

Summary
Cadherin-mediated interactions are integral to synapse formation and potentiation. Here we show that N-cadherin is required for memory formation and regulation of a subset of underlying biochemical processes. N-cadherin antagonistic peptide containing the His-Ala-Val motif (HAV-N) transiently disrupted hippocampal N-cadherin dimerization and impaired the formation of long-term contextual fear memory while sparing short-term memory, retrieval and extinction. HAV-N impaired the learning-induced phosphorylation of a distinctive, cytoskeletally-associated fraction of hippocampal Erk-1/2 and altered the distribution of IQGAP1, a scaffold protein linking cadherin-mediated cell adhesion to the cytoskeleton. This effect was accompanied by diminished of N-cadherin/IQGAP1/Erk-2 interactions. Similarly, in primary neuronal cultures, HAV-N prevented NMDA-induced dendritic Erk-1/2 phosphorylation and caused relocation of IQGAP1 from dendritic spines into the shafts. The data suggest that the newly identified role of hippocampal N-cadherin in memory consolidation may be mediated, at least in part, by cytoskeletal IQGAP1/Erk signaling.
Keywords: N-cadherin, HAV peptide, fear conditioning, Erk, IQGAP1, cytoskeleton
Introduction
Classic type I cadherins, including neuronal (N)-cadherin, play a critical role in the developmental organization of the brain (Redies, 2000) and synapse formation in the adult CNS (Junghans et al., 2005). These processes are regulated by the extracellular domains mediating cell-cell adhesion in a calcium-dependent, predominantly homophilic manner (Shapiro and Colman, 1998). Intracellularly, cadherins are anchored to the actin cytoskeleton by multiprotein complexes including beta- and alpha-catenin (Angst et al., 2001) and associated with docking proteins to intracellular signaling pathways (Husi et al., 2000) enabling the translation of cell adhesion signals into long-term cellular responses (Marambaud et al., 2003; Perron and Bixby, 1999; Widelitz, 2005).
Earlier studies reported a role of N-cadherin in synaptic potentiation. By applying blocking antibodies or antagonistic peptides containing the His-Ala-Val (HAV) motif for classic type I cadherin dimerization, Tang et al. (1998) observed a significant impairment of hippocampal long-term potentiation (LTP). Bozdagi et al. (2000) subsequently reported similar findings with N-cadherin blocking antibodies in a model of late-LTP, and additionally demonstrated that synaptic potentiation increases the synthesis and recruitment of N-cadherin to newly formed synapses. These findings suggested that N-cadherin–mediated cell adhesion may contribute to learning and memory (Hagler and Goda, 1998; Murase and Schuman, 1999).
A role of cadherins in memory has not yet been documented. Contrary to expectation, mice lacking the classical type II cadherin-11 displayed enhanced LTP and normal spatial learning (Manabe et al., 2000). On this basis, it was suggested that cadherins might be functionally redundant thereby masking the effects of individual members (Togashi et al., 2002). The role of classical type I cadherins, such as N-cadherin, could not be studied with constitutive gene knockouts due to embryonic lethality (Radice et al., 1997). By using an alternative approach, conditional expression of dominant negative protein in the adult brain, (Edsbagge et al., 2004) attempted to nonselectively impair the function of type I cadherins. Surprisingly, these mice did not show any alterations of synaptic potentiation or spatial learning. It should be noted, however, that N-cadherin was produced normally in the brain of transgenic mice, and that the expected functional consequences of the transgene could not be demonstrated due to its expression later in life.
To overcome possible compensatory or developmental effects of genetic manipulations, we employed acute injection of antagonistic peptides containing the His-Ala-Val motif (HAV-N) to examine the role of N-cadherin in learning and memory. HAV-N containing aspartic (D) amino acid flanking the HAV sequence was shown to specifically bind to N-cadherin when compared to other classic cadherins (Williams et al., 2000). On this basis as well as their thorough characterization in electrophysiological experiments (Tang et al., 1998), we selected HAV-N and a scrambled peptide (HAV-S) to perform these studies. The hippocampally-dependent contextual fear conditioning paradigm served to establish the effect of HAV-N on memory formation, retrieval and extinction. Furthermore, we investigated the interference of HAV-N with postadhesion signaling associated with N-cadherin complexes, including extracellular signal-regulated kinases 1 and 2 (Erk-1/2) and IQGAP1 (Derycke and Bracke, 2004).
We demonstrated a role of hippocampal N-cadherin in early mechanisms leading to consolidation of long-term contextual fear memory. Biochemical findings suggested that these mechanisms include the activation of a discrete fraction of cytoskeletal Erk-1/2 and its interaction with IQGAP1. N-cadherin-mediated cytoskeletal IQGAP1/Erk-1/2 signaling may be important for synaptic remodelling associated with memory.
Results
HAV-N impairs memory formation without affecting retrieval or extinction
Mice were implanted with microcannula into the dorsal hippocampus and subjected to context and tone-dependent fear conditioning. Dorsohippocampal injection of HAV-N immediately after training dose-dependently impaired contextual freezing (F (4,45) = 4.57, p < 0.05) during the memory test performed 24 hr later (Figure 1A). Tone-dependent fear conditioning (Figure 1A) was not affected by any of the applied treatments (F (4,45) = 1.43, p = 1.22). The specificity of the most effective dose (750 ng/mouse) was compared to a group receiving the same amount of a scrambled peptide (HAV-S) or vehicle. Only HAV-N impaired long-term memory of contextual fear, as indicated by a significant reduction of freezing (F (2,33) = 6.16, p < 0.01) and increase of mean activity (F (2,33) = 3.251, p < 0.05) during the contextual memory test performed 24 hr later (Figure 1B). HAV-N injected in separate groups of mice immediately posttraining did not affect freezing determined 1 hr later (F(2,30) = 0.57, p = 0.75), indicating that HAV-N did not affect short-term memory (Figure 1C). Additional time-course studies showed that HAV-N impaired long-term memory formation when injected 15 min before training (F (2,28) = 4.31, p < 0.01), however injections performed 60 min posttraining did not significantly affect consolidation, as indexed by freezing behavior (F (2,29) = 1.98, p = 0.93; Figure 2A). Pretraining injections of HAV-N did not reveal any effects of the peptide on mean activity (F (2,28) = 0.62, p = 0.87) or activity burst to the shock (F(2,28) = 0.34, p = 0.43). The group values (in cm/s) for activity were: vehicle 11.5 ± 1.3; HAV-S 11.8 ± 0.8; HAV-N 11.1 ± 0.9; and for shock response: vehicle 41.9 ± 5.6, HAV-S 43.3 ± 4.7, HAV-N 39.7 ± 4.9.
Figure 1.
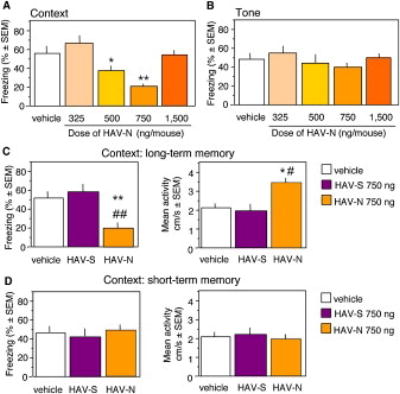
HAV-N Impairs Contextual Fear Conditioning
(A) Dose-dependent impairment of contextual freezing by immediate posttraining injections of HAV-N (left panel). The same doses did not affect tone-dependent freezing (right panel).
(B) A dose of 750 ng/mouse of HAV-N but not a scrambled peptide, HAV-S, resulted in a significant decrease of freezing behavior (left) and increase of mean activity (right) during the test of long-term contextual memory performed 24 hr after training.
(C) The same dose did not affect freezing (left) or activity (right) during the test of short-term contextual memory performed 1 hr after training.
Statistically significant differences: *p < 0.05, **p < 0.01 vs vehicle; #p < 0.05, ##p < 0.01 vs HAV-S. The number of mice per group was 10 (A), 12 (B) and 11 (C).
Figure 2.
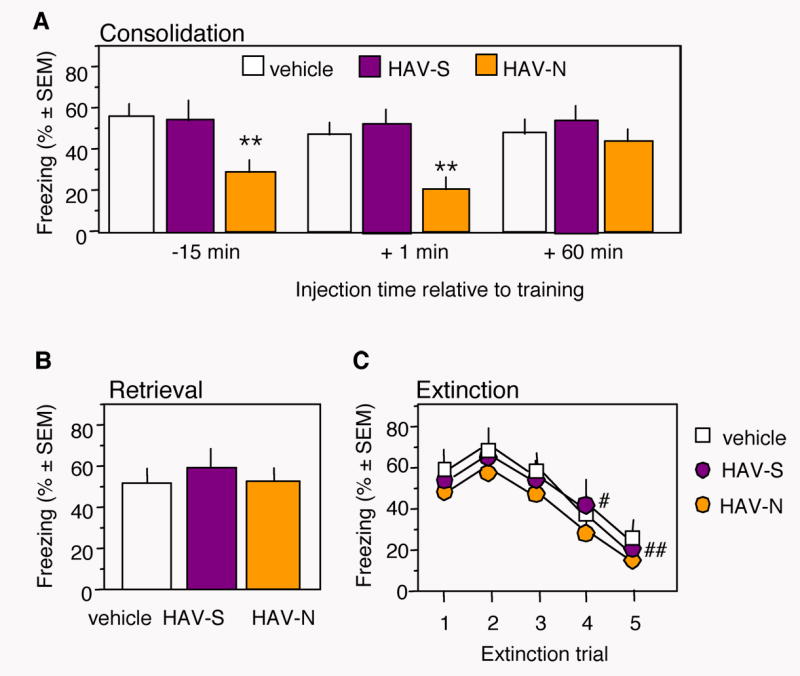
Effect of HAV-N on Memory Formation, Retrieval and Extinction
(A) Injections of HAV-N 15 min before or immediately after training impaired contextual freezing during the memory test whereas injections 1 hr posttraining were ineffective.
(B) HAV-N administered 15 min before the memory test did not affect freezing, indicating normal contextual memory retrieval.
(C) HAV-N injected immediately after individual nonreinforced trials did not affect extinction of contextual fear, as shown by a similar decrease of freezing behavior in all experimental groups. HAV-N and HAV-S were injected at a dose of 750 ng/mouse.
Statistically significant differences: **p < 0.01 vs vehicle and HAV-S; #p < 0.05, ##p < 0.01 vs test 1. The number of mice per group was 10-11.
To test the effect of N-cadherin signaling on memory retrieval and extinction, HAV-N was injected 15 min before the test (Figure 2B) or immediately after nonreinforced trials (Figure 2C), respectively. Neither treatment affected freezing behavior when compared to the control injections (pre-test: F (2,27) = 1.79, p = 0.98; extinction: F (8,135) = 0.56, p = 1.85). These results indicated that the HAV-N antagonistic peptide significantly impaired memory consolidation without affecting retrieval or extinction of contextual fear.
HAV-N blocks N-cadherin dimerization
HAV-N is expected to interfere with N-cadherin function by decreasing the dimerization of N-cadherin molecules. By employing one way ANOVA with factor Group (naïve, vehicle, HAV-S, and HAV-N) for each dose and time point or two way ANOVA with Treatment and Time as factors, we analysed the dorsohippocampal levels of N-cadherin monomers and dimers after HAV-N treatment (Figure 3A-3D). All mice except for those of the naïve group were exposed to fear conditioning and injected immediately thereafter with vehicle, HAV-S or HAV-N. Immunoblot signals of total dorsohippocampal lysates showed a significant increase of N-cadherin monomers after fear conditioning (Figure 3A, 3B) in all groups when compared to the naïve (Group: F(18, 76) = 18.7, p < 0.01) group independently of treatment. Considering the very short lifetime (Klingelhofer et al., 2002) and low level (Bozdagi et al., 2000) of cadherin dimers, the effect of HAV-N on dimerization was examined under nonreducing conditions ((-β-mercaptoethanol; -β-ME). This approach, selected to preserve disulfide bonds promoting and stabilizing cadherin dimerization (Makagiansar et al., 2002), enhanced dimer detection in the control groups (Figure 3C). N-cadherin dimers in the membrane fractions were also significantly elevated after fear conditioning (vehicle and HAV-S groups) when compared to naïve mice. HAV-N significantly impaired N-cadherin dimer formation depending on the dose (F(3, 56) = 116.446, p < 0.001) and postinjection time (F(2,56) = 63.067, p < 0.001). Post-hoc analyses revealed significant effects of 500 ng/mouse (Figure 3D) and 750 ng/mouse (Figure 3C-3D) of HAV-N at the time points 15 and 30 min posttraining when compared to the other groups. Analyses of membrane fractions of dorsohippocampal lysates obtained from naïve, vehicle-, or HAV-S-treated mice showed weak but clear bands at 230 kDa (Bozdagi et al., 2000) under nonreducing but not under reducing (+βME) conditions, indicating N-cadherin dimers (Figure 3C). This band was not detectable in lysates obtained 15 and 30 min after HAV-N injection and reappeared, although with diminished intensity in the 500 (F(2,12) = 4.65, p < 0.05) and 750 ng groups (F(2,12) = 4.12, p < 0.05), after 1 hr. The decrease of N-cadherin dimerization by HAV-N (Figure S1C) was confirmed by a mouse monoclonal N-cadherin antibody (clone G-C4, Sigma). These results showed that the behaviorally effective doses of HAV-N prevented N-cadherin dimerization in a restricted time window posttraining. Notably, the effect of HAV-N on N-cadherin dimerization was dissociable from N-cadherin production. Thus, N-cadherin dimerization early after fear conditioning and its later up-regulation may independently contribute to the early and late biochemical processes leading to long-term memory formation. Attenuated N-cadherin cell-cell adhesion activates the small GTP-ase Rac1 (Charrasse et al., 2002), whereas disruption of cell-cell adhesion may alter the subcellular distribution of N-cadherin and beta-catenin (Rhee et al., 2002). To examine whether HAV-N affected these parameters associated with N-cadherin function, we determined the levels of N-cadherin and beta-catenin as well as the localization and activity of Rac1 (see Supplemental Procedures) in several subcellular hippocampal fractions. Immunoblot analyses indicated that HAV-N did not significantly affect beta-catenin-mediated signaling or N-cadherin redistribution under these conditions (Figure S1A, S1B). However, the activity of Rac1, a protein recruited to the membrane when cell adhesion is weakened (Kaibuchi et al., 1999), was elevated in the membrane fractions (Figure S1D). Thus, some effects of decreased N-cadherin dimerization, such as activation of Rac1, have been reproduced by HAV-N. Expectedly, the peptide did not mimic the alterations accompanying severely disrupted cell-cell adhesion typically involving N-cadherin and beta-catenin redistribution.
Figure 3.

HAV-N transiently disrupts N-cadherin dimerization
(A) The levels of N-cadherin increase after fear conditioning, as revealed by immunoblot analyses performed under reducing conditions in total dorsohippocampal lysates. Under these conditions, N-cadherin is detected as a monomer. The levels of N-cadherin were not affected by HAV-N treatment.
Statistically significant differences: *p < 0.001 vs naïve. The number of samples/group was 5.
(B) Representative immunoblot showing up-regulation of N-cadherin monomers.
(C) Representative immunoblots of N-cadherin in membrane dorsohippocampal fractions resolved on 5% SDS gels show distinctive bands at 230 kDa under nonreducing conditions indicating N-cadherin dimers. These bands were not observed in samples obtained from dorsal hippocampi of mice injected with 500 and 750 ng/mouse HAV-N and sacrificed 15 and 30 min later. One hr after HAV-N injection, the band was detectable although weaker than in vehicle-and HAV-S-treated mice. The dimer band was not detectable under reducing conditions.
(D) Fear conditioning triggers up-regulation of N-cadherin dimers in the vehicle, HAV-S, HAV-N (325 ng and 1500 ng) groups. One way ANOVA revealed significantly stronger signals in these groups (p < 0.001) when compared to the naïve group. The behaviorally effective doses of HAV-N (500 and 750 ng/mouse) significantly impaired N-cadherin dimerization 15 and 30 min posttraining.
Statistically significant differences: *p < 0.01 vs naïve; #p < 0.01 vs all other experimental groups except for naïve (this group was not included in two way ANOVA analysis because it represents a single time point). The number of samples per group was 4.
Hippocampal N-cadherin levels increase after contextual fear conditioning
Increased levels of N-cadherin may contribute to LTP (Bozdagi et al., 2000) and possibly memory consolidation by strengthening cell-cell adhesion. On this basis, we examined the levels of N-cadherin at different time points after training. In order to provide a maximal signal/noise ratio, we employed a dilution of anti-N-cadherin antibody that gave just detectable signals. N-cadherin levels significantly increased after fear conditioning (F (7,40) = 5.97, p < 0.01). An up-regulation was observed shortly after training, peaking 1 hr later and exhibiting a persistent elevation, particularly in the CA2 subfield, up to 5 days posttraining (Figure 4A-4C). The specificity of N-cadherin production after fear conditioning was shown in two experiments. To control for the persistent N-cadherin elevation after conditioning, a group of mice exposed to daily extinction trials leading to fear reduction was introduced (Fischer et al., 2004). Nonreinforced trials abolished the increase of N-cadherin levels observed 5 days posttraining (Figure 4A,4C), suggesting that the up-regulation of N-cadherin levels were specific for conditioning relative to extinction. Whether the observed down-regulation of N-cadherin contributes to fear extinction remains to be established.
Figure 4.

Fear Conditioning Up-regulates N-cadherin in the Hippocampus
(A) N-cadherin levels significantly increased after contextual fear conditioning up to 120 hr (5 days) posttraining. Extinction (E) training abolished the increase of N-cadherin observed at the 120 hr time point (120 + E, light green).
(B) Representative micrographs of the CA1 subfield of naïve mice and 1 hr posttraining.
(C) Representative micrographs of the CA2 subfield of naïve mice, 1 hr posttraining, 120 hr posttraining and 120 hr with extinction (120 hr + E). Statistically significant differences: *p < 0.05, **p < 0.01, ***p < 0.01 vs time point 0 (naïve mice).
(D) Freezing behavior was induced only by paired (CTS, context-tone-shock) but not reverse, unpaired (STC, shock-tone-context) presentation of the conditioned and unconditioned stimuli. Statistically significant differences: *p < 0.05 vs naïve, ***p < 0.01 vs naïve, ###p < 0.001 vs STC. The number of mice per group was 7.
(E) N-cadherin levels were significantly higher in mice of the CTS when compared to the STC and naïve groups (left). Representative micrographs (right).
Statistically significant differences: *p < 0.05, ***p < 0.01 vs vehicle; ###p < 0.001 vs STC. The number of mice per group was 6.
In a separate experiment, a group of mice was exposed to an inverse presentation of training stimuli so that an immediate shock was followed by tone and context. This group exhibited significantly lower freezing than the paired group (F (2,27) = 8.45, p < 0.01, Figure 4D) as observed in our earlier work (Fischer et al., 2002; Sananbenesi et al., 2002), and did not show up-regulation of N-cadherin levels comparable to the paired context-tone-shock group (F (2,15) = 4.78, p < 0.01, Figure 4E).
Taken together, these findings demonstrated a long lasting increase of N-cadherin levels in hippocampal neurons specifically accompanying consolidation of associative learning.
HAV-N selectively disrupts the activation of Erk-1/2 associated with the cytoskeleton
N-cadherins are associated with a number of signaling molecules within multiprotein complexes linking the cell membrane to the actin cytoskeleton (Husi et al., 2000). On this basis we hypothesized the memory-impairing actions of HAV-N may be due to interference with signal transduction pathways involved in memory formation. We tested this possibility by monitoring the activation of Erk-1/2, cAMP-dependent protein kinase binding protein (CREB), p38 mitogen-activated protein kinase (p38MAPK) and stress-activated protein kinase (SAPK) in hippocampal subcellular fractions prepared 1 hr after fear conditioning and immediate posttraining injection with HAV-N, HAV-S or vehicle. In all subcellular fractions pErk-1/2 levels increased (vehicle vs naïve: membrane: t (7) = 5.17, p = 0.01; cytosol: t (7) = 3.95, p = 0.05; cytoskeleton: t (7) = 4.82, p < 0.01) after training (Figure 5). Analyses of immunoblots demonstrated significant effects of HAV-N on Erk-1/2-mediated signaling (Figure 5A-5C). Whereas the levels of total Erk-1/2, but not the pErk/Erk ratio (F (2,10) = .929, p = 0.426), were increased in the membrane fraction (total Erk: F (2,10) = 7.42, p < 0.01; Figure 5A, lower panel), the phosphorylation of Erk-1/2 within the cytoskeletal protein pool (Figure 5C, upper panel) was significantly reduced (F (2,10) = 6.975, p < 0.01). In contrast to pErk-1/2, the activation of p38MAPK and pSAPK in the cytoskeleton, and CREB in the nucleus were not significantly affected (Figure S2A-S2C) at the corresponding time point.
Figure 5.

HAV-N Affects Erk-1/2 Phosphorylation and Distribution
(A) HAV-N did not affect pErk-1/2 (top) but increased total Erk-1/2 (bottom) levels in membrane fractions of hippocampal extracts.
(B) HAV-N did not exhibit significant alterations of pErk-1/2 and total Erk-1/2 in cytosolic fractions.
(C) HAV-N disrupted the phosphorylation of Erk-1/2 without affecting total Erk-1/2 in cytoskeletal fractions.
HAV-N, HAV-S (750 ng/mouse each) and vehicle were used for treatment. Middle panels: representative immunoblots of hippocampal extracts obtained 1 hr posttraining. The levels of total Erk-1/2 were normalized to actin in individual extracts. The number of individual samples per treatment was 4-5. Statistically significant differences: *p < 0.05, **p < 0.01 vs naive; #p < 0.05, ###p < 0.01 vs vehicle and HAV-S.
Given that both pErk-1/2 (Sananbenesi et al., 2002, 2003) and N-cadherin levels peak 1 hr after fear conditioning, we selected this time point to examine by immunofluorescence whether these molecules colocalize within the hippocampus. When compared to naïve mice (Figure 6A), mice exposed to fear conditioning exhibited a marked up-regulation of pErk-1/2 and N-cadherin in the pyramidal cell layer and str. radiatum (Figure 6B). Analyses of high magnification captures revealed that 65% ± 8 of pyramidal neuron soma within the hippocampal CA1 subfield were positive for both N-cadherin and pErk-1/2 (Figure C). N-cadherin signals typically representing synaptic puncta (Bozdagi et al., 2000) were also observed in 75% ± 11 of pErk-1/2-positive apical dendrites (Figure 6 lower pannel). It should be noted that pErk-1/2 showed 53% ± 7 and 57% ± 5 overlap in the soma and dendrites, respectively (data not shown). Thus, most N-cadherin signals colocalized with pErk-1/2 whereas a subset of pErk signals colocalized with N-cadherin. This finding was expected given the coupling of Erk-1/2 to a vast number of receptor molecules (Fukunaga and Miyamoto, 1998).
Figure 6.
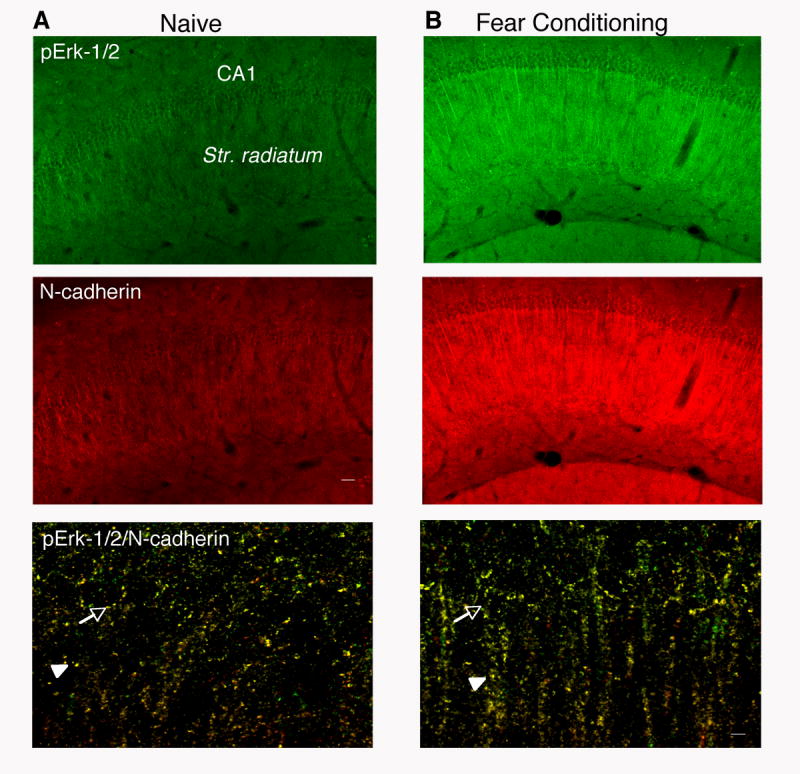
Up-regulation of pErk-1/2 in N-Cadherin-Positive Hippocampal Neurons
(A) pErk-1/2 (green) and N-cadherin (red) in hippocampi of naïve mice.
(B) Up-regulation of pErk-1/2 (green) and N-cadherin (red) in CA1 pyramidal cell soma and dendrites after fear conditioning.
The top and middle panels represent 10x images of the CA1 region. Scale bar = 40 μm. The bottom panel represents single z stacks of merged N-cadherin and pErk-1/2 high magnification (63x) images of the CA1 subfield and adjacent str. radiatum. showing colocalization within the soma (arrow) as well as apical dendrites (arrowhead). Scale bar = 5 μm.
Taken together, these data showed that although learning activated Erk-1/2 in all subcellular fractions, HAV-N selectively impaired the fear conditioning-induced phosphorylation of cytoskeletally-bound Erk-1/2. Notably, this delayed effect (1 hr posttraining) probably resulted from an early disruption of N-cadherin interactions, given that newly synthesized N-cadherin did not rescue the phosphorylation of Erk-1/2 at the examined time point. The colocalization of pErk-1/2 and N-cadherin support the view that these proteins may interact, probably indirectly, within individual pyramidal cells.
HAV-N triggers redistribution of IQGAP1
Several docking proteins compartmentalize Erk-1/2 with upstream regulators thereby tightly controlling its activity. On the basis of its ability to bridge cadherin-mediated adhesion to cytoskeletal alterations and Erk-1/2 signaling (Brown and Sacks, 2006; Roy et al., 2004), we hypothesized that IQGAP1 may be involved in the effects of HAV-N on pErk-1/2. HAV-N injections differentially affected IQGAP1 levels in hippocampal subcellular fractions (Figure 7A-7C). The significant decrease and increase of IQGAP-1 in the cytosolic (F (2,10) = 4.17, p < 0.05) and cytoskeletal (F (2,10) = 4.302, p < 0.05) fractions (Figure 7B,C), respectively, indicated redistribution of IQGAP1 from the cytosol to the cytoskeleton in response to HAV-N. However, the peptide did not affect (F (2,10) = 1.085, p = 0.374) membrane levels of IQGAP1 (Figure 7A). In response to fear conditioning, the distribution of IQGAP1 between subcellular compartments of vehicle controls was not altered (Figure 7D; membrane: t (7) = 0.177, p = 0.65; cytosol: t (7) = 0.251, p = 0.73; cytoskeleton: t (7) = 0.213, p = 0.69), suggesting that maintaining the localization of IQGAP1 during learning may be an important role of N-cadherin. Such effects were not observed in identically treated but non-trained (Figure S3), confirming that HAV-N exerts maximal activity when applied concurrently with fear conditioning.
Figure 7.

HAV-N Alters the Distribution of IQGAP1
(A) HAV-N did not affect membrane levels of IQGAP1.
(B) HAV-N significantly decreased the level of IQGAP1 in the cytosol.
(C) HAV-N caused an accumulation of IQGAP1 in the cytoskeleton.
Statistically significant differences: *p < 0.05, **p < 0.01 vs vehicle; #p < 0.05, ##p < 0.01 vs HAV-S. The number of individual samples per group was 4-5.
(D) Fear conditioning did not affect significantly the distribution of IQGAP1 in subcellular fractions of hippocampal extracts. The optical density for IQGAP1 was normalized to the level of β-actin.
(E) Dose-response and time course curves demonstrating the effects of HAV-N on IQGAP1 (left) and pErk-1/2 (right) levels in cytoskeletal preparations. Data are expressed as percent of mean O.D. in naïve mice. The number of individual samples per group was 4-5. Statistically significant differences: *p < 0.001 vs vehicle, HAV-N 325 and 1500 ng/mouse; #p < 0.01 vs naïve; ap < 0.001 vs time point 0 (immediately post-injection).
To study in more detail the relationship between cytoskeletal IQGAP1 and pErk-1/2, we performed time course and dose-response studies with HAV-N. In the same cytoskeletal preparations, HAV-N decreased and increased the levels of pErk-1/2 and IQGAP1, respectively, with maximal effects observed 1 hr after training/injection and only with the behaviorally effective doses of 500 and 750 ng/mouse (Figure 7E). Two-way ANOVA revealed significant effects of dose (pErk-1/2: F (4,80) = 7.446, p < 0.001; IQGAP1: F (4,80) = 6.756, p < 0.001) and time (pErk-1/2: F (3,80) = 3.97, p < 0.05; IQGAP1: F (3,80) = 13.114, p < 0.001). The behaviorally ineffective doses of 325 and 1500 ng/mouse did not produce significant biochemical alterations. Given that increased concentrations of IQGAP1 negatively regulate Erk-1/2 phosphorylation (Roy et al., 2004), these results suggest that HAV-N may disrupt the activity of cytoskeletal Erk-1/2 by increasing the levels of IQGAP1 bound to the cytoskeleton.
HAV-N attenuates the interactions between N-cadherin, IQGAP1 and Erk
We further examined whether HAV-N affects the functional interactions between N-cadherin, IQGAP1 and Erk. As cytoskeletal preparations contain high levels of denaturating agents and low protein amount, we performed coimmunoprecipitation studies using total dorsohippocampal lysates. The samples were prepared from fear conditioned mice injected posttraining with vehicle, HAV-S (750 ng) or HAV-N (750 ng) and euthanized 1 hr later. N-cadherin, IQGAP1, Erk-1/2 and pErk-1/2 were coimmunoprecipitated and detected by each of the employed antibodies, showing that these proteins interact in the dorsohippocampal tissue (Figure 8A-8D). We detected only Erk-2 in the immunoprecipitates with N-cadherin and IQGAP1 (Figure 8A, 8B), despite the presence of the Erk-1 isoform in the input samples (Figure 8E). Analyses of N-cadherin bound complexes demonstrated a significant decrease of pErk-2 (p < 0.01), Erk2 (p < 0.01) and IQGAP1 (p < 0.01) in response to HAV-N treatment. Similarly, significantly lower levels of N-cadherin (p < 0.01) were detected in immunoprecipitates performed with IQGAP1, pErk-1/2, and Erk-1/2 (Figure 8B-8D). IQGAP1 complexes with pErk-2 were significantly increased after fear conditioning in vehicle and HAV-S samples (p < 0.05), whereas HAV-N caused a reduction of pErk-2 bound to IQGAP1 (p < 0.01) despite an increased interaction with total Erk-2 (p < 0.01). These observations were confirmed with immunoprecipitates employing pErk-1/2 and Erk-1/2 antibodies (Figure 8C, 8D). Consistent with increased pErk levels in all cellular fractions, input pErk levels in the total lysates was expectedly higher. Supporting specific interaction with IQGAP1, pErk was not enhanced in N-cadherin (Figure 8A) or control IgG (Figure 8E) coimmunoprecipitates. Furthermore, the level of total Erk was not increased in the IQGAP1 complexes (Figure 8B).
Figure 8.

N-cadherin/IQGAP1/pErk-1/2 interactions are disrupted by HAV-N Dorsohippocampal lysates were prepared from naïve mice or mice trained in the fear conditioning paradigm and injected immediately afterwards with vehicle (white bars), HAV-S (yellow bars) or HAV-N (purple bars) (750 ng/mouse each). Dashed line represent values of the naïve group (100%). Data are expressed as percentage of naïve.
(A) Decreased levels of pErk-2, Erk-2 and IQGAP1 in dorsohippocampal lysates of HAV-N-treated mice after immunoprecipitation with N-cadherin.
(B) Decreased levels of pErk-2 and N-cadherin, and increased level of Erk-2 in the same HAV-N lysates after immunoprecipitation with IQGAP1.
(C) Decreased levels of IQGAP1 and N-cadherin in the HAV-N lysates after immunoprecipitation with pErk-1/2.
(D) Increased levels of IQGAP1 and decreased levels of N-cadherin in the HAV-N lysates after immunoprecipitation with Erk-1/2.
(E) Levels of N-cadherin, IQGAP1, pErk-1/2 and Erk-1/2 in the input samples or after immunoprecipitation with a corresponding anti-rabbit or anti-mouse immunoglobulin (IgG) serving as a negative control.
Statistically significant differences: *p < 0.01 vs naïve, #p < 0.01 vs vehicle and HAV-S. The number of individual samples/group was 3. Labels: 1, naïve; 2, vehicle; 3, HAV-S; 4, HAV-N.
These findings indicated that N-cadherin, IQGAP1 and Erk-2 form complexes in the dorsal hippocampus. Notably, although IQGAP1 levels were not affected by fear conditioning, its interaction with Erk-2 was significantly enhanced. The composition of N-cadherin/IQGAP1/Erk-2 complexes was disrupted by HAV-N, possibly as a result of an impaired N-cadherin dimerization.
HAV-N inhibits Erk-1/2 phosphorylation and removes IQGAP1 from dendritic spine heads
To study in more detail the localization of N-cadherin, Erk-1/2 and IQGAP1 and their relationship to N-cadherin function we employed primary cortical cultures. We first established that N-cadherin and Erk-1/2 colocalize with IQGAP1 in this cultures (Figure 9A, 9B). NMDA-induced activation of these cells resulted in a significant (p < 0.001) rise of pErk-1/2 levels that was completely blocked by pre-treatment with HAV-N but not HAV-S (Figure 9C, 9D). Under the same conditions, HAV-N did not affect total IQGAP1 levels, as indicated by fluorescence intensity, but resulted in decreased IQGAP1 in dendritic spines and an accumulation in dendritic shafts (Figure 9E-9G), revealed by a decrease of spine/shaft ratio (p < 0.01) of IQGAP1 distribution. These findings replicated our key in vivo observations indicating a role of N-cadherin in IQGAP1/Erk-1/2 signaling, and suggested that the decrease of pErk-1/2 by HAV-N may be associated with the removal of IQGAP1 from dendritic spine heads and its increase in the dendritic shafts. Owing to the similarities in N-cadherin function and its interactions with synaptic proteins in hippocampal and cortical neurons (Nuriya and Huganir, 2006), as well as consistencies between the in vitro and in vivo data, we hypothesize that the identified role of N-cadherin in IQGAP1/Erk signaling encompasses both cortical and hippocampal plasticity.
Figure 9.

HAV-N Inhibits Erk-1/2 Phosphorylation and Removes IQGAP1 from Spine Heads.
(A) IQGAP1 and N-cadherin colocalize at the plasma membrane and spine heads in cortical pyramidal neurons. Scale bars: 5 μm.
(B) A similar colocalization pattern (arrow: plasma membrane; arrowhead: spine heads) is observed for IQGAP1 and Erk.
(C) Erk-1/2 phosphorylation in pyramidal neurons with or without chemical activation of NMDA receptors for 30 min in the presence or absence of HAV-S or HAV-N. Images are displayed in pseudocolor to demonstrate changes in p-Erk-1/2 levels.
(D) Quantification of pERK1/2 levels (n= 9 neurons, 3 experiments). Statistically significant differences: * p < 0.001 vs all other groups.
(E) IQGAP1 immunofluorescence in pyramidal neurons with or without chemical activation of NMDA receptors for 30 min in the presence or absence of HAV-S or HAV-N: white arrows show immunofluorescence in spine heads of control neurons and after NMDA receptor activation in presence or absence of HAV-S or HAV-N peptide, solid white arrow head shows IQGAP1 immunofluorescence in the dendritic shaft. Dashed boxes indicate spine head and dendritic shaft used in line scan quantification.
(F) HAV-N decreases the ratio of IQGAP1 signals in the dendritic spines vs dendritic shafts. Statistically significant differences: * p < 0.001 vs all other groups.
(G) Line scan of IQGAP1 immunofluorescence intensity along a spine head and adjacent dendritic shaft. Blue/green line: 30 min activation + HAV-S; red/white line: 30 min activation + HAV-N. Statistically significant differences: *p < 0.01 vs control, activated and HAV-S.
Discussion
We demonstrated an important role of N-cadherin in the consolidation of hippocampally dependent memory and associated neuronal signaling mechanisms. Biochemically, N-cadherin was identified as a regulator of the distribution of IQGAP1 and activation of a distinctive, learning-induced, cytoskeletal fraction of pErk-1/2. In turn, learning mechanisms led to long-term up-regulation of hippocampal N-cadherin levels. Above all, this increase persisted in the CA2 hippocampal subfield known to transmit delayed excitatory signals to CA1 neurons (Sekino et al., 1997) and maintain long-range axonal connectivity (Zaidel, 1999), the link to the basolateral amygdala being particularly significant for the integration of emotional and cognitive processing (Benes et al., 2004; Nakao et al., 2004). The N-cadherin antagonistic peptide HAV-N disrupted fear conditioning-induced N-cadherin dimerization and cytoskeletal Erk-1/2 signaling but not N-cadherin up-regulation. This observation is not surprising given that plasticity-induced increase of N-cadherin requires de novo protein synthesis mediated by nuclear effects of cAMP-dependent protein kinase signaling (Bozdagi et al., 2000), a pathway not affected by the employed HAV-N treatment.
Disulfide bonds (Makagiansar et al., 2002) and other interactions (Troyanovski et al., 2006) promote and stabilize cadherin dimerization. HAV peptides are thought to prevent these interactions, thereby disrupting N-cadherin dimerization and postadhesion signaling (Harrison et al., 2005; Renaud-Young and Gallin, 2002; Shapiro et al., 1995). Accordingly, it was demonstrated that the formation of cadherin dimers is required for cadherin-mediated signaling (Kim et al., 2005). HAV-N may also attenuate N-cadherin dimerization, at least in part, by causing redistribution of the N-cadherin/Erk-1/2 docking protein IQGAP1 known to stabilize surface N-cadherin molecules (Noritake et al., 2005). We confirmed those observations in vivo, by showing that selected doses of HAV-N transiently impaired N-cadherin dimerization, pErk-1/2-mediated signal transduction and IQGAP1 distribution in hippocampal neurons. Importantly, only the doses of HAV-N that disrupted fear conditioning caused these biochemical effects, suggesting a link between the observed molecular and behavioral alterations.
HAV-N significantly impaired memory formation of contextual fear at selected doses relative to training. U-shaped dose-response curves are typically observed with in vivo pharmacological studies employing biologically active peptides (Calabrese and Baldwin, 2003). Although the mechanisms of such effects are not known in detail, it may be speculated that at higher doses the HAV-N molecules might interact with one another, with other related cadherins, or with unrelated proteins containing N-cadherin motifs, such as fibroblast growth factor receptors (Williams et al., 2001).
The involvement of N-cadherin in LTP induction but not LTP maintenance (Tang et al., 1998), when N-cadherin production and synaptic recruitment significantly increase (Bozdagi et al., 2000), is consistent with our in vivo experiments. Despite this early involvement, N-cadherin mediated the development of late but not early LTP, as revealed by the use of blocking antibodies (Bozdagi et al., 2000). Similarly, in our study HAV-N impaired long- but not short-term memory. These results indicated a role of N-cadherin in early biochemical events initiating memory consolidation (Miyamoto, 2006). Lack of effects of HAV peptides at later time points after training, during retrieval or extinction does not exclude a role of learning-induced N-cadherin in these processes as posttraining actions of HAV-N may be limited by several factors. First, in response to synaptic activity, N-cadherin levels increase in the form of molecularly modified, stable, protease-resistant dimers (Tanaka et al., 2000). Such modifications could limit the actions of HAV-N on dimerization of newly synthesized N-cadherin and behavior. Second, the effects of HAV-N critically and inversely depend on the concentration of extracellular Ca2+ (Tang et al., 1998). The transition of calcium ions from extracellular spaces into the cell during early phases of memory formationmay enhance the efficiency of HAV peptides during these phases, as suggested earlier (Tang et al., 1998). Thus, elevated N-cadherin levels, stabilization of N-cadherin molecules, or weaker calcium ion fluctuations may result in diminished efficiency of HAV-N. Consistent with the latter possibility, extinction mechanisms, although shared to some extent with those involved in conditioning, appear to recruit a more limited set of biochemical pathways (Berman and Dudai, 2001; Lin et al., 2003). Furthermore, processes other than dimerization, including cleavage or decreased N-cadherin production, could contribute to extinction. Such alterations may be required to weaken the synaptic contacts established during fear conditioning, so that acquired fear would not persist without aversive reinforcement. The potential roles of N-cadherin in the later phases of memory formation, retrieval and extinction, remaining to be established by more potent interfering agents or transient genetic manipulations, are likely to reveal additional mechanisms associated with the function of newly synthesized N-cadherin.
The significant involvement of N-cadherin dimerization in the activation of Erk-1/2, a key kinase involved in memory consolidation (Atkins et al., 1998), was not surprising. However, the selective role of N-cadherin dimerization in the phosphorylation of a segregated, cytoskeletal fraction of Erk-1/2 (but not pCREB, p38MAPK, pSAPK or beta-catenin signaling at the same time-points) was unexpected, given that signal transduction by PKA (Dell’acqua et al., 2006) as well as beta-catenin (Yu and Malenka, 2003) also depends on their interactions with N-cadherin. Possibly, the functions of the latter pathways are predominantly regulated by N-cadherin cleavage (Reiss et al., 2005), or alternative cascades including the Wnt pathway (De Ferrari et al., 2003).
Contrary to the significant decrease of Erk-1/2 activation in cytoskeletal preparations of HAV-N injected hippocampi, pErk-1/2 levels in the membrane extracts increased proportionally to the levels of total Erk-1/2 protein in this fraction. Weakening of cell-cell interactions by the antagonistic peptide may trigger the activation of Rac1 (Charrasse et al., 2002), as confirmed here, or redistribution of other signaling molecules towards the cell membrane (Retta et al., 2006). Although membrane interactions and docking of Erk-1/2 significantly contribute to synaptic plasticity and learning (Morozov et al., 2003; Shalin et al., 2006), it appears that they could not compensate for the memory impairing effect of HAV-N. Selective activation of cytoskeletal Erk-1/2 by N-cadherin may thus be required for memory formation in a nonredundant manner to membrane-docked Erk-1/2. Because the threshold for intracellular Erk-1/2 phosphorylation is markedly higher than for membrane-bound Erk-1/2 (Harding et al., 2005), N-cadherin interactions may selectively lower the activation threshold for Erk-1/2 bound to the cytoskeleton. Such effects may be mediated by IQGAP1, a scaffold for cadherins and cytoskeletal elements, that has recently emerged as a main regulator of cell adhesion-induced cytoskeletal rearrangement and signal transduction (Fukata et al., 2001; Noritake et al., 2005). Fear conditioning did not affect IQGAP1 levels, however its interaction with pErk-2, the main isoform activated by fear conditioning (Atkins et al., 1998), was significantly enhanced. HAV-N triggered redistribution of IQGAP1 from the cytosol to the cytoskeleton and diminished N-cadherin binding to IQGAP1 and pErk, suggesting that normal N-cadherin function was required for optimal interaction of these molecules. Relatively small increases or decreases of IQGAP1 and other scaffold proteins are known to decrease Erk-1/2 activity by altering the stoichiometry of the mitogen-activated protein kinases serving as upstream Erk activators (Levchenko et al., 2000; Roy et al., 2004). In agreement with the model proposed by Roy et al. (2005), the enhanced binding of IQGAP1 to Erk-2 was accompanied by significantly decreased Erk-2 phosphorylation in response to HAV-N treatment. Thus, IQGAP1 may be a strong candidate for linking N-cadherin and Erk-1/2 signaling during contextual fear memory formation.
The cytoskeletal preparations employed here contained both actin and microtubules that are predominantly localized in dendritic spines and shafts, respectively (Kaech et al., 1997; Widelitz, 2005; Wilson and Keith, 1998). To more closely determine the redistribution of IQGAP1 and its relationship to Erk-1/2 phosphorylation we employed neuronal cultures activated by NMDA with or without pre-treatment with HAV-S and HAV-N. Consistent with the in vivo generated data, HAV-N abolished the phosphorylation of pErk-1/2. At the same time, IQGAP1 signals increased in the dendritic shafts while decreasing in the spines, suggesting that N-cadherin may be required for maintaining IQGAP1/Erk-1/2 interactions in the dendritic spines. In view of the well established role of IQGAP1 in cellular polarization in developing cells (Noritake et al., 2005), similar interactions between N-cadherin and IQGAP1 in adult neurons may lead to localized changes in potentiated synapses underlying associative neuronal plasticity (Frey and Morris, 1997).
In conclusion, we demonstrated that N-cadherin is required for memory formation of contextual fear. Data generated both in vivo and in vitro suggest that these behavioral effects may be based on N-cadherin-mediated interactions of IQGAP1 and Erk-1/2 within the cytoskeleton of dendritic spines. The N-cadherin-regulated cytoskeletal pool of Erk-1/2 may be an important mediator of activity-induced synaptic remodelling (Okamura et al., 2004) and memory formation mechanisms involving actin rearrangement (Fischer et al., 2004).
Experimental Procedures
Animals
Nine-week old C57BL/6J mice were obtained from Jackson Laboratories. The mice were individually housed in a satellite facility provided with a separate ventilation system (15 air exchanges/hr), a 12/12 dark light cycle (7 am-7 pm), 40-50% humidity, and 20 ± 2°C temperature adjacent to the behavioral room. All studies were approved by Northwestern University the Animal Care and Use Committee in compliance with National Institutes of Health standards. The number of mice per group was 10-12 for behavioral, 4-5 for immunoblot and 6 for immunohistochemical experiments.
Peptides
The HAV-N peptide, Ala-Arg-Phe-His-Leu-Arg-Ala-His-Ala-Val-Asp-Ile-Asn-Gly-Asn-Gln-Val, and a scrambled peptide (HAV-S), Val-Ala-Val-Leu-Tyr-Glu-Lys-Ser-Gly-Ile-Ala-Tyr-His-Asn-Ser-Ala-Ser, were synthesized and kindly provided by Lars van Werven, Max Planck Institute for Experimental Medicine, Goettingen. The peptides were dissolved in artificial cerebrospinal fluid immediately before injections at the indicated concentrations.
Cannulation and injections
Double cannulae were placed into the dorsal hippocampus (anteroposterior - 1.5 mm, mediolateral 1 mm, dorsoventral 2 mm) as described earlier (Radulovic et al., 1999). Injections were delivered bilaterally (0.25 μl/side) over a 30 s period at the indicated doses and times relative to the behavioral task. The cannula position was determined for each mouse by methylene-blue injection after the end of experiments and only data obtained from mice with correctly inserted cannula were analyzed. For all biochemical experiments, the peptides were injected immediately after fear conditioning.
Fear conditioning
Contextual and tone-dependent fear conditioning was performed in an automated system (TSE Inc.) and consisted of a single exposure to context (3 min) followed by a 30-s tone (10 kHz, 75 dB SPL) and a footshock (2s, 0.7 mA, constant current) as described previously (Radulovic et al., 1998). Context-dependent freezing was measured 24 hr later every 10th second over 180 s by two observers unaware of the experimental conditions and expressed as percentage of total number of observations. Freezing to the tone was similarly scored in a novel context during a 3-min exposure. Extinction trials were performed at 24 hr intervals and consisted of nonreinforced 3-min exposures to the context (Fischer et al., 2004).
Immunohistochemistry
Mice were anesthetized with an intraperitoneal injection of 240 mg/kg of Avertin at indicated times after training, and transcardially perfused with ice-cold 4% paraformaldehyde in phosphate buffer (pH 7.4, 150 ml/mouse). Brains were processed for immunihistochemistry with specific antibodes as described in detail in the Supplemental Procedures. Quantification of immunostaining signals was performed as described previously (Winder et al., 1999; Sananbenesi et al., 2002). Digital images were captured with a cooled color CCD camera (RTKE Diagnostic Instruments) and SPOT software for Macintosh. Adobe Photoshop 5.0 for Macintosh was used for image processing. The background corresponding to areas without immunopositive cells was subtracted. Images were binarized and digitally contrast stretched for maximum resolution of N-cadherin-positive cells. The optical density within the CA1 and CA2 subfield and stratum radiatum were determined for each image with the ImageJ software (NIH). The average pixel intensity reflecting the mean value of these three areas, was then divided by the total area to determine integrated pixel density.
For coimmunolabeling studies the TSA Fluorescence System (NEN Life Science Products) was employed, using fluorescein as substrate for pErk-1/2 and rhodamine for N-cadherin. Multicolor immunofluorescence was captured using a Zeiss LSM5 Pascal confocal microscope and Z-stacks of images were taken using the 10x and 63x objectives and analyzed with a Metamorph software (Macintosh Universal Imaging). The number of coimmunolabeled somas and dendrites was determined as percentage of a total of 100 cells obtained from 3 captured images/mouse.
Protein extraction and immunoblot
Mice were injected with vehicle, HAV-N or HAV-S immediately after training, and euthanized by cervical dislocation 1 hr later. Individual dorsal hippocampi (the rostral 2.5 mm septal pole) were dissected, frozen in liquid nitrogen and kept at -80°C. Cytoplasmic, membrane, cytoskeletal and nuclear fractions were prepared by using the ProteoExtract kit for subcellular proteome extraction (EMD Biosciences) according to the instructions. Aliquots of individual samples were stored in loading buffers with or without β-mercaptoethanol (β-ME) for analyses under reducing or nonreducing conditions. The purity of the nuclear, cytoplasmic and cytoskeletal samples was confirmed by immunoblot with membrane (Na/K ATP-ase), nuclear (CREB), cytoskeletal (β-catenin) and cytoplasmic (LDH) markers (Figure S2D). After determining the protein concentration (Bio-Rad), the lysates (5-20 μg/well) were subjected to 10% SDS polyacrylamide gel electrophoresis and subsequently blotted to PVDF membranes (Millipore) as described previously (Sananbenesi et al., 2002). The membranes were saturated with I-block (Tropix) and then incubated with the primary (Supplemental Procedures) and corresponding secondary antibodies, enhancer (Nitro-block II, Tropix) and chemiluminescent substrate (CDP Star, Tropix). Blots were exposed to X-ray films and developed in the range of maximal chemiluminescence emission (10 min). Molecular weight and densitometric calculations of pErk-1/2 were performed with the computer software ImageJ (NIH). Inter-assay variability between blots was determined by a standard control sample for each individual fraction obtained from pooled hippocampi of five naïve mice. The levels of individual proteins were normalized to β-actin. The signals of pErk-1/2 were additionally normalized to those of total Erk-1/2 in corresponding individual samples.
Immunoprecipitation
Naive mice or mice exposed to fear conditioning were injected i.h. immediately afterwards with vehicle, HAV-S or HAV-N (n = 9/group). Individual dorsal hippocampi were collected 1 hour later, when HAV-N typically causes a significant decrease of pErk. The hippocampi were lysed in a modified radioimmunoprecipitation buffer, incubated 15 min on ice and centrifuged for 15 min, 15000 × g, 4°C as described (Fischer et al., 2007). After determining the protein concentration for each lysate (Bio-Rad protein assay), 2 mg of total protein/lysate was prepared by pooling 3 samples of each group. Five hundred μg/sample/group was incubated for 1h at 4°C with 4 μg anti-N-cadherin, IQGAP1, Erk or pErk-1/2 antibodies. Immunoprecipitation was performed by the Catch and Release kit (Upstate) as described in the user’s manual. Subsequent immunoblot and analyses were performed as described above.
Neuronal culture and treatments
Medium density cortical neuron cultures were prepared from rat E18 embryos as described previously (Liao et al., 1999; see Supplemental Procedures). Chemical activation of NMDA receptors was preformed at DIV 28 as follows. Neuron cultures were maintained in presence of 200 μM D,L-amino-phosphonovalerate (D,L-APV). For treatments, neurons were pre-incubated in aCSF (in mM: 125 NaCl, 2.5 KCl, 26.2 NaHCO3, 1 NaH2PO4, 11 glucose and 2.5 CaCl2 ± 200 μM APV and 1.25 MgCl2) for 30 min at 37°C. Coverslips were also pre-treated or not, with the peptides HAV-S or HAV-N at a concentration of 200 μM for 2 hours. Following a wash in treatment medium (aCSF plus 10 μM glycine, 100 μM picrotoxin and 1 μM strychnine), coverslips were transferred to the treatment chambers in aCSF ± peptides. Treatments were allowed to proceed for the indicated times after which cells were fixed for 10 minutes with methanol chilled to -20°C. Coverslips were then processed for immunostaining and analyses as previously described (Penzes et al., 2003; See supplemental Procedures)
Data analysis
Statistically significant differences were determined by Student’s t-test, or one (factor Treatment) and two way ANOVA (factors Dose × Time) followed by Scheffe’s test for post-hoc comparisons. The results are presented as mean ± S.E.M.
Supplementary Material
Acknowledgments
We thank Dr. Deanna Benson (Mount Sinai School of Medicine) for helpful discussion. This work was supported by the NIMH grant MH073669 to J.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angst BD, Marcozzi C, Magee AI. The cadherin superfamily: diversity in form and function. J Cell Sci. 2001;114:629–641. doi: 10.1242/jcs.114.4.629. [DOI] [PubMed] [Google Scholar]
- Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- Benes FM, Burke RE, Walsh J, Berretta S, Matzilevich D, Minns M, Konradi C. Acute amygdalar activation induces an upregulation of multiple monoamine G protein coupled pathways in rat hippocampus. Mol Psychiatry. 2004;9:932–945. 895. doi: 10.1038/sj.mp.4001524. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y. Memory extinction, learning anew, and learning the new: dissociations in the molecular machinery of learning in cortex. Science. 2001;291:2417–2419. doi: 10.1126/science.1058165. [DOI] [PubMed] [Google Scholar]
- Bozdagi O, Shan W, Tanaka H, Benson DL, Huntley GW. Increasing numbers of synaptic puncta during late-phase LTP: N-cadherin is synthesized, recruited to synaptic sites, and required for potentiation. Neuron. 2000;28:245–259. doi: 10.1016/s0896-6273(00)00100-8. [DOI] [PubMed] [Google Scholar]
- Brown MD, Sacks DB. IQGAP1 in cellular signaling: bridging the GAP. Trends Cell Biol. 2006 doi: 10.1016/j.tcb.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Calabrese EJ, Baldwin LA. Toxicology rethinks its central belief. Nature. 2003;421:691–692. doi: 10.1038/421691a. [DOI] [PubMed] [Google Scholar]
- Charrasse S, Meriane M, Comunale F, Blangy A, Gauthier-Rouviere C. N-cadherin-dependent cell-cell contact regulates Rho GTPases and beta-catenin localization in mouse C2C12 myoblasts. J Cell Biol. 2002;158:953–965. doi: 10.1083/jcb.200202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Ferrari GV, Chacon MA, Barria MI, Garrido JL, Godoy JA, Olivares G, Reyes AE, Alvarez A, Bronfman M, Inestrosa NC. Activation of Wnt signaling rescues neurodegeneration and behavioral impairments induced by beta-amyloid fibrils. Mol Psychiatry. 2003;8:195–208. doi: 10.1038/sj.mp.4001208. [DOI] [PubMed] [Google Scholar]
- Dell’acqua ML, Smith KE, Gorski JA, Horne EA, Gibson ES, Gomez LL. Regulation of neuronal PKA signaling through AKAP targeting dynamics. Eur J Cell Biol. 2006 doi: 10.1016/j.ejcb.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Derycke LD, Bracke ME. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int J Dev Biol. 2004;48:463–476. doi: 10.1387/ijdb.041793ld. [DOI] [PubMed] [Google Scholar]
- Edsbagge J, Zhu S, Xiao MY, Wigstrom H, Mohammed AH, Semb H. Expression of dominant negative cadherin in the adult mouse brain modifies rearing behavior. Mol Cell Neurosci. 2004;25:524–535. doi: 10.1016/j.mcn.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Cyclin-dependent kinase 5 is required for associative learning. J Neurosci. 2002;22:3700–3707. doi: 10.1523/JNEUROSCI.22-09-03700.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Schrick C, Spiess J, Radulovic J. Distinct roles of hippocampal de novo protein synthesis and actin rearrangement in extinction of contextual fear. J Neurosci. 2004;24:1962–1966. doi: 10.1523/JNEUROSCI.5112-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga K, Miyamoto E. Role of MAP kinase in neurons. Mol Neurobiol. 1998;16:79–95. doi: 10.1007/BF02740604. [DOI] [PubMed] [Google Scholar]
- Frey U, Morris RG. Synaptic tagging and long-term potentiation. Nature. 1997;385:533–536. doi: 10.1038/385533a0. [DOI] [PubMed] [Google Scholar]
- Fukata M, Nakagawa M, Itoh N, Kawajiri A, Yamaga M, Kuroda S, Kaibuchi K. Involvement of IQGAP1, an effector of Rac1 and Cdc42 GTPases, in cell-cell dissociation during cell scattering. Mol Cell Biol. 2001;21:2165–2183. doi: 10.1128/MCB.21.6.2165-2183.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagler DJ, Jr, Goda Y. Synaptic adhesion: the building blocks of memory? Neuron. 1998;20:1059–1062. doi: 10.1016/s0896-6273(00)80486-9. [DOI] [PubMed] [Google Scholar]
- Harding A, Tian T, Westbury E, Frische E, Hancock JF. Subcellular localization determines MAP kinase signal output. Curr Biol. 2005;15:869–873. doi: 10.1016/j.cub.2005.04.020. [DOI] [PubMed] [Google Scholar]
- Harrison OJ, Corps EM, Kilshaw PJ. Cadherin adhesion depends on a salt bridge at the N-terminus. J Cell Sci. 2005;118:4123–4130. doi: 10.1242/jcs.02539. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- Junghans D, Haas IG, Kemler R. Mammalian cadherins and protocadherins: about cell death, synapses and processing. Curr Opin Cell Biol. 2005;17:446–452. doi: 10.1016/j.ceb.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Kaech S, Fischer M, Doll T, Matus A. Isoform specificity in the relationship of actin to dendritic spines. J Neurosci. 1997;17:9565–9572. doi: 10.1523/JNEUROSCI.17-24-09565.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaibuchi K, Kuroda S, Fukata M, Nakagawa M. Regulation of cadherin-mediated cell-cell adhesion by the Rho family GTPases. Curr Opin Cell Biol. 1999;11:591–596. doi: 10.1016/s0955-0674(99)00014-9. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Johnson KR, Wheelock MJ. N-cadherin-mediated cell motility requires cis dimers. Cell Commun Adhes. 2005;12:23–39. doi: 10.1080/15419060500305971. [DOI] [PubMed] [Google Scholar]
- Klingelhofer J, Laur OY, Troyanovsky RB, Troyanovsky SM. Dynamic interplay between adhesive and lateral E-cadherin dimers. Mol Cell Biol. 2002;22:7449–7458. doi: 10.1128/MCB.22.21.7449-7458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levchenko A, Bruck J, Sternberg PW. Scaffold proteins may biphasically affect the levels of mitogen-activated protein kinase signaling and reduce its threshold properties. Proc Natl Acad Sci U S A. 2000;97:5818–5823. doi: 10.1073/pnas.97.11.5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Zhang X, O’Brien R, Ehlers MD, Huganir RL. Regulation of morphological postsynaptic silent synapses in developing hippocampal neurons. Nat Neurosci. 1999;2:37–43. doi: 10.1038/4540. [DOI] [PubMed] [Google Scholar]
- Lin CH, Yeh SH, Leu TH, Chang WC, Wang ST, Gean PW. Identification of calcineurin as a key signal in the extinction of fear memory. J Neurosci. 2003;23:1574–1579. doi: 10.1523/JNEUROSCI.23-05-01574.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makagiansar IT, Nguyen PD, Ikesue A, Kuczera K, Dentler W, Urbauer JL, Galeva N, Alterman M, Siahaan TJ. Disulfide bond formation promotes the cis- and trans-dimerization of the E-cadherin-derived first repeat. J Biol Chem. 2002;277:16002–16010. doi: 10.1074/jbc.M200916200. [DOI] [PubMed] [Google Scholar]
- Manabe T, Togashi H, Uchida N, Suzuki SC, Hayakawa Y, Yamamoto M, Yoda H, Miyakawa T, Takeichi M, Chisaka O. Loss of cadherin-11 adhesion receptor enhances plastic changes in hippocampal synapses and modifies behavioral responses. Mol Cell Neurosci. 2000;15:534–546. doi: 10.1006/mcne.2000.0849. [DOI] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–645. doi: 10.1016/j.cell.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Miyamoto E. Molecular mechanism of neuronal plasticity: induction and maintenance of long-term potentiation in the hippocampus. J Pharmacol Sci. 2006;100:433–442. doi: 10.1254/jphs.cpj06007x. [DOI] [PubMed] [Google Scholar]
- Morozov A, Muzzio IA, Bourtchouladze R, Van-Strien N, Lapidus K, Yin D, Winder DG, Adams JP, Sweatt JD, Kandel ER. Rap1 couples cAMP signaling to a distinct pool of p42/44MAPK regulating excitability, synaptic plasticity, learning, and memory. Neuron. 2003;39:309–325. doi: 10.1016/s0896-6273(03)00404-5. [DOI] [PubMed] [Google Scholar]
- Murase S, Schuman EM. The role of cell adhesion molecules in synaptic plasticity and memory. Curr Opin Cell Biol. 1999;11:549–553. doi: 10.1016/s0955-0674(99)00019-8. [DOI] [PubMed] [Google Scholar]
- Nakao K, Matsuyama K, Matsuki N, Ikegaya Y. Amygdala stimulation modulates hippocampal synaptic plasticity. Proc Natl Acad Sci U S A. 2004;101:14270–14275. doi: 10.1073/pnas.0405709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noritake J, Watanabe T, Sato K, Wang S, Kaibuchi K. IQGAP1: a key regulator of adhesion and migration. J Cell Sci. 2005;118:2085–2092. doi: 10.1242/jcs.02379. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Huganir RL. Regulation of AMPA receptor trafficking by N-cadherin. J Neurochem. 2006;97:652–661. doi: 10.1111/j.1471-4159.2006.03740.x. [DOI] [PubMed] [Google Scholar]
- Okamura K, Tanaka H, Yagita Y, Saeki Y, Taguchi A, Hiraoka Y, Zeng LH, Colman DR, Miki N. Cadherin activity is required for activity-induced spine remodeling. J Cell Biol. 2004;167:961–972. doi: 10.1083/jcb.200406030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Beeser A, Chernoff J, Schiller MR, Eipper BA, Mains RE, Huganir RL. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron. 2003;37:263–274. doi: 10.1016/s0896-6273(02)01168-6. [DOI] [PubMed] [Google Scholar]
- Perron JC, Bixby JL. Distinct neurite outgrowth signaling pathways converge on ERK activation. Mol Cell Neurosci. 1999;13:362–378. doi: 10.1006/mcne.1999.0753. [DOI] [PubMed] [Google Scholar]
- Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO. Developmental defects in mouse embryos lacking N-cadherin. Dev Biol. 1997;181:64–78. doi: 10.1006/dbio.1996.8443. [DOI] [PubMed] [Google Scholar]
- Radulovic J, Kammermeier J, Spiess J. Relationship between fos production and classical fear conditioning: effects of novelty, latent inhibition, and unconditioned stimulus preexposure. J Neurosci. 1998;18:7452–7461. doi: 10.1523/JNEUROSCI.18-18-07452.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovic J, Ruhmann A, Liepold T, Spiess J. Modulation of learning and anxiety by corticotropin-releasing factor (CRF) and stress: differential roles of CRF receptors 1 and 2. J Neurosci. 1999;19:5016–5025. doi: 10.1523/JNEUROSCI.19-12-05016.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redies C. Cadherins in the central nervous system. Prog Neurobiol. 2000;61:611–648. doi: 10.1016/s0301-0082(99)00070-2. [DOI] [PubMed] [Google Scholar]
- Reiss K, Maretzky T, Ludwig A, Tousseyn T, de Strooper B, Hartmann D, Saftig P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. Embo J. 2005;24:742–752. doi: 10.1038/sj.emboj.7600548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud-Young M, Gallin WJ. In the first extracellular domain of E-cadherin, heterophilic interactions, but not the conserved His-Ala-Val motif, are required for adhesion. J Biol Chem. 2002;277:39609–39616. doi: 10.1074/jbc.M201256200. [DOI] [PubMed] [Google Scholar]
- Retta SF, Balzac F, Avolio M. Rap1: A turnabout for the crosstalk between cadherins and integrins. Eur J Cell Biol. 2006;85:283–293. doi: 10.1016/j.ejcb.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Rhee J, Mahfooz NS, Arregui C, Lilien J, Balsamo J, VanBerkum MF. Activation of the repulsive receptor Roundabout inhibits N-cadherin-mediated cell adhesion. Nat Cell Biol. 2002;4:798–805. doi: 10.1038/ncb858. [DOI] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB. IQGAP1 binds ERK2 and modulates its activity. J Biol Chem. 2004;279:17329–17337. doi: 10.1074/jbc.M308405200. [DOI] [PubMed] [Google Scholar]
- Roy M, Li Z, Sacks DB. IQGAP1 is a scaffold for mitogen-activated protein kinase signaling. Mol Cell Biol. 2005;25:7940–7952. doi: 10.1128/MCB.25.18.7940-7952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Phosphorylation of hippocampal Erk-1/2, Elk-1, and p90-Rsk-1 during contextual fear conditioning: interactions between Erk-1/2 and Elk-1. Mol Cell Neurosci. 2002;21:463–476. doi: 10.1006/mcne.2002.1188. [DOI] [PubMed] [Google Scholar]
- Sekino Y, Obata K, Tanifuji M, Mizuno M, Murayama J. Delayed signal propagation via CA2 in rat hippocampal slices revealed by optical recording. J Neurophysiol. 1997;78:1662–1668. doi: 10.1152/jn.1997.78.3.1662. [DOI] [PubMed] [Google Scholar]
- Shalin SC, Hernandez CM, Dougherty MK, Morrison DK, Sweatt JD. Kinase suppressor of Ras1 compartmentalizes hippocampal signal transduction and subserves synaptic plasticity and memory formation. Neuron. 2006;50:765–779. doi: 10.1016/j.neuron.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Colman DR. Structural biology of cadherins in the nervous system. Curr Opin Neurobiol. 1998;8:593–599. doi: 10.1016/s0959-4388(98)80086-x. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, Legrand JF, Als-Nielsen J, Colman DR, Hendrickson WA. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Tanaka H, Shan W, Phillips GR, Arndt K, Bozdagi O, Shapiro L, Huntley GW, Benson DL, Colman DR. Molecular modification of N-cadherin in response to synaptic activity. Neuron. 2000;25:93–107. doi: 10.1016/s0896-6273(00)80874-0. [DOI] [PubMed] [Google Scholar]
- Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron. 1998;20:1165–1175. doi: 10.1016/s0896-6273(00)80497-3. [DOI] [PubMed] [Google Scholar]
- Togashi H, Abe K, Mizoguchi A, Takaoka K, Chisaka O, Takeichi M. Cadherin regulates dendritic spine morphogenesis. Neuron. 2002;35:77–89. doi: 10.1016/s0896-6273(02)00748-1. [DOI] [PubMed] [Google Scholar]
- Troyanovsky RB, Sokolov EP, Troyanovsky SM. Endocytosis of cadherin from intracellular junctions is the driving force for cadherin adhesive dimer disassembly. Mol Biol Cell. 2006;17:3484–3493. doi: 10.1091/mbc.E06-03-0190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widelitz R. Wnt signaling through canonical and non-canonical pathways: recent progress. Growth Factors. 2005;23:111–116. doi: 10.1080/08977190500125746. [DOI] [PubMed] [Google Scholar]
- Williams E, Williams G, Gour BJ, Blaschuk OW, Doherty P. A novel family of cyclic peptide antagonists suggests that N-cadherin specificity is determined by amino acids that flank the HAV motif. J Biol Chem. 2000;275:4007–4012. doi: 10.1074/jbc.275.6.4007. [DOI] [PubMed] [Google Scholar]
- Williams EJ, Williams G, Howell FV, Skaper SD, Walsh FS, Doherty P. Identification of an N-cadherin motif that can interact with the fibroblast growth factor receptor and is required for axonal growth. J Biol Chem. 2001;276:43879–43886. doi: 10.1074/jbc.M105876200. [DOI] [PubMed] [Google Scholar]
- Wilson MT, Keith CH. Glutamate modulation of dendrite outgrowth: alterations in the distribution of dendritic microtubules. J Neurosci Res. 1998;52:599–611. doi: 10.1002/(SICI)1097-4547(19980601)52:5<599::AID-JNR12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Winder DG, Martin KC, Muzzio IA, Rohrer D, Chruscinski A, Kobilka B, Kandel ER. ERK plays a regulatory role in induction of LTP by theta frequency stimulation and its modulation by beta-adrenergic receptors. Neuron. 1999;24:715–726. doi: 10.1016/s0896-6273(00)81124-1. [DOI] [PubMed] [Google Scholar]
- Yu X, Malenka RC. Beta-catenin is critical for dendritic morphogenesis. Nat Neurosci. 2003;6:1169–1177. doi: 10.1038/nn1132. [DOI] [PubMed] [Google Scholar]
- Zaidel DW. Quantitative morphology of human hippocampus early neuron development. Anat Rec. 1999;254:87–91. doi: 10.1002/(SICI)1097-0185(19990101)254:1<87::AID-AR11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.