

Abstract
The twin-arginine translocation (Tat) pathway in Escherichia coli transports fully folded and assembled proteins across the energy-transducing periplasmic membrane. In chloroplasts, Tat transport requires energy input only from the proton motive force. To elucidate the mechanism and energetics of bacterial Tat protein transport, we developed an efficient in vitro transport assay using TatABC-enriched inverted membrane vesicles and the physiological precursor pre-SufI. We report transport efficiencies of 60–80% for nanomolar pre-SufI concentrations. Dissipation of the pH gradient does not reduce pre-SufI transport efficiency. Instead, pre-SufI transport requires at least two electrical potential (Δψ)–dependent steps that differ in both the duration and minimum magnitude of the required Δψ. The data are consistent with a model in which a substantial Δψ of short duration is required for an early transport step, and in which a small Δψ of long duration is necessary to drive a later transport step.
Introduction
Protein targeting and transport across lipid bilayers is a fundamental energy-requiring process in all organisms. Up to approximately half of the proteins in an organism's proteome are inserted into or transported across membranes by protein translocation systems, or translocons (Schatz and Dobberstein, 1996; Wallin and von Heijne, 1998). Most bacterial proteins are transported using the conserved Sec translocation pathway (de Keyzer et al., 2003). However, a distinct set of proteins are transported in fully folded and assembled form by the twin-arginine translocation (Tat) pathway (Berks et al., 2000; Palmer et al., 2005). Tat substrates are characterized by a twin-arginine–containing consensus motif (SRRxFLK) present in the N-terminal signal peptide of precursor proteins. In Escherichia coli, approximately two thirds of Tat substrates contain prosthetic groups, which are inserted into the proteins in the cytoplasm (Berks et al., 2005). A poorly understood proofreading mechanism prevents transport of substrates until they are properly folded and assembled (Musser and Theg, 2000; Jack et al., 2005).
The E. coli Tat translocation system contains four identified protein components: TatA, TatB, TatC, and TatE. TatA, TatB, and TatE each contain a single N-terminal transmembrane domain and a C-terminal cytoplasmic domain; the transmembrane domain is followed by an amphipathic helix that could preferentially interact with the lipid–water interface (Settles et al., 1997; Berks et al., 2000). TatC, which contains part of the signal sequence binding site (Alami et al., 2003; Holzapfel et al., 2007), has six transmembrane domains with both N and C termini facing the cytoplasm (Behrendt et al., 2004; Ki et al., 2004). Mutational analyses have shown that a functional Tat system minimally requires TatB, TatC, and either TatA or TatE (Sargent et al., 1998, 1999; Weiner et al., 1998). Thus, TatA and TatE are structural and functional homologues.
Three main oligomeric Tat complexes have been found in the E. coli periplasmic membrane. TatA forms oligomers from <100 kD to >500 kD that have been characterized as ring-like structures by electron microscopy (Porcelli et al., 2002; Oates et al., 2003, 2005; Gohlke et al., 2005). TatBC oligomers have an average molecular mass of ∼500 kD (McDevitt et al., 2006) wherein the TatB/TatC ratio is ∼1:1 (Bolhuis et al., 2001). The average molecular mass of TatABC complexes as estimated by gel-filtration (Bolhuis et al., 2001; Sargent et al., 2001) and blue-native gel electrophoresis (Oates et al., 2005) is ∼600 kD and ∼370 kD, respectively. TatA is found in large molar excess (as much as ∼20-fold) over TatB and TatC (Bolhuis et al., 2000), suggesting that the TatA complexes outnumber the TatBC complexes. It has been hypothesized that a pore composed of TatA oligomers allows the mature domain of the precursor protein to cross the membrane (Sargent et al., 2001). In such a model, the mature domain of a precursor protein bound to a TatBC complex through its signal sequence would have to be transferred through the TatA pore, perhaps as a result of oligomerization of a TatBC complex and a TatA complex.
The Tat system was first identified in plant thylakoids as a translocation system that requires the proton motive force (PMF), and not ATP, for transport. The energy stored in the PMF has two components, the electric field gradient (Δψ) and the pH gradient (ΔpH). From early experiments on thylakoids, it was concluded that the Tat system is energetically driven by the ΔpH alone (Mould and Robinson, 1991; Cline et al., 1992). This basic conceptual finding was recently challenged (Finazzi et al., 2003), and more recent work indicates that the Δψ can also contribute to driving Tat transport in thylakoids (Braun et al., 2007). Energetic studies of the bacterial Tat machinery have been hampered by the lack of an efficient in vitro assay. The first reported in vitro assay yielded a transport efficiency of <1% (Yahr and Wickner, 2001). Subsequently, it was found that precursors can be transported with up to ∼20% transport efficiency if they are synthesized via in vitro translation in the presence of inverted membrane vesicles (IMVs) (Alami et al., 2002). Here, we report the development of an efficient in vitro assay for the E. coli Tat machinery using purified overexpressed precursors. We show that two distinct Δψ-dependent steps are required for Tat transport. We did not detect a role for the ΔpH in influencing transport efficiency, despite the presence of substantial pH gradients. Our data are consistent with a model in which a relatively large Δψ of brief duration is required for an initial step in the transport process, and in which a small Δψ of long duration is required for a later step.
Results
An efficient in vitro pre-SufI transport assay
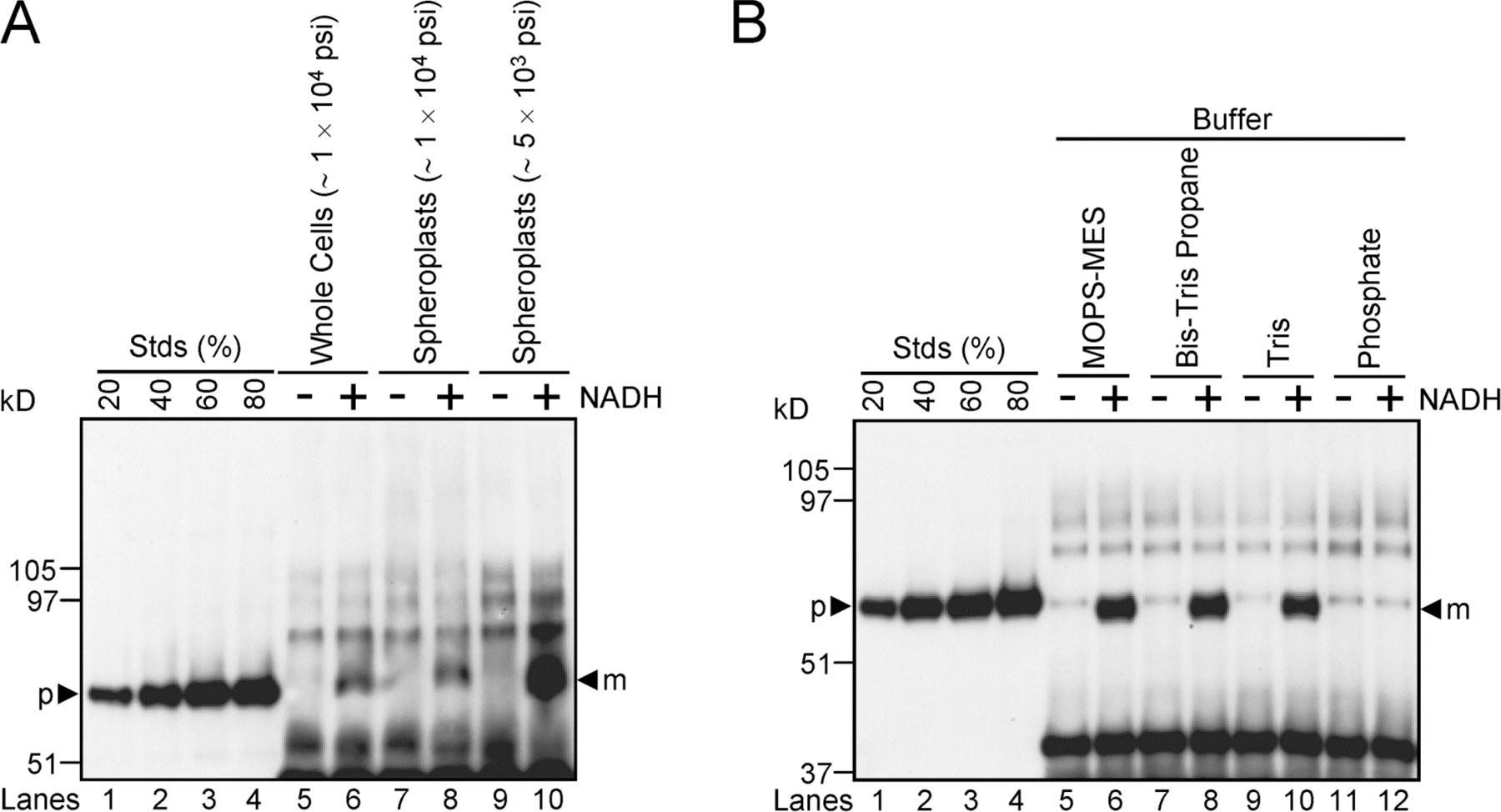
To avoid the assembly and folding complications inherent in using a Tat substrate that contains a cofactor, we have used pre-SufI, spSufI-GFP, and spTorA-GFP (Fig. 1 A). Although SufI is homologous to proteins of the multi-copper oxidase family, SufI does not appear to bind Cu2+ (Stanley et al., 2000) or possess a cofactor binding site (Berks, 1996). Pre-SufI was isolated under native conditions. The artificial GFP substrates were isolated under denaturing conditions and folded in vitro. Proper folding of the GFP substrates was assumed based on their green fluorescence emission upon UV excitation (Fig. 1 B).
Figure 1.

Precursor proteins, transport efficiencies, TatABC expression levels, and membrane orientation in in vitro Tat transport assays. (A) The precursor proteins used in the transport assays. The twin-arginine motifs (bold), signal peptidase recognition sequences (underlined) and signal sequence cleavage sites (arrow) are identified. Lowercase, italicized letters are residues of the mature SufI and TorA domains that have been retained in the GFP fusions. (B) Purity and fluorescence of the precursor proteins. Purified precursor proteins were visualized in SDS-PAGE gels by Coomassie blue staining (left) and the fluorescence emission upon UV excitation (right). (C) A standard in vitro transport assay. In this anti-SufI immunoblot, known amounts of pre-SufI (lanes 1–4), marked as a percentage of the precursor protein (p) added to the transport reactions, allowed quantification of the transport efficiency. Samples were proteinase K treated after 30 min at 37°C. NADH (4 mM) promoted the appearance of protease-protected mature-length protein (m). Higher molecular weight bands result from immuno-crossreactivity with unknown endogenous proteins (see G). Under some conditions, minute quantities of precursor were detectable in the absence of NADH; control experiments indicate that this was likely due to incomplete protease digestion, and this amount was always subtracted from that observed in the presence of NADH for accurate quantification of transport. Efficient transport required overexpression of TatABC, and was typically higher in JM109(pTatABC) than in MC4100(pTatABC), here quantified as 73 and 45%, respectively. Lane 8 was considered standard assay conditions. [pre-SufI] = 50 nM; [IMV] = 5 (A 280). (D) Transport efficiencies of different MC4100-derived strains. This anti-SufI immunoblot demonstrates that overexpression of TatABC promoted efficient protein transport (compare lanes 4 and 8), and that TatD and TatE were not required for efficient transport (compare lanes 6 and 10). Same conditions as panel C. (E) TatABC overexpression. Expression of TatA, TatB, and TatC proteins was induced from the pTatABC plasmid in JM109 (J) and MC4100 (M) strains with 0.7% arabinose, as indicated. All three proteins were retained in IMVs. Five microliters of total cell extracts (A 600 = 4.4) were loaded per lane; 1 μl of IMVs (A 280 = 50) was loaded per lane. (F) TatB accessibility assay. The percentage of inside-out oriented vesicles in IMV preparations was determined by the protease accessibility of the TatB cytoplasmic domain. This anti-TatB immunoblot shows different amounts of control IMVs for quantification purposes (lanes 1–7), where the 100% lane represents the amount of TatB present before proteinase K (PK) treatment. After protease treatment (lane 8), ∼10% of the original TatB remained, and upon protease treatment in the presence of 0.05% TX-100 (lane 9), all the TatB was digested. These data indicate that the vesicles in this IMV preparation were ∼90% inside-out. (G) Protease digestion of mature SufI after membrane permeabilization. The left anti-SufI immunoblot shows that addition of 0.05% Triton X-100 (TX-100) during proteinase K (PK) treatment under standard assay conditions (see panel C) resulted in complete loss of the mature SufI protein band (compare lanes 9 and 12). The right anti-SufI immunoblot shows that PK addition resulted in complete digestion of pre-SufI. (H) Same experiment as panel G, but using 6xHis antibodies. Due to the lower detection efficiency of the 6xHis antibodies, samples were sedimented and gel loads (based on IMV amount) were 16 times higher than in panel G.
A typical in vitro transport assay consisted of the addition of TatABC-enriched IMVs to prewarmed (37°C) tubes containing 50 nM pre-SufI. NADH was used to generate a PMF. Reactions were incubated at 37°C for 30 min and then treated with protease (proteinase K) to digest any remaining untransported pre-SufI. The proteins in each reaction were resolved by SDS-PAGE and immunoblotted using SufI antibodies (see Materials and methods). Overexpression of TatA, TatB, and TatC was essential for detecting transport, and only these three Tat proteins were required (Fig. 1, C–E). In general, IMVs from JM109 were easier to work with and yielded better transport efficiencies than those from MC4100 (Fig. 1 C). Membrane orientation in vesicle preparations was estimated by the accessibility of the TatB cytoplasmic domain to protease digestion. Typical membrane preparations consisted of ∼90% IMVs and ∼10% right-side-out vesicles (Fig. 1 F). Addition of 0.05% Triton X-100 during protease treatment resulted in complete digestion of mSufI (Fig. 1 G), indicating that pre-SufI that had translocated into the vesicle lumen could be digested by protease after membrane permeabilization. Control experiments lacking added pre-SufI demonstrate that the high molecular weight bands observed on anti-SufI immunoblots arose from endogeneous proteins within the IMV preparations, and not from pre-SufI aggregates (Fig. 1 G, lanes 4 and 7). Detection with 6xHis antibodies confirms this result (Fig. 1 H). Mature- and precursor-length SufI were not always resolvable due to their small difference in molecular weights (e.g., compare Fig. 1, C, G, and H).
Transport of GFP fused with TorA and SufI signal peptides
Having developed an efficient in vitro Tat transport assay, we then tested whether a common model substrate, spTorA-GFP (Santini et al., 2001; Thomas et al., 2001; DeLisa et al., 2004), could be efficiently transported under the conditions that yielded efficient pre-SufI transport. The spTorA-GFP protein was transported at a much lower efficiency (up to ∼15%; Fig. 2 A) than pre-SufI (72 ± 7%). We considered the possibility that proteins with the TorA signal peptide could not be efficiently transported in our standard assay for unknown reasons. Therefore, we changed the signal peptide on spTorA-GFP to that of pre-SufI yielding spSufI-GFP (Fig. 1 A). No transport of spSufI-GFP could be detected (Fig. 2 B).
Figure 2.

Transport of spTorA-GFP and spSufI-GFP and their ability to compete for transport with pre-SufI. (A) Transport efficiency of spTorA-GFP. This anti-GFP immunoblot shows that transport of spTorA-GFP (50 nM) in the presence of 4 mM NADH requires TatABC overexpression. Based on known amounts of spTorA-GFP (lanes 1–4), the transport efficiency of this precursor in this experiment was ∼15%. Because proteinase K (PK) treatment digests the precursor only to a mature-sized fragment, reactions were sedimented before running the gel. Mature-length protein generated by PK treatment was lost due to this sedimentation procedure (lane 14). Thus, the mature-length protein observed in lanes 8 and 12 was generated by transport into the vesicle lumen. The protein present in lane 13 represents the amount of precursor-length protein that aggregated or bound to the walls of the reaction tube. Siliconized tubes were used to minimize adsorption of precursor to the walls of the tubes. (B) Transport efficiency of spSufI-GFP. This anti-GFP immunoblot shows that transport of spSufI-GFP (50 nM) was undetectable. Experiments were performed identically to those in panel A. (C) Competition between spSufI-GFP and pre-SufI. This anti-SufI immunoblot shows the pre-SufI transport efficiency under standard conditions (Fig. 1 C) in the presence of a 0- to 50-fold molar excess of spSufI-GFP. IMVs were preincubated with both precursor proteins for 10 min at 37°C and transport reactions were initiated with 4 mM NADH. (D) Competition between spTorA-GFP and pre-SufI. Transport reactions were performed identically as in panel C, except that spTorA-GFP was used instead of spSufI-GFP.
spTorA-GFP is a competitive inhibitor for pre-SufI transport
We expected that protein chimeras containing Tat signal peptides should at least interact with the signal peptide binding site of the Tat translocation machinery. If so, such chimeras should competitively inhibit transport of pre-SufI. Surprisingly, even a 50-fold molar excess of spSufI-GFP had no effect on transport of pre-SufI (Fig. 2 C). These data indicate that spSufI-GFP cannot bind (or only very weakly binds) to the Tat pathway receptor complex. In contrast, pre-SufI transport was almost completely inhibited by a 10-fold molar excess of spTorA-GFP (Fig. 2 D). These data likely indicate that spTorA-GFP was recognized by the Tat translocation machinery through the TorA signal peptide, and therefore, that spTorA-GFP competitively inhibited pre-SufI transport.
Transport kinetics
Having established optimum conditions for pre-SufI transport, we then examined the transport kinetics. Transport reactions were quenched at various times by plunging reaction tubes into an ice bath. Two notable features were immediately apparent: (1) transport was a relatively lengthy process, occurring on the timescale of many minutes; and (2) the transport kinetics were complex (i.e., not a single exponential), exhibiting a lag period before the appearance of transported protein (Fig. 3). A lag period was observed earlier for the thylakoid Tat system (Musser and Theg, 2000).
Figure 3.

Transport kinetics. All assays were conducted under standard transport conditions (Fig. 1 C). Reactions were initiated with IMVs and quenched in an ice bath (n = 7). The transport efficiency is scaled relative to the amount of precursor added.
Tat transport is completed in the absence of a detectable PMF
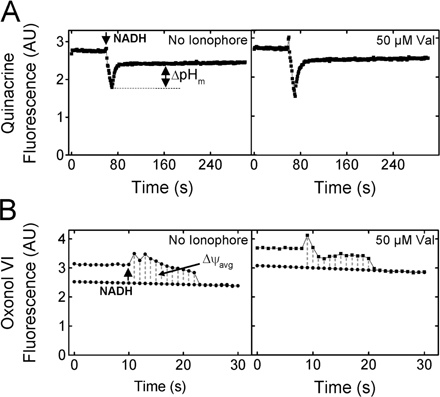
When NADH is added to IMVs, the electron transport chain generates a PMF using dioxygen (O2) as the final electron acceptor. Thus, the duration of the resultant pH gradient and electrical potential is limited by how rapidly the dissolved oxygen is consumed. Steady-state fluorescence spectroscopy was used to monitor the presence of a ΔpH and a Δψ (see Materials and methods). Because the detectable gradients (as reported by the dyes) decayed in seconds to tens of seconds after oxygen consumption, the duration that detectable gradients existed (ΔpHd and Δψd) was somewhat longer than the time to anaerobiosis (ΔpHt and Δψt), although the latter were more easily measurable due to discrete inflection points. ΔpHt and Δψt were identical (within error) for a given set of conditions; ΔpHd and Δψd were not identical. At least part of these differences can be attributed to the inherent slow response of the dye distributions to the gradients. As expected, both ΔpHt and Δψt were inversely affected by an increase in IMV concentration, consistent with a faster enzymatic consumption of dissolved oxygen at higher IMV concentrations (Fig. 4, A and B). At the concentration of TatABC-enriched IMVs used in our standard transport assay (A 280 = 5), the solutions became anaerobic in 10 ± 4 s (Fig. 4, C and D), and detectable ΔpH and Δψ gradients were observed for ∼15 s and ∼10 s, respectively (Fig. 4, A and B). However, mature SufI continued to accumulate for at least ∼12 min after the reaction became anaerobic (Fig. 3), indicating that Tat pathway–mediated transport of precursor proteins can be completed in the absence of a detectable ΔpH and Δψ.
Figure 4.

Duration of the detectable PMF and its effect on pre-SufI transport yield. (A) ΔpHt dependence on the IMV concentration. ΔpH generation was measured by quinacrine fluorescence in the presence (red) and absence (black) of 10 μM nigericin at the indicated IMV concentrations. The time span between NADH (4 mM) addition (20 s time point; identified by the open arrowhead) and the time at which the next inflection point occurred (identified by the filled arrowhead), presumably the point at which the solution became anaerobic, was termed the ΔpHt. The time span between NADH addition and the time at which the detectable gradient ceased to exist was termed the ΔpHd. (B) Δψt dependence on the IMV concentration. Δψ generation was measured by oxonol VI fluorescence in the presence (red) and absence (black) of 10 μM nigericin and 10 μM valinomycin at the indicated IMV concentrations. Δψt and Δψd were defined similarly to ΔpHt and ΔpHd in (A), respectively, as indicated. (C) Mean ΔpHt plotted against the IMV concentration (n = 3). (D) Mean Δψt plotted against the IMV concentration (n = 3). (E) Dependence of transport yield per vesicle on vesicle concentration. Transport yield per vesicle was measured at a high pre-SufI concentration (250 nM) at various IMV concentrations (n = 3). A measure of the transport yield per vesicle was obtained by dividing the transport yield (pmol pre-SufI) by the IMV concentration (A 280). Though not the actual transport yield per vesicle, these values are expected to be directly proportional to the true values.
We next tested whether the duration of the detectable ΔpH and Δψ influenced pre-SufI transport efficiency by varying the concentration of the IMVs. As the IMV concentration was decreased from A 280 = 5 to A 280 = 0.5, the Δψt and ΔpHt increased from ∼10 s to ∼42 s (Fig. 4, C and D). At a high precursor concentration (presumably saturating the Tat translocons over all the IMV concentrations tested), the amount of precursor transported per IMV was invariable (Fig. 4 E). These data indicate that the approximately fourfold increase in the duration of the detectable Δψ and ΔpH did not affect pre-SufI transport efficiency under these conditions.
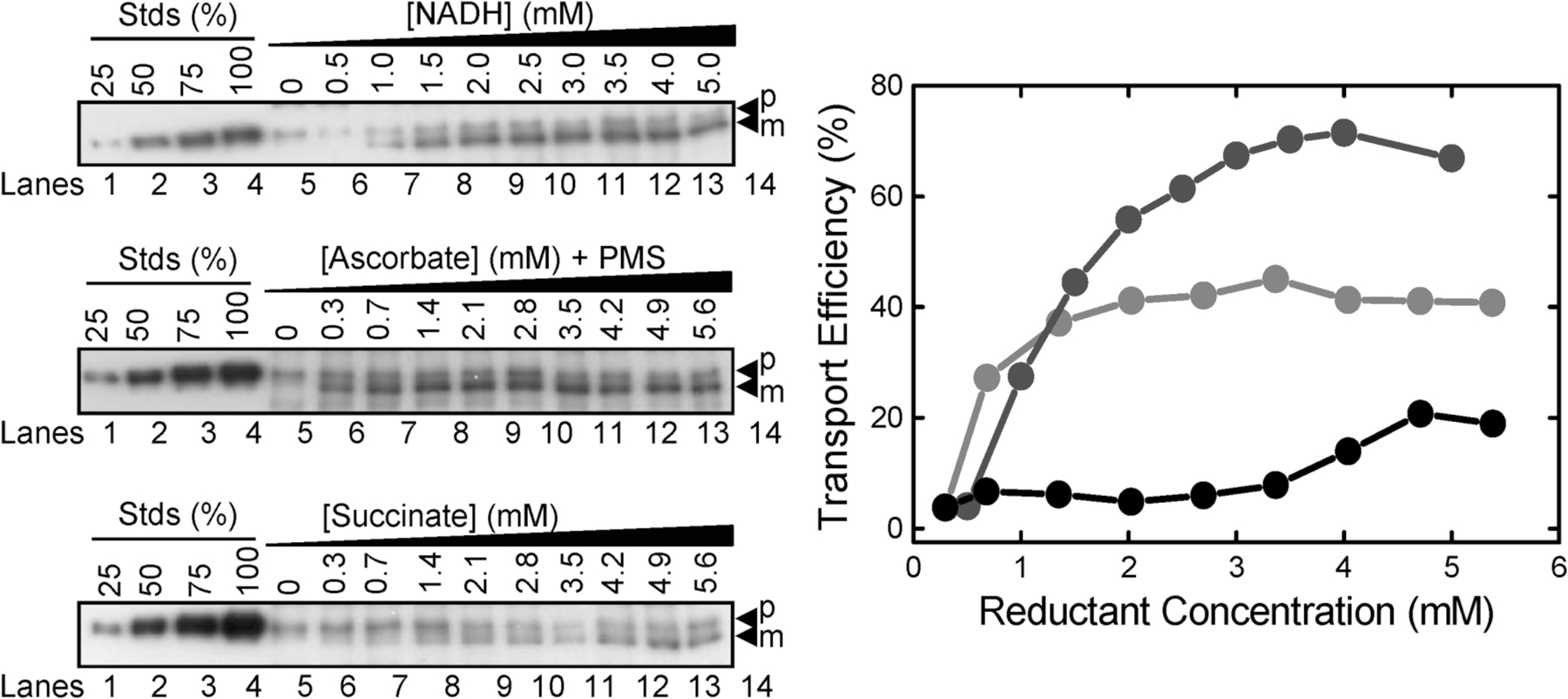
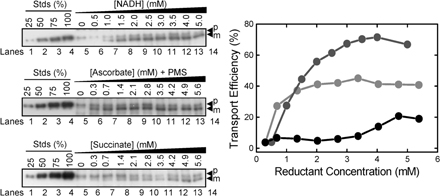
The Δψ alone provides the energetic driving force for pre-SufI transport
We next examined the effect of the magnitude of the detectable Δψ and ΔpH on pre-SufI transport efficiency. Because ΔpH generation takes many seconds due to the large number of ions that must be translocated, the point at which the maximum ΔpH (ΔpHm) was generated was easily estimated (Fig. 5 A). In contrast, Δψ generation is very fast due to the much lower number of ions that must be translocated, and it was not always clear if observed signals were due to the injection needle, mixing artifacts, or Δψ generation. Instead, we estimated the average Δψ (Δψavg; Fig. 5 B). We compared the pre-SufI transport efficiencies obtained with various uncoupler concentrations. When the ΔpH was selectively reduced with various concentrations of nigericin (an electroneutral K+/H+ exchanger), pre-SufI transport was unaffected or slightly increased. Increased transport efficiencies correlated with an increased Δψavg (Fig. 5 C). When the Δψ was selectively reduced with valinomycin (a K+ ionophore), the ΔpHm remained high and transport efficiency again correlated with Δψavg (Fig. 5 D). The Δψ could not be completely dissipated with valinomycin alone (see Fig. S3 for an explanation, available at http://www.jcb.org/cgi/content/full/jcb.200702082/DC1). However, in the presence of both valinomycin and nigericin, the detectable ΔpH and the Δψ were both completely collapsed and pre-SufI transport was completely inhibited (Fig. 5 E). For complete dissipation of the Δψ by valinomycin (at A 280 = 5), a low concentration of nigericin was necessary (Fig. 5 B). When the detectable ΔpH was first completely dissipated by a low concentration of nigericin and the Δψ was progressively dissipated by increased concentrations of valinomycin, the pre-SufI transport efficiency was highly correlated with Δψavg (Fig. 6 A). When the Δψ was dramatically reduced by 25 mM NaSCN without decreasing the ΔpH gradient (Fig. 5, A and B), pre-SufI transport was almost completely inhibited (Fig. 6 B). Transport reactions with spTorA-GFP confirmed the pre-SufI results that Tat transport requires a Δψ (Fig. 6 C). The increased spTorA-GFP transport observed when nigericin was present (Fig. 6 C) is likely explained by the increased Δψavg observed under these conditions (Fig. 5, B and C), which likely results from compensation for the PMF decrease that results from loss of the ΔpH. The NaSCN data support the hypothesis that inhibition of pre-SufI transport by valinomycin was through collapse of the Δψ, rather than by a direct effect of valinomycin on the Tat translocation machinery. In total, these data indicate that pre-SufI transport was largely, if not completely, independent of the ΔpH and strongly dependent on the Δψ.
Figure 5.

Pre-SufI transport efficiency dependence on ΔpHm and Δψavg. (A) Determination of the ΔpHm under various conditions. The ΔpH was measured as in Fig. 4 A by the addition of 4 mM NADH (open arrowhead). The ΔpHm was defined to be the difference between the baseline after the solution became anaerobic and the minimum fluorescence observed. [IMV] = 5 (A 280). (B) Determination of the Δψavg under various conditions. The Δψ was measured as in Fig. 4 B. The Δψavg was defined to be the area bounded by the fluorescence curve and the extrapolated baseline after the solution became anaerobic between the point of reaction initiation and the point at which the detectable gradient disappeared. Note that the time axes are different in panels A and B. [IMV] = 5 (A 280). (C) Effect of nigericin on ΔpHm, Δψavg and pre-SufI transport efficiency. Values are given as mean ± SD (n ≥ 3), where 100% = the values obtained in the absence of ionophores. (black) ΔpHm; (red) Δψavg; (blue) pre-SufI transport efficiency. (D) Effect of valinomycin on ΔpHm, Δψavg and pre-SufI transport efficiency. Same key as panel C. (E) Effect of nigericin and valinomycin on ΔpHm, Δψavg and pre-SufI transport efficiency. Both ionophores were always present at the same concentration. Same key as panel C.
Figure 6.

Effect of selective Δψ and ΔpH gradient collapse on pre-SufI and spTorA-GFP transport efficiency. (A) Correlation between Δψavg and pre-SufI transport efficiency. This anti-SufI immunoblot shows standard transport assays (Fig. 1 C) with different concentrations of nigericin (Nig.) alone (lanes 6–8), or with both nigericin and valinomycin (Val.) (lanes 11–14). The control reaction (lane 4) had no NADH added. Transport efficiency was calculated based on the transport observed in the absence of ionophores (lanes 5 and 9). Same key as Fig. 5 C. (B) Effect of selective gradient collapse on pre-SufI transport efficiency. This anti-SufI immunoblot shows standard transport assays with 10 μM nigericin (N), 10 μM valinomycin (V), and/or 25 mM NaSCN (S) added before reaction initiation (0) or 30 s after reaction initiation (30 s), as indicated. The control reactions (C) had no uncouplers. Values are given in the bar graph as mean ± SD (n = 3). Transport efficiency was calculated based on the transport observed in the absence of uncouplers (lane 6). (C) Effect of selective gradient collapse on spTorA-GFP transport efficiency. The same experiment as panel B using spTorA-GFP instead of pre-SufI.
A long duration, undetectable Δψ is required for pre-SufI transport
The data discussed in the previous two sections indicate that pre-SufI transport required a Δψ, yet transport occurred on a much longer timescale (many minutes) than the time a detectable Δψ (∼10 s) was maintained across IMV membranes. One possible explanation for these results is that the Δψ is required only for an early step in the transport process and that later steps of transport do not require a Δψ. To investigate this possibility, we added ionophores to dissipate any nonmeasurable gradients at various times after reaction initiation. As expected based on earlier results (Fig. 5 and Fig. 6), pre-SufI transport efficiency was not affected by dissipation of the ΔpH by 10 μM nigericin either early or late in the transport process (Fig. 7 A). In contrast, addition of 10 μM nigericin and 10 μM valinomycin early in the transport process (e.g., at ∼1–5 min after reaction initiation) almost completely inhibited pre-SufI transport (Fig. 7 B), despite the fact that the detectable gradients had collapsed when the ionophores were added (Fig. 4, A and B). Our interpretation of these data is that undetectable ΔpH and Δψ gradients existed after the reaction solutions became anaerobic. One possibility is that the solutions were not completely anaerobic due to gas exchange at the aqueous–air interface, thereby allowing low level respiratory activity. Alternatively, a small Δψ was maintained by an anaerobic pathway. Regardless, our interpretation of these data is that a small, undetectable Δψ was maintained after collapse of the short duration but substantial Δψ spike (Fig. 4 B), and that this undetectable Δψ was essential for driving pre-SufI transport. The threshold sensitivity of our Δψ measurements is unknown. Other investigators have estimated a threshold sensitivity of ∼50 mV, but this value is strongly dependent on the lipid to dye ratio (Ghelli et al., 1997).
Figure 7.

Effect of the delayed addition of ionophores and pre-SufI on SufI transport efficiency. All assays were conducted under standard transport conditions (Fig. 1 C). (A) Effect of ΔpH collapse time on pre-SufI transport efficiency. This anti-SufI immunoblot shows the mean amount of pre-SufI transported when nigericin (10 μM) was added at various times after reaction initiation (n = 3). The control lanes show the amount of pre-SufI transported after 30 min in the presence (+) and absence (−) of NADH. Transport efficiency was calculated based on the transport observed in the 30-min lane (lane 14). (B) Effect of PMF collapse time on pre-SufI transport efficiency. This anti-SufI immunoblot shows the mean amount of pre-SufI transported when both nigericin (10 μM) and valinomycin (10 μM) were added at various times after reaction initiation (n = 3). The control lanes are the same as in panel A. (C) Effect of delayed pre-SufI addition on pre-SufI transport yield. This anti-SufI immunoblot shows the mean amount of pre-SufI transported when the precursor (50 nM) was added at various times after reaction initiation (n = 3). The control lane shows the amount of pre-SufI transported after 30 min in the absence of NADH. Transport efficiency was calculated based on the transport observed when pre-SufI was present at the time of NADH addition (0 min).
Efficient transport does not require the precursor protein to be present during the Δψ spike period
To address whether a precursor protein must be present during the Δψ spike period, pre-SufI was added to IMVs at various times after addition of NADH. Efficient Tat transport (> ∼90% of control) occurred even if pre-SufI was added up to ∼5 min after membrane energization, long after collapse of the short, initial Δψ spike (Fig. 7 C). Transport efficiency decreased by ∼90% with an ∼30 min delay between precursor addition and membrane energization (Fig. 7 C), possibly due to consumption of NADH.
A second Δψ spike increases transport yield only after a time delay
Because efficient pre-SufI transport resulted when the precursor was added after collapse of the Δψ spike, we considered the possibility that the undetectable Δψ gradient was sufficient to drive the entire Tat translocation cycle. To test this hypothesis, we measured the translocation yield after producing a second Δψ spike by addition of O2-saturated buffer. We observed that the pre-SufI translocation yield increased by ∼60% when a second Δψ spike was generated ∼12.5–20 min after the first. A second Δψ spike had little to no effect if it was generated < ∼5 min after reaction initiation (Fig. 8, A and B).
Figure 8.

Effect of a second Δψ pulse on transport yield and transport kinetics at different IMV concentrations. All assays were conducted under standard transport conditions (Fig. 1 C), unless otherwise noted. (A) Effect of delayed addition of oxygenated buffer on transport yield. This anti-SufI immunoblot shows the mean amount of pre-SufI transported when the reaction volume was doubled with prewarmed (37°C) oxygenated buffer (35 μL; no NADH) at various times after reaction initiation in the presence of 8 mM NADH and 5 μM nigericin (n = 3). The control lane shows the amount of pre-SufI transported after 30 min in the absence of NADH. Note that the ordinate shows transport yield, i.e., the amount of precursor transported (in pmol) rather than the percentage of precursor transported relative to some control. [pre-SufI] = 250 nM; [IMV] = 2 (A 280). (B) Generation of a second Δψ pulse at the indicated times (arrowheads) after the initial Δψ pulse. Time delays correspond to the points in panel A. Identical conditions as panel A. ΔF (100%) corresponds to the total fluorescence observed at the beginning of the experiment (before NADH addition). (C) Kinetics of pre-SufI translocation at different IMV concentrations, normalized to the 30-min time point. [pre-SufI] = 50 nM; [IMV] = 2 (black), 4 (red), and 6 (blue) (A 280).
The transport kinetics are similar over a range of IMV concentrations
The fact that a second Δψ spike generated within 5 min of the first had little to no influence on transport yield indicates that the translocons were predominantly at a stage in the precursor translocation cycle where the energy of the second Δψ spike could not be used. This suggested to us that perhaps the kinetics shown in Fig. 7 B reflected a single translocon turnover cycle, and that a Δψ spike might be required to initiate a second turnover cycle. To test this possibility, we reasoned that the observed kinetics should be essentially invariable with IMV concentration if they reported a single translocation cycle. In contrast, if the kinetics in Fig. 7 B reflected numerous enzyme turnovers, we reasoned that faster transport would be observed at higher IMV concentrations. We observed that the transport kinetics were largely independent of IMV concentration (Fig. 8 C). According to the predictions just discussed, these data are consistent with the picture that the observed transport kinetics reflect a single turnover cycle.
spTorA-GFP can competitively inhibit pre-SufI transport long after transport initiation
Considering the conclusion from the previous section that the transport kinetics are consistent with a single turnover cycle, we hypothesized that if spTorA-GFP was added after pre-SufI transport was initiated, it might no longer be competitive for transport. We found that pre-SufI transport was sensitive to the addition of spTorA-GFP competitor more than 5 min after reaction initiation. In fact, the transport inhibition versus the time delay of competitor addition curve (Fig. 9) looks surprisingly similar to the transport kinetics of the reaction as determined by ionophore quenching (Fig. 7 B).
Figure 9.

Effect of delayed addition of spTorA-GFP on pre-SufI transport efficiency. This anti-SufI immunoblot shows the amount of pre-SufI transported when spTorA-GFP (500 nM) was added at various times after reaction initiation (n = 3). The control lanes show the amount of pre-SufI transported after 30 min in the presence (+) and absence (−) of NADH with no spTorA-GFP. Transport efficiency was calculated based on the transport observed in the +NADH control lane. All assays were conducted under standard transport conditions (Fig. 1 C).
Translocation speed is dependent on the magnitude of the steady-state Δψ
To maintain a high magnitude steady-state Δψ, ATP was used to generate a PMF through catalytic reversal of the F0F1 ATPase. With this approach, the Δψ is independent of the dissolved O2 concentration and depends only on the ATP energy charge. Because ATP can be solubilized at higher concentrations than O2 and regenerated in situ, the Δψ gradient can be maintained for a longer period. When ATP (plus an ATP regenerating system) was used to energize IMVs, both ΔpH and Δψ remained detectable for at least 9 min (Fig. 10, A and B), and pre-SufI translocation was approximately threefold faster (Fig. 10 C) without the detectable lag phase observed when IMVs were energized by NADH (Fig. 7 B). Despite the ability of ATP to establish a long-lived high-magnitude ΔpH, pre-SufI transport was, once again, largely, if not completely, independent of the ΔpH and strongly dependent on the Δψ (Fig. 10D). These data support the hypothesis that translocation speed is dependent on the magnitude of the steady-state Δψ. When NADH was used to generate a PMF, the length of the lag period was observed to vary for different IMV preparations (not depicted). In light of the above ATP results, we surmise that the length of the lag period reflects the differential ability of different IMV preparations to support a Δψ.
Figure 10.

Effect of ATP on pre-SufI transport. (A) Generation of ΔpH by ATP. IMVs (A 280 = 5) were energized with 4 mM NADH (black) or 4 mM ATP + regenerating system (red). The ATP-induced ΔpH was collapsed by 5 μM nigericin (blue). ATP regenerating system: 2.9 mM phosphocreatine, 0.29 mg/ml creatine kinase. (B) Generation of Δψ by ATP. IMVs (A 280 = 5) were energized with 4 mM NADH (black) or 4 mM ATP + regenerating system (red). The ATP-induced Δψ was collapsed by 25 mM NaSCN (blue). Note that the final baseline is significantly higher in the presence of ATP than NADH due to a direct effect of NADH on the fluorescence emission yield of oxonol VI (compare with the traces in Fig. 4 B in the presence of ionophores). (C) Effect of ATP on pre-SufI transport kinetics. This anti-SufI immunoblot shows the mean amount of pre-SufI transported as a function of time when the PMF was generated by 4 mM ATP + regenerating system (n = 3). (D) Effect of selective gradient collapse on pre-SufI transport efficiency using ATP to generate a PMF. The same experiment as Fig. 6 B using 4 mM ATP + regenerating system instead of NADH.
Discussion
The development of an efficient in vitro transport assay for the E. coli Tat machinery has allowed us to decipher a number of important fundamental features of the transport mechanism: (1) the energetic driving force for transport comes largely, if not entirely, from the Δψ alone—we have no evidence that the ΔpH assists with promoting transport (Figs. 5, 6, and 10); (2) at least two distinct transport steps require a Δψ—one Δψ requiring step occurs early in the transport process and requires a Δψ of relatively high magnitude that may be short-lived (Fig. 8, A and B), and a second Δψ requiring step minimally requires a long duration, but relatively low magnitude Δψ (Fig. 7 B); (3) transport speed is increased if the steady-state Δψ is increased (Fig. 10); (4) transport efficiency is decreased if the average Δψ is decreased (Fig. 6 A); (5) the first Δψ requiring step can occur in the absence of precursor protein (Fig. 7 C); and (6) transport can be competitively inhibited long after transport initiation (Fig. 9). These findings provide important constraints for the transport mechanism. Some of the implications are discussed in the following paragraphs.
A natural question to arise from these studies is whether the E. coli Tat transport system is fundamentally unique or different than the Tat transport system found in photosynthetic organisms. Early in vitro studies in higher plant thylakoid systems support a picture wherein it is the ΔpH alone that provides the driving force for Tat precursor transport (Mould and Robinson, 1991; Cline et al., 1992). More recent data suggest a minimum substrate-specific ΔpH (Alder and Theg, 2003). In contrast, in vivo studies in Chlamydomonas reinhardtii and barley leaves support a picture where it is the Δψ alone that provides the driving force for Tat precursor transport (Finazzi et al., 2003). Precursor maturation in tobacco protoplasts does not require either a ΔpH or a Δψ, but it is not clear whether the mature domain can be transported under these conditions (Di Cola et al., 2005). Possible explanations for the apparent discrepancies are that something is missing from in vitro assays and/or that both the Δψ and the ΔpH can support Tat transport under certain conditions (Theg et al., 2005). The hypothesis that both the ΔpH and Δψ components of the PMF can energetically contribute to driving precursor transport is supported by recent in vitro studies in thylakoids (Braun et al., 2007). The studies reported here, however, raise some additional possibilities: (1) under some conditions the Δψ is not completely collapsed by valinomycin alone; and (2) only a brief, yet relatively large, Δψ is necessary to initiate a transport cycle. In thylakoids, a brief but substantial Δψ does exist upon photoillumination, which may be sufficient to drive the first step of Tat transport, and a relatively low Δψ does exist during steady state under in vivo conditions and under some in vitro conditions (Cruz et al., 2001), which may be sufficient to drive the completion of Tat transport.
According to current models, an early step in Tat transport is precursor binding to the TatBC complex. In some models, recruitment of TatA molecules leads to formation of a highly selective pore that allows the precursor protein to cross the membrane (Palmer et al., 2005; Lee et al., 2006). In thylakoids, transport can occur without migration of the signal peptide from the TatBC binding site (Gerard and Cline, 2006). We show here, however, that transport was blocked by an excess of a precursor that was added many minutes after transport initiation. If transport was inhibited by competitive binding, which seems likely, one interpretation is that the signal sequence binding interaction must be readily reversible. This conclusion is somewhat puzzling because it contradicts the thylakoid data, and because it would seem to lead to slow and inefficient transport. However, if different types of translocons existed (e.g., arising from different oligomeric states of TatA), a reversible signal sequence binding interaction on TatBC would be an effective way for the precursor to sample many different structures. One way that this could happen without losing the precursor to the bulk solution is if the signal peptide binds to the lipid alone, as has been recently demonstrated (Shanmugham et al., 2006). This picture could explain how spTorA-GFP can compete with pre-SufI for transport long after reaction initiation (Fig. 9). On the other hand, if multiple TatBC complexes can bind to the same TatA oligomer, multiple precursors could compete for translocation through the same gated pore. Hence, spTorA-GFP added after transport of pre-SufI has begun could bind to TatBC oligomers that are a part of TatABC–pre-SufI complexes, and thereby reduce the efficiency of pre-SufI transport. According to this picture, the signal sequence binding interaction could be relatively strong and not readily reversible, consistent with the thylakoid data that indicate that transport can occur without migration of the signal peptide from the TatBC binding site (Gerard and Cline, 2006).
There are at least two interpretations of the transport kinetics shown in Fig. 7 B. We discussed the single turnover cycle model in the Results section. According to this interpretation, a second turnover cycle cannot initiate without a second high-magnitude Δψ pulse, and this pulse must occur at, or near, the end of the first turnover cycle (compare Fig. 8 A with Fig. 7 B). An alternative interpretation is that each high-magnitude Δψ pulse is sufficient to drive numerous turnover cycles. The initial lag in the observed kinetics would then be consistent with the hypothesis that the first turnover cycle is relatively slow, and subsequent turnover cycles are faster. The existence of “slow” and “fast” enzymatic forms is not a novel concept (Shoji et al., 2000). In contrast, as discussed earlier, if the translocons function with only one translocation speed under a given set of conditions, then it is expected that higher translocon concentrations would yield faster overall translocation kinetics, which was not observed (Fig. 8 C). In conclusion, both the single and multiple turnover cycle models (with the slow and fast caveat for the latter) are consistent with our data.
The nature of the coupling between the Δψ and protein transport by the E. coli Tat machinery remains unclear. We have not ruled out the possibility that a concentration gradient other than the ΔpH is coupled to Tat transport—e.g., a sodium or other ion gradient. The coupling of ion flow to protein transport would imply that a portion of the energy stored in a potential gradient is “consumed” to drive transport because ion flow would actually reduce the gradient. However, energy “consumption” is not strictly required. E. coli Tat protein transport occurs down a concentration gradient because precursor proteins are only found on the cytoplasmic side of the cytoplasmic membrane (the concentration of mature protein in the periplasm can be much higher, but this does not affect the thermodynamics of transport because these proteins cannot be transported after signal peptide cleavage). Thus, instead of being consumed to energetically drive transport, it is possible that the Δψ is instead coupled to protein conformational changes (“gating reactions”) that are required for transport. In the case of voltage-gated ion channels, the membrane Δψ is coupled to the opening and closing of pores required for ion transport. Current models postulate that a charged region of voltage-gated ion channels moves in response to a Δψ, thereby causing the channel to open (Tombola et al., 2005). In the case of the Tat pathway, it is unlikely that the Δψ would simply open large pores allowing folded proteins to diffuse across the membrane because this would result in the immediate collapse of the PMF and other ion gradients. However, a reasonable scenario is that movement of certain charged regions within Tat proteins could be induced by a Δψ. For example, the movement of Δψ sensing domains could lead to the oligomerization or rearrangements of subunits, leading the translocon to be primed and ready for catalyzing transport. We emphasize that this picture still requires additional gating reactions (e.g., signal peptide recognition) for controlling access to the translocation channel.
In summary, we have demonstrated an efficient in vitro transport assay for the E. coli Tat machinery. When NADH was used to produce a PMF, transport was relatively slow and occurred with a half-time of ∼10 min. Because faster translocation could be achieved with a higher magnitude and longer lasting PMF, we predict that in vivo transport rates are faster, and that our current transport rates are limited by the leakiness of IMVs. Nonetheless, under our conditions, precursor transport is ΔpH independent and there exist at least two Δψ-dependent transport steps.
Materials and methods
Bacterial strains, growth conditions, and plasmids
E. coli strains MC4100, MC4100ΔTatABCDE, JM109, and BL21(λDE3) have been described earlier (Casadaban and Cohen, 1979; Yanisch-Perron et al., 1985; Studier et al., 1990; Wexler et al., 2000). Overexpression cultures were grown in Luria-Bertani (LB) medium at 37°C supplemented with appropriate antibiotics (Sambrook and Russell, 2001), unless otherwise noted.
The spTorA-GFP protein was expressed from plasmid pTorA-GFP. Plasmid pTorA-GFP was constructed by QuikChange site-directed mutagenesis (Stratagene) from pJDT1 (Thomas et al., 2001) using primers prTorAHis6C-F and prTorAHis6C-R as forward and reverse primers, respectively (Table S1, available at http://www.jcb.org/cgi/content/full/jcb.200702082/DC1). This process added a C-terminal HHHHHHC tag to the encoded protein.
The spSufI-GFP protein was expressed from plasmid pSufI-GFP, which was constructed in two steps by replacing the TorA signal peptide in pTorA-GFP with the SufI signal peptide. First, a unique SacII restriction endonuclease site was generated 130 bp downstream of the translation start site encoding spTorA-GFP in pTorA-GFP by the QuikChange protocol using prTorA-ScII-F and prTorA-ScII-R as forward and reverse primers, respectively (Table S1), generating plasmid pTorA-GFP2. Then, the PCR product generated from amplification of pET-SufI (Yahr and Wickner, 2001) with primers prSufI-sp-F and prSufI-sp-R (Table S1) was digested with EcoRI and SacII and inserted into pTorA-GFP2 digested with the same two restriction enzymes, generating plasmid pSufI-GFP. Coding regions were confirmed by DNA sequencing.
Western blotting
TatA, TatB, and TatC were detected by Western blotting using rabbit polyclonal TatA, TatB, and TatC antibodies (1:5,000 dilutions) (Yahr and Wickner, 2001) and GFP was detected by using rabbit polyclonal GFP antibodies (1:10,000; Santa Cruz Biotechnology, Inc.) in 1× PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 2 mM KH2PO4, pH 7.4) with 2% nonfat dry milk, 0.1% Triton X-100, and 0.1% Tween. SufI was detected as described above by using rabbit 6xHis antibodies (1:1,000; Santa Cruz Biotechnology, Inc.), or by using SufI antibodies (1:15,000) (Yahr and Wickner, 2001) in 1× PBS as described above except with higher detergent concentrations (0.5% Triton X-100 and 0.5% Tween 20). Goat polyclonal anti–rabbit IgG-HRP conjugate (1:15,000; Santa Cruz Biotechnology, Inc.) was used as the secondary antibody, and bands were visualized by chemiluminescence (Harlow and Lane, 1999). Band intensities were quantified with a PhosphorImager (model FX; Bio-Rad Laboratories).
Isolation of inverted membrane vesicles (IMVs)
Cells for IMV isolation were grown in low-salt LB medium (1% bactotryptone, 0.5% yeast extract, and 0.25% NaCl) supplemented with 5% glycerol. Overnight cultures were subcultured (1:100; 4 × 750 ml) and grown at 37°C for 3–4 h until the A 600 reached ∼1. TatABC expression from pTatABC (Yahr and Wickner, 2001) was induced with 0.7% arabinose and growth was continued for another 4 h. Cultures were plunged into an ice bath and cells were harvested by centrifugation at 4,000 g for 8 min at 4°C. The cell pellet (10–12 g) was suspended in 50 ml ice-cold Buffer A (1 mM MgSO4, 0.5% polyvinylpyrrolidine [MW 360,000], 450 mM mannitol, 2 mM DTT, 50 μg/ml DNase I, 10 μg/ml RNAase, 1 mM KCl, and 100 mM Tricine, pH 7.5) with 0.4 mg/ml lysozyme, 0.5 mM EDTA, and protease inhibitors (10 mM PMSF, 100 μg/ml trypsin inhibitor, 20 μg/ml leupeptin, and 100 μg/ml pepstatin), and incubated on ice for 20 min to produce spheroplasts. The spheroplasts were sedimented by centrifugation at 4,000 g for 10 min at 4°C, resuspended in Buffer A, and passed through a French pressure cell once at ∼6,000 psi to produce IMVs. The IMV solution was centrifuged at 4,000 g for 10 min at 4°C to remove debris. The supernatant (8-ml portions) was layered over 6-ml sucrose cushions (Buffer B [1 mM KCl, 1 mM MgSO4, 2 mM DTT, and 10 mM Hepes, pH 7.0] with 2.2 M sucrose) and centrifuged for 75 min at 108,000 g. The band from the top of the sucrose cushion in each tube was collected. IMVs were pooled, diluted 1:4 with Buffer B, and centrifuged for 45 min at 108,000 g. The pellet was resuspended in 4–6 ml of Buffer B with 50% glycerol, and frozen immediately at −80°C. Total protein was quantified as the A 280 in 2% SDS. Typical IMV stock solutions had an A 280 ≈ 50–60.
Protein expression and purification
Pre-SufI was overexpressed from pET-SufI (Yahr and Wickner, 2001) in BL21(λDE3) and purified under native conditions by Ni-NTA chromatography. LB cultures (500 ml) were incubated at 37°C until the A 600 reached ∼3. The pH of the cultures was raised with 25 ml of 0.5 M CAPS buffer (pH 9.0), and induced with 0.5 mM IPTG for 2.5 h. Cultures were chilled in an ice bath and centrifuged at 5,000 g for 8–12 min at 4°C. Pellets were rapidly resuspended on ice in 50 ml Buffer C (100 mM Tris and 25 mM CAPS, pH 9.0) containing 250 mM NaCl, 20 mM imidazole, 0.2% Triton X-100, and protease inhibitors. Cells were then passed through a French pressure cell once at 16,000 psi. The cell lysate was cleared of cellular debris by centrifugation at 50,000 g for 10 min at 4°C, and then stirred with 2 ml Ni-NTA Superflow resin (QIAGEN) that had been pre-equilibrated with Buffer C for 10 min on ice. The resin was loaded onto a 10 × 1 cm column, and sequentially washed with: (1) 100 ml of Buffer D (10 mM Tris-HCl, 1 M NaCl, and 20 mM imidazole, pH 8.0) with 0.1% Triton X-100; (2) 20 ml of Buffer D; (3) 20 ml of 100 mM NaCl and 10 mM imidazole, pH 8.0; and (4) 10 ml of 100 mM NaCl, 10 mM imidazole, and 50% glycerol, pH 8.0. Pre-SufI was eluted with 100 mM NaCl, 250 mM imidazole, and 50% glycerol, pH 8.0 as 1-ml fractions and stored at −80°C. Typical yield was 8–10 mg protein/L of culture.
The spSufI-GFP and spTorA-GFP proteins were overexpressed from plasmids pSufI-GFP and pTorA-GFP, respectively, in MC4100ΔTatABCDE. These GFP proteins were purified under denaturing conditions and refolded by dilution/dialysis from 9 M urea. Overnight LB cultures with 1% glucose were subcultured into fresh LB medium with 1% glucose (1:100 dilution; 3 × 500 ml) and grown at 37°C until the A 600 reached ∼5 (∼4 h). Cells were harvested by centrifugation, resuspended in fresh LB medium with 1.5% arabinose to induce protein expression, and incubated at 37°C for 2.5 h. Cells (∼7 g) were harvested by centrifugation, resuspended in 50 ml Buffer E (20 mM CAPS, 2 mM DTT, and 0.2% Triton X-100, pH 9.0) with protease inhibitors, and stirred on ice for 30 min. The cell suspension was passed through a French pressure cell once at 16,000 psi. The cell lysate was diluted to 400 ml with Buffer E, and centrifuged at 15,000 g for 30 min. Pellets containing inclusion bodies were suspended in Buffer F (10 mM CAPS, 5 mM DTT, and 9 M urea, pH 9.0), and stirred at room temperature (RT) for 30 min. The resuspension was centrifuged at 50,000 g for 30 min. The supernatant was stirred with 4 ml Ni-NTA Superflow resin equilibrated with Buffer F for 30 min at RT, and then loaded onto a 10 × 1 cm column. The resin was washed sequentially with: (1) 20 ml Buffer F; (2) 20 ml Buffer F with 0.1% Triton X-100; and (3) 10 ml Buffer G (50 mM MOPS, 9 M urea, 100 mM KCl, 10% glycerol, 5 mM Mg acetate, 50 mM glycine-glycine, and 5 mM DTT, pH 8.0) with 10 mM imidazole. Proteins were eluted with Buffer G with 250 mM imidazole. Protein concentration was quantified using ɛ280 = 2.06 × 104 M−1 cm−1 (Enoki et al., 2004), and solutions were diluted to 50 ng/ml with Buffer G. Proteins were folded by dialyzing (10 kD molecular weight cut-off) out the urea using a four-step gradient (9 M → 7 M → 5 M → 3 M; ∼3 h per step) in the dark (typical yield was 95–99% folded protein). The dialyzate was centrifuged at 15,000 g for 30 min. Folded protein was recovered from the supernatant using 5 ml Ni-NTA Superflow resin equilibrated with 50 mM MOPS, 100 mM KCl, 5 mM Mg acetate, and 5 mM DTT, pH 8.0. The Ni-NTA–adsorbed GFP proteins were washed and eluted identically as described above for the purification of pre-SufI. Typical yield was ∼2 mg protein/L of culture.
Protein concentrations of all Tat substrates were quantified by SDS-PAGE using bovine serum albumin (BSA) as the standard and quantified with a PhosphorImager (model FX; Bio-Rad Laboratories).
In vitro translocation assay
Standard in vitro translocation assays used a 35-μl reaction volume containing 50 nM pre-SufI and 4 mM NADH in Translocation Buffer (TB; 5 mM MgCl2, 50 mM KCl, 200 mM sucrose, 57 μg/ml BSA, 25 mM MOPS, and 25 mM MES, pH 7.0). Solutions were prewarmed at 37°C for 5 min before the addition of IMVs (to a typical final concentration of A 280 = 5). After a 30 min incubation at 37°C, reactions were quenched in an ice bath for 2 min. Samples were digested with 0.73 mg/ml proteinase K for 40 min at RT. Digestions were quenched with 68 mM PMSF, diluted twofold with 2× Gel Buffer (4% SDS, 10% glycerol, 0.04% bromophenol blue, 0.4% β-mercaptoethanol, 10 M urea, and 200 mM Tris, pH 6.8), and incubated in a boiling water bath for 10 min. Samples were centrifuged briefly at 16,000 g, and then were resolved by 8% SDS-PAGE with known standards. Gels were electroblotted onto PVDF membranes and immunoblotted with SufI antibodies. For spSufI-GFP and spTorA-GFP translocation assays, both protease-treated and untreated IMVs were sedimented at 100,000 g at 4°C for 15 min (or 16,100 g at 4°C for 45 min). The IMVs (pellets) were washed with 500 μl TB with 20 μg/ml BSA, resuspended in 35 μl TB containing 68 mM PMSF, resolved by 10% SDS-PAGE with known standards, and immunoblotted using GFP antibodies.
The sidedness of IMV preparations
The TatB protein has a single transmembrane domain near the N terminus and a C-terminal cytoplasmic domain (Berks et al., 2000). TatB is unlikely to undergo topology inversion (Bolhuis et al., 2001), as has been reported for TatA (Gouffi et al., 2004; Chan et al., 2007). The percentage of IMVs with an inside-out orientation was determined based on the protease accessibility of the TatB C-terminal domain using TatB antibodies raised against peptides within this domain (Yahr and Wickner, 2001). IMVs in TB without BSA were incubated with 2 mg/ml proteinase K at RT for 30 min. Digestions were quenched with 20 mM PMSF, diluted twofold with 2× Gel Buffer, and incubated in a boiling water bath for 10 min. Samples were centrifuged briefly at 16,000 g, and then were resolved by 18% SDS-PAGE with known standards. Gels were electroblotted onto PVDF membranes and immunoblotted with TatB antibodies.
Determination of ΔpH and Δψ gradients
The presence of ΔpH and Δψ gradients across IMV membranes was determined by fluorescence spectroscopy using 2.5 μM quinacrine (EX = 420 nm, EM = 510 nm) and 100 nM oxonol VI (EX = 610 nm, EM = 645 nm), respectively (Kawasaki et al., 1993). IMVs were preincubated in TB at 37°C for ∼5 min before the addition of 4 mM NADH or 4 mM ATP. Conditions were identical to those used for gel-based transport assays. Control experiments indicated that the effects observed when ionophores were added were not due to dilution or solvent.
Data analysis
All errors are standard deviations.
Online supplemental material
Table S1 summarizes the DNA primers used for plasmid construction. Fig. S1 shows the effect of IMV formation method and transport buffer on transport efficiency. Fig. S2 shows the transport efficiency dependence on energy source. Fig. S3 shows the effect of high valinomycin concentration on ΔpH and Δψ. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200702082/DC1.
Supplementary Material
Acknowledgments
We thank T.L. Yahr for pET-SufI, pTatABC, and the TatA, TatB, TatC, and SufI antibodies; C. Robinson for pJDT1; T. Palmer for MC4100 and MC4100ΔTatABCDE; Meng Chen and Kelly Soltysiak for technical assistance; and C. Koehler and N. Whitaker for critical evaluation of the manuscript.
This work was supported by the National Institutes of Health (GM065534) and the Welch Foundation (BE-1541).
Abbreviations used in this paper: IMV, inverted membrane vesicle; PMF, proton motive force; Tat, twin-arginine translocation.
References
- Alami, M., D. Trescher, L.F. Wu, and M. Muller. 2002. Separate analysis of twin-arginine translocation (Tat)-specific membrane binding and translocation in Escherichia coli. J. Biol. Chem. 277:20499–20503. [DOI] [PubMed] [Google Scholar]
- Alami, M., I. Luke, S. Deitermann, G. Eisner, H.G. Koch, J. Brunner, and M. Muller. 2003. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol. Cell. 12:937–946. [DOI] [PubMed] [Google Scholar]
- Alder, N.N., and S.M. Theg. 2003. Energetics of protein transport across biological membranes. A study of the thylakoid ΔpH-dependent/cpTat pathway. Cell. 112:231–242. [DOI] [PubMed] [Google Scholar]
- Behrendt, J., K. Standar, U. Lindenstrauss, and T. Brüser. 2004. Topological studies on the twin-arginine translocase component TatC. FEMS Microbiol. Lett. 234:303–308. [DOI] [PubMed] [Google Scholar]
- Berks, B. 1996. A common export pathway for proteins binding complex redox factors? Mol. Microbiol. 22:393–404. [DOI] [PubMed] [Google Scholar]
- Berks, B.C., F. Sargent, and T. Palmer. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260–274. [DOI] [PubMed] [Google Scholar]
- Berks, B.C., T. Palmer, and F. Sargent. 2005. Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr. Opin. Microbiol. 8:174–181. [DOI] [PubMed] [Google Scholar]
- Bolhuis, A., E.G. Bogsch, and C. Robinson. 2000. Subunit interactions in the twin-arginine translocase complex of Escherichia coli. FEBS Lett. 472:88–92. [DOI] [PubMed] [Google Scholar]
- Bolhuis, A., J.E. Mathers, J.D. Thomas, C.M. Barrett, and C. Robinson. 2001. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J. Biol. Chem. 276:20213–20219. [DOI] [PubMed] [Google Scholar]
- Braun, N.A., A.W. Davis, and S.M. Theg. 2007. The chloroplast Tat pathway utilizes the transmembrane electrical potential as an energy source. Biophys. J. 93:1993–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban, M.J., and S.N. Cohen. 1979. Lactose genes fused to exogenous promoters in one step using a Mu-lac bacteriophage: in vivo probe for transcriptional control sequences. Proc. Natl. Acad. Sci. USA. 76:4530–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, C.S., M.R. Zlomislic, D.P. Tieleman, and R.J. Turner. 2007. The TatA subunit of Escherichia coli twin-arginine translocase has an N-in topology. Biochemistry. 46:7396–7404. [DOI] [PubMed] [Google Scholar]
- Cline, K., W.F. Ettinger, and S.M. Theg. 1992. Protein-specific energy requirements for protein transport across or into thylakoid membranes. Two lumenal proteins are transported in the absence of ATP. J. Biol. Chem. 267:2688–2696. [PubMed] [Google Scholar]
- Cruz, J.A., C.A. Sacksteder, A. Kanazawa, and D.M. Kramer. 2001. Contribution of electric field (Δψ) to steady-state transthylakoid proton motive force (pmf) in vitro and in vivo. Control of pmf parsing into Δψ and ΔpH by ionic strength. Biochemistry. 40:1226–1237. [DOI] [PubMed] [Google Scholar]
- de Keyzer, J., C. van der Does, and A.J. Driessen. 2003. The bacterial translocase: a dynamic protein channel complex. Cell. Mol. Life Sci. 60:2034–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa, M.P., P. Lee, T. Palmer, and G. Georgiou. 2004. Phage shock protein PspA of Escherichia coli relieves saturation of protein export via the Tat pathway. J. Bacteriol. 186:366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cola, A., S. Bailey, and C. Robinson. 2005. The thylakoid delta pH/delta psi are not required for the initial stages of Tat-dependent protein transport in tobacco protoplasts. J. Biol. Chem. 280:41165–41170. [DOI] [PubMed] [Google Scholar]
- Enoki, S., K. Saeki, K. Maki, and K. Kuwajima. 2004. Acid denaturation and refolding of green fluorescent protein. Biochemistry. 43:14238–14248. [DOI] [PubMed] [Google Scholar]
- Finazzi, G., C. Chasen, F.A. Wollman, and C. de Vitry. 2003. Thylakoid targeting of Tat passenger proteins shows no delta pH dependence in vivo. EMBO J. 22:807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard, F., and K. Cline. 2006. Efficient twin arginine translocation (Tat) pathway transport of a precursor protein covalently anchored to its initial cpTatC binding site. J. Biol. Chem. 281:6130–6135. [DOI] [PubMed] [Google Scholar]
- Ghelli, A., B. Benelli, and M.D. Esposti. 1997. Measurement of the membrane potential generated by complex I in submitochondrial particles. J. Biochem. (Tokyo). 121:746–755. [DOI] [PubMed] [Google Scholar]
- Gohlke, U., L. Pullan, C.A. McDevitt, I. Porcelli, E. de Leeuw, T. Palmer, H.R. Saibil, and B.C. Berks. 2005. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc. Natl. Acad. Sci. USA. 102:10482–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouffi, K., F. Gerard, C.L. Santini, and L.F. Wu. 2004. Dual topology of the Escherichia coli TatA protein. J. Biol. Chem. 279:11608–11615. [DOI] [PubMed] [Google Scholar]
- Harlow, E., and D. Lane. 1999. Using Antibodies: A Laboratory Manual. Cold Spring Harbor Laboratory Press, New York. 495 pp.
- Holzapfel, E., G. Eisner, M. Alami, C.M.L. Barrett, G. Buchanan, I. Lüke, J.-M. Betton, C. Robinson, T. Palmer, M. Moser, and M. Müller. 2007. The entire N-terminal half of TatC is involved in twin-arginine precursor binding. Biochemistry. 46:2892–2898. [DOI] [PubMed] [Google Scholar]
- Jack, R.L., A. Dubini, T. Palmer, and F. Sargent. 2005. Common principles in the biosynthesis of diverse enzymes. Biochem. Soc. Trans. 33:105–107. [DOI] [PubMed] [Google Scholar]
- Kawasaki, S., S. Mizushima, and H. Tokuda. 1993. Membrane vesicles containing overproduced SecY and SecE exhibit high translocation ATPase activity and countermovement of protons in a SecA- and presecretory protein-dependent manner. J. Biol. Chem. 268:8193–8198. [PubMed] [Google Scholar]
- Ki, J.J., Y. Kawarasaki, J. Gam, B.R. Harvey, B.L. Iverson, and G. Georgiou. 2004. A periplasmic fluorescent reporter protein and its application in high-throughput membrane protein topology analysis. J. Mol. Biol. 341:901–909. [DOI] [PubMed] [Google Scholar]
- Lee, P.A., D. Tullman-Ercek, and G. Georgiou. 2006. The bacterial twin-arginine translocation pathway. Annu. Rev. Microbiol. 60:373–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDevitt, C.A., G. Buchanan, F. Sargent, T. Palmer, and B.C. Berks. 2006. Subunit composition and in vivo substrate-binding characteristics of Escherichia coli Tat protein complexes expressed at native levels. FEBS J. 273:5656–5668. [DOI] [PubMed] [Google Scholar]
- Mould, R.M., and C. Robinson. 1991. A proton gradient is required for the transport of two lumenal oxygen-evolving proteins across the thylakoid membrane. J. Biol. Chem. 266:12189–12193. [PubMed] [Google Scholar]
- Musser, S.M., and S.M. Theg. 2000. Characterization of the early steps of OE17 precursor transport by the thylakoid ΔpH/Tat machinery. Eur. J. Biochem. 267:2588–2598. [DOI] [PubMed] [Google Scholar]
- Oates, J., J. Mathers, D. Mangels, W. Kühlbrandt, C. Robinson, and K. Model. 2003. Consensus structural features of purified bacterial TatABC complexes. J. Mol. Biol. 330:277–286. [DOI] [PubMed] [Google Scholar]
- Oates, J., C.M. Barrett, J.P. Barnett, K.G. Byrne, A. Bolhuis, and C. Robinson. 2005. The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J. Mol. Biol. 346:295–305. [DOI] [PubMed] [Google Scholar]
- Palmer, T., F. Sargent, and B.C. Berks. 2005. Export of complex cofactor-containing proteins by the bacterial Tat pathway. Trends Microbiol. 13:175–180. [DOI] [PubMed] [Google Scholar]
- Porcelli, I., E. de Leeuw, R. Wallis, E. van den Brink-van der Laan, B. de Kruijff, B.A. Wallace, T. Palmer, and B.C. Berks. 2002. Characterization and membrane assembly of the TatA component of the Escherichia coli twin-arginine protein transport system. Biochemistry. 41:13690–13697. [DOI] [PubMed] [Google Scholar]
- Sambrook, J., and D.W. Russell. 2001. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor Press, Cold Spring Harbor, New York.
- Santini, C., A. Bernadac, M. Zhang, A. Chanal, B. Ize, C. Blanco, and L. Wu. 2001. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic shock. J. Biol. Chem. 276:8159–8164. [DOI] [PubMed] [Google Scholar]
- Sargent, F., E.G. Bogsch, N.R. Stanley, M. Wexler, C. Robinson, B.C. Berks, and T. Palmer. 1998. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 17:3640–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent, F., N.R. Stanley, B.C. Berks, and T. Palmer. 1999. Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J. Biol. Chem. 274:36073–36082. [DOI] [PubMed] [Google Scholar]
- Sargent, F., U. Gohlke, E. de Leeuw, N.R. Stanley, T. Palmer, H.R. Saibil, and B.C. Berks. 2001. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur. J. Biochem. 268:3361–3367. [DOI] [PubMed] [Google Scholar]
- Schatz, G., and B. Dobberstein. 1996. Common principles of protein translocation across membranes. Science. 271:1519–1526. [DOI] [PubMed] [Google Scholar]
- Settles, A.M., A. Yonetani, A. Baron, D.R. Bush, K. Cline, and R. Martienssen. 1997. Sec-independent protein translocation by the maize Hcf106 protein. Science. 278:1467–1470. [DOI] [PubMed] [Google Scholar]
- Shanmugham, A., H.W.W.F. Sang, Y.J.M. Bollen, and H. Lill. 2006. Membrane binding of twin arginine preproteins as an early step in translocation. Biochemistry. 45:2243–2249. [DOI] [PubMed] [Google Scholar]
- Shoji, K., A. Giuffè, E. D'Itri, K. Hagiwara, T. Yamanaka, M. Brunori, and O. Sarti. 2000. The ratio between the fast and slow forms of bovine cytochrome c oxidase is changed by cholate or nucleotides bound to the cholate-binding site close to the cytochrome a3/CuB binuclear centre. Cell. Mol. Life Sci. 57:1482–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley, N.R., T. Palmer, and B.C. Berks. 2000. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J. Biol. Chem. 275:11591–11596. [DOI] [PubMed] [Google Scholar]
- Studier, F.W., A.H. Rosenberg, J.J. Dunn, and J.W. Dubendorff. 1990. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185:60–89. [DOI] [PubMed] [Google Scholar]
- Theg, S.M., K. Cline, G. Finazzi, and F.A. Wollman. 2005. The energetics of the chloroplast Tat protein transport pathway revisited. Trends Plant Sci. 10:153–154. [DOI] [PubMed] [Google Scholar]
- Thomas, J.D., R.A. Daniel, J. Errington, and C. Robinson. 2001. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol. Microbiol. 39:47–53. [DOI] [PubMed] [Google Scholar]
- Tombola, F., M.M. Pathak, and E.Y. Isacoff. 2005. How far will you go to sense voltage? Neuron. 48:719–725. [DOI] [PubMed] [Google Scholar]
- Wallin, E., and G. von Heijne. 1998. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7:1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner, J.H., P.T. Bilous, G.M. Shaw, S.P. Lubitz, L. Frost, G.H. Thomas, J.A. Cole, and R.J. Turner. 1998. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell. 93:93–101. [DOI] [PubMed] [Google Scholar]
- Wexler, M., F. Sargent, R.L. Jack, N.R. Stanley, E.G. Bogsch, C. Robinson, B.C. Berks, and T. Palmer. 2000. TatD is a cytoplasmic protein with DNase activity. J. Biol. Chem. 275:16717–16722. [DOI] [PubMed] [Google Scholar]
- Yahr, T.L., and W.T. Wickner. 2001. Functional reconstitution of bacterial Tat translocation in vitro. EMBO J. 20:2472–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 33:103–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}