

Abstract
Testosterone and structurally related anabolic steroids have been used to treat hypogonadism, muscle wasting, osteoporosis, male contraception, cancer cachexia, anemia, and hormone replacement therapy in aging men or age-related frailty; while antiandrogens may be useful for treatment of conditions like acne, alopecia (male-pattern baldness), hirsutism, benign prostatic hyperplasia (BPH) and prostate cancer. However, the undesirable physicochemical and pharmacokinetic properties of steroidal androgen receptor (AR) ligands limited their clinical use. Nonsteroidal AR ligands with improved pharmacological and pharmacokinetic properties have been developed to overcome these problems. This review focuses on the pharmacokinetics, metabolism, and pharmacology of clinically used and emerging nonsteroidal AR ligands, including antagonists, agonists, and selective androgen receptor modulators.
Keywords: androgen receptor, antiandrogen, pharmacokinetics, prostate cancer, selective androgen receptor modulator, testosterone
INTRODUCTION
Androgens are essential for male development and the maintenance of male secondary characteristics, such as bone mass, muscle mass, body composition, and spermatogenesis. Testosterone and structurally related anabolic steroids have been used to treat hypogonadism, muscle wasting, osteoporosis, cancer cachexia, and anemia, and been used for male contraception, and hormone replacement therapy in aging men or age-related frailty; while antiandrogens may be useful for treatment of conditions like acne, alopecia (male-pattern baldness), hirsutism, benign prostatic hyperplasia (BPH) and prostate cancer. However, the clinical application of the steroidal AR ligands has been limited by poor oral bioavail-ability, potential hepatotoxicity, lack of tissue selectivity, and occasionally, cross reaction with other steroid receptors. Also, structural modification of the steroidal ligands is somewhat limited by the steroid skeleton. Therefore, nonsteroidal AR ligands with improved pharmacological and pharmacokinetic properties have been developed to overcome these problems.
The known AR ligands can be classified as steroidal or nonsteroidal based on the structure or as agonist and antagonist (antiandrogen) based on their ability to activate or inhibit transcription of AR target genes. Endogenous androgens, including testosterone and dihydrotestosterone (DHT), are steroidal agonists. Structural modifications of the endogenous steroids led to the development of various synthetic steroids, including agonists and antagonists. Nonsteroidal ligands were also proposed to achieve 1) high AR specificity, 2) improved oral bioavailability, 3) improved tissue selectivity, and 4) more flexible structural modifications. Nonsteroidal antiandrogens were first developed for the treatment of prostate cancer, and further structural modifications of these nonsteroidal anti-androgens led to the discovery of several different structural classes of nonsteroidal AR agonists.
The latest developments in nonsteroidal AR ligands have been extensively reviewed in last five years, including the structural biology and structure–activity relationships (1), medicinal chemistry (2), and potential clinical applications (3). This review focuses on the in vivo pharmacokinetic and pharmacologic properties of nonsteroidal AR ligands, including antagonists, agonists, and selective androgen receptor modulators (SARMs). Ligands in current clinical use are emphasized due to the greater availability of relevant literature.
ANDROGEN RECEPTOR
The AR is a member of the steroid and nuclear receptor superfamily, and is a soluble protein that functions as an intracellular transcriptional factor. The structural biology and ligand chemistry of the AR were reviewed recently (1). Structurally, AR contains three major functional domains, N-terminal domain (NTD), DNA-binding domain (DBD), and ligand binding domain (LBD), as demonstrated in Fig. 1. AR ligands regulate receptor function through binding to the LBD, which initiates sequential conformational changes of the receptor. Upon agonist binding, the receptor then undergoes dissociation from the chaperones, dimerization, phosphorylation, translocation to the nucleus, and binding to the androgen response element (4). Recruitment of other transcription co-regulators and transcriptional machinery further ensures the transactivation of the AR-regulated gene expression upon agonist activation.
Fig. 1.

Structural organization of the human androgen receptor (AR).
AR is mainly expressed in androgen target tissues, such as the prostate, skeletal muscle, liver, and central nervous system (CNS), with the highest expression level observed in the prostate, adrenal gland, and epididymis as determined by real-time PCR (5). AR can be activated by the binding of endogenous androgens, including testosterone and DHT. Physiologically, functional AR is responsible for male sexual differentiation in utero and for male pubertal changes. In adult males, androgen is mainly responsible for maintaining libido, spermatogenesis, muscle mass and strength, bone mineral density, and erythropoiesis (6,7). The actions of androgen in reproductive tissues, including prostate, seminal vesicle, testis, and accessory structures, are known as androgenic effects, while the nitrogen-retaining effects of androgen in muscle and bone are known as anabolic effects. Gonadal production of testosterone is under the feedback regulation of circulating testosterone through the hypothalamo–pituitary–gonadal axis.
STEROIDAL LIGANDS
Testosterone and DHT are endogenous androgens. There are three modes of action of testosterone (Fig. 2). It may directly act through AR in target tissues where 5α-reductase is not expressed, be converted to 5α-DHT (5–10%) by 5α-reductase before binding to AR, or be aromatized to estrogen (0.2%) and act through the estrogen receptor (6). The formation of 5α-DHT is a natural way for the ‘DHT-dependent’ tissues, such as prostate and seminal vesicle, to amplify the androgenic activity of testosterone since 5α-DHT is more potent than testosterone. DHT binds to AR with higher affinity, and has two to ten fold higher potency than testosterone in androgen-responsive tissues (8). On the other hand, estrogen plays a major role in regulating metabolic processes (9,10), mood and cognition (11), cardiovascular disease (12,13), sexual function including libido (14), and bone turnover in men (15,16). Testosterone is the major androgen that acts in ‘DHT-independent’ tissues, such as skeletal muscle, where 5α-reductase is not expressed or expressed at a very low level (17) and it directly regulates skeletal muscle growth, bone formation, fat distribution, and sexual function.
Fig. 2.

Physiological actions of testosterone.
Following oral administration, the plasma half life of testosterone is less than 30 min, due to extensive metabolism. Approximately 90% of an oral dose of testosterone is metabolized before it reaches the systemic circulation. To improve the bioavailability, most of the testosterone preparations are delivered through transdermal patch or intramuscular injections. Alkylation or esterification at the 17 position (Fig. 3) was widely used in structural modification of the steroid skeleton to markedly slow down the hepatic metabolism and increase the oral bioavailability or duration of testosterone action. However, 17α-alkylated steroidal androgens are more likely to cause hepatotoxicity, the most serious side effect of the synthetic steroids. On the other hand, complete separation of androgenic and anabolic activity has not been accomplished with synthetic steroids. The androgenic activities of the synthetic steroids often cause undesirable side effects during therapy. Due to the structural similarity in the steroid skeleton, steroidal AR ligands also tend to cross react with other steroid receptors, which is also associated with adverse effects (i.e., gynecomastia).
Fig. 3.
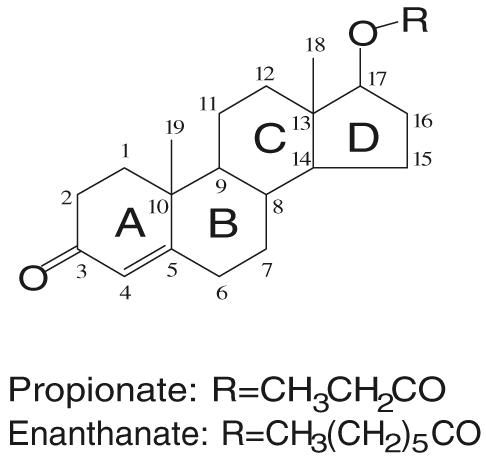
Chemical structure of testosterone and commonly used testosterone esters.
CLINICAL APPLICATIONS OF STEROIDAL AR LIGANDS
Classically, testosterone is used to treat male hypogonadism, protein wasting diseases associated with cancer, burns, traumas, or Acquired Immunodeficiency Syndrome (AIDS), anemia secondary to chronic renal failure, aplastic anemia, hereditary angioedema, or as a component of hormonal male contraception (6). Recently, hormone replacement therapy (HRT) in aging males has also been proposed to improve body composition, bone and cartilage metabolism, certain domains of brain function, and even decrease cardiovascular risk (8). For most clinical applications, testosterone is usually given as longer acting esters through intramuscular injections, surgical implantation for implants and pellets, or transdermal delivery, such as patches and gels. In general, these administration routes are not very convenient, and are sometimes associated with fluctuation in serum testosterone levels, skin rashes and irritation.
Pharmacologically, exogenous testosterone works well for male hypogonadism (18) related to deficiency of endogenous hormone production, including primary (testicular), secondary (hypothalamic or pituitary), and age-related hypogonadism. However, when testosterone is used for age-related hypogonadism (HRT in aging men), the potential risk in the prostate becomes a major concern of long term treatment. Besides hypogonadism, testosterone is mainly used for the treatment of disease related muscle wasting and male hormonal contraception. When supraphysiologic concentrations of testosterone is used for male contraception, steroid-related side effects, including decreases in HDL cholesterol, increases in hematologic parameters such as hemoglobin and hematocrit, increased body weight, and acne, are the major drawbacks of the treatment.
Androgen can also be used as anabolic reagent to treat muscle wasting. Commonly used anabolic steroids include nandrolone decanoate and oxandrolone, although nandro-lone decanoate is known to be associated with hepatotoxicity and side effects on the blood lipid profile. Muscle is not the only anabolic tissue. It has also been proposed that testosterone can be used as an anabolic reagent to treat osteoporosis, since androgens seem to have direct anabolic effects in bone, and the anabolic effects in skeletal muscle mass and strength could also be beneficial to the treatment of osteoporosis. However, the androgenic effects associated with most steroidal androgens become major concerns for therapy, particularly in aging men and women.
On the other hand, both antiandrogens and 5α-reductase inhibitors are used to block androgen action in prostate cancer, BPH, and acne. The application of these steroidal antiandrogens, like cyproterone acetate and spironolactone, has been limited by the weak antagonist activities or cross-reaction with other steroid receptors. Also, due to the lack of tissue selectivity, complete androgen blockage with antiandrogens also cause severe side effects related to androgen deficiency (e.g., loss of libido, hot flashes, impotence, and increased incidence of osteoporosis). In comparison, 5α-reductase inhibitors are considered more ‘tissue-selective’ due to the tissue-specific expression of 5α-reductase, even though the inhibition of 5α-reductase in male could cause gynecomastia due to the increase in estrogen production (Fig. 2).
In summary, steroidal AR ligands, including agonists and antagonists, are used in the treatment of a variety of androgen disorders. However, the side effects related to the lack of tissue selectivity, hepatotoxicity, and inconvenience of delivery limits the more widespread therapeutic applications of androgens. A variety of nonsteroidal AR ligands have been or are being developed to overcome these limitations with 1) improved pharmacokinetic profile and oral bioavailability; 2) improved tissue selectivity; 3) higher specificity for AR; and 4) less heptatotoxicity as major goals.
IN VITRO AND IN VIVO MODELS FOR EVALUATION OF AR LIGANDS
Generally, novel AR ligands are first identified by in vitro receptor binding assay, using either rat prostate cytosolic AR (rat AR and human AR share identical LBD), recombinant AR protein, or cells that express AR (endogenously expressed or transiently transfected), as summarized by Fang et al. (19). Although the binding affinity of the ligands were determined by different research groups using slightly different methods (detailed binding assay methods can be retrieved from corresponding references), the AR binding affinity of all ligands discussed in this review will be presented as the relative binding affinity (RBA) compared to the synthetic steroid R1881 (Tables II and IV), which has a Kd value of 0.53 nM as determined by Kelce et al. (20) and Waller et al. (21) using rat prostate cytosolic AR. The agonist or antagonist activity of the ligand is often examined in vitro using reporter assays in which a hormone-dependent reporter gene is transiently expressed in a cell line that contains AR.
Table II.
PK and PD of Nonsteroidal Antiandrogens
| ID | Compound structure |
RBAa (%) | Orally available |
|---|---|---|---|
| R-Bicalutamide |  |
0.4 (73) | F = 0.7 (rat) (1 mg/kg) (74) F = 0.1 (rat) (250 mg/kg) F≈1 (human) (50 mg) (31) |
| Hydroxy-flutamide |  |
0.1 (73) | Yes |
| Nilutamide |  |
0.08 (22) | F = 1 (rat) (26) |
| RU58642 |  |
6 (52) | Yes |
| RU58841 |  |
5 (42) | No |
| BMS-25 |  |
N/A | Yes |
| BMS-7 |  |
N/A | Yes |
| BMS-9 |  |
N/A | Yes |
| LG120907 |  |
14 (45) | Yes |
| LG105 |  |
11 (45) | Yes |
| Intact male rat model |
|||
|---|---|---|---|
| In vivo half life | Prostate | L.A. | Serum T |
| 17–21 h (rats) (30) 6 days (human) (29) | 35% intact level @ 25 mg/kg, po (33) | 55% intact level @ 8 mg/kg, sc | 3 ng/ml @ 25 mg/kg, po (33) |
| 4–22 h (human) (23) | 45% intact level @ 25 mg/kg, sc (53) | 70% intact level @ 25 mg/kg, sc (53) | 18 ng/ml @ 25 mg/kg, sc (53) |
| 6 h (rat) (26) 6 days (human) (26) | 60% intact level @ 30 mg/kg, po (35) | N/A | 6 ng/ml @ 25 mg/kg, po (35) |
| N/A | 30% intact level @ 30 mg/kg, po (35) | N/A | 7 ng/ml @ 30 mg/kg, po (35) |
| N/A | N/A | N/A | N/A |
| N/A | Modest inhibition @ 100 mg/kg, po (40) | Modest inhibition @ 100 mg/kg, po (40) | N/A |
| N/A | Modest inhibition @ 100 mg/kg, po (40) | Modest inhibition @ 100 mg/kg, po (40) | N/A |
| N/A | Modest inhibition @ 100 mg/kg, po (40) | Modest inhibition @ 100 mg/kg, po (40) | N/A |
| N/A | 65% intact level @ 30 mg/kg, po (75) | N/A | Intact level @ 40 mg/kg, po (76) |
| N/A | 33% intact level @ 30 mg/kg, po (75) | N/A | N/A |
RBA: Relative binding affinity compared to DHT.
Table IV.
PK and PD of Nonsteroidal Androgen Receptor Agonists
| ID | Compound structure | RBAa (%) |
Orally available |
In vivo half life |
|---|---|---|---|---|
| Testosterone |  |
28 (73) | No | <30 min |
| LG121071 |  |
18 (48) | Yes | N/A |
| LGD2226 |  |
170 (77) | Yes | N/A |
| S4 | 6 (52) | F = 1 (rat) (58) (10 mg/kg) | 4 h (rat) (58) 4 h (human) | |
| S1 | 4 (52) | F = 0.6 (rat) (57) (10 mg/kg) | 4 h (rat) (57) | |
| C6 | 11 (56) | F = 0.8 (rat) (56) (10 mg/kg) | 6 h (rat) (56) | |
| S22 |  |
6 (57) | Yes | 6 h (rat) (57) |
| BMS 564929 |  |
3 (67) | Yes | 8Y14 h (human) |
| S-40503 |  |
0.3 (68) | Yes | N/A |
| Castrated rat modelb | |||
|---|---|---|---|
| Prostate | L.A. | Bone | LH/FSH |
| ED50 = 0.5 mg/kg Emax = 120% (52) | ED50 = 0.6 mg/kg Emax = 104% (52) | Full agonist | ID50 = 0.26 mg/kg (67) |
| N/A | N/A | N/A | ↓ LH to intact level @ 20 mg/kg, po (48) |
| 100% intact level @ 100 mg/kg | 100% intact level @ 3 mg/kg | Full agonist (49) | N/A |
| ED50 = 1.6 mg/kg Emax = 35% (52) | ED50 = 0.6 mg/kg Emax = 101% (52) | Full agonist (55) | Partial agonist (52) |
| ED50 = 1.7 mg/kg Emax = 15% (52) | ED50 = 1.6 mg/kg Emax = 75% (52) | N/A | Partial agonist (52) |
| ED50 = 3.9 mg/kg Emax = 130% (56) | ED50 = 0.85 mg/kg Emax = 130% (56) | N/A | ↓ LH to intact level @ 1.5 mg/kg (56) |
| ED50 = 0.5 mg/kg Emax = 51% (57) | ED50 = 0.12 mg/kg Emax = 136% (57) | Full agonist | N/A |
| ED50 = 141 μg/kg Emax = 105% (67) | ED50 = 0.9 μg/kg Emax = 120% (67) | N/A | ID50 = 8 μg/kg (67) |
| 80% intact level @ 30 mg/kg | 115% intact level @ 30 mg/kg | Full agonist | N/A |
RBA: Relative binding affinity compared to DHT.
All reagents were administered via daily subcutaneous injections unless noted otherwise.
However, the in vitro models cannot accurately predict the in vivo PK and PD profiles of the ligands. As such, ligands with high binding affinity and potent intrinsic activity in stimulating transcription activation are normally further evaluated in vivo. AR agonist activity is usually tested in castrated rats (treatment starts the day after castration for immediate treatment), in which endogenous testosterone is depleted. Thus, the inherent anabolic and androgenic activity of a compound of interest can be evaluated in the absence of the endogenous agonist. Antagonist activity is often tested in an intact male rat model, which contains normal level of endogenous testosterone, affording the potential to discern the ability of an investigational agent to inhibit the actions of the endogenous agonist. Treatment generally lasts for two weeks, with androgen responsive tissues weighed at the end of the study to evaluate the androgenic (i.e., prostate and seminal vesicle) and anabolic (i.e., levator ani muscle, bone mineral density) activity of the ligand. Serum LH, FSH, and testosterone levels are often measured as well to examine the effects of the ligands on the hypothalamic–pituitary–gonadal axis. Sometimes, the antagonist activity of the ligand is also tested as its ability to suppress androgen-dependent prostate cancer cell growth. Alternatively, antiandrogen activity can also be tested in androgen sensitive prostate cancer xenograft models.
NONSTEROIDAL ANDROGEN RECEPTOR ANTAGONISTS
Several structural classes of nonsteroidal AR antagonists have been discovered. In this review, we will focus our discussion on the ones that have been characterized, such as toluidide, hydantoin, and quinolinone antiandrogens.
Substituted toluidides, including bicalutamide, flutamide, and nilutamide (Table I), were the first nonsteroidal AR ligands developed and act as nonsteroidal antiandrogens. Unlike the steroidal antiandrogens, these toluidides are considered pure antiandrogens since they possess little if any intrinsic androgenic activity when bound to wild type AR, and have high specificity for AR without cross-reaction with any of the other steroid receptors. As such, these nonsteroidal antiandrogens are mainly used to treat androgen sensitive prostate cancer or hyperplasia (BPH). Besides their pure antagonist activity, these ligands are orally available with in vivo half lives ranging from 8 h to 6 days in humans (Table II).
Table I.
Nonsteroidal Antiandrogens
| Chemotype | General chemical structure |
Lead compound | Company (stage of development) |
||
|---|---|---|---|---|---|
| Aryl propionamide analogs |  |
Bicalutamide |  |
AstraZeneca Pharmaceuticals LP | Casodex™ |
| Flutamide |  |
Schering Corp. | Eulexin™ | ||
| Hydantoin analogs |  |
Nilutamide |  |
Aventis | Nilandron™ |
| RU58642 |  |
Roussel-Uclaf SA | Preclinical | ||
| RU58841 |  |
Roussel-Uclaf SA | Preclinical | ||
| BMS-25 |  |
 |
Preclinical | ||
| BMS-7 |  |
||||
| BMS-9 |  |
||||
| Quinoline analogs (Tri-cyclic) |  |
LG120907 |  |
Ligand Pharmaceuticals | Preclinical |
| LG105 |  |
Ligand Pharmaceuticals | Preclinical | ||
After oral administration, flutamide is completely absorbed from the gastrointestinal tract, and undergoes extensive first pass metabolism to its major metabolite 2-hydroxyflutamide and hydrolysis product, 3-trifluoromethyl-4-nitroaniline (Fig. 4A). 2-Hydroxyflutamide is a more powerful antiandrogen in vivo, with higher binding affinity for the AR than flutamide (22). In humans, hydroxyflutamide has an elimination half life of about 8 h (23). Hydrolysis of the amide bond represents the major metabolic pathway for this active metabolite (24). Due to its relatively low binding affinity to AR, flutamide is generally used at high doses of 750 mg/day in order to achieve complete AR blockage in therapy. Extensive hepatic metabolism of the drug generates a large amount of hydrolysis product, 3-trifluoromethyl-4-nitroaniline, which might be related to the hepatotoxicity sometimes observed with flutamide (25).
Fig. 4.

Major phase I metabolic pathways of flutamide (24) (A) and bicalutamide (30,31) (B).
As a hydantoin analog of flutamide, nilutamide is also eliminated exclusively by metabolism (26), mainly reduction of the aromatic nitro group (Fig. 5). Although the hydrolysis of one of the carbonyl functions of the imidazolinedione was also identified, it is much less susceptible to hepatic metabolism than the amide bond in hydroxyflutamide, which results in a much longer half life of two days in humans. Even so, the nitro anion-free radical formed during nitro reduction might still be associated with hepatotoxicity (27,28) in humans, especially when using the relatively high dosage (150–300 mg/day) employed for androgen blockage.
Fig. 5.

Major phase I metabolic pathways of nilutamide (26).
Currently, bicalutamide has replaced flutamide and nilutamide as the antiandrogen of choice for prostate cancer treatment, since it has less hepatotoxicity and longer half life (6 days in humans) (29) that allows once a day administration at relatively lower dosage (50 mg/day). As a structural analog, bicalutamide shares the amide bond structure with flutamide. However, amide bond hydrolysis was observed in rat, but not in humans (30,31) (Fig. 4B), which could explain the prolonged half life of bicalutamide in humans. Bicalutamide is mainly metabolized by hydroxylation and glucuronidation in humans. Also, the replacement of the nitro group with a cyano group avoids the nitro reduction observed in nilutamide. With the presence of the chiral carbon in structure, bicalutamide is administered as racemate. However, the in vivo antiandrogenic activity of bicalutamide arises almost entirely from its R-isomer, which has approximately 30-fold greater binding affinity and is cleared at a rate 1/100th of the S-isomer.
The greatly improved specificity and favorable pharmacokinetic profile of nonsteroidal antiandrogens, as compared to steroidal antiandrogens, affords much more efficient androgen blockage for prostate cancer treatment. Even so, at therapeutic doses, due to the complete blockage of AR in both the prostate and pituitary, these drugs often trigger significant increases in luteinizing hormone (32) release, which further stimulates higher serum testosterone concentrations. Therefore, these antiandrogens are used primarily in combination with a gonadotropin releasing hormone analog, which shuts down the testicular but not adrenal testosterone production. The treatments are similarly effective as surgical castration (32). However, this so-called ‘chemical castration’ also abolishes libido and the anabolic activity of androgens in the muscle and bone, causing undesirable side effects.
Animal studies suggested that bicalutamide might work as a tissue-selective AR ligand, since bicalutamide appeared to be peripherally selective in rats (33) with less antiandrogen activity in the pituitary (i.e., less suppression of gonadotropin release, and less increase in testosterone production, Table II), which could be related to its low tissue distribution in the CNS in rats. However, similar tissue selectivity was not observed in humans (29). On the other hand, Lefort et al. (34) showed that bicalutamide treatment does not cause significant decreases in BMD or bone mechanical strength as surgical castration in 16-week old male rats. However, bicalutamide was given at 5 mg/kg/day dose rate through oral gavage, which failed to demonstrate antiandrogen activity in the prostate (35). More importantly, higher incidence of osteoporosis related to androgen deprivation therapy is well recognized clinically. To minimize the side effects caused by complete androgen blockage, tissue-selective AR antagonists, agents that work as antagonist in the prostate with little or no effects in the anabolic tissues or CNS, become one of the major features for the next generation of antiandrogens to achieve.
Another major drawback of the existing antiandrogens is the development of antiandrogen withdrawal syndrome (20) in prostate cancer patients. Although antiandrogens are particularly useful for the treatment of prostate cancer during its early stages, prostate cancer often advances to a “hormone-refractory” state in which the disease progresses in the presence of continued androgen ablation or antiandrogen therapy, suggesting the development of androgen-independent prostate cancer cells or the ability of adrenal androgens to support tumor growth. AWS has often been reported after prolonged treatment with antiandrogens (36), and is defined in terms of the tumor regression or symptomatic relief observed upon cessation of antiandrogen therapy. The mechanism of AWS is not well understood, but it is believed that AR mutations, which could result in receptor promiscuity and the ability of some antiandrogens to exhibit agonist activity, might at least partially account for this phenomenon. For example, hydroxyflutamide and bicalutamide actually act as AR agonists in T877A and W741L/W741C AR mutants (37,38), respectively; and these mutations were developed after long term exposure to antiandrogen therapy (37,39). Therefore, more research efforts have been devoted to the development of new generation of ‘pure antiandrogens’ that would work in both wild-type and mutant AR.
A variety of hydantoin derivatives are in preclinical development (Table I). These compounds have not yet been evaluated clinically, but demonstrate potent antiandrogenic activity both in vitro and in vivo (Table II). In 2002 and 2003, Bristol-Myers Squibb disclosed a new series of bicyclic-1H-isoindole-1,3(2H)-dione analogs that act as ‘selective’ anti-androgens (40), by combining the structural features of bicalutamide and their previous bicyclic hydantoin analogs (41). Lead compounds 7 {(3aα,4β,7β,7aα)-4-[2-(4-fluorophenoxy)ethyl]hexahydro-7-methyl-2-(4-nitro-1-naphthalenyl)-4, 7-epoxy-1H-isoindole-1,3(2H)-dione}, 9 {(3aα,4β,7β,7aα)-4-[octahydro-4-methyl-1,3-dioxo-7-[2-[4-(trifluoromethyl) phenoxy]ethyl]-4,7-epoxy-2H-isoindole-2-yl]-2-(trifluromethyl)benzonitrile}, and 25 {[3aR-(3aα,4β,5β,7β,7aα)-4-[7-[2-(4-acetylphenoxy)ethyl]-octahydro-5-hydroxy-4-methyl-1, 3-dioxo-4,7-epoxy-2H-isoindol-2-yl]-1-naphthalenecarbonitrile]} (Table I) were reported to have antagonist activity against hormone-dependent tumors while exhibiting no agonist activity in other androgen target tissues, including the prostate, seminal vesicle, levator ani muscle, and pituitary.
These compounds were able to inhibit DHT-stimulated transcription activation in an MDA MB-453 (breast cancer cell line that expresses wild type AR) reporter assay with an IC50 less than 0.8 μM, while they were much less efficient in stimulating transcriptional activation in the same system, with ED50 values greater than 5 μM. The racemate of these compounds were also tested in vivo. In intact mature male rats, these compounds showed modest inhibitory effects in the prostate, seminal vesicle, and levator ani muscle at 100 mg/kg/day after oral administration for 14 days. The serum LH level in these animals was not significantly affected by the treatment, suggesting that these ligands had little, if any effects in the pituitary. On the other hand, the agonist activity of these ligands was tested in castrated rats at high dose (90 mg/kg/day) through oral administration. Compound 7 showed moderate agonist activity in the levator ani muscle by increasing the tissue weight by 27% compared to castrated control without stimulating the prostate growth. Compound 9 showed no effects in neither of these tissues. These in vivo data suggest that these compounds are weak AR antagonist with little or no intrinsic agonist activity in normal androgen target tissues.
The antagonist activities of these compounds were further evaluated in a CWR-22 prostate carcinoma xenograft model in nude mice. At 75 mg/kg/day, compounds 7 and 9 inhibited tumor growth to a similar extent as bicalutamide (150 mg/kg/day). Separated antipode of compound 25 was more potent as it achieved similar inhibition at an even lower dose rate of 19 mg/kg/day. However, the effects of these compounds in normal androgen target tissues in the xenograft nude mice were not reported. It is unclear if there is differential distribution of these ligands between normal androgen target tissues and the tumor, which could be responsible for any ‘tissue selectivity’ that exists. Nevertheless, ‘tissue selectivity,’ particularly for the prostate tumor, has become one of the major pharmacological features to achieve in the development of novel nonsteroidal antiandrogens.
Another series of hydantoin analogs are nilutamide derivatives like RU58642 [4-(3-Cyanomethyl-4,4-dimethyl-2,5-dioxo-imidazolidin-1-yl)-2-trifluoromethyl-benzonitrile] and RU58841 [1-(4-Hydroxy-butyl)-3-(4-isocyano-3-trifluoromethyl-phenyl)-5,5-dimethyl-imidazolidine-2,4-dione]. RU58841 (Tables I and II) was developed in Europe for topical treatment of acne and alopecia (42,43), due to its short half life in vivo (less than one hour). Topical application not only avoids extensive hepatic metabolism (N-dealkylation) but also provides for effective regional treatment without systemic antiandrogen activity due to the formation of active metabolite (43,44). In comparison, structural analog RU58642 was shown to be orally active (35), and could significantly reduce prostate and seminal vesicle weights in intact male rats at dose rates from 1 to 30 mg/kg/day. It also dramatically increased serum testosterone levels in these animals by blocking the feedback regulation of LH release. The overall pharmacological profile of this compound is very similar to that of nilutamide. Although RU58642 was more potent than bicalutamide, hydroxyflutamide, and nilutamide, which could be related to its high binding affinity to AR (Table II), no further development of this ligand has been reported since 1998 (35).
Different from the bicalutamide and nilutamide derivatives, Ligand Pharmaceuticals developed a series of quinolinone derivatives (Table I), with a linear tricyclic pharmacophore, 2(1H)-piperidino[3,2-g]quinolinone, that bind to the AR in the nanomolar range and work as ‘selective’ AR antagonists. In intact male rats, lead compound LG120907 {1,2,3,4-tetrahydro-2,2-dimethyl-6-trifluoromethyl-8-pyridono[5,6-g]quinoline} showed antagonist activity in the prostate and seminal vesicle without raising the plasma levels of LH and testosterone (45). Compound LG105 {7-fluoro-1,2,3,4-tetra-hydro-2,2-dimethyl-6-trifluoromethyl-8-pyridono[5,6-g]quinoline} also binds to the AR with high affinity (Table II), and demonstrated strong antagonist activity in the prostate, which seemed to be more potent than LG120907. Both LG120907 and LG105 are orally available (as evidenced by animal studies), although detailed pharmacokinetic data is not available.
Despite the dramatic differences in structural features, the pharmacological profiles of these compounds appeared to be very similar to that of bicalutamide. However, it is important to note that, the tissue selectivity observed in these quinolinone compounds has not yet been demonstrated in humans; and even if they could selectively avoid feedback regulation, the anabolic effects of androgens in the muscle and bone will still likely be abolished. Further characterization of these compounds would be necessary to confirm their tissue selectivity.
NONSTEROIDAL ANDROGEN RECEPTOR AGONISTS
In the past several years, the successful development and marketing of selective estrogen receptor modulators (SERMs) has raised the possibility of developing selective ligands for other members of the nuclear receptor superfamily. The concept of selective androgen receptor modulators (SARMs) (46,47) also emerged: a compound that is an antagonist or weak agonist in the prostate, but agonist in the pituitary, muscle, and bone; and orally available with low hepatotoxicity. For an ideal SARM, the antagonist or weak agonist activity in the prostate would reduce concern for the risk to stimulate nascent or undetected prostate cancer, particularly in aging male; while the strong agonist activity in the muscle and bone can be used as anabolic agent to treat muscle-wasting conditions, age-related hypogonadism and/or frailty, and even osteoporosis in both men and women.
As discussed above, several pharmacophores possessing high binding affinity to AR were identified during the development of nonsteroidal antiandrogens (Table I). Further structural modifications of these pharmacophores led to the discovery of several classes of nonsteroidal AR agonists, including the quinolones, tetrahydroquinolone, hydantoin, and bicalutamide derivatives (Table III). These nonsteroidal AR ligands are not substrates of 5α-reductase or aromatase (Fig. 2), they maintained the full agonist activity of testosterone in the DHT-independent tissues (i.e., muscle, bone, and pituitary), but only possess weak agonist (lower potency (ED50) or efficacy (Emax)) activity in DHT-dependent tissues (i.e., prostate). Therefore, most active compounds demonstrated various degree of tissue selectivity in castrated rat model, and are defined as tissue selective AR modulators. The pharmacokinetic and pharmacodynamic profiles of the characterized nonsteroidal AR agonists are summarized in Table IV, although some of the in vivo data is not available due to their early stage of development and limitations of the released information.
Table III.
Nonsteroidal Androgen Receptor Agonists
| Chemotype | General chemical structure | Lead compound | Company (stage of development) |
||
|---|---|---|---|---|---|
| Quinoline analogs (Tri-cyclic) |  |
LG121071 |  |
Ligand Pharmaceuticals | Preclinical |
| (Bi-cyclic) |  |
LGD2226 |  |
Ligand & TAP Pharmaceuticals | Terminated |
| Aryl propionamide analogs | S4 |  |
Preclinical Phase I/II | ||
| S1 |  |
||||
| C6 | |||||
| S22 | |||||
| Bi-cyclic hydantoin analogs |  |
BMS 564929 |  |
Bristol-Myers Squibb | Phase I |
| Tetrahydro-quinoline analogs |  |
S-40503 |  |
Kaken (Japan) | Preclinical |
SELECTIVE ANDROGEN RECEPTOR MODULATORS (SARMS)
The tri-cyclic quinoline derivative (Table III), LG121071 {4-Ethyl-1,2,3,4-tetrahydro-6-(trifluoromethyl)-8-pyridono[5,6-g]quinoline}, was disclosed by Ligand Pharmaceuticals in 1999 as the first orally active nonsteroidal AR agonist (48), with binding affinity in the nanomolar range (Ki = 17 nM, Table IV). LG121071 (20 mg/kg/day) successfully suppressed LH release in castrated rats after 2 weeks treatment through oral administration, suggesting that it works as a full agonist in the pituitary and is orally bioavailable. Testosterone propionate (TP, 1 mg/kg/day) was included as a control and administered subcutaneously. Both treatments restored serum LH levels to that observed in intact control animals. However, the in vivo androgenic and anabolic activities, and detailed PK profiles of LG121071 were not discussed in published data.
An orally available bi-cyclic quinoline derivative, LGD2226 {6-[Bis-(2,2,2-trifluoro-ethyl)-amino]-4-trifluoromethyl-1H-quinolin-2-one}, was later developed by Ligand and TAP Pharmaceuticals in 2001. LGD2226 was shown to be tissue selective after two weeks treatment in castrated rat model. Levator ani muscle weight was returned to intact control level at the dose rate of 3 mg/kg/day, while the prostate weight was returned to intact control level only at higher dose of 100 mg/kg/day, suggesting that LGD2226 was much less potent than DHT in the prostate. The anabolic effects of LGD2226 in bone were also reported (49). During a four-month treatment period, LGD2226 prevented castration-induced bone loss and maintained bone quality by stimulating bone formation and inhibiting bone turnover. However, the detailed experimental data was not available.
Both LG121071 and LGD2226 bind to the receptor with high affinity. Computer modeling (50) suggested that the A-ring keto group and C-ring ethyl group in LG121071 mimic the A-ring keto group and the 17β-OH group in testosterone (Fig. 3), respectively, which could explain the relatively high binding affinity of LG121071. Comparing the structure of LG120907 (antagonist, Table I) and LG121071 (agonist, Table III), it is clear that C-ring substituents play an important role in determining the agonist or antagonist activity in tri-cyclic quinoline molecules (51). The A-ring and B-ring in LGD2226 may very likely mimic the steroid skeleton plane in a similar way as LG121071, while it is unclear if one of the trifluoroethyl groups mimics the 17β-OH group in testosterone.
Structural modifications of bicalutamide led to the discovery of the first generation of the aryl propionamide analogs (Table III). Lead compounds S1 [3-(4-Fluoro-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide] and S4 [3-(4-Acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide] bind AR with high affinity (low nanomolar range), and demonstrate much improved PK profile and tissue selectivity in animal models (52-54) (Table IV). In castrated rats, S4 prevented castration caused tissue weight loss during the two-week treatment, and behaved as partial agonists in the prostate (ED50 = 1.6 mg/kg/day), but full agonists in the levator ani muscle (ED50 = 0.6 mg/kg/day). Furthermore, prolonged treatment (8 weeks) with S4 selectively restored the tissue weight loss three months after castration. At a dose rate of 3 mg/kg/day, S4 only partially restored the prostate weight to less than 20% of intact level, but fully restored the levator ani muscle weight to control level (54), significantly increased the total body bone mineral density, improved the body composition by increasing lean mass, and suppressed LH (32) and FSH release (54), suggesting its potential application in the treatment of disease-related muscle wasting and HRT. Besides restoring levator ani muscle weight, S4 (3 mg/kg/day) was also able to restore skeletal muscle (i.e., soleus muscle) strength in castrated rats, which is important for the treatment of muscle wasting and male HRT. On the other hand, improved muscle strength can indirectly contribute to the anabolic effects of androgens on bone, which could be beneficial to the treatment of osteoporosis as well.
The anabolic effects of S4 in bone were further investigated in ovariectomized rat model for osteoporosis (55). S4 (3 and 10 mg/kg/day, 8 weeks treatment) was able to prevent (immediate treatment model, treatment initiates right after ovariectomy) and restore (delayed treatment model, treatment initiated two months after ovariectomy) whole body and trabecular BMD, cortical content, and increased bone strength while decreasing body fat in ovariectomized rats. Mechanistic studies using primary culture of bone marrow osteoprogenitor cells showed that S4 was more anabolic in promoting osteoblast formation than DHT, but less potent in inhibiting osteoclast formation than DHT, which further conformed that SARM can be used to treat osteoporosis as anabolic agents.
In the presence of full agonists, partial agonists could behave as competitive antagonists. The partial agonist activity of S1 and S4 were further characterized in intact male rats (53). Both S1 (2 mg/kg/day) and S4 (2 mg/kg/day) worked as antagonists in the prostate without abolishing the anabolic effects of androgens in the levator ani muscle. In more detailed dose response studies, S1 (5, 10, and 25 mg/kg) selectively decreased the prostate weight with similar efficacy to finasteride (5 mg/kg, 5α-reducatese inhibitor), without affecting the levator ani muscle or increasing the plasma levels of testosterone, LH, and FSH. However, the antiandrogen hydroxyflutamide (0.5, 1, 5, 10, and 25 mg/kg) decreased both the prostate and levator ani muscle weights without any selectivity and increased plasma hormone levels in a dose-dependent manner, suggesting that SARMs with low intrinsic activity in the prostate might serve as an alternative therapy for BPH or even prostate cancer.
Another important target tissue for androgens is the pituitary. For most clinical applications, strong agonist activity in the pituitary is not desirable considering the danger of ‘chemical castration,’ except for male contraceptives. For ‘oral male contraceptive’ to replace testosterone agents, strong agonist activity to suppress gonadotropin release is necessary to shut down endogenous testosterone production while maintaining the beneficial anabolic actions of androgens. Compound C6 [3-(4-Chloro-3-fluoro-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide] (56) was identified as a strong agonist in the pituitary that is orally available (Table IV). In castrated rats, C6 suppressed LH release to intact control level at a dose rate as low as 1.5 mg/kg/day, but maintained tissue selectivity for anabolic (levator ani muscle, ED50 0.85 mg/kg/day) over androgenic tissues (prostate, ED50 3.9 mg/kg/day). Ten weeks treatment with C6 (2.5 mg/kg/day) significantly decreased sperm count in adult male rats to less than 30% of intact level in the testis, suggesting that potent nonsteroidal androgens that affect the pituitary could potentially be used as an oral male contraceptive.
Even though S1, S4, and C6 were administered subcutaneously in the pharmacological studies, the pharmacokinetic profiles of these compounds were examined in detailed PK studies (56-58) as well. All three compounds are bioavailable after oral administration at pharmacologically relevant doses, with an in vivo half life of 3–6 h in rats and dogs (59). S1, S4, and C6 share similar metabolic labile sites, including the amide bond and the A-ring nitro group, as amide bond hydrolysis and nitro reduction were identified as the major metabolic pathway (Fig. 6) both in vivo (data not published) and in vitro (60,61). All three derivatives are eliminated exclusively through hepatic metabolism.
Fig. 6.

Major phase I metabolic pathways of S4 (60).
Extensive SAR studies (57,62-64) show that both the ether linkage and B-ring para-position substituents are critical for the agonist activity of these bicalutamide derivatives (62). Based on available crystal structures, compounds with the ether linkage adopt a more compact conformation than bicalutamide due to the establishment of an intramolecular H bond (65), allowing the B-ring to avoid steric conflict with the side chain of W741 in the wild type AR (as is observed with bicalutamide) (66) and potentially explaining the agonist activity observed in compounds incorporating ether or thio-ether linkages. On the other hand, the interaction between the B-ring para-position substituents and the LBD also contributes significantly to the various binding affinity and intrinsic activity of these ligands (65), which explains the stronger intrinsic activity of C6 compared to S1, despite the significant similarity in structure.
Besides solving the binding mechanism of these ligands, the crystal structures also solved the conformation of the bound ligand (65,66), showing an intra-molecular hydrogen bond between the oxygen atom from the ether linkage and the hydrogen atom from the amide bond, which might be related to the susceptibility of the amide bond to hydrolysis. Furthermore, the electron density of the oxygen atom is greatly affected by the para-substituents on the B-ring, suggesting that compounds incorporating different para-substituents might show differing degrees of hydrolysis by altering the electron density of the intra-molecular bond, which could result in differences in the pharmacokinetic profile of these compounds. in vivo pharmacokinetic studies with a series of halogen derivatives (57) of S1 showed that the presence of a weaker electron withdrawing group at B-ring para-position significantly prolonged the in vivo half life of these analogs, from 4 h (fluoro- (S1), nitro- (S19), or cyano- (S20)) to 15 h (iodo- (S11)) in rats, which could be a direct result of increased stability of the intra-molecular bond as discussed above. In the case of bicalutamide, the oxygen atom from the sulfonyl linkage could also form a similar intra-molecular hydrogen bond, which could be related to the extensive hydrolysis of its amide bond in rats. However, it is important to point out that the amide bond hydrolysis reaction is much more significant in rats than in humans, suggesting that aryl propionamide SARMs with prolonged half-lives in humans can be identified.
Based on the established SAR and metabolic profile, a second generation of aryl propionamide was developed, in which: 1) the ethyl linkage was kept to maintain the agonist activity of these ligands; 2) different B-ring substituents were introduced to achieve various intrinsic activity (57,63) and reduce the chance of metabolism; 3) the A-ring nitro group was replaced by a cyano group to improve the metabolism and pharmacokinetic profile of these compounds. Compound S22 [3-(4-Cyano-phenoxy)-N-(4-cyano-3-trifluoromethyl-phenyl)-2-hydroxy-2-methyl-propionamide] (Tables III and IV) contains two cyano groups, which eliminated the metabolically labile sites of previous analogs, and maintained the tissue-selective pharmacological activity of this class of ligands (57). Since cyano group does not undergo reduction as the nitro group, the in vivo half life of S22 in rat was prolonged to 6 h.
This class of bicalutamide derivatives has been well characterized both in vitro and in vivo. All lead compounds are orally bioavailable, with various in vivo half life, intrinsic activity, and tissue selectivity. Most ligands have high specificity for the AR, and are potent anabolic agents in the muscle and bone with weak androgenic activity in the reproductive tissues and little, if any, effects on the serum lipid profiles, suggesting that these ligands could be used for male HRT, treatment of osteoporosis in both men and women, and male contraception without causing the undesirable side effects that are often observed with testosterone therapy.
Another important structural class of SARMs is the hydantoin derivatives (Table III) (67) developed by Bristol-Myers Squibb. Lead compound BMS-564929 [3-(4-Cyano-phenoxy)-N-(4-cyano-3-trifluoromethyl-phenyl)-2-hydroxy-2-methyl-propionamide] binds AR with high affinity (RBA 3%) and high specificity. Crystallography and molecular modeling studies suggest that the five-membered ring in BMS-564929 creates the optimal geometry for the hydroxyl group to H bond with T877, which could contribute to the high binding affinity and agonist activity of the ligand.
In castrated rats, BMS-564929 demonstrated tissue selectivity, with ED50 = 141 μg/kg/day in the prostate, ED50 = 0.9 μg/kg/day in the levator ani muscle, and ID50 = 8 μg/kg/day in suppressing LH release. The compound is orally available in humans, with an in vivo half life of 8–14 h. As hydantoin analogs, BMS-564929 and nilutamide share more structural similarities, which might explain the long half life of this compound in human: the imidazole structure reduces the risk of amide bond hydrolysis (Fig. 5) that was observed in bicalutamide derivative, and the cyano group attached to the bezene ring is not as susceptible to reduction as the nitro group. Compared to the bicalutamide derivatives, the prolonged in vivo half life of these ligands could explain the lower dose needed to achieve its pharmacological activities in animal models, since the in vivo activities seem to be more related to the tissue exposure of the ligands when they share similar binding affinity and intrinsic activity (57). However, the potent suppression of LH observed with these compounds may have implications for their use in male HRT. Studies regarding the effects of BMS-564929 on bone or other androgenic and anabolic tissues have not been reported to date.
Kaken Pharmaceuticals (68,69) developed a series of tetrahydroquinolin (THQ) derivatives as tissue selective AR agonist for bone by combining the structural features of steroidal androgens and nonsteroidal antiandrogens, bicalutamide and hydroxyflutamide. Leading compound S-40503 (2-(4-Dimethylamino-6-nitro-1,2,3,4-tetrahydroquinolin-2-yl)-2-methylpropan-1-ol) binds AR with high affinity and specificity (68). The pharmacological activity of S-40503 was further evaluated in vivo. Since the drug was administered subcutaneously in these studies, it's unclear if it is orally available, and its in vivo PK profile is not available according to currently released information. However, considering that the nitro group on the benzene ring is susceptible to reduction as that has been observed with the bicalutamide derivatives, this compound is not likely to have a very long half life in vivo.
In castrated rats, S-40503 showed similar potency in maintaining prostate and levator ani muscle weights after four-week immediate treatment, with significant increases in tissue weights observed in higher dose groups (10 and 30 mg/kg/day). In comparison, the femoral BMD tended to increase at lower doses from 1 to 10 mg/kg/day, but a statistically significant increase was only observed in the 30 mg/kg/day group. DHT showed very similar pharmacological activities in these tissues, except that lower doses were used (0.01 to 10 mg/kg). A significant increase in femoral BMD was only observed at highest dose of DHT tested (10 mg/kg/day). Comparing S-40503 (30 mg/kg/day) and DHT (10 mg/kg/day) at the highest doses tested in castrated rats, both ligands showed similar anabolic effects in bone (femoral BMD) and muscle (levator ani muscle), but S-40503 showed less stimulatory effects in the prostate (about 80% of intact control level) than DHT (about 140% of intact control level), suggesting that S-40503 is more selective for anabolic tissues than DHT. Similar tissue selectivity of S-40503 was also observed in intact animals (4 weeks treatment). At 30 mg/kg/ day, S-40503 increased levator ani muscle weight by 30% after four weeks treatment without significantly changing the prostate weight, while DHT (10 mg/kg/day) increased the levator ani muscle by 50% and significantly increased the size of the prostate to twice of the intact level. It is unclear how S-40503 affects the endocrine parameters (i.e., LH and FSH) in these animals. The direct effects of S-40503 on bone were also confirmed using sciatic neurectomized castrated rats. Again, both S-40503 and DHT demonstrated similar anabolic activity in restoring BMD, in cortical bone particularly, with S-40503 returning the prostate weight to intact control level, while DHT stimulated the prostate growth to almost twice of the intact level.
Besides characterizing the tissue selectivity of S-40503 in different male rat models, its anabolic activity in bone was further characterized in ovariectomized rats (delayed treatment model). Both S-40503 and DHT restored femoral BMD, and increased bone formation rate (measured as mineral apposition rate) and mechanical strength. Since S-40503 has very little cross activity with other steroid receptors, its anabolic effects on bone is considered solely through its interaction with the AR. These studies further demonstrated that S-40503 stimulates bone formation as an anabolic agent, which is suggested by the significant increase in cortical bone mineral density as compared to cancellous bone, similar to that observed with aryl propionamide derivative S4 (55). In comparison, an anti-resorptive agent, like estrogen or SERM, is more effective in preventing cancellous bone loss after ovariectomy or castration. Therefore, combination therapy of SARM plus SERM might provide a novel strategy for the treatment of osteoporosis. To date, no further pharmacological data about this compound has been published by Kaken Pharmaceuticals.
CONCLUSIONS AND FUTURE PERSPECTIVES
Several different structural classes of nonsteroidal AR ligands were summarized in this review. In general, most of these nonsteridal ligands demonstrate much improved PK profile as compared to steroidal ligands, with reasonable oral bioavailability and in vivo half life (as summarized in Tables II and IV). Also, with the flexibility of the nonsteroidal structure, and better understanding of the SAR and metabolic profiles, most of these ligands could be further modified to achieve more desirable PK and PD profiles if necessary. Pharmacologically, most of the nonsteroidal ligands developed so far bind to the AR with high affinity (low nanomolar range) and specificity, which would help avoid the undesirable side effects of the steroidal ligands caused by the cross reactivity with other steroid receptors.
Unlike testosterone, the androgenic activity of the nonsteroidal agonists cannot be amplified by conversion to DHT through 5α-reductase, and most of the ligands demonstrated varying degrees of tissue selectivity (i.e., strong agonists in the anabolic tissues, while weak agonist in the androgenic tissue). The tissue selectivity of the nonsteroidal ligands is certainly advantageous for androgen therapy by reducing the risk of androgens in the prostate and lipid profiles, but the mechanism of the tissue selectivity is not well understood. Although the lack of interaction between the nonsteroidal ligands and 5α-reductase seemed to explain the weak agonist activity observed with most of the nonsteroidal agonists (Table IV), it is unclear if other more complicated mechanisms are also involved, like those observed with SERMs (70-72).
Besides the successful clinical development of the nonsteroidal agonists, tissue-selective nonsteroidal antagonists also attracted much attention in the last several years. Antiandrogens that demonstrate selectivity for the prostate tumor tissue, and can block both wild type and mutant AR; become the major goals to achieve for the next generation of nonsteroidal AR antagonist.
In summary, the development of nonsteroidal AR ligands will continue, with particular focus on the search for ligands that are AR specific, metabolically stable, safe, and tissue selective. A better understanding of the mechanism of action of the known nonsteroidal AR ligands will help design the next generation of ligands with improved target specificity and tissue selectivity that could greatly benefit the treatment of many diseases.
REFERENCES
- 1.Gao W, Bohl CE, Dalton JT. Chemistry and structural biology of androgen receptor. Chem. Rev. 2005;105:3352–3370. doi: 10.1021/cr020456u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buijsman RC, Hermkens PH, van Rijn RD, Stock HT, Teerhuis NM. Non-steroidal steroid receptor modulators. Curr. Med. Chem. 2005;12:1017–1075. doi: 10.2174/0929867053764671. [DOI] [PubMed] [Google Scholar]
- 3.Chen J, Kim J, Dalton JT. Discovery and therapeutic promise of selective androgen receptor modulators. Mol. Interv. 2005;5:173–188. doi: 10.1124/mi.5.3.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venturoli S, Marescalchi O, Colombo FM, Macrelli S, Ravaioli B, Bagnoli A, Paradisi R, Flamigni C. A prospective randomized trial comparing low dose flutamide, finasteride, ketoconazole, and cyproterone acetate-estrogen regimens in the treatment of hirsutism. J. Clin. Endocrinol. Metab. 1999;84:1304–1310. doi: 10.1210/jcem.84.4.5591. [DOI] [PubMed] [Google Scholar]
- 5.Keller ET, Ershler WB, Chang C. The androgen receptor: a mediator of diverse responses. Front Biosci. 1996;1:d59–d71. doi: 10.2741/a116. [DOI] [PubMed] [Google Scholar]
- 6.Goodman LS, Hardman JG, Limbird LE, Gilman AG. Goodman & Gilman's the Pharmacological Basis of Therapeutics. McGraw-Hill Medical Pub. Division; New York: 2001. [Google Scholar]
- 7.Johansen KL. Testosterone metabolism and replacement therapy in patients with end-stage renal disease. Semin. Dial. 2004;17:202–208. doi: 10.1111/j.0894-0959.2004.17307.x. [DOI] [PubMed] [Google Scholar]
- 8.Oettel M. Testosterone metabolism, dose-response relationships and receptor polymorphisms: selected pharmacological/toxicological considerations on benefits versus risks of testosterone therapy in men. Aging Male. 2003;6:230–256. doi: 10.1080/13685530312331309772. [DOI] [PubMed] [Google Scholar]
- 9.Oettel M. Is there a role for estrogens in the maintenance of men's health? Aging Male. 2002;5:248–257. [PubMed] [Google Scholar]
- 10.Rondede W, Pols HA, van Leeuwen JP, de Jong FH. The importance of oestrogens in males Jongde. Clin. Endocrinol. (Oxf) 2003;58:529–542. doi: 10.1046/j.1365-2265.2003.01669.x. [DOI] [PubMed] [Google Scholar]
- 11.Barrett-Connor E, Goodman-Gruen D, Patay B. Endogenous sex hormones and cognitive function in older men. J. Clin. Endocrinol. Metab. 1999;84:3681–3685. doi: 10.1210/jcem.84.10.6086. [DOI] [PubMed] [Google Scholar]
- 12.Van Pottelbergh I, Braeckman L, De Bacquer D, De Backer G, Kaufman JM. Differential contribution of testosterone and estradiol in the determination of cholesterol and lipoprotein profile in healthy middle-aged men. Atherosclerosis. 2003;166:95–102. doi: 10.1016/s0021-9150(02)00308-8. [DOI] [PubMed] [Google Scholar]
- 13.Mukherjee TK, Dinh H, Chaudhuri G, Nathan L. Testosterone attenuates expression of vascular cell adhesion molecule-1 by conversion to estradiol by aromatase in endothelial cells: implications in atherosclerosis. Proc. Natl. Acad. Sci. USA. 2002;99:4055–4060. doi: 10.1073/pnas.052703199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Toda K, Okada T, Takeda K, Akira S, Saibara T, Shiraishi M, Onishi S, Shizuta Y. Oestrogen at the neonatal stage is critical for the reproductive ability of male mice as revealed by supplementation with 17beta-oestradiol to aromatase gene (Cyp19) knockout mice. J. Endocrinol. 2001;168:455–463. doi: 10.1677/joe.0.1680455. [DOI] [PubMed] [Google Scholar]
- 15.Falahati-Nini A, Riggs BL, Atkinson EJ, O'Fallon WM, Eastell R, Khosla S. Relative contributions of testosterone and estrogen in regulating bone resorption and formation in normal elderly men. J. Clin. Invest. 2000;106:1553–1560. doi: 10.1172/JCI10942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orwoll ES. Men, bone and estrogen: unresolved issues. Osteoporos. Int. 2003;14:93–98. doi: 10.1007/s00198-002-1332-9. [DOI] [PubMed] [Google Scholar]
- 17.Thigpen AE, Silver RI, Guileyardo JM, Casey ML, McConnell JD, Russell DW. Tissue distribution and ontogeny of steroid 5 alpha-reductase isozyme expression. J. Clin. Invest. 1993;92:903–910. doi: 10.1172/JCI116665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Isidori AM, Greco EA, Aversa A. Androgen deficiency and hormone-replacement therapy. BJU Int. 2005;96:212–216. doi: 10.1111/j.1464-410X.2005.05603.x. [DOI] [PubMed] [Google Scholar]
- 19.Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H, Xie Q, Perkins R, Owens W, Sheehan DM. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem. Res. Toxicol. 2003;16:1338–1358. doi: 10.1021/tx030011g. [DOI] [PubMed] [Google Scholar]
- 20.Kelce WR, Monosson E, Gamcsik MP, Laws SC, Gray LE., Jr Environmental hormone disruptors: evidence that vinclozolin developmental toxicity is mediated by antiandrogenic metabolites. Toxicol. Appl. Pharmacol. 1994;126:276–285. doi: 10.1006/taap.1994.1117. [DOI] [PubMed] [Google Scholar]
- 21.Waller CL, Juma BW, Gray LE, Jr., Kelce WR. Three-dimensional quantitative structure–activity relationships for androgen receptor ligands. Toxicol. Appl. Pharmacol. 1996;137:219–227. doi: 10.1006/taap.1996.0075. [DOI] [PubMed] [Google Scholar]
- 22.Wakeling AE, Furr BJ, Glen AT, Hughes LR. Receptor binding and biological activity of steroidal andc non-steroidal antiandrogens. J. Steroid Biochem. 1981;15:355–359. doi: 10.1016/0022-4731(81)90297-1. [DOI] [PubMed] [Google Scholar]
- 23.Schulz M, Schmoldt A, Donn F, Becker H. The pharmacokinetics of flutamide and its major metabolites after a single oral dose and during chronic treatment. Eur. J. Clin. Pharmacol. 1988;34:633–636. doi: 10.1007/BF00615229. [DOI] [PubMed] [Google Scholar]
- 24.Katchen B, Buxbaum S. Disposition of a new, nonsteroid, antiandrogen, alpha,alpha,alpha-trifluoro-2-methyl-4′-nitro-m-propionotoluidide (Flutamide), in men following a single oral 200 mg dose. J. Clin. Endocrinol. Metab. 1975;41:373–379. doi: 10.1210/jcem-41-2-373. [DOI] [PubMed] [Google Scholar]
- 25.Fau D, Eugene D, Berson A, Letteron P, Fromenty B, Fisch C, Pessayre D. Toxicity of the antiandrogen flutamide in isolated rat hepatocytes. J. Pharmacol. Exp. Ther. 1994;269:954–962. [PubMed] [Google Scholar]
- 26.Creaven PJ, Pendyala L, Tremblay D. Pharmacokinetics and metabolism of nilutamide. Urology. 1991;37:13–19. doi: 10.1016/0090-4295(91)80096-p. [DOI] [PubMed] [Google Scholar]
- 27.Berson A, Wolf C, Berger V, Fau D, Chachaty C, Fromenty B, Pessayre D. Generation of free radicals during the reductive metabolism of the nitroaromatic compound, nilutamide. J. Pharmacol. Exp. Ther. 1991;257:714–719. [PubMed] [Google Scholar]
- 28.Fau D, Berson A, Eugene D, Fromenty B, Fisch C, Pessayre D. Mechanism for the hepatotoxicity of the antiandrogen, nilutamide. Evidence suggesting that redox cycling of this nitroaromatic drug leads to oxidative stress in isolated hepatocytes. J. Pharmacol. Exp. Ther. 1992;263:69–77. [PubMed] [Google Scholar]
- 29.Cockshott ID. Bicalutamide: clinical pharmacokinetics and metabolism. Clin. Pharmacokinet. 2004;43:855–878. doi: 10.2165/00003088-200443130-00003. [DOI] [PubMed] [Google Scholar]
- 30.Boyle GW, McKillop D, Phillips PJ, Harding JR, Pickford R, McCormick AD. Metabolism of Casodex in laboratory animals. Xenobiotica. 1993;23:781–798. doi: 10.3109/00498259309166784. [DOI] [PubMed] [Google Scholar]
- 31.McKillop D, Boyle GW, Cockshott ID, Jones DC, Phillips PJ, Yates RA. Metabolism and enantioselective pharmacokinetics of Casodex in man. Xenobiotica. 1993;23:1241–1253. doi: 10.3109/00498259309059435. [DOI] [PubMed] [Google Scholar]
- 32.Schellhammer P, Sharifi R, Block N, Soloway M, Venner P, Patterson AL, Sarosdy M, Vogelzang N, Jones J, Kolvenbag G. A controlled trial of bicalutamide versus flutamide, each in combination with luteinizing hormone-releasing hormone analogue therapy, in patients with advanced prostate cancer. Casodex Combination Study Group. Urology. 1995;45:745–752. doi: 10.1016/s0090-4295(99)80077-6. [DOI] [PubMed] [Google Scholar]
- 33.Furr BJ, Tucker H. The preclinical development of bicalutamide: pharmacodynamics and mechanism of action. Urology. 1996;47:13–25. doi: 10.1016/s0090-4295(96)80003-3. discussion 29–32. [DOI] [PubMed] [Google Scholar]
- 34.Lefort M, Diaz Curiel M, Carrascal MT, Mendez-Davila C, de la Piedra C. Comparative effects of bicalutamide (Casodex) versus orchidectomy on bone mineral density, bone remodelling, and bone biomechanics in healthy rats. Urol. Int. 2005;74:301–307. doi: 10.1159/000084427. [DOI] [PubMed] [Google Scholar]
- 35.Battmann T, Branche C, Bouchoux F, Cerede E, Philibert D, Goubet F, Teutsch G, Gaillard-Kelly M. Pharmacological profile of RU 58642, a potent systemic antiandrogen for the treatment of androgen-dependent disorders. J. Steroid Biochem. Mol. Biol. 1998;64:103–111. doi: 10.1016/s0960-0760(97)00151-9. [DOI] [PubMed] [Google Scholar]
- 36.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, Small EJ. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J. Clin. Oncol. 2003;21:2673–2678. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: relation to antiandrogen withdrawal syndrome. Prostate. 1996;29:153–158. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 38.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- 39.Yoshida T, Kinoshita H, Segawa T, Nakamura E, Inoue T, Shimizu Y, Kamoto T, Ogawa O. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65:9611–9616. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 40.Salvati ME, Gottardis M, Krystek SR, Attar RM, Sack J. Patent WO0200617 Selective androgen receptor modulators and methods for their identification, design, and use. 2002
- 41.Salvati ME, Balog A, Shan W, Wei DD, Pickering D, Attar RM, Geng J, Rizzo CA, Gottardis MM, Weinmann R, Krystek SR, Sack J, An Y, Kish K. Structure based approach to the design of bicyclic-1H-isoindole-1,3(2H)-dione based androgen receptor antagonists. Bioorg. Med. Chem. Lett. 2005;15:271–276. doi: 10.1016/j.bmcl.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 42.Van Dort ME, Jung YW. Synthesis and structure–activity studies of side-chain derivatized arylhydantoins for investigation as androgen receptor radioligands. Bioorg. Med. Chem. Lett. 2001;11:1045–1047. doi: 10.1016/s0960-894x(01)00146-9. [DOI] [PubMed] [Google Scholar]
- 43.Battmann T, Bonfils A, Branche C, Humbert J, Goubet F, Teutsch G, Philibert D. RU 58841, a new specific topical antiandrogen: a candidate of choice for the treatment of acne, androgenetic alopecia and hirsutism. J. Steroid Biochem. Mol. Biol. 1994;48:55–60. doi: 10.1016/0960-0760(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 44.Cousty-Berlin D, Bergaud B, Bruyant MC, Battmann T, Branche C, Philibert D. Preliminary pharmacokinetics and metabolism of novel non-steroidal antiandrogens in the rat: relation of their systemic activity to the formation of a common metabolite. J. Steroid Biochem. Mol. Biol. 1994;51:47–55. doi: 10.1016/0960-0760(94)90114-7. [DOI] [PubMed] [Google Scholar]
- 45.Hamann LG, Higuchi RI, Zhi L, Edwards JP, Wang XN, Marschke KB, Kong JW, Farmer LJ, Jones TK. Synthesis and biological activity of a novel series of nonsteroidal, peripherally selective androgen receptor antagonists derived from 1,2-dihydropyridono[5,6-g]quinolines. J. Med. Chem. 1998;41:623–639. doi: 10.1021/jm970699s. [DOI] [PubMed] [Google Scholar]
- 46.Zhi L, Martinborough E. Selective androgen receptor modulators (SARMs) Annual Reports of Medicinal Chemistry. 2001;36:169–180. [Google Scholar]
- 47.Negro-Vilar A. Selective androgen receptor modulators (SARMs): a novel approach to androgen therapy for the new millennium. J. Clin. Endocrinol. Metab. 1999;84:3459–3462. doi: 10.1210/jcem.84.10.6122. [DOI] [PubMed] [Google Scholar]
- 48.Hamann LG, Mani NS, Davis RL, Wang XN, Marschke KB, Jones TK. Discovery of a potent, orally active, nonsteroidal androgen receptor agonist: 4-ethyl-1,2,3,4-tetrahydro-6-(trifluoromethyl)-8-pyridono[5,6-g]-quinoline ( LG121071) J. Med. Chem. 1999;42:210–212. doi: 10.1021/jm9806648. [DOI] [PubMed] [Google Scholar]
- 49.Rosen J, Negro-Vilar A. Novel, non-steroidal, selective androgen receptor modulators (SARMs) with anabolic activity in bone and muscle and improved safety profile. J. Musculoskelet. Neuronal. Interact. 2002;2:222–224. [PubMed] [Google Scholar]
- 50.Bohl CE, Chang C, Mohler ML, Chen J, Miller DD, Swaan PW, Dalton JT. A ligand-based approach to identify quantitative structure-activity relationships for the androgen receptor. J. Med. Chem. 2004;47:3765–3776. doi: 10.1021/JM0499007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhi L, Tegley CM, Marschke KB, Jones TK. Switching androgen receptor antagonists to agonists by modifying C-ring substituents on piperidino[3,2-g]quinolinone. Bioorg. Med. Chem. Lett. 1999;9:1009–10012. doi: 10.1016/s0960-894x(99)00119-5. [DOI] [PubMed] [Google Scholar]
- 52.Yin D, Gao W, Kearbey JD, Xu H, Chung K, He Y, Marhefka CA, Veverka KA, Miller DD, Dalton JT. Pharmacodynamics of selective androgen receptor modulators. J. Pharmacol. Exp. Ther. 2003;304:1334–1340. doi: 10.1124/jpet.102.040840. [DOI] [PubMed] [Google Scholar]
- 53.Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Comparison of the pharmacological effects of a novel selective androgen receptor modulator, the 5alpha-reductase inhibitor finasteride, and the antiandrogen hydroxyflutamide in intact rats: new approach for benign prostate hyperplasia. Endocrinology. 2004;145:5420–5428. doi: 10.1210/en.2004-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao W, Reiser PJ, Coss CC, Phelps MA, Kearbey JD, Miller DD, Dalton JT. Selective androgen receptor modulator treatment improves muscle strength and body composition and prevents bone loss in orchidectomized rats. Endocrinology. 2005;146:4887–4897. doi: 10.1210/en.2005-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kearbey JD, Gao W, Miller DD, Dalton JT. Selective androgen receptor modulators inhibit bone resorption in rats. AAPS PharmSci. 2003;5 [Google Scholar]
- 56.Chen J, Hwang DJ, Bohl CE, Miller DD, Dalton JT. A selective androgen receptor modulator for hormonal male contraception. J. Pharmacol. Exp. Ther. 2005;312:546–553. doi: 10.1124/jpet.104.075424. [DOI] [PubMed] [Google Scholar]
- 57.Kim J, Wu D, Hwang DJ, Miller DD, Dalton JT. The para substituent of S-3-(phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-prop ionamides is a major structural determinant of in vivo disposition and activity of selective androgen receptor modulators. J. Pharmacol. Exp. Ther. 2005;315:230–239. doi: 10.1124/jpet.105.088344. [DOI] [PubMed] [Google Scholar]
- 58.Kearbey JD, Wu D, Gao W, Miller DD, Dalton JT. Pharmacokinetics of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide in rats, a non-steroidal selective androgen receptor modulator. Xenobiotica. 2004;34:273–280. doi: 10.1080/0049825041008962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao W, Johnston JS, Miller DD, Dalton JT. Inter-Species Differences in Pharmacokinetics and Metabolism of S-3-(4-acetylamino-phenoxy)-2-hydroxy-2-methyl-N-(4-nitro-3-trifluoromethyl-phenyl)-propionamide: the role of N-Acetyltransferase. Drug. Metab. Dispos. 2005 doi: 10.1124/dmd.105.007120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao W, Wu Z, Bohl CE, Yang J, Miller DD, Dalton JT. Characterization of the in vitro metabolism of selective androgen receptor modulator (SARM) using human, rat and dog liver enzyme preparations. Drug Metab. Dispos. 2005 doi: 10.1124/dmd.105.007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao W, Wu Z, Chung K, Miller DD, Dalton JT. Phase I Metabolism Study of Selective Androgen Receptor Modulators (SARMs) with human liver microsomes. AAPS PharmSci. 2003;5 [Google Scholar]
- 62.Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Key structural features of nonsteroidal ligands for binding and activation of the androgen receptor. Mol. Pharmacol. 2003;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- 63.Chen J, Hwang DJ, Chung K, Bohl CE, Fisher SJ, Miller DD, Dalton JT. In vitro and in vivo structure–activity relationships of novel androgen receptor ligands with multiple substituents in the B-ring. Endocrinology. 2005;146:5444–5454. doi: 10.1210/en.2005-0732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. Design, synthesis, and biological characterization of metabolically stable selective androgen receptor modulators. J. Med. Chem. 2004;47:993–998. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 2005;280:37747–37754. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc. Natl. Acad. Sci. USA. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hamann LG. Discovery and preclinical profile of a highly potent and muscle selective androgen receptor modulator (SARM); 227th National Meeting of the American Chemical Society Medicinal Chemistry Division; 2004. [Google Scholar]
- 68.Hanada K, Furuya K, Yamamoto N, Nejishima H, Ichikawa K, Nakamura T, Miyakawa M, Amano S, Sumita Y, Oguro N. Bone anabolic effects of S-40503, a novel non-steroidal selective androgen receptor modulator (SARM), in rat models of osteoporosis. Biol. Pharm. Bull. 2003;26:1563–1569. doi: 10.1248/bpb.26.1563. [DOI] [PubMed] [Google Scholar]
- 69.Miyakawa M, Oguro N, Hanada K, Furuya K, Yamamoto N. Patent WO 2004013104 Preparation of novel tetrahydroquinoline derivatives as androgen receptor agonists. 2004
- 70.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat. Rev. Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 71.Smith CL, O'Malley BW. Coregulator function: a key to understanding tissue specificity of selective receptor modulators. Endocr. Rev. 2004;25:45–71. doi: 10.1210/er.2003-0023. [DOI] [PubMed] [Google Scholar]
- 72.Katzenellenbogen BS, Katzenellenbogen JA. Biomedicine. Defining the “S” in SERMs. Science. 2002;295:2380–2381. doi: 10.1126/science.1070442. [DOI] [PubMed] [Google Scholar]
- 73.Christiansen RG, Bell MR, D'Ambra TE, Mallamo JP, Herrmann JL, Ackerman JH, Opalka CJ, Kullnig RK, Winneker RC, Snyder BW, et al. Antiandrogenic steroidal sulfonylpyrazoles. J. Med. Chem. 1990;33:2094–2100. doi: 10.1021/jm00170a008. [DOI] [PubMed] [Google Scholar]
- 74.Cockshott ID, Plummer GF, Cooper KJ, Warwick MJ. The pharmacokinetics of Casodex in laboratory animals. Xenobiotica. 1991;21:1347–1355. doi: 10.3109/00498259109043209. [DOI] [PubMed] [Google Scholar]
- 75.Edwards JP, Higuchi RI, Jones TK, Hamann LG. Androgen receptor modulator compounds and methods. Ligand Pharmaceuticals Incorporated; United States: 2000. [Google Scholar]
- 76.Allan GF, Sui Z. Therapeutic androgen receptor ligands. NURSA e-Journal. 2003;1 doi: 10.1621/nrs.01009. ID# 3.09172003.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhi L, Tegley CM, Pio B, Arjan VOC, Motamedi M, Martinborough E, West S, Higuchi RI, Hamann LG, Farmer LJ. Patent WO0116108 Bicyclic androgen and progesterone receptor modulator compounds and methods. 2001