

Abstract
The Notch ligands Dll1 and Dll3 are coexpressed in the presomitic mesoderm of mouse embryos. Despite their coexpression, mutations in Dll1 and Dll3 cause strikingly different defects. To determine if there is any functional equivalence, we replaced Dll1 with Dll3 in mice. Dll3 does not compensate for Dll1; DLL1 activates Notch in Drosophila wing discs, but DLL3 does not. We do not observe evidence for antagonism between DLL1 and DLL3, or repression of Notch activity in mice or Drosophila. In vitro analyses show that differences in various domains of DLL1 and DLL3 individually contribute to their biochemical nonequivalence. In contrast to endogenous DLL1 located on the surface of presomitic mesoderm cells, we find endogenous DLL3 predominantly in the Golgi apparatus. Our data demonstrate distinct in vivo functions for DLL1 and DLL3. They suggest that DLL3 does not antagonize DLL1 in the presomitic mesoderm and warrant further analyses of potential physiological functions of DLL3 in the Golgi network.
Introduction
The Notch signaling pathway is of central importance for the regulation of developmental processes by mediating direct cell-to-cell communication between cells in a wide variety of developmental contexts in different species (Campos-Ortega, 1994; Muskavitch, 1994; Blaumueller and Artavanis-Tsakonas, 1997; Gridley, 1997; Artavanis-Tsakonas et al., 1999). Products of the Notch, Delta, and Serrate (called Jagged in mammals) genes are crucial for this direct interaction between neighboring cells. Notch genes encode large transmembrane proteins that, at the extracellular surface of a cell, act as receptors for proteins encoded by the Delta and Serrate genes. Like Notch, Delta and Serrate are transmembrane ligands with a variable number of EGF-like repeats in their extracellular domains (Wharton et al., 1985; Vässin et al., 1987; Thomas et al., 1991). In addition, the Delta and Serrate proteins contain in their extracellular portion a conserved cysteine-rich region known as DSL domain (Delta, Serrate, lag-2), which is essential for ligand binding (Shimizu et al., 1999, 2000). Generally, vertebrates contain several copies of genes encoding particular Notch pathway components. In the mouse, there are four genes encoding Notch proteins, three genes coding for Delta (Dll1, Dll3, and Dll4), and two for Serrate (Jagged1 and Jagged2) proteins, respectively. Little is known about how these ligands interact with various Notch receptors in vivo, and whether the signals elicited by these interactions are quantitatively or qualitatively different.
Studies in zebrafish, Xenopus, chicken, and mouse embryos have demonstrated an essential requirement for Notch signaling during somite formation and patterning. In mice, mutational analyses have shown that two ligands, Dll1 (Hrabe de Angelis et al., 1997) and Dll3 (Kusumi et al., 1998; Dunwoodie et al., 2002), are essential for normal somite formation and patterning. Dll1 and Dll3 are coexpressed throughout most of the presomitic mesoderm (PSM) and differentially expressed in the anterior and posterior compartments of newly formed somites (Dunwoodie et al., 1997). However, despite the overlapping mRNA expression in the PSM, loss of Dll1 or Dll3 function leads to clearly distinct phenotypes. Somites in Dll1 mutant embryos lack any detectable anterior–posterior (A-P) polarity, as indicated by the loss of Uncx4.1 expression. Loss of segment polarity is already evident in the anterior PSM. Somites are not fully epithelialized, their borders are not maintained, and Lfng expression in the PSM is down-regulated to barely detectable levels (Hrabe de Angelis et al., 1997; Morales et al., 2002). In contrast, null alleles of Dll3 (Kusumi et al., 1998; Dunwoodie et al., 2002) disrupt somite polarity such that Uncx4.1 expression appears randomized throughout somites instead of being restricted to the posterior compartment, and Lfng expression is readily detected, although transcriptional oscillation appears abnormal (Dunwoodie et al., 2002; Kusumi et al., 2004). These qualitatively different phenotypes suggest that these ligands are not functionally equivalent in vivo. Support for this notion comes from a recent study that showed that DLL3 cannot activate Notch in vitro and suggested that DLL3 acts as an antagonist to DLL1 on the cell surface (Ladi et al., 2005).
Here, we demonstrate that the DLL1 and DLL3 proteins are not equivalent in mouse embryos. Replacement of the Notch ligand Dll1 with Dll3 in mice resulted in a phenotype indistinguishable from the Dll1-null phenotype, although Dll3 expressed from the Dll1 locus is functional. Similarly, in transgenic flies, Dll1, as well as Dll4, acted as a bona fide activator of Drosophila melanogaster Notch in the wing imaginal disc, whereas Dll3 did not. Changing the ratios of Dll1and Dll3 in vivo in mice or ectopic expression in flies did not provide genetic evidence for antagonism between DLL1 and DLL3 or repression of Notch activity by DLL3. Also, NICD was not up-regulated in the PSM of embryos lacking DLL3. In vitro analyses using chimeric DLL1-DLL3 proteins showed that differences in the DSL domains, EGF repeats, and intracellular domains (ICDs), respectively, contribute to their biochemical nonequivalence. In contrast to DLL1, DLL3 protein was predominantly detected inside the cell, including in the Golgi network. Our data prove that DLL1 and DLL3 have distinct functions in vivo; under physiological conditions, the proteins are differentially localized in the cell, and DLL3 might not act simply by antagonizing DLL1.
Results
Generation of Dll1 and Dll3 knockin alleles
To express the Dll3 coding region from the Dll1 locus and simultaneously eliminate Dll1 function, we generated mice that carried a chimeric Dll3 “minigene” fused in frame into the ATG of the endogenous Dll1 gene, analogous to the Dll1lacZ-null allele generated previously (Hrabe de Angelis et al., 1997). In the Dll3 minigene, the Dll3 coding sequence, either with or without a C-terminal HA tag, was linked at the 3′ end to genomic sequences of the Dll1 gene containing exons 9–11 (Fig. 1 A). After processing of the primary transcript, the Dll3 coding sequence is thus flanked by the Dll1 5′ and 3′ UTRs, which should generate Dll3 transcripts with stability and properties similar to those of the genuine Dll1 mRNA. As a control to ensure that this structural alteration at the Dll1 locus had no adverse effects on expression of the Dll3 minigene, we also generated mice that carried an analogous minigene version of Dll1 targeted to the Dll1 locus (Fig. 1 A).
Figure 1.

Generation of Dll1Dll3Haki, Dll1Dll3ki, and Dll1Dll1ki mice. (A) Targeting strategy for introduction of Dll3 and Dll1 into the Dll1 locus. White and black boxes indicate noncoding and coding regions, respectively. The blue box indicates a Dll1, Dll3, or Dll3HA cDNA, respectively. DT, diphtheria toxin A chain. (B) E10.5 embryos. (C) Verification of Dll3HA expression by Western blot analysis with anti-HA antibodies. Lysates of CHO cells transfected with Dll3HA cDNA (a) and of wild type (b) and Dll1Dll3HA/Dll3HA (c) E9.5 embryos. Embryo lysates represent half of one embryo, respectively.
The Dll3 knockin (Dll1Dll3HAki and Dll1Dll3ki) and the Dll1 control alleles (Dll1 Dll1ki) were passed through the germ line of ZP3∷Cre females (de Vries et al., 2000) to remove the floxed neo cassette that was included in the targeting vectors (Fig. 1 A). Heterozygous mice carrying either knockin allele did not show obvious external phenotypes or obvious malformations of the axial skeleton (Fig. 2 A, a–c; and not depicted). Likewise, homozygous mice carrying the Dll1 Dll1ki allele were viable and fertile without any apparent phenotype (Fig. 2 A, m-o; and not depicted), indicating that the Dll1 minigene was sufficient to compensate for the disrupted endogenous gene. In contrast, no homozygous Dll1Dll3HAki or Dll1Dll3ki offspring were obtained from matings of heterozygous Dll1Dll3HAki or Dll1Dll3ki mice, respectively, although the HA-tagged DLL3 protein was expressed (Fig. 1 C). This suggested that Dll3 was unable to functionally replace Dll1.
Figure 2.

Phenotypes and somite patterning of embryos with various Dll1 and Dll3 allele combinations. (A) Embryo appearance (top) and skeletal preparations (bottom) of E18.5 embryos with the genotypes indicated on top. Dll1 Dll3HA/+ embryos (a–c) have an essentially normal axial skeleton despite an increased gene dosage ratio of Dll3 to Dll1. Skeletal defects of homozygous Dll3-null mutants (d–f) are rescued by Dll3HA expressed from the Dll1 locus (g–i) except for minor residual defects (i, arrowheads). Increased gene dosage ratio of Dll3 to Dll1 does not enhance a hypomorphic Dll1 phenotype (compare p–r with s–u). Note that modification of the Dll1 locus does not lead to any phenotypic alterations in the skeleton (m–o). (B) A-P somite patterning at E9.5 indicated by Uncx4.1 expression and dynamic Lfng expression patterns are indistinguishable in wild-type (a–c) and Dll1 Dll3HA/+ embryos (g–i). Abnormal expression of Uncx4.1 and Lfng in homozygous Dll3pu-null mutants (d–f) is restored by Dll3HA expressed from the Dll1 locus (m–o) except for minor irregularities of Uncx4.1 expression (m, arrowhead). Expression of Dll3HA instead of Dll1 (j–l) does not rescue the patterning defects of Dll1-null mutants (p–r).
DLL3 generated from the knockin alleles is functional
To test whether the Dll3 and Dll3HA cDNAs expressed from the Dll1 locus generate functional proteins, we crossed heterozygous Dll1Dll3ki and Dll1Dll3HAki mice, respectively, to mice carrying a null mutation of Dll3, Dll3pu (Kusumi et al., 1998). Dll3pu disrupts A-P somite patterning and leads to severe malformations of the axial skeleton (Fig. 2 A, d–f). Compound heterozygous mice carrying one copy of the Dll1lacZ and the Dll3pu-null allele, respectively, i.e., mice that have only one functional copy of either gene are normal (unpublished data). Mice that are homozygous mutant for Dll3pu but are heteroallelic for the Dll1 wild type and the Dll1Dll3 knockin alleles also have one functional copy of Dll1 and one copy of Dll3 or Dll3HA expressed from the Dll1 locus. Therefore, we reasoned that functional DLL3 protein generated from the knockin alleles should at least partially rescue the phenotype of homozygous Dll3pu mice. Indeed, homozygous Dll3pu mice that carried one copy of the Dll1Dll3ki or Dll1Dll3HAki allele (n = 2 and 4, respectively) were normal or showed only subtle defects of the vertebral column (Fig. 2 A, g–i; and not depicted), in contrast to homozygous Dll3pu mice (Fig. 2 A, d–f). Consistent with normal axial skeleton development, cyclic Lfng expression and stripy expression of Uncx4.1 was restored in Dll1 Dll3HAki/+; Dll3pu/pu embryos (Fig. 2 B, m–o). This unambiguously demonstrated that the Dll3 and Dll3HA cDNA expressed from the Dll1 locus generated fully functional DLL3 protein and indicates that the C-terminal HA tag does not interfere with the physiological functions of DLL3.
DLL3 does not rescue the loss of DLL1 in vivo
To address whether the DLL3 protein can rescue some aspects of the loss of DLL1 function, we analyzed homozygous Dll1Dll3ki and Dll1Dll3HAki embryos. Embryos homozygous for the Dll1lacZ-null allele die around embryonic day (E) 11.5 and can be readily identified at E10.5 by large hemorrhages (Fig. 1 B b). Homozygous embryos with either knockin allele were virtually identical to null mutants (Fig. 1 B, c and d), and A-P somite patterning, as well as cyclic gene expression, was similarly disrupted in homozygous Dll1Dll3HAki and Dll1lacZ embryos (Fig. 2 B, compare j–l with p–r), indicating that Dll3 cannot functionally replace Dll1 in vivo. Collectively, our analyses demonstrate unequivocally that the DLL1 and DLL3 proteins are biochemically not equivalent and have divergent functions in vivo.
DLL3 does not repress DLL1-mediated Notch activation in vivo
Based on in vitro experiments and overexpression in Xenopus embryos, it was suggested that Dll3 functions as an inhibitor of Notch signaling in a cell-autonomous manner (Ladi et al., 2005). If this reflects the physiological role of Dll3 in the PSM of mouse embryos, where Dll1 and Dll3 are coexpressed in the majority of cells (Dunwoodie et al., 1997), changes in the ratio of Dll3 to Dll1 might lead to either reduced or enhanced Notch signaling and defects in somite patterning and axial skeleton development. Indeed, about one third of mice heterozygous for the Dll1-null allele showed defects in individual vertebrae, such as minor fusions or reduction of laminae, split vertebral bodies, and reduced pedicles (Cordes et al., 2004). This indicates that somite patterning is sensitive to Dll1 dosage (i.e., Notch activity) and raises the possibility that the increased gene dosage ratio of Dll3/Dll1 (2:1) in Dll1lacZ heterozygotes contributes to the haploinsufficiency phenotype. We therefore analyzed whether penetrance and expressivity of axial skeleton defects are increased in Dll1Dll3HAki embryos, which carry three copies of Dll3 but only one copy of Dll1, and thus have a Dll3/Dll1 ratio of 3:1. In 10 out of 28 Dll1Dll3HAki skeletons, we found minor vertebral malformations similar to those observed in Dll1lacZ heterozygotes (unpublished data), indicating that the increase of Dll3 dose did not enhance expressivity or penetrance of defects found in Dll1lacZ heterozygotes. In addition, we made use of the Dll1 Dll1ki allele that still contained the neo cassette. The presence of the neo cassette in the Dll1 Dll1ki allele (Dll1 Dll1ki+neo) attenuates Dll1 expression and generates a hypomorphic Dll1 allele that is lethal at birth in homozygotes (Schuster-Gossler et al., 2007). Heterozygous Dll1lacZ mice, which have only one functional copy of Dll1, and thus most likely half the level of Dll1 mRNA and protein, survive. This suggests that total DLL1 protein in homozygous Dll1 Dll1ki+neo hypomorphs is below this level, and one hypomorphic Dll1 Dll1ki+neo allele thus generates less than half of one wild-type allele (i.e., <25% of the total amount in wild type). We compared the phenotypes of mice that were heteroallelic for the hypomorphic and the null allele (Dll1 Dll1ki+neo/lacZ; <25% Dll1; two copies of Dll3) with mice heteroallelic for the hypomorphic and the Dll3 knockin allele (Dll Dll1ki/Dll3HAki; <25% Dll1; three copies of Dll3) and observed no obvious enhancement of the phenotype (Fig. 2 A, compare p–r with s–u). Thus, over the range of gene doses that we tested, we obtained no genetic evidence for antagonism of Dll1 and Dll3 during somitogenesis under physiological conditions.
To address more directly how DLL3 affects Notch activation in vivo, we analyzed the formation of the activated ICD of Notch1, NICD, in mouse embryos by immunohistochemistry. In wild-type embryos, Notch1 is activated in a sharp band in the anterior PSM, and posterior in variable patterns that reflect cyclic Notch activity (Fig. 3, A–C; Morimoto et al., 2005). Loss of DLL1 function abolishes formation of NICD in the PSM (n = 4; Fig. 3 D and not depicted; Morimoto et al., 2005), indicating that DLL1 is the major activator of Notch1 in the PSM. If DLL3 acts as an antagonist of DLL1, one would expect increased levels or expression of activated Notch1 throughout the PSM in Dll3 mutants, similar to embryos without Lfng function (Morimoto et al., 2005). In embryos lacking DLL3 (n = 14), NICD was detected in a fuzzy stripe in the anterior PSM and at the posterior end, but not throughout the PSM, and levels appeared reduced rather than increased (Fig. 3, E–G; and not depicted).
Figure 3.

NICD expression in the PSM of Dll3 mutant embryos. Whole-mount immunohistochemistry for activated Notch 1 (NICD) readily detected oscillating activity in E10.5 embryo wild-type tails (A–C; n = 13). In Dll1-null mutant tails (D), NICD was not detected in the PSM (n = 4), whereas in Dll3-null mutant tails (E–G), the pattern of Notch1 activity is static (n = 14) with no obvious up-regulation of NICD protein levels.
DLL1 and DLL4, but not DLL3, activates Notch in D. melanogaster
To further analyze the activities of Dll1 and Dll3 in vivo, we generated transgenic flies carrying UAS constructs of both Dll genes and expressed them with help of the Gal4 system during wing development using ptcGal4. The activity of Dll1 and Dll3 was monitored by analyzing the activation of the Notch target Wg. During wing development, Wg is expressed in a stripe of cells along the dorsoventral boundary under control of the Notch pathway (Klein, 2001). Expression of D. melanogaster Dl with ptcGal4 induces ectopic Wg expression along the A-P boundary (Doherty et al., 1996) and inhibited Notch activation in the region of highest Dl expression (Fig. 4, B and C, arrowheads), consistent with the known inhibition of Notch by coexpression of high levels of Dl (Doherty et al., 1996). Similarly, Dll1flag expression induced Wg along the A-P boundary (Fig. 4, D and E), indicating that the mouse DLL1 protein can activate D. melanogaster Notch in imaginal discs in vivo. This was also observed in the absence of endogenous Dl (unpublished data). Likewise, DLL4, another mammalian ligand known to activate Notch (Iso et al., 2006; Diez et al., 2007), induced Wg expression (Fig. 4, F and G), suggesting that mammalian DSL ligands that activate mammalian Notch in general can activate the D. melanogaster Notch receptor. In contrast, Wg expression was not induced in wing discs expressing Dll3 or Dll3flag (five independent lines tested for each construct; Fig. 4, H and I; and not depicted), consistent with the inability of Dll3 to substitute for Dll1 in mice. In addition, a chimeric DLL1-DLL3 ligand that did not activate mammalian Notch (Fig. 5, construct A; see the following paragraph) did not activate D. melanogaster Notch in the wing disc (Fig. 4, J and K), supporting the idea that the inability of DLL3 to activate D. melanogaster Notch does not simply reflect species differences, although formally we cannot exclude this possibility. Dll3flag did not affect the normal expression of Wg induced along the dorsoventral border by endogenous Dl and Ser in the ptc domain overlapping with the dorsoventral border (Fig. 4, H and I, arrowheads), indicating that expression of Dll3 does not block Notch activation by the endogenous ligands. Identical results were obtained with HA-tagged or untagged versions of Dll1 and Dll3 (unpublished data).
Figure 4.

Activity of Dll1 and Dll3 in D. melanogaster wing discs. Expression of D. melanogaster Dl and vertebrate orthologues with ptcGal4 in the wing imaginal discs. (A) Expression of Wg and ptcGal4 in a wing imaginal disc of the late third larval instar. Wg (red) is induced along the dorsoventral (d-v) compartment boundary by Notch signaling. Expression of UAS GFP (green) reveals the stripe-like expression domain of ptcGal4, which runs perpendicular to the Wg domain at the anterior side of the A-P compartment border. Expression within the ptcGal4 domain increases toward the posterior (right). (B and C) Ectopic Wg activation along the A-P boundary (arrows) induced by ectopic expression of D. melanogaster Dl. Two ectopic stripes of Wg expression are induced (arrows). The broader, anterior-located stripe is in the region of low Dl expression. The second thinner is induced in cells adjacent to the ptc domain. In the region with highest expression of Dl, expression of Wg is not induced because of the cis-inhibitory effect of Dl at high levels of expression. Note that high levels of Dl also suppress Notch activity at the dorsoventral boundary as indicated by down-regulation of Wg (arrowheads). Expression of Dll1flag (D and E) or Dll4flag (F and G) along the A-P boundary activates ectopic expression of Wg (arrows) in a pattern similar to Dl. However, the cis-inhibitory effect is weaker than in the case of Dl. (H and I) Expression of Dll3flag has no effect on the activity of the Notch pathway in D. melanogaster. In addition, endogenous expression of Wg along the dorsoventral border is not affected (arrowheads). (J and K) Expression of construct A (Dll1/Dll3 chimera; Fig. 5) does not activate the Notch pathway in D. melanogaster. (L and M) Ectopic Wg activation along the A-P wing border (arrow) by ectopic expression of construct C (Dll1/Dll3 chimera; Fig. 5). In the regions with highest expression, construct C slightly suppresses Notch activity, as indicated by the slight down-regulation of Wg in its normal expression domain (arrowheads). (N and O) Ectopic Wg activation along the A-P wing border (arrows) by ectopic expression of construct G (Dll1/Dll3 chimera; Fig. 5). In the regions with highest expression, construct G slightly suppresses Notch activity, as indicated by the slight down-regulation of Wg in its normal expression domain (arrowheads). Note that construct C, as well as construct G, signals to cells adjacent to the posterior domain boundary (L–O, arrows).
Figure 5.

Analysis of Dll1-Dll3 chimeric ligands. (A) Schematic overview of wild-type DLL1 and DLL3 and chimeric constructs used to generate stably expressing CHO cell lines. DLL1 protein is shown in black and DLL3 in red. Numbers indicate the amino acid residue numbers. DSL, DSL domain; E1–E8, EGF-like repeats; the flag tag is indicated by gray ovals and the HA tag in construct H by a black oval. Corresponding EGF repeats of DLL1 and DLL3 are connected by black lines. (B) Western blot analysis of cell lysates (input) and streptavidin immunoprecipitated protein after surface biotinylation (IP). CHO cells stably expressing chimeric ligands show similar (input A and B) or even more (input C–H) expression compared with DLL1-expressing cells. All chimeric ligands are present on the cell surface (IP), chimeric ligands A, C, and G at lower levels and chimeric ligands B, D–F, and H at similar or even higher levels compared with DLL1. (C) Notch transactivation assays. CHO cells stably expressing DLL1 and chimeric ligands as shown in panel A were cocultivated with Notch1-HeLa cells transfected with the (RbpJ)6-luciferase reporter gene. Luciferase activity (percentage of activation) of chimeric ligands A–H was measured against negative (CHO wild-type cells) and positive (CHO-Dll1 cells) controls set to 0 and 100% relative activation, respectively. Four cocultivations were performed per construct and analyzed in two independent experiments each, including negative and positive controls.
DLL1 domains required for Notch activation
The DLL1 and DLL3 proteins show considerable differences in their amino acid sequences. Compared with DLL1, the DLL3 protein has a divergent DSL domain, fewer EGF repeats, and altered spacing between some EGF repeats, and it lacks Lysine residues and a PDZ binding domain in its ICD (see Discussion). To analyze which of these structural differences contribute to the functional divergence, we generated various C-terminally flag-tagged chimeric Dll1-Dll3 cDNAs (Fig. 5 A) and cell lines stably expressing the chimeric proteins. Cell lines were analyzed for expression by Western blotting to identify clones expressing similar levels of different protein variants for further analyses. Chimeric ligands A–H but not DLL3 (see the following section) were readily detected on the cell surface after surface biotinylation, followed by immunoprecipitation and Western blotting (Fig. 5 B).
A chimeric ligand, in which the N-terminal portion of DLL3, including the DSL domain, was replaced by the corresponding DLL1 sequence (Fig. 5 A, construct A) did not activate Notch neither in vitro (Fig. 5 C) nor in D. melanogaster wing discs (Fig. 4, J and K), indicating that the N terminus and DSL domain of DLL1 are not sufficient to confer Notch activating properties on DLL3. The inability of construct A to activate Notch could be due to the presence of the ICD of DLL3, or specific EGF repeats, or both. To distinguish between these possibilities, we tested a chimeric ligand consisting of the extracellular domain of DLL1 and the transmembrane domain (TM) and ICD of DLL3 (Fig. 5 A, construct B). This construct also did not activate Notch (Fig. 5 C), indicating that the ICD of DLL1 is essential, which was further supported by the inability of a DLL1 variant that lacked the ICD (Fig. 5 A, construct H) to activate Notch (Fig. 5 C). To further define features in the extracellular domain of DLL1 that are essential for Notch activation, we tested various constructs that contained the ICD of DLL1 and combinations of portions of the extracellular domains of DLL1 and DLL3.
EGF repeats 3–6 of DLL3 closely resemble repeats 5–8 in DLL1. We first tested a DLL1 variant that had EGF repeats 5–8 replaced by EGF 3–6 from DLL3. This ligand (Fig. 5 A, construct C) effectively activated Notch in vitro (Fig. 5 C) and in D. melanogaster wing discs (Fig. 4, L and M), indicating that the four proximal EGF repeats of DLL1 and DLL3 are functionally equivalent. We thus focused on the distal EGF repeats for further analyses. A DLL1 version that contained EGF repeats 1 and 2 from DLL3 (Fig. 5 A, construct D) did not activate, suggesting that either the spacing of DLL3 EGF repeats or their sequence is important. To distinguish these possibilities, we inserted the DLL3 spacer sequence between EGF repeat 1 and 2 of DLL1 (Fig. 5 A, construct E) or deleted the spacer between EGF repeat 1 and 2 of DLL3 (Fig. 5 A, construct F). Both ligands did not activate Notch, indicating that both spacing and sequence of the first EGF repeats are critical for DLL1 function. To test whether the DSL domain and distal two EGF repeats of DLL1 are sufficient to activate Notch, we replaced the six proximal EGF repeats of DLL1 by the EGF repeats of DLL3 (Fig. 5 A, construct G). This ligand activated Notch in vitro, though not at maximal levels (Fig. 5 C), and in D. melanogaster wing discs (Fig. 4, N and O), suggesting that EGF repeats 1 and 2 of DLL1, or the spacing between these EGF repeats, are also essential for full activity.
DLL3 is predominantly located intracellularly
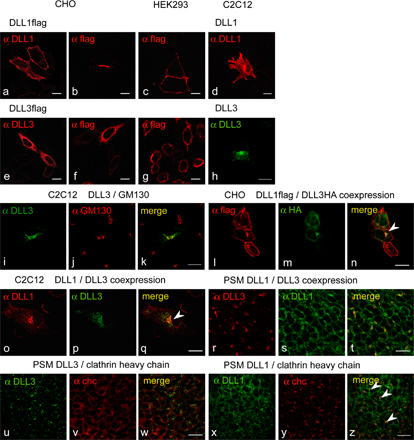
In the course of analyzing chimeric ligands, we also generated CHO cell lines stably expressing DLL3. Surprisingly, it proved difficult to obtain cells that expressed DLL3 efficiently on the surface, as determined by surface biotinylation and subsequent immunoprecipitation and Western blotting, and several clones expressing DLL3 had only minor amounts on the surface. Even when DLL3 was detected on the surface, relative amounts of DLL3 were always significantly lower than those of DLL1 (Fig. 6 A, compare lane a with lanes b and c). Likewise, we detected no or minor amounts of biotinylated DLL3 on the cell surface of C2C12, HEK293, and CHO cells after transient transfection, although DLL3 protein was readily detected in cell lysates (Fig. S1, available at http://www.jcb.org/cgi/content/full/jcb.200702009/DC1). To further analyze the apparent difference in surface presentation, we transiently expressed DLL3 in cells stably expressing DLL1, and vice versa, and analyzed the surface expression of both proteins. In either case, DLL1 was readily detected on the surface, whereas DLL3 was not or only at minor amounts (Fig. 6 A, lanes d–i). Consistent with the surface biotinylation data, DLL1 was readily detected by immunohistochemistry on the cell surface of CHO cells stably expressing DLL1 (Fig. 6 B a), as well as on the surface of transiently transfected CHO, HEK293, and C2C12 cells (Fig. S2, a–d; and not depicted), and on the apical surface of D. melanogaster wing disc cells (Fig. 6 B, i and j), although surface expression patterns were variable, and DLL1 protein was also detected in vesicular structures in the cytoplasm. In contrast, DLL3 staining was mostly perinuclear (Fig. 6 B b and Fig. S2, e–h). In the perinuclear region, DLL3 colocalized with GM130, a marker for the cis-Golgi network (Fig. S2, i–k). When coexpressed, DLL1 and DLL3 colocalized in perinuclear structures but essentially not at the membrane (Fig. 6 B e and Fig. S2, n and q). Collectively, our data suggested that in cells expressing DLL1 and/or DLL3, DLL1 is largely on the cell surface and DLL3 is largely intracellular, including the Golgi apparatus. Similarly, in embryos in PSM cells, endogenous DLL1 was present on the surface and colocalized with membrane proteins detected by an anti-pancadherin antibody (Fig. 6 B, k–m), in addition to some intracellular DLL1 that colocalized mainly with GM130 (Fig. 6 B, q–s). Importantly, endogenous DLL3 was not detected on the membrane of PSM cells (Fig. 6 B, n–p) but in intracellular punctae largely overlapping with GM130 (Fig. 6 B, t–v), indicating that the localization of DLL3 in the Golgi network occurs under physiological conditions. Both DSL proteins colocalized in some areas but were otherwise essentially nonoverlapping (Fig. S2, r–t). We also observed in PSM cells colocalization of DLL1 but not DLL3 with clathrin heavy chain that marks clathrin-coated vesicles and early endosomes (Fig. S2, u–z), further supporting differential subcellular localization of DLL1 and DLL3. Collectively, our data suggest that in vivo DLL3 accumulates in the Golgi network and only minor amounts, if any, are present on the surface of PSM cells.
Figure 6.

Localization of DLL1 and DLL3 proteins. (A) Western blot analysis of cell lysates (input) and streptavidin-immunoprecipitated protein after surface biotinylation (IP). CHO cells stably expressing DLL3 (b and c) at amounts similar to cells expressing DLL1 (a) present significantly less DLL3 on the surface. L cells (mouse fibroblast cell line) coexpressing DLL3flag at significantly higher levels than DLL1HA (compare input lanes d and g) present DLL1 efficiently on the surface but not DLL3 (compare IP lanes d and g). CHO cells coexpressing DLL3HA and DLL1flag (compare input lanes e and f with h and i) present DLL1 efficiently on the surface but DLL3 only in trace amounts (compare IP lanes e and f with h and i). (B) Detection of DLL1 and DLL3 by immunofluorescence. (a–e) Localization of DLL1 and DLL3 in overexpressing CHO cells. CHO cells expressing DLL1 (a and c) show a clear cell surface staining, whereas DLL3 (b and d) is detected almost exclusively inside the cell. DLL1 and DLL3 colocalize only in some vesicular structures (e, arrowheads) but not significantly at the membrane. (f–j) Localization of DLL1 and DLL3 in D. melanogaster wing disc cells. (f) Overview of a wing disc stained for the apical cell membrane marker aPKC and DLL3flag, and Hoechst staining to visualize nuclei. (g and h) Confocal images of two opposed apical cell membranes (red) of an epithelial fold in panel f. DLL3 is found in intracellular granules or vesicles. (i and j) Confocal images of two opposed apical cell membranes (red) of a wing disc stained for aPKC and DLL1flag. DLL1 outlines cell membranes and colocalizes at the apical membrane with aPKC (j, arrowheads). (k–v) Immunofluorescent detection of DLL1 and DLL3 in PSM cells of E9.5 embryos. Endogenous DLL1 is present at the surface (k) and colocalizes with the membrane (m) and in vesicular structures with the cis-Golgi marker GM130 (s). DLL3 does not localize to the membrane (n) and does not colocalize with anti-pancadherin staining (p) but is detected in vesicular structures in the cytoplasm (t), mostly overlapping with GM130 (v). Bars, 10 μm.
Protein domains affecting subcellular localization
The surface biotinylation results suggested that chimeric ligands differ with respect to their propensity to localize to the surface. To analyze in more detail how different portions of DLL1 and DLL3 affect the distribution of stably overexpressed chimeric ligands in the cell, we studied their localization on the cellular level by indirect immunofluorescence (Fig. 7). Chimeric ligands that contained the TM and ICD (TM-ICD) of DLL1 and at least the DLL1 N-terminal portion including the DSL domain fused to extracellular DLL3 sequences were detected on the cell surface, in addition to variable intracellular expression (Fig. 7 B, c–e). In contrast, the DLL1 N terminus alone (chimera J) was not sufficient to direct detectable surface expression (Fig. 7 B f), similar to the extracellular domain of DLL3 fused to DLL1 TM-ICD, (chimera K; Fig. 7 B g). Because DLL1 lacking the ICD was also detected predominantly on the surface (Fig. 7 B b), the DSL domain of DLL1 appears to be necessary to direct surface expression of these chimeric ligands. A chimera containing the DLL1 extracellular domain juxtaposed to the DLL3 TM-ICD (chimera B) was predominantly on the surface, although not evenly distributed (Fig. 7 B h). Chimeras that contained the DLL1 N-terminal portion, including the DSL domain, and the DLL3 TM in the context of juxtaposed DLL3 intra- and extracellular sequences were found predominantly intracellular (Fig. 7 B, i and j). In contrast to cells expressing DLL1, or construct H or B, that consistently showed clear expression on the surface, surface presentation of most chimeric ligands was variable and not detected in all expressing cells. As expected, a chimera containing the DLL1 N-terminal portion without the DSL domain fused to DLL3 (chimera M) showed no detectable membrane localization (Fig. 7 B k). Collectively, it appears that the DLL3 TM and adjacent extra- and intracellular sequences contribute to retention of chimeric ligands in intracellular compartments and localization of DLL3 in the Golgi network.
Figure 7.

Localization of chimeric DLL1 and DLL3 proteins. (A) Schematic representation of chimeric proteins containing the ICD of DLL1 (left) or DLL3 (right) arranged according to the extent of extracellular DLL1 sequences (top to bottom). Gray parts and red parts indicate DLL1 and DLL3 sequences, respectively. White filling indicates DSL domains, light gray or red shading indicates TMs, and filled boxes indicate EGF repeats. (B) Confocal images of CHO cells stably expressing chimeric ligands and stained by indirect immunofluorescence. Similar to DLL1 (a) and DLL1 lacking the ICD (b), chimeric ligands that contained the TM-ICD of DLL1 and the DLL1 N-terminal portion including the DSL domain were detected on the cell surface (c–e), in addition to some variable intracellular expression. Presence of the DLL1 N terminus alone was not sufficient to direct detectable surface expression (f), similar to the extracellular domain of DLL3 fused to DLL1 TM-ICD (g). Surface presentation of a chimera containing the DLL1 extracellular domain juxtaposed to the DLL3 TM-ICD (h). Chimeras that contain the DLL1 N-terminal portion including the DSL domain, and the DLL3 TM in the context of juxtaposed DLL3 intra- and extracellular sequences were retained intracellularly (i and j). Intracellular localization of DLL3 with the N terminus replaced by the corresponding DLL1 sequence, and of DLL3, respectively (k and l). A–C, G, and H refer to chimeras shown in Fig. 5 A and I–M to additional ones. Chimera H (b) was detected with anti-DLL1 and all other chimeras with anti-flag antibodies. Bar, 10 μm.
Discussion
This study addresses the biochemical and functional equivalence of the Notch ligands DLL1 and DLL3 and shows that, under physiological conditions, in vivo DLL3 cannot substitute for DLL1 in mice, and DLL1 (and DLL4, but not DLL3) can function as an activating Notch ligand in mice and D. melanogaster. In addition, we have defined regions in DLL1 that are essential for its function as activating Notch ligand. In contrast to the reported potent inhibitory functions of DLL3 overexpressed in vitro, and in Xenopus embryos (Ladi et al., 2005), increased ratios of Dll3/Dll1 in mouse embryos or overexpression in D. melanogaster did not provide evidence for antagonism between Dll1 (or Dl or Ser) and Dll3. In addition, our data show an unanticipated accumulation of DLL3 in the Golgi network that warrants further investigation of potential intracellular functions of DLL3.
DLL3 cannot substitute for DLL1 in vivo
The rescue of Dll3pu mutant phenotype by Dll3 expressed from the Dll1 locus demonstrated that sufficient amounts of functional DLL3 protein are generated from the knockin allele. However, homozygous Dll1Dll3HAki or Dll1Dll3ki embryos were virtually indistinguishable from Dll1-null mutant embryos (Fig. 1), indicating that DLL3 cannot substitute for DLL1 and activate Notch in vivo. Given that <25% of normal Dll1 levels are sufficient to significantly improve the phenotype of Dll1-null mutants, and that DLL3 was not found on the cell surface in appreciable amounts (see below), it is highly unlikely that DLL3 has the ability to activate Notch under physiological conditions. Our results provide definitive proof that the DLL1 and DLL3 proteins are functionally highly diverged and are biochemically not equivalent in vivo and support recent in vitro data showing that DLL3 cannot bind to and activate Notch in trans (Ladi et al., 2005).
Regions in DLL1 required for activation of Notch
The DLL1 and DLL3 proteins differ in various respects. The DSL domain, which is essential for ligand binding to Notch (Shimizu et al., 1999), is highly divergent in DLL3 and lacks a YY and a GWXG motif that is present in the DSL domains of other Notch ligands in various species. These features appear to be essential, as mutation of either of these motifs abolishes Dll1 function (unpublished data). However, the transfer of the DLL1 N terminus and DSL domain to the DLL3 EGF repeats (Fig. 5, construct A) was not sufficient to confer activating properties to DLL3, which was also reported by Ladi et al. (2005), indicating that other portions of DLL1 are essential for its activating properties. The C-terminal amino acid sequence of DLL1 and Jagged1, but not DLL3, constitutes a potential PDZ ligand, which was recently shown to be required for Jagged1-mediated cellular transformation (Ascano et al., 2003). The ICD of DLL3 also lacks lysine residues that are required for ubiquitin conjugation and ubiquitin-dependent processing and internalization of DLL1 (Pavlopoulos et al., 2001; Barsi et al., 2005; Koo et al., 2005; Pitsouli and Delidakis, 2005). The exchange of the TM-ICD of DLL1 with the corresponding region from DLL3 (Fig. 5, construct B) abolished the ability to activate Notch, indicating that properties of this region are critical for DLL1 function. The C-terminal tag is likely to interfere with binding to PDZ binding proteins. Because C-terminal tagging of the full-length DLL1 protein does not interfere with Notch activation in trans in cell culture experiments and flies, PDZ binding is unlikely to be required for Notch activation. This is supported by the observation that a zebrafish DeltaD variant that cannot interact with PDZ domains functions normally as a Notch ligand (Wright et al., 2004). Thus, the inability of construct B to activate Notch might be due to loss of ubiquitin-dependent processing (Le Borgne et al., 2005) or loss of other as-yet-undefined modifications (Ilagan and Kopan, 2007) that require the presence of the ICD. This is further supported by the results obtained with construct H, which indicate that mere presentation of the extracellular domain of DLL1 on the surface is not sufficient for Notch activation in trans. Activating properties of DLL1 were maintained when the N-terminal region of DLL1, including EGF repeats 1 and 2, was fused to EGF repeats 1–6 from DLL3, followed by the TM-ICD of DLL1 (construct G). This was not true of constructs that contained DLL3 EGF repeats 1 and 2 with or without correct spacing (constructs D and F) or DLL1 EGF repeats 1 and 2 with changed spacing (construct E). This indicates that EGF repeats 1 and 2 of DLL1 have specific properties that are not present in the corresponding EGF repeats of DLL3 and is consistent with a requirement for the DSL domain and EGF repeats 1 and 2 in Jagged1 for Notch binding in vitro (Shimizu et al., 1999). Collectively, DLL3 has acquired several alterations, each of which individually appears to be sufficient to abolish its function as an activating Notch ligand.
Is DLL3 an intracellular component of the Notch pathway?
Surprisingly, in contrast to DLL1, we found that the majority of DLL3 overexpressed in cell lines resided within the cell and was virtually absent from the cell surface. The same was true for DLL3 expressed in D. melanogaster wing disc cells and for endogenous DLL3 expressed in the PSM. Thus, DLL1 and DLL3 appear to differ significantly in their propensity to localize to the cell membrane. This is further supported by the distinct subcellular localization of DLL1/DLL3 chimeras depending on the presence of particular regions of DLL1 and DLL3 (Fig. 7). The transmembrane region of DLL3, together with flanking sequences on either side, appears to prevent efficient surface presentation and might be important for Golgi retention, as has been found for other Golgi-retained proteins, such as glycosyltransferases (Colley, 1997). The presence of a retention signal could explain why significant amounts of DLL3 can only be detected on the surface when DLL3 is highly overexpressed. In this context, it is worth mentioning that it is not clear if the majority of DLL3 was located intracellularly in the study of Ladi et al. (2005), as they did not report immunohistochemical data. Importantly, endogenous DLL3, in contrast to DLL1, did not colocalize with a membrane marker in PSM cells but was found to colocalize with GM130. Thus, under physiological conditions, the majority of DLL3 appears to reside in the Golgi apparatus (Fig. 6), suggesting that it can exert its function inside the cell. However, we cannot exclude the possibility that minor amounts of DLL3 act on the cell surface. Binding of DLL3 to Notch in cis (when coexpressed in the same cells), but not in trans (when expressed in adjacent cells; Ladi et al., 2005) would also be consistent with Notch interaction and DLL3 function inside the cell.
Is DLL3 an antagonist of DLL1 in vivo?
It has been suggested that DLL3 acts as a Notch antagonist during somitogenesis (Ladi et al., 2005). This is because in vitro DLL3 did not activate Notch in trans, but rather inhibited Notch signaling when expressed in the same cell as Notch. In addition, overexpression of Dll3 in Xenopus embryos enhanced neuronal differentiation. Cell-autonomous inhibition and intracellular association with Notch is also a property of ligands that physiologically activate Notch in trans when they are overexpressed in cis (Doherty et al., 1996; Henrique et al., 1997; Klein et al., 1997; Sakamoto et al., 2002; Itoh et al., 2003). Thus, inhibition of Notch by overexpressed DLL3 in cis is not necessarily indicative of an antagonistic function in vivo, a notion that is supported by our results. Heterozygous mice that carry the Dll1Dll3HAki or Dll1Dll3ki alleles (i.e., mice that overexpress Dll3 and simultaneously have only one copy of Dll1 were indistinguishable from heterozygous Dll1-null mice). In these mice, Dll3 is also expressed ectopically in the posterior somite compartment with no apparent effect on Dll1 function in somite patterning. Also, the phenotypes of embryos with further reduced Dll1 levels with or without additional Dll3 were virtually identical. Thus, reducing the activating ligand DLL1 and simultaneously increasing DLL3 did not lead to observable enhancements of Dll1 phenotypes, suggesting that overexpression at physiological levels does not significantly impair Notch activation. If DLL3 inhibits Notch in vivo, loss of Dll3 function should lead to increased Notch activity and, thus, up-regulation of Notch target genes. However, known Notch1 targets, like Nrarp or Hes5, are either not significantly deregulated or down-regulated in Dll3 mutants (Krebs et al., 2001; Dunwoodie et al., 2002), which suggests a stimulatory function of DLL3 in Notch signaling. Also, we did not observe evidence for increased NICD production in the PSM. This implies that, if DLL3 acts to antagonize DLL1, this effect would have to occur downstream of NICD formation (i.e., Notch activation). A recent study indicates that in order for cis inhibition to occur, the ligands have to come to the cell surface (Glittenberg et al., 2006). However, we found that the vast majority of DLL3 resides in the Golgi apparatus. This finding further argues against an inhibitory function of DLL3, at least by a mechanism similar to the other ligands. Also, the similarity of the Dll3- and Lfng-null phenotypes does not allow one to conclude that DLL3 acts as an inhibitor similar to LFNG, as loss of Lfng function and overexpression of Lfng cause virtually identical phenotypes (Serth et al., 2003).
Collectively, our in vivo data unambiguously demonstrate that DLL1 and DLL3 have distinct functions under physiological conditions in vivo and open the possibility that these proteins function at different sites in the cell. In addition, our in vivo data do not support an antagonistic function of DLL1 and DLL3 in PSM cells and point toward a potential, thus far undefined intracellular function.
Materials and methods
Generation of Dll3 knockin mice
The Dll3 ORF (with or without a C-terminal HA tag) was fused to a genomic Dll1 SacI–EcoRI fragment containing part of exons 9, 10, and 11. A PGK-neomycin expression cassette flanked by loxP sites was introduced 3′ to the Dll3-Dll1 fusion. A 4.6-kb BamHI–KpnI fragment of Dll1 genomic DNA upstream of the ATG fused in frame to Dll3, and ∼3 kb of Dll1 genomic DNA downstream of the SalI site in exon 2 were included as regions of 5′ and 3′ homology, respectively. A diphtheria toxin A expression cassette was cloned upstream and downstream of the homology arms, respectively. The analogous Dll1ki targeting vector was cloned as previously described (Schuster-Gossler et al., 2007). Linearized vector DNA was electroporated into 129Sv/ImJ embryonic stem cells and selected as described previously (Abdelkhalek, 2004). Correctly targeted clones were identified by PCR using primers derived from the neo sequence (TGTCACGTCCTGCACGACG) and genomic sequences downstream of the targeting vector (GGTATCGGATGCACTCATCGC). PCR-positive clones were verified by Southern blot analysis using external probes located 3′ and 5′ to the regions of homology in the vector and used to generate chimeric mice. The neo cassette was removed in the female germ line using ZP3∷Cre mice (de Vries et al., 2000; backcross generation N6 to 129Sv/ImJ).
Genotyping of mice and embryos
Genomic DNA isolated from tail biopsies or yolk sacs was genotyped by PCR. Used primers were as follows: for Dll1 wild-type allele, Dll1F2 (CTGAAGCGACCTGGCCCTGATAGCAC) and Dll1R1 (GGAGTCGACACCCAGCACTGGCG; 425 bp); for Dll1lacZ allele, Melta38 (ATCCCTGGGTCTTTGAAGAAG) and LacZ1/Dll1ko (CAAATTCAGACGGCAAAC; 578bp); for Dll1Dll3HA, Dll1Dll3, and Dll1Dll1 Δneo alleles, EGF-neoFOR (ATGGACAGCATTTCCTCCTGCCTC) and EGF-neoREV (GCCAGTCAGTTCCCAGTAAGAAGTC; 280 bp); for Dll1 Dll1+neo allele, neoF (TGGATGTGGAATGTGTGCGAG) and Dll1h3′B6 (AAGGGGAGAAGATGCTTGATAACC); for Dll3pu allele, Dll3pu1 (ACGAGCGTCCCGGTCTATAC) and Dll3pu2 (AGGTGGAGGTTGGACTCACC). After amplification, PCR products were cleaved with HaeIII and separated on 3% agarose gels (Dll3pu/pu, 100 bp; and wild type, 65 bp).
Skeletal preparations of E18.5 embryos
E18.5 embryos were eviscerated and skinned, and skeletons were stained as described previously (Serth et al., 2003) with slightly longer incubation periods. Stained skeletons were stored and photographed in ethanol/glycerol (1:1) using a microscope (M420; Leica) with Apozoom 1:6 and Photograb-300Z version 2.0 software (Fujiflm).
Whole-mount in situ hybridization
Whole-mount in situ hybridization was performed following a standard procedure with digoxygenin-labeled antisense riboprobes (Wilkinson, 1992) with minor modifications. Pictures were taken using the Leica M420 microscope with Apozoom 1:6 and Photograb-300Z version 2.0 software.
Whole-mount immunohistochemistry
E10.5 embryos were collected in PBS, immediately fixed in MeOH/DMSO/30% H2O2 (1:1:1) for 1 h on ice, and washed 3× 10 min and 2× 1 h in 50 mM NH4Cl at room temperature, followed by an incubation in TS-PBS (PBS, 10% FCS, and 1% Triton X-100) for 3× 10 min and 2× 1 h at 4°C. Embryos were then successively incubated with anti–cleaved Notch1 antibody (Val1744; Cell Signaling), biotinylated anti-rabbit antibody (Vector Laboratories), and streptavidin-HRP (NEL750; Perkin-Elmer) at a dilution of 1:100 in TS-PBS overnight at 4°C, respectively. Between antibody incubations, embryos were washed repeatedly with TS-PBS during the day at room temperature. For the color reaction, embryos were incubated 2× 10 min in solution A (100 mM Tris-HCl, pH 7.5, 0.1% Triton X-100, and 0.04% 4 Chloro-1 naphthol), 2× 5 min and 1× 10 min in solution B (solution A without Triton X-100), 1× 10 min in solution C (2 parts of 0.125% 4 Chloro-1 naphtol in 100% ethanol mixed with 3 parts of distilled water) followed by incubation in solution D (solution C with 0.006% H2O2), and stopped with 4% paraformaldehyde. Pictures were taken using the Leica M420 microscope with Apozoom 1:6 and Photograb-300Z version 2.0 software.
Whole-mount immunofluorescence
Embryos were dissected at E9.5, fixed in 4% paraformaldehyde overnight at 4°C, and stored in methanol at −20°C. Rehydrated embryos were washed three times in antigen unmasking solution (Vector Laboratories), heated to 100°C for 10 min, and allowed to cool to room temperature. Embryos were washed in water and cracked for 8 min in 100% acetone prechilled to −20°C and then rehydrated in water. Embryos were blocked overnight in 1% BSA dissolved in PBS-TR (PBS containing 0.1% Triton X-100) at 4°C. Primary antibodies diluted in block were incubated with embryos at 4°C for 2–3 d with gentle agitation. Embryos were washed six times in PBS-TR for 30 min each and then reblocked for 1–2 h at room temperature. Fluorochrome-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories and Invitrogen) were diluted in block and incubated with embryos at 4°C overnight with gentle agitation. The embryos were washed six times in PBS-TR for 30 min each, cleared by successive 10-min washes in 25% glycerol, 50% glycerol, and 70% glycerol. The posterior third of the embryos was dissected and flat-mounted sagittally in ProLong Gold antifade (Invitrogen). Fluorochromes used were Texas red, Alexa Fluor 488, FITC, Cy2, Cy3, and Cy5. Labeled cells were analyzed at room temperature by confocal laser-scanning microscopy using the LSM 510 Meta (Carl Zeiss MicroImaging, Inc.) connected to the inverted microscope (Axiovert 200M; Carl Zeiss MicroImaging, Inc.) with a Plan Apochromat 63×/1.4 oil differential interference contrast objective or using a TCS SP confocal microscope (Leica) using a PL APO 100×/1.4 objective (Leica). For image acquisition, LSM 510 and Leica Confocal Software v2.5 were used, respectively. For images acquired on the TCS SP confocal microscope, ImageJ was used to add scale bars. Pictures were processed and assembled using Photoshop and Illustrator CS (Adobe).
Immunofluorescence staining of cells
Immunocytochemistry was performed as described by Dahlqvist et al. (2003) and visualized at room temperature using a TCS SP confocal microscope or as follows. Cells grown on gelatin-coated coverslips were rinsed twice with PBS and fixed with methanol for 10 min at 4°C. After three washes with PBS, the cells were blocked with 5% donkey serum in PBS for 30 min at room temperature. Cells were incubated with the primary antibody for 1 h at room temperature and, after three washes with PBS, with the fluorochrome-conjugated secondary antibody (Dianova; Invitrogen). After washing, the coverslips were mounted in Gel/Mount (Biomeda) or ProLong Gold antifade. Texas red–, FITC- and/or Alexa Fluor 488–labeled cells were analyzed at room temperature by confocal laser-scanning microscopy using the LSM 510 Meta connected to the inverted microscope Axiovert 200M with a Plan Apochromat 63×/1.4 oil differential interference contrast objective. Images were processed using LSM 510 software. Pictures were processed and assembled using Photoshop and Illustrator CS.
Generation of expression constructs
The pTracer-Dll1Flag plasmid (a gift from S. Chiba, University of Tokyo, Tokyo, Japan) was modified by inserting an IRES-neo cassette after the Dll1Flag ORF and served as a vector (pTracer-IRESneo) for expression of flag-tagged Dll1, Dll3, and chimeric ligands. Chimeric ligands were generated by conventional cloning methods. Junctions between the Dll1 and Dll3 sequences were created without changing the amino acid sequence by PCR mutagenesis using primers with a restriction site–containing overhang. In the case of the chimeric ligands D and E, two gene fragments containing a deletion or an insertion between EGF1 and -2 were synthesized (GenScript). In addition, HA-tagged versions of Dll1 and Dll3 were cloned into pTracer. The integrity of all constructs was verified by sequencing. The junctions of Dll1 and Dll3 sequences in chimeric ligands are shown in Table S1 (available at http://www.jcb.org/cgi/content/full/jcb.200702009/DC1).
Cell lines
L-Dl19 cells stably expressing rDll1HA were provided by G. Weinmaster (University of California, Los Angeles, Los Angeles, CA). CHO cells stably expressing Dll1-Dll3 chimeric ligands were generated by transfection of CHO cells using Jetpei (BIOMOL Research Laboratories, Inc.) according to the manufacturer's instructions followed by neomycin selection. HeLa cells stably expressing Notch1 (Jarriault et al., 1998) were provided by A. Israël (Institut Pasteur, Paris, France).
Notch transactivation assay
HeLaN1 cells were transiently transfected with the Rbp-J luciferase reporter construct (Rbp)6-luc (Minoguchi et al., 1997) using Jetpei, following the manufacturer's instructions. 106 transfected HeLaN1 cells were cocultivated on 6-well plates for 24 h with 106 CHO cells expressing ligands. Each CHO cell line was cocultivated four times in two independent experiments. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System (Promega). Firefly luciferase activity was normalized to cotransfected Renilla luciferase activity (pRL-TK; Promega). Expression of chimeric ligands was verified by Western blot analysis.
Cell surface biotinylation
Biotinylation-streptavidin pull down was performed essentially as described previously (Bush et al., 2001) or as described below. Cells were plated on 6-cm dishes and grown to confluence. Plates were washed three times with cold PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.4 mM KH2PO4, pH 7.3, 1 mM MgCl2, and 0.1 mM CaCl2) and placed on ice with 500 μl PBS. 10 μl Sulfo-NHS-LC-Biotin solution (5 mg/ml in 0.1 M sodium phosphate buffer, pH 7; Pierce Chemical Co.) were added three times in 10-min intervals. After 30 min, the biotin solution was aspirated, and the plates were washed once with 50 mM glycine in DME and incubated for 30 min to quench the biotinylation reaction. Cells were washed twice with PBS and lysed with 400 μl RIPA (50 mM Tris/HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% DOC, and 0.1% SDS, supplemented with 2.8 μg/ml aprotinin, 0.15 mM benzamidine, 2.5 μg/ml leupeptin, and 2.5 μg/ml pepstatin A). Lysates were incubated for 30 min on ice and centrifuged for 10 min at 12,000 g to remove cellular debris. The biotinylated proteins were precipitated with streptavidin agarose (Sigma-Aldrich) overnight at 4°C. The streptavidin agarose beads were washed three times with RIPA before resuspension in 2× sample buffer. Equivalent amounts of lysates and precipitates were subjected to SDS-PAGE and analyzed by Western blotting as described.
Antibodies
Antibodies used were as follows: HA (rat; clone 3F10; Boehringer), Wg (mouse; clone 4D4; Developmental Studies Hybridoma Bank), β-gal (rabbit, Cappel Research Products), Flag (mouse; clone M2; Sigma-Aldrich), PKCζ C20 (rabbit; Santa Cruz Biotechnology, Inc.), DLL1 (rabbit; Santa Cruz Biotechnology, Inc.), GM130 (mouse; clone 35; BD Biosciences), pancadherin (mouse; clone CH-19; Sigma-Aldrich), and clathrin heavy chain (BD Biosciences). Monoclonal antibodies against DLL1 were generated by immunization of rats with a peptide comprising amino acids 524–540 (PGPMVVDLSERHMESQG) of mouse DLL1 coupled to KLH or ovalbumin (Peptide Specialty Laboratories) subcutaneously and intraperitoneally with a mixture of 50 μg peptide-KLH, 5 nmol CPG oligonucleotide (Tib Molbiol), 500 μl PBS, and 500 μl IFAs. After a 6-wk interval, a final boost without adjuvant was given 3 d before fusion of the rat spleen cells with the murine myeloma cell line P3X63-Ag8.653. Hybridoma supernatants were tested in ELISA using the specific peptide or an irrelevant peptide coupled to ovalbumin. Peptide-specific mAbs were further characterized in Western blotting. mAb PGPM-1F9 reacted specifically with the DLL1 protein and was used for this study. In addition, guinea pig antisera were raised against the peptide CSPEHGYCEEPDE mapping to residues 222–234 of mouse DLL3 and affinity purified according to the manufacturer's instructions (Peptide Specialty Laboratories).
Transgenic fly lines
All constructs were cloned into the pUAST vector (Brand and Perrimon, 1993) and used to generate transgenic flies by P-element–mediated transformation of D. melanogaster embryos (Rubin and Spradling, 1982). mDLL1-Flag was cloned through EcoRI + XbaI digestion from pTracerCMVDll1. mDLL3-Flag was cloned with EcoRI + NotI digestion from pTracerCMVDll3. pUAST–construct G plasmid was generated by a three-fragment ligation with the EcoRI–NotI and NotI–NotI fragments from pTracerCMV construct G into EcoRI + NotI digested pUAST vector. pUAST–construct C was cloned by an EcoRI + XbaI digestion from the pTracerCMVconstruct C. ratDLL1HA was cloned with XbaI from the pEF-Bos vector (pEF-Bos-ratDLL1HA vector was provided by G. Weinmaster). mDLL4-cDNA was received from Amgen and cloned by EcoRI digestion from pCR2.1 (Invitrogen) into the pUAST vector. DLL4HA was generated by replacing the PshAI–KpnI fragment of the DLL4-cDNA with a 351-bp PCR product generated by using following oligonucleotides: A-5′-CCAGCTCAAAAACACAAACCAGAAG-3′ and B-5′ AATTCTCTAGATCAAGCGTAATCTGGCACATCGTATGGGTAAGCTACCTCTGTGGCAATCACACA-3′. Activity of the Notch signaling pathway was revealed by monitoring the expression of Wg and the synthetic reporter construct Gbe+Su(H)m8-lacZ with antibody staining (Furriols and Bray, 2001). Immunostainings of wing imaginal discs were performed as described by Jaekel and Klein (2006) using Alexa 488, 568, and 647 goat anti–mouse, Alexa 568 and 647 goat anti–rat, and Alexa 568 and 647 goat anti–rabbit antibodies, respectively (Invitrogen). Discs were mounted in VectaShield H-1000 (Vector Laboratories), and fluorochromes were visualized using an Axioplan2 with ApoTome (Carl Zeiss MicroImaging, Inc.), 10×/0.30 Plan-NEOFLUAR, 25×/0.80 Imm Korr Plan-NEOFLUAR, and 63×/1.4 Oil Plan-Apochromat lenses (Carl Zeiss MicroImaging, Inc.) at 25°C. Pictures were taken with an AxioCam HRm camera (Carl Zeiss MicroImaging, Inc.) and AxioVision (versions 4.4 and 4.6) software (Carl Zeiss MicroImaging, Inc.) and processed using Photoshop CS.
Online supplemental material
Fig. S1 shows surface biotinylation data of DLL3 expressed in HEK293, CHO, and C2C12 cells. Fig. S2 shows immunofluorescence detection of DLL3 and DLL1 in cell lines and PSM cells. Table S1 shows the junctions of Dll1 and Dll3 sequences in chimeric ligands. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200702009/DC1.
Supplementary Material
Acknowledgments
We would like to thank Drs. A. Israël, S. Chiba, K. Shimizu, S. Bray, and G. Weinmaster, as well as Amgen, for providing cell lines and reagents; Drs. O. Prall and R. Bryson-Richardson for technical advice; P. Delany-Heiken and H. Burkhardt for technical assistance; and N. Wise for assistance with mice.
G. Chapman was an National Health and Medical Research Council CJ Martin Fellow (158043) and is a Cancer Institute NSW Fellow. D.B. Sparrow is a Westfield-Belconnen Fellow. S.L. Dunwoodie is a Pfizer Foundation of Australia Senior Research Fellow. This work was supported by National Health and Medical Research Council project grant 404804 (S.L. Dunwoodie) and Deutsche Forschungsgemeinschaft grant Go449/9 (A. Gossler).
I. Geffers and K. Serth contributed equally to this paper.
Abbreviations used in this paper: A-P, anterior–posterior; E, embryonic day; ICD, intracellular domain; PSM, presomitic mesoderm; TM, transmembrane domain.
References
- Abdelkhalek, H.B., A. Beckers, K. Schuster-Gossler, M.N. Pavlova, H. Burkhardt, H. Lickert, J. Rossant, R. Reinhardt, L.C. Schalkwyk, I. Müller, et al. 2004. The mouse homeobox gene Not is required for caudal notochord development and affected by the truncate mutation. Genes Dev. 18:1725–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artavanis-Tsakonas, S., M.D. Rand, and R.J. Lake. 1999. Notch signaling: cell fate control and signal integration in development. Science. 284:770–776. [DOI] [PubMed] [Google Scholar]
- Ascano, J.M., L.J. Beverly, and A.J. Capobianco. 2003. The C-terminal PDZ- ligand of JAGGED1 is essential for cellular transformation. J. Biol. Chem. 278:8771–8779. [DOI] [PubMed] [Google Scholar]
- Barsi, J.C., R. Rajendra, J.I. Wu, and K. Artzt. 2005. Mind bomb1 is a ubiquitin ligase essential for mouse embryonic development and Notch signaling. Mech. Dev. 122:1106–1117. [DOI] [PubMed] [Google Scholar]
- Blaumueller, C.M., and S. Artavanis-Tsakonas. 1997. Comparative aspects of Notch signaling in lower and higher eukaryotes. Perspect. Dev. Neurobiol. 4:325–343. [PubMed] [Google Scholar]
- Brand, A.H., and N. Perrimon. 1993. Targeted gene-expression as a means of altering cell fates. Development. 118:401–415. [DOI] [PubMed] [Google Scholar]
- Bush, G., G. diSibio, A. Miyamoto, J.B. Denault, R. Leduc, and G. Weinmaster. 2001. Ligand-induced signaling in the absence of furin processing of Notch1. Dev. Biol. 229:494–502. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega, J.A. 1994. Genetic mechanisms of early neurogenesis in Drosophila melanogaster. J. Physiol. (Paris). 88:111–122. [DOI] [PubMed] [Google Scholar]
- Colley, K.J. 1997. Golgi localization of glycosyltransferases: more questions than answers. Glycobiology. 7:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes, R., K. Schuster-Gossler, K. Serth, and A. Gossler. 2004. Specification of vertebral identity is coupled to Notch signalling and the segmentation clock. Development. 131:1221–1233. [DOI] [PubMed] [Google Scholar]
- Dahlqvist, C., A. Blokzijl, G. Chapman, A. Falk, K. Dannaeus, C.F. Ibanez, and U. Lendahl. 2003. Functional Notch signaling is required for BMP4-induced inhibition of myogenic differentiation. Development. 130:6089–6099. [DOI] [PubMed] [Google Scholar]
- de Vries, W.N., L.T. Binns, K.S. Fancher, J. Dean, R. Moore, R. Kemler, and B.B. Knowles. 2000. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis. 26:110–112. [PubMed] [Google Scholar]
- Diez, H., A. Fischer, A. Winkler, C.J. Hu, A.K. Hatzopoulos, G. Breier, and M. Gessler. 2007. Hypoxia-mediated activation of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and adoption of arterial cell fate. Exp. Cell Res. 313:1–9. [DOI] [PubMed] [Google Scholar]
- Doherty, D., G. Feger, S. Younger-Shepherd, L.Y. Jan, and Y.N. Jan. 1996. Delta is a ventral to dorsal signal complementary to Serrate, another Notch ligand, in Drosophila wing formation. Genes Dev. 10:421–434. [DOI] [PubMed] [Google Scholar]
- Dunwoodie, S.L., D. Henrique, S.M. Harrison, and R.S. Beddington. 1997. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development. 124:3065–3076. [DOI] [PubMed] [Google Scholar]
- Dunwoodie, S.L., M. Clements, D.B. Sparrow, X. Sa, R.A. Conlon, and R.S. Beddington. 2002. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development. 129:1795–1806. [DOI] [PubMed] [Google Scholar]
- Furriols, M., and S. Bray. 2001. A model Notch response element detects Suppressor of Hairless-dependent molecular switch. Curr. Biol. 11:60–64. [DOI] [PubMed] [Google Scholar]
- Glittenberg, M., C. Pitsouli, C. Garvey, C. Delidakis, and S. Bray. 2006. Role of conserved intracellular motifs in Serrate signalling, cis-inhibition and endocytosis. EMBO J. 25:4697–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gridley, T. 1997. Notch signaling in vertebrate development and disease. Mol. Cell. Neurosci. 9:103–108. [DOI] [PubMed] [Google Scholar]
- Henrique, D., E. Hirsinger, J. Adam, I. Le Roux, O. Pourquie, D. Ish-Horowicz, and J. Lewis. 1997. Maintenance of neuroepithelial progenitor cells by Delta-Notch signalling in the embryonic chick retina. Curr. Biol. 7:661–670. [DOI] [PubMed] [Google Scholar]
- Hrabe de Angelis, M., J. McIntyre II, and A. Gossler. 1997. Maintenance of somite borders in mice requires the Delta homologue DII1. Nature. 386:717–721. [DOI] [PubMed] [Google Scholar]
- Ilagan, M.X., and R. Kopan. 2007. SnapShot: notch signaling pathway. Cell. 128:1246. [DOI] [PubMed] [Google Scholar]
- Iso, T., T. Maeno, Y. Oike, M. Yamazaki, H. Doi, M. Arai, and M. Kurabayashi. 2006. Dll4-selective Notch signaling induces ephrinB2 gene expression in endothelial cells. Biochem. Biophys. Res. Commun. 341:708–714. [DOI] [PubMed] [Google Scholar]
- Itoh, M., C.H. Kim, G. Palardy, T. Oda, Y.J. Jiang, D. Maust, S.Y. Yeo, K. Lorick, G.J. Wright, L. Ariza-McNaughton, et al. 2003. Mind bomb is a ubiquitin ligase that is essential for efficient activation of Notch signaling by Delta. Dev. Cell. 4:67–82. [DOI] [PubMed] [Google Scholar]
- Jaekel, R., and T. Klein. 2006. The Drosophila Notch inhibitor and tumor suppressor gene lethal (2) giant discs encodes a conserved regulator of endosomal trafficking. Dev. Cell. 11:655–669. [DOI] [PubMed] [Google Scholar]
- Jarriault, S., O. Le Bail, E. Hirsinger, O. Pourquié, F. Logeat, C.F. Strong, C. Brou, N.G. Seidah, and A. Isra. 1998. Delta-1 activation of notch-1 signaling results in HES-1 transactivation. Mol. Cell. Biol. 18:7423–7431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein, T. 2001. Wing disc development in the fly: the early stages. Curr. Opin. Genet. Dev. 11:470–475. [DOI] [PubMed] [Google Scholar]
- Klein, T., K. Brennan, and A.M. Arias. 1997. An intrinsic dominant negative activity of serrate that is modulated during wing development in Drosophila. Dev. Biol. 189:123–134. [DOI] [PubMed] [Google Scholar]
- Koo, B.K., H.S. Lim, R. Song, M.J. Yoon, K.J. Yoon, J.S. Moon, Y.W. Kim, M.C. Kwon, K.W. Yoo, M.P. Kong, et al. 2005. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development. 132:3459–3470. [DOI] [PubMed] [Google Scholar]
- Krebs, L.T., M.L. Deftos, M.J. Bevan, and T. Gridley. 2001. The Nrarp gene encodes an ankyrin-repeat protein that is transcriptionally regulated by the notch signaling pathway. Dev. Biol. 238:110–119. [DOI] [PubMed] [Google Scholar]
- Kusumi, K., E.S. Sun, A.W. Kerrebrock, R.T. Bronson, D.C. Chi, M.S. Bulotsky, J.B. Spencer, B.W. Birren, W.N. Frankel, and E.S. Lander. 1998. The mouse pudgy mutation disrupts Delta homologue Dll3 and initiation of early somite boundaries. Nat. Genet. 19:274–278. [DOI] [PubMed] [Google Scholar]
- Kusumi, K., M.S. Mimoto, K.L. Covello, R.S. Beddington, R. Krumlauf, and S.L. Dunwoodie. 2004. Dll3 pudgy mutation differentially disrupts dynamic expression of somite genes. Genesis. 39:115–121. [DOI] [PubMed] [Google Scholar]
- Ladi, E., J.T. Nichols, W. Ge, A. Miyamoto, C. Yao, L.T. Yang, J. Boulter, Y.E. Sun, C. Kintner, and G. Weinmaster. 2005. The divergent DSL ligand Dll3 does not activate Notch signaling but cell autonomously attenuates signaling induced by other DSL ligands. J. Cell Biol. 170:983–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne, R., A. Bardin, and F. Schweisguth. 2005. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development. 132:1751–1762. [DOI] [PubMed] [Google Scholar]
- Minoguchi, S., Y. Taniguchi, H. Kato, T. Okazaki, L.J. Strobl, U. Zimber-Strobl, G.W. Bornkamm, and T. Honjo. 1997. RBP-L, a transcription factor related to RBP-Jkappa. Mol. Cell. Biol. 17:2679–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales, A.V., Y. Yasuda, and D. Ish-Horowicz. 2002. Periodic lunatic fringe expression during segmentation is controlled by a cyclic transcriptional enhancer responsive to Notch signalling. Dev. Cell. 3:63–74. [DOI] [PubMed] [Google Scholar]
- Morimoto, M., Y. Takahashi, M. Endo, and Y. Saga. 2005. The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity. Nature. 435:354–359. [DOI] [PubMed] [Google Scholar]
- Muskavitch, M.A. 1994. Delta-notch signaling and Drosophila cell fate choice. Dev. Biol. 166:415–430. [DOI] [PubMed] [Google Scholar]
- Pavlopoulos, E., C. Pitsouli, K.M. Klueg, M.A. Muskavitch, N.K. Moschonas, and C. Delidakis. 2001. neuralized encodes a peripheral membrane protein involved in delta signaling and endocytosis. Dev. Cell. 1:807–816. [DOI] [PubMed] [Google Scholar]
- Pitsouli, C., and C. Delidakis. 2005. The interplay between DSL proteins and ubiquitin ligases in Notch signaling. Development. 132:4041–4050. [DOI] [PubMed] [Google Scholar]
- Rubin, G.M., and A.C. Spradling. 1982. Genetic transformation of Drosophila with transposable element vectors. Science. 218:348–353. [DOI] [PubMed] [Google Scholar]
- Sakamoto, K., O. Ohara, M. Takagi, S. Takeda, and K. Katsube. 2002. Intracellular cell-autonomous association of Notch and its ligands: a novel mechanism of Notch signal modification. Dev. Biol. 241:313–326. [DOI] [PubMed] [Google Scholar]
- Schuster-Gossler, K., R. Cordes, and A. Gossler. 2007. Premature myogenic differentiation and depletion of progenitor cells cause severe muscle hypotrophy in Delta1 mutants. Proc. Natl. Acad. Sci. USA. 104:537–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serth, K., K. Schuster-Gossler, R. Cordes, and A. Gossler. 2003. Transcriptional oscillation of lunatic fringe is essential for somitogenesis. Genes Dev. 17:912–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu, K., S. Chiba, K. Kumano, N. Hosoya, T. Takahashi, Y. Kanda, Y. Hamada, Y. Yazaki, and H. Hirai. 1999. Mouse jagged1 physically interacts with notch2 and other notch receptors. Assessment by quantitative methods. J. Biol. Chem. 274:32961–32969. [DOI] [PubMed] [Google Scholar]
- Shimizu, K., S. Chiba, T. Saito, K. Kumano, and H. Hirai. 2000. Physical interaction of Delta1, Jagged1, and Jagged2 with Notch1 and Notch3 receptors. Biochem. Biophys. Res. Commun. 276:385–389. [DOI] [PubMed] [Google Scholar]
- Thomas, U., S.A. Speicher, and E. Knust. 1991. The Drosophila gene Serrate encodes an EGF-like transmembrane protein with a complex expression pattern in embryos and wing discs. Development. 111:749–761. [DOI] [PubMed] [Google Scholar]
- Vässin, H., K.A. Bremer, E. Knust, and J.A. Campos-Ortega. 1987. The neurogenic gene Delta of Drosophila melanogaster is expressed in neurogenic territories and encodes a putative transmembrane protein with EGF-like repeats. EMBO J. 6:3431–3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton, K.A., K.M. Johansen, T. Xu, and S. Artavanis-Tsakonas. 1985. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell. 43:567–581. [DOI] [PubMed] [Google Scholar]
- Wilkinson, D.G. 1992. Whole mount in situ hybridization of vertebrate embryos. In In situ hybridization: A practical approach. D.G. Wilkinson, editor. Oxford University Press, Oxford, England. 75–84.
- Wright, G.J., J.D. Leslie, L. Ariza-McNaughton, and J. Lewis. 2004. Delta proteins and MAGI proteins: an interaction of Notch ligands with intracellular scaffolding molecules and its significance for zebrafish development. Development. 131:5659–5669. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}