75cyt-expressing cells, 13.2 ± 1.6 μM; OMM-IP3R-LBD224-605 /grp75cyt–expressing cells, 18.8 ± 2.48. *, P < 0.05; **, P < 0.01.
Discussion
Based on previous observations (Gincel et al., 2001; Csordas et al., 2002; Rapizzi et al., 2002; Israelson et al., 2005), we used VDAC1 as the start point for proteomic search of interacting proteins and for unraveling the molecular basis of mitochondrial Ca2+ homeostasis. An unexpected, but intriguing, finding of our biochemical studies was the central location of the chaperone grp75 in the interaction between ER and mitochondrial Ca2+ channels. grp75, a conserved chaperone, has a well studied role in protein import through the IMM. Still, in yeast mitochondria, mtHsp70/Ssc1 was shown to be significantly more abundant than the translocase (TIM23 complex). Thus, only a small fraction of the protein appears to be involved directly in preprotein translocation (Dekker et al., 1997; Sanjuan Szklarz et al., 2005), suggesting the existence of different pools of the protein. Previous work also reported extramitochondrial localization of grp75 (Ran et al., 2000), and its interaction with extramitochondrial proteins such as the cytosolic p53 or the ER luminal grp94 (Takano et al., 2001; Wadhwa et al., 2002b), although the mechanisms that control the differential sorting of the protein are still completely unknown. According to our immunofluorescence and GFP-tagging studies in HeLa cells grp75 shows complete mitochondrial localization, but obviously cannot be discriminated from an OMM-associated pool. Biochemical studies, however, demonstrate that a matrix-localized pool participates in forming complexes in the 200–400-kD range and represents the major fraction of the total mitochondrial grp75 content, whereas a minor grp75 pool resides in the low-density (MAM) mitochondrial fraction, participating in complexes in the megaDalton range and comprising OMM and ER membrane proteins. To further support an independent function of the nonmatrix pool, we constructed a grp75 mutant lacking the mitochondrial presequence, and thus incompetent for import in the matrix. This protein retained the capacity to enhance mitochondrial Ca2+ accumulation, strongly arguing for the notion that this role of grp75 is not only independent from its chaperone activity in the matrix but also depends on a physically separated protein pool.
How is the newly identified regulatory activity on mitochondrial Ca2+ uptake exerted? In principle, two different mechanisms can be envisioned. In the first, grp75 could be involved in scaffolding the ER–mitochondria contacts, and thus determines the number of sites in which mitochondria are exposed to the high [Ca2+] microdomains generated at the mouth of IP3Rs. Fluorescent labeling studies of the ER and mitochondria revealed a partial (5–20%) colocalization, reflecting these interactions. However, no increase in colocalization has been observed by overexpression of grp75 (or of the IP3R-LBD224-605; unpublished data), suggesting that they do not directly function as structural determinants of the contacts. In a second scenario, grp75 could control the interaction of ER and mitochondrial proteins at the existing organelle contacts, and thus allow cross-talk between signaling partners, e.g., the ion channels of the two membranes. Indeed, grp75, as shown by its knockdown and overexpression models, was necessary and sufficient for the stimulatory effect of the IP3R-LBD224-605 on mitochondrial Ca2+ uptake. Moreover, the proteomic data also highlight the central role of grp75 in this interaction. VDAC and IP3Rs coprecipitate with grp75, and the chaperone is coimmunoprecipitated by both anti-IP3R and -VDAC antibodies, indicating that it is the key assembling molecule in the loose interaction between the two ion channels.
Within the IP3R–grp75–VDAC complex, potentiation of mitochondrial Ca2+ accumulation by the IP3R-LBD224-605 does not require IP3 binding, as demonstrated by the fact that it is retained by the K508A mutant, which is unable to bind IP3 (Varnai et al., 2005). Although the mutant shows the same stimulatory effect (Fig. 2), one should remember that wild-type IP3R-LBD224-605, because of IP3 buffering, reduces ER Ca2+ release, and thus conclude that the wild type is somewhat more effective than the mutant. To further confirm independence from IP3 buffering, we measured mitochondrial Ca2+ uptake after capacitative influx through the plasma membrane (Figs. 4 and 6). Also, under those experimental conditions, the IP3R-LBD224-605 potently stimulated mitochondrial Ca2+ uptake.
As for the molecular mechanism of the effect on the mitochondrial Ca2+ machinery, different scenarios could be envisioned. In the first, the recombinantly expressed IP3R-LBD, both from the OMM and ER side, could interact with the endogenous IP3R itself, and modify the probability of its interaction with grp75/VDAC. Indeed, it was previously shown that intramolecular interactions between different domains of the IP3R, such as the 224–605 minimal IP3-binding domain and the 1–223 N-terminal repression domain, regulate IP3R channel opening upon IP3 binding. Thus, one could hypothesize that the high expression levels of IP3R-LBD224-605 represses an interaction between the extreme N-terminal of the endogenous receptor and grp75/VDAC. To clarify this issue, we expressed the whole (aa 1–604) IP3R-LBD, which is targeted to the OMM. The IP3R-LBD1-604 had the same effect as IP3R-LBD224-605, thus, excluding competition of these two cytoplasmic, N-terminal domains of the IP3R. In the second, simpler scenario, the IP3R-LBD224-605 mimics the effect of the endogenous IP3R. Thus, it directly enhances mitochondrial Ca2+ uptake by maximizing, within the macromolecular complex, the interaction with the mitochondrial VDAC channel. Indeed, the density of the exogenous IP3R-LBD224-605, based on fluorescence labeling (Varnai et al., 2005) and Scatchard plot analysis of IP3 binding (Wibo and Godfraind, 1994), can be assumed to be at least one order of magnitude higher than the endogenous receptor, and indeed, high expression levels were necessary for the effect of IP3R on mitochondrial Ca2+ uptake.
The central role of grp75 in the IP3R-LBD–induced augmentation of Ca2+ uptake was clearly shown by the siRNA-driven silencing of the protein, leading to the abolition of the effect. Conversely, high-level expression of grp75 induced a compound effect involving at least three different locations, as follows: (1) the ER, decreasing the steady [Ca2+]er level; (2) the OMM, interacting with VDAC, whose permeability/ion selectivity was shown to be modified by grp-75 binding (Schwarzer et al., 2002); and (3) the mitochondrial matrix, modifying mitochondrial parameters, such as pH or Ca2+ buffering capacity. Expression of the cytosolic grp75 and measurement of Ca2+ influx–induced mitochondrial Ca2+ uptake allowed us to eliminate the intramitochondrial effect and changes of ER Ca2+ handling. Importantly, mitochondrial Ca2+ uptake in this approach was markedly increased, and grp75cyt potentiated the effect of OMM-IP3R-LBD, clarifying the effect of the OMM-associated pool of grp75.
In conclusion, we demonstrated that the IP3R is part of a signaling complex that directly controls Ca2+ uptake into mitochondria. Much remains to be understood, but by these results the concept of macromolecular assembly of signaling elements, previously put forward for several plasma membrane channels, can be extended to defined microdomains at the ER–mitochondrial interface. Such an arrangement highlights novel routes for pharmacological intervention that may be used for the modulation of downstream events such as metabolism and apoptosis.
Materials and methods
Yeast two-hybrid screening
Yeast two-hybrid screening was carried out using the pLexA system according to the protocol of Gyuris et al. (1993). For details see Supplemental materials and methods (available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1).
Subcellular fractionation and proteomic analysis
HeLa cells and rat liver were homogenized, and crude mitochondrial fraction (8,000-g pellet) was subjected to separation on a 30% self-generated Percoll gradient, as previously described (Vance, 1990). A low-density band (denoted as the MAM fraction) and a high-density band (denoted as Mito P) were collected and analyzed by immunoblotting and Blue native/SDS-PAGE 2D separation, which are described in detail in the Supplemental materials and methods. Proteinase K (Sigma-Aldrich) digestion was performed with 50 μg enzyme in the presence of 50 μg proteins (10 min, on ice) in solution A used to resuspend subcellular fractions (250 mM mannitol, 5 mM Hepes, and 0.5 mM EGTA, pH 7.4). Hyposmotic shock (50 mM mannitol, 5 mM Hepes, and 0.1 mM EGTA, pH 7.4, for 30 min at room temperature) was applied to induce mitochondrial swelling.
IP3R and grp75 expression constructs
Mouse grp75, cloned into the expression vector pTOPO (Invitrogen), was provided by R. Wadhwa (University of Tokyo, Tokyo, Japan; Wadhwa et al., 1993). Full-length mouse IP3R-1 was obtained from K. Mikoshiba (RIKEN Brain Science Institute, Wako City, Saitama, Japan). The constructs encoding the fusion proteins of the PH domain of the p130 protein (from GenBank/EMBL/DDBJ under accession no.D45920; residues 95–233) and the IP3R-LBD domain (residues 224–605) of the human IP3R-1 with monomeric red fluorescent protein (mRFP1), GFP, or YFP, as well as the strategies for ER targeting, have been previously described (Lin et al., 2005; Varnai et al., 2005). For OMM tethering, the N-terminal mitochondrial localization sequence of the mouse AKAP1 protein (from GenBank/EMBL/DDBJ under accession no. V84389; residues 34–63) was fused to the N termini of the IP3R-LBD and p130PH constructs through a short linker (DPTRSR). The OMM-IP3-LBD1-604-mRFP1 construct was obtained by amplification of the 1–604 fragment of IP3R-1 cDNA and insertion into the AKAP1/mRFP1 vector. The GRP75cyt cDNA was amplified from a human liver cDNA library (Origene) using the primers 5′-CCCAAGCTTATGAAGGGAGCAGTTGTTGGTATTG-3′ and 5′-CGCGGATCCTTACTGTTTTTCCTCCTTTTGATC-3′. After digestion with HindIII and BamHI, the product was ligated into the pcDNA3 plasmid (Invitrogen) digested with the same restriction enzymes. The construct was verified with bidirectional sequencing.
Transient transfection was done by the Ca2+-phosphate precipitation technique. Experiments were performed 24–36 h after transfection.
Dynamic in vivo [Ca2+] measurements with targeted aequorin probes
cytAEQ-, mtAEQmut-, or erAEQmut-expressing cells were reconstituted with coelenterazine and transferred to the perfusion chamber, and light signal was collected in a purpose-built luminometer and calibrated into [Ca2+] values, as previously described (Chiesa et al., 2001). All aequorin measurements were performed in Krebs-Ringer bicarbonate (KRB) containing 1 mM CaCl2 (KRB/Ca2+; Krebs-Ringer modified buffer: 135 mM NaCl, 5 mM KCl, 1 mM MgSO4, 0.4 mM K2HPO4, 1 mM CaCl2, 5.5 mM glucose, and 20 mM Hepes, pH 7.4). [Ca2+]c after capacitative Ca2+ influx was measured by preincubating HeLa cells with the SERCA blocker tBHQ (100 μM) in a KRB solution containing no Ca2+ and 100 μM EGTA. Cytoplasmic Ca2+ signal and mitochondrial Ca2+ uptake were evoked by adding 2 mM CaCl2 to the medium. For [Ca2+]er measurements, erAEQmut-transfected cells were reconstituted with coelenterazine n, after ER Ca2+ depletion in a solution containing 0 [Ca2+], 600 μM EGTA, and 1 μM ionomycin, as previously described (Szabadkai et al., 2004). Experiments in permeabilized HeLa cells were performed as previously described (Rapizzi et al., 2002), except that 25 μM digitonin was used to preserve ER–mitochondrial contacts.
Imaging techniques
For 3D morphological image acquisition, the cells were transfected with mRFP1-fused IP3R-LBD224-605 constructs and loaded with 50 nM MitoTracker Green (Invitrogen) for 20 min at 37°C. For morphological studies, cells were placed in a thermostatted chamber at 37°C in KRB/Ca2+ solution and imaged using an inverted microscope (Axiovert 200M; Carl Zeiss MicroImaging, Inc.) using a 63×/1.4 Plan-Apochromat objective, a CoolSNAP HQ interline charge-coupled device camera (Roper Scientific) and the MetaMorph 5.0 software (Universal Imaging Corp.). Z-series images were deconvolved using the PSF-based Exhaustive Photon Reassignment deconvolution software (Carrington et al., 1995; Rizzuto et al., 1998a), running on a Linux-based PC. For colocalization analysis, thresholded images were 3D rendered using the Data Analysis and Visualization Environment software (Lifshitz, 1998; Rapizzi et al., 2002). To approximate real colocalization, and to exclude artificial ones produced by the noise of the signal, only the voxels with <50% difference in their normalized intensity were taken into account.
Online supplemental material
Table S1 shows [Ca2+]m and [Ca2+]c responses of HeLa cells expressing the constructs in this study. Fig. S1 shows the proteomic analysis of molecular components of the MAM fraction. Fig. S2 shows the effects of IP3R-LBD224-605 on cytoplasmic Ca2+ responses and ER Ca2+ homeostasis. Fig. S3 shows the effect of cytosolic- and OMM-targeted p130-PH domain on mitochondrial Ca2+ uptake. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200608073/DC1.
Supplementary Material
Acknowledgments
We thank Drs. R. Wadhwa and P. Csermely for helpful discussions and M. Negrini and C. Schwienbacher for help with yeast two-hybrid studies.
This work was supported by grants from Telethon-Italy, the Italian Association for Cancer Research, the Italian University Ministry (Programmi di Ricerca di Rilevante Interesse Nazionale, Italian Investment Fund for Basic Research, and local research grants), the Emilia-Romagna Programma Regionale per la Ricerca Industriale, l'Innovazione e il Trasferimento Tecnologico program, the Ferrara Objective 2 funds, and the Italian Space Agency to R. Rizzuto. P. Várnai was supported by the Hungarian Scientific Research fund, the Medical Research Council, and the Hungarian National Committee for Technological Development. M.R. Wieckowski was a recipient of a European Molecular Biology Laboratory short-term fellowship. Part of the work by G. Szabadkai was supported by a Marie-Curie individual fellowship.
G. Szabadkai, K. Bianchi, and P. Várnai contributed equally to this paper.
Abbreviations used in this paper: grp, glucose-regulated protein; IP3, inositol 1,4,5-trisphosphate; IP3R, IP3 receptor; KRB, Krebs-Ringer bicarbonate; MAM, mitochondria-associated membrane; OMM, outer mitochondrial membrane; SERCA, sarcoplasmic reticulum Ca2+ ATPase; tBHQ, tert-butyl-benzohydroquinone; VDAC, voltage-dependent anion channel.
References
- Bassik, M.C., L. Scorrano, S.A. Oakes, T. Pozzan, and S.J. Korsmeyer. 2004. Phosphorylation of BCL-2 regulates ER Ca(2+) homeostasis and apoptosis. EMBO J. 23:1207–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge, M.J., M.D. Bootman, and H.L. Roderick. 2003. Calcium signalling: dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 4:517–529. [DOI] [PubMed] [Google Scholar]
- Boehning, D., and S.K. Joseph. 2000. Direct association of ligand-binding and pore domains in homo- and heterotetrameric inositol 1,4,5-trisphosphate receptors. EMBO J. 19:5450–5459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosanac, I., T. Michikawa, and K. Mikoshiba. 2004. Structural insights into the regulatory mechanism of IP3 receptor. Biochim. Biophys. Acta. 1742:89–102. [DOI] [PubMed] [Google Scholar]
- Brough, D., M.J. Schell, and R.F. Irvine. 2005. Agonist-induced regulation of mitochondrial and endoplasmic reticulum motility. Biochem. J. 392:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultynck, G., P. de Smet, D. Rossi, G. Callewaert, L. Missiaen, V. Sorrentino, H. De Smedt, and J.B. Parys. 2001. Characterization and mapping of the 12 kDa FK506-binding protein (FKBP12)-binding site on different isoforms of the ryanodine receptor and of the inositol 1,4,5-trisphosphate receptor. Biochem. J. 354:413–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrington, W.A., R.M. Lynch, E.D. Moore, G. Isenberg, K.E. Fogarty, and F.S. Fay. 1995. Superresolution three-dimensional images of fluorescence in cells with minimal light exposure. Science. 268:1483–1487. [DOI] [PubMed] [Google Scholar]
- Cheng, E.H., T.V. Sheiko, J.K. Fisher, W.J. Craigen, and S.J. Korsmeyer. 2003. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 301:513–517. [DOI] [PubMed] [Google Scholar]
- Chiesa, A., E. Rapizzi, V. Tosello, P. Pinton, M. de Virgilio, K.E. Fogarty, and R. Rizzuto. 2001. Recombinant aequorin and green fluorescent protein as valuable tools in the study of cell signalling. Biochem. J. 355:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini, M. 2004. VDAC: the channel at the interface between mitochondria and the cytosol. Mol. Cell. Biochem. 256-257:107–115. [DOI] [PubMed] [Google Scholar]
- Csermely, P. 2004. Strong links are important, but weak links stabilize them. Trends Biochem. Sci. 29:331–334. [DOI] [PubMed] [Google Scholar]
- Csordas, G., M. Madesh, B. Antonsson, and G. Hajnoczky. 2002. tcBid promotes Ca(2+) signal propagation to the mitochondria: control of Ca(2+) permeation through the outer mitochondrial membrane. EMBO J. 21:2198–2206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danial, N.N., C.F. Gramm, L. Scorrano, C.Y. Zhang, S. Krauss, A.M. Ranger, S.R. Datta, M.E. Greenberg, L.J. Licklider, B.B. Lowell, et al. 2003. BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature. 424:952–956. [DOI] [PubMed] [Google Scholar]
- Dekker, P.J., F. Martin, A.C. Maarse, U. Bomer, H. Muller, B. Guiard, M. Meijer, J. Rassow, and N. Pfanner. 1997. The Tim core complex defines the number of mitochondrial translocation contact sites and can hold arrested preproteins in the absence of matrix Hsp70-Tim44. EMBO J. 16:5408–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri, K.F., and G. Kroemer. 2001. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 3:E255–E263. [DOI] [PubMed] [Google Scholar]
- Filippin, L., P.J. Magalhaes, G. Di Benedetto, M. Colella, and T. Pozzan. 2003. Stable interactions between mitochondria and endoplasmic reticulum allow rapid accumulation of calcium in a subpopulation of mitochondria. J. Biol. Chem. 278:39224–39234. [DOI] [PubMed] [Google Scholar]
- Forte, M., and P. Bernardi. 2005. Genetic dissection of the permeability transition pore. J. Bioenerg. Biomembr. 37:121–128. [DOI] [PubMed] [Google Scholar]
- Frey, T.G., C.W. Renken, and G.A. Perkins. 2002. Insight into mitochondrial structure and function from electron tomography. Biochim. Biophys. Acta. 1555:196–203. [DOI] [PubMed] [Google Scholar]
- Gilabert, J.A., and A.B. Parekh. 2000. Respiring mitochondria determine the pattern of activation and inactivation of the store-operated Ca(2+) current I(CRAC). EMBO J. 19:6401–6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gincel, D., H. Zaid, and V. Shoshan-Barmatz. 2001. Calcium binding and translocation by the voltage-dependent anion channel: a possible regulatory mechanism in mitochondrial function. Biochem. J. 358:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, S., and A.A. Knowlton. 2005. HSP60, Bax, apoptosis and the heart. J. Cell. Mol. Med. 9:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyuris, J., E. Golemis, H. Chertkov, and R. Brent. 1993. Cdi1, a human G1 and S phase protein phosphatase that associates with Cdk2. Cell. 75:791–803. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., R. Hager, and A.P. Thomas. 1999. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J. Biol. Chem. 274:14157–14162. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., L.D. Robb-Gaspers, M.B. Seitz, and A.P. Thomas. 1995. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 82:415–424. [DOI] [PubMed] [Google Scholar]
- He, L., and J.J. Lemasters. 2003. Heat shock suppresses the permeability transition in rat liver mitochondria. J. Biol. Chem. 278:16755–16760. [DOI] [PubMed] [Google Scholar]
- Israelson, A., L. Arzoine, S. Abu-Hamad, V. Khodorkovsky, and V. Shoshan-Barmatz. 2005. A photoactivable probe for calcium binding proteins. Chem. Biol. 12:1169–1178. [DOI] [PubMed] [Google Scholar]
- Lemasters, J.J., and E. Holmuhamedov. 2006. Voltage-dependent anion channel (VDAC) as mitochondrial governator-Thinking outside the box. Biochim. Biophys. Acta. 1762:181–190. [DOI] [PubMed] [Google Scholar]
- Levine, T., and C. Rabouille. 2005. Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr. Opin. Cell Biol. 17:362–368. [DOI] [PubMed] [Google Scholar]
- Lifshitz, L.M. 1998. Determining data independence on a digitized membrane in three dimensions. IEEE Trans. Med. Imaging. 17:299–303. [DOI] [PubMed] [Google Scholar]
- Lin, X., P. Varnai, G. Csordas, A. Balla, T. Nagai, A. Miyawaki, T. Balla, and G. Hajnoczky. 2005. Control of calcium signal propagation to the mitochondria by inositol 1,4,5-trisphosphate-binding proteins. J. Biol. Chem. 280:12820–12832. [DOI] [PubMed] [Google Scholar]
- Liu, Y., W. Liu, X.D. Song, and J. Zuo. 2005. Effect of GRP75/mthsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 268:45–51. [DOI] [PubMed] [Google Scholar]
- Mannella, C.A., K. Buttle, B.K. Rath, and M. Marko. 1998. Electron microscopic tomography of rat-liver mitochondria and their interaction with the endoplasmic reticulum. Biofactors. 8:225–228. [DOI] [PubMed] [Google Scholar]
- Marsh, B.J., D.N. Mastronarde, K.F. Buttle, K.E. Howell, and J.R. McIntosh. 2001. Organellar relationships in the Golgi region of the pancreatic beta cell line, HIT-T15, visualized by high resolution electron tomography. Proc. Natl. Acad. Sci. USA. 98:2399–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak, M., J.M. Robert Parker, and M. Opas. 2002. Ca2+ signaling and calcium binding chaperones of the endoplasmic reticulum. Cell Calcium. 32:269–278. [DOI] [PubMed] [Google Scholar]
- Neupert, W., and M. Brunner. 2002. The protein import motor of mitochondria. Nat. Rev. Mol. Cell Biol. 3:555–565. [DOI] [PubMed] [Google Scholar]
- Pinton, P., D. Ferrari, P. Magalhaes, K. Schulze-Osthoff, F. Di Virgilio, T. Pozzan, and R. Rizzuto. 2000. Reduced loading of intracellular Ca(2+) stores and downregulation of capacitative Ca(2+) influx in Bcl-2-overexpressing cells. J. Cell Biol. 148:857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinton, P., D. Ferrari, E. Rapizzi, F.D. Di Virgilio, T. Pozzan, and R. Rizzuto. 2001. The Ca2+ concentration of the endoplasmic reticulum is a key determinant of ceramide-induced apoptosis: significance for the molecular mechanism of Bcl-2 action. EMBO J. 20:2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran, Q., R. Wadhwa, R. Kawai, S.C. Kaul, R.N. Sifers, R.J. Bick, J.R. Smith, and O.M. Pereira-Smith. 2000. Extramitochondrial localization of mortalin/mthsp70/PBP74/GRP75. Biochem. Biophys. Res. Commun. 275:174–179. [DOI] [PubMed] [Google Scholar]
- Rapizzi, E., P. Pinton, G. Szabadkai, M.R. Wieckowski, G. Vandecasteele, G. Baird, R.A. Tuft, K.E. Fogarty, and R. Rizzuto. 2002. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 159:613–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto, R., W. Carrington, and R.A. Tuft. 1998. a. Digital imaging microscopy of living cells. Trends Cell Biol. 8:288–292. [DOI] [PubMed] [Google Scholar]
- Rizzuto, R., P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M. Lifshitz, R.A. Tuft, and T. Pozzan. 1998. b. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766. [DOI] [PubMed] [Google Scholar]
- Sanjuan Szklarz, L.K., B. Guiard, M. Rissler, N. Wiedemann, V. Kozjak, L.M. van der Laan, C. Lohaus, K. Marcus, H.E. Meyer, A. Chacinska, et al. 2005. Inactivation of the mitochondrial heat shock protein zim17 leads to aggregation of matrix hsp70s followed by pleiotropic effects on morphology and protein biogenesis. J. Mol. Biol. 351:206–218. [DOI] [PubMed] [Google Scholar]
- Schwarzer, C., S. Barnikol-Watanabe, F.P. Thinnes, and N. Hilschmann. 2002. Voltage-dependent anion-selective channel (VDAC) interacts with the dynein light chain Tctex1 and the heat-shock protein PBP74. Int. J. Biochem. Cell Biol. 34:1059–1070. [DOI] [PubMed] [Google Scholar]
- Simmen, T., J.E. Aslan, A.D. Blagoveshchenskaya, L. Thomas, L. Wan, Y. Xiang, S.F. Feliciangeli, C.H. Hung, C.M. Crump, and G. Thomas. 2005. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 24:717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soti, C., C. Pal, B. Papp, and P. Csermely. 2005. Molecular chaperones as regulatory elements of cellular networks. Curr. Opin. Cell Biol. 17:210–215. [DOI] [PubMed] [Google Scholar]
- Szabadkai, G., and R. Rizzuto. 2004. Participation of endoplasmic reticulum and mitochondrial calcium handling in apoptosis: more than just neighborhood? FEBS Lett. 567:111–115. [DOI] [PubMed] [Google Scholar]
- Szabadkai, G., A.M. Simoni, M. Chami, M.R. Wieckowski, R.J. Youle, and R. Rizzuto. 2004. Drp-1 dependent division of the mitochondrial network blocks intraorganellar Ca2+ waves and protects against Ca2+ mediated apoptosis. Mol. Cell. 16:59–68. [DOI] [PubMed] [Google Scholar]
- Takano, S., R. Wadhwa, Y. Mitsui, and S.C. Kaul. 2001. Identification and characterization of molecular interactions between glucose-regulated proteins (GRPs) mortalin/GRP75/peptide-binding protein 74 (PBP74) and GRP94. Biochem. J. 357:393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance, J.E. 1990. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 265:7248–7256. [PubMed] [Google Scholar]
- Vance, J.E. 2003. Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog. Nucleic Acid Res. Mol. Biol. 75:69–111. [DOI] [PubMed] [Google Scholar]
- Varadi, A., L.I. Johnson-Cadwell, V. Cirulli, Y. Yoon, V.J. Allan, and G.A. Rutter. 2004. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin- related protein-1. J. Cell Sci. 117:4389–4400. [DOI] [PubMed] [Google Scholar]
- Varnai, P., A. Balla, L. Hunyady, and T. Balla. 2005. Targeted expression of the inositol 1,4,5-triphosphate receptor (IP3R) ligand-binding domain releases Ca2+ via endogenous IP3R channels. Proc. Natl. Acad. Sci. USA. 102:7859–7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendelin, M., M. Lemba, and V.A. Saks. 2004. Analysis of functional coupling: mitochondrial creatine kinase and adenine nucleotide translocase. Biophys. J. 87:696–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura-Clapier, R., A. Kaasik, and V. Veksler. 2004. Structural and functional adaptations of striated muscles to CK deficiency. Mol. Cell. Biochem. 256-257:29–41. [DOI] [PubMed] [Google Scholar]
- Wadhwa, R., S.C. Kaul, Y. Ikawa, and Y. Sugimoto. 1993. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J. Biol. Chem. 268:6615–6621. [PubMed] [Google Scholar]
- Wadhwa, R., K. Taira, and S.C. Kaul. 2002. a. An Hsp70 family chaperone, mortalin/mthsp70/PBP74/Grp75: what, when, and where? Cell Stress Chaperones. 7:309–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa, R., T. Yaguchi, M.K. Hasan, Y. Mitsui, R.R. Reddel, and S.C. Kaul. 2002. b. Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp. Cell Res. 274:246–253. [DOI] [PubMed] [Google Scholar]
- Wadhwa, R., S. Takano, K. Kaur, S. Aida, T. Yaguchi, Z. Kaul, T. Hirano, K. Taira, and S.C. Kaul. 2005. Identification and characterization of molecular interactions between mortalin/mthsp70 and hsp60. Biochem. J. 391:185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, L., and G. Hajnoczky. 2005. Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J. Bioenerg. Biomembr. 37:191–206. [DOI] [PubMed] [Google Scholar]
- Wibo, M., and T. Godfraind. 1994. Comparative localization of inositol 1,4,5-trisphosphate and ryanodine receptors in intestinal smooth muscle: an analytical subfractionation study. Biochem. J. 297:415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, M., D. Weaver, and G. Hajnoczky. 2004. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J. Cell Biol. 167:661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young, J.C., J.M. Barral, and H.F. Ulrich. 2003. More than folding: localized functions of cytosolic chaperones. Trends Biochem. Sci. 28:541–547. [DOI] [PubMed] [Google Scholar]
- Zahedi, R.P., A. Sickmann, A.M. Boehm, C. Winkler, N. Zufall, B. Schonfisch, B. Guiard, N. Pfanner, and C. Meisinger. 2006. Proteomic Analysis of the Yeast Mitochondrial Outer Membrane Reveals Accumulation of a Subclass of Preproteins. Mol. Biol. Cell. 17:1436–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}