

Abstract
The mechanism by which the androgen receptor (AR) distinguishes between agonist and antagonist ligands is poorly understood. AR antagonists are currently used to treat prostate cancer. However, mutations commonly develop in patients that convert these compounds to agonists. Recently, our laboratory discovered selective androgen receptor modulators, which structurally resemble the nonsteroidal AR antagonists bicalutamide and hydroxyflutamide but act as agonists for the androgen receptor in a tissue-selective manner. To investigate why subtle structural changes to both the ligand and the receptor (i.e. mutations) result in drastic changes in activity, we studied structure-activity relationships for nonsteroidal AR ligands through crystallography and site-directed mutagenesis, comparing bound conformations of R-bicalutamide, hydroxyflutamide, and two previously reported nonsteroidal androgens, S-1 and R-3. These studies provide the first crystallographic evidence of the mechanism by which nonsteroidal ligands interact with the wild type AR. We have shown that changes induced to the positions of Trp-741, Thr-877, and Met-895 allow for ligand accommodation within the AR binding pocket and that a water-mediated hydrogen bond to the backbone oxygen of Leu-873 and the ketone of hydroxyflutamide is present when bound to the T877A AR variant. Additionally, we demonstrated that R-bicalutamide stimulates transcriptional activation in AR harboring the M895T point mutation. As a whole, these studies provide critical new insight for receptor-based drug design of nonsteroidal AR agonists and antagonists.
The androgen receptor (AR)3 is a ligand-inducible nuclear hormone receptor involved in regulation of prostate growth, muscle and bone mass, and spermatogenesis in males. Endogenous ligands for the AR include the steroid, testosterone, and its more potent metabolite, dihydrotestosterone (DHT). Agonist compounds for the AR provide therapeutic potential in the treatment of osteoporosis, cachexia, contraception, and androgen deficiency (1). Antagonist AR ligands are commonly used in the treatment of androgen-dependent prostate cancer. The clinically available nonsteroidal antiandrogens, flutamide and bicalutamide, are advantageous over steroidal drug treatments because of their oral bioavailability and lack of cross-reactivity with other steroid receptors. Mutations to the AR that cause resistance to antiandrogens by converting the compounds to agonists exist and represent a significant problem with currently prescribed prostate cancer drugs that target the AR (2-4). Different AR mutations cause agonism for hydroxyflutamide (HF, active form of flutamide) as compared with bicalutamide, suggesting that these ligands do not antagonize the AR by the same mechanism. Our recent report of the W741L-R-bicalutamide crystal structure (5) demonstrated that R-bicalutamide induces the same overall fold of the AR ligand binding domain (LBD) observed in steroidal androgen-bound AR LBD crystal structures when bound to a mutant AR associated with resistance. Additionally, mutations to Thr-877 (4, 6) have been shown to result in agonist activity for HF as well as steroidal AR antagonists (7). Subtle differences in the AR-ligand interaction therefore are responsible for the activity of androgen antagonists. The recent discovery of nonsteroidal AR agonists by structural modification of bicalutamide and HF further reinforces this finding (8-10).
To date, multiple docking solutions have been reported to explain how the extra bulk on bicalutamide as compared with steroidal androgens can be accommodated in the AR ligand binding pocket (7, 11-14). None of these models resembled the bound conformation of R-bicalutamide in the W741L mutant (5). Thus, questions regarding how R-bicalutamide and structurally related selective androgen receptor modulators could be accommodated in the wild type (WT) AR remained unanswered. The presence of the Trp-741 side chain would seemingly preclude the B-ring of nonsteroidal selective androgen receptor modulators from binding in the same position occupied by the B-ring of R-bicalutamide in the W741L AR. Although our efforts to obtain crystal structures of the AR LBD complexed to pure antiandrogens have been unsuccessful due to tight association with the groEL chaperone protein, crystal structures of the WT AR bound to agonists that closely resemble bicalutamide and HF in structure were obtained. Herein we present the first evidence as to how nonsteroidal ligands are accommodated in the WT AR by inducing changes to the positions of residues in the binding pocket. We also report the crystal structure of HF bound to the well known resistance mutation, T877A. Lastly, we present evidence that M895T represents a novel resistance mutation for R-bicalutamide and may also be involved in bicalutamide withdrawal syndrome similar to mutations to Trp-741 (2, 5).
MATERIALS AND METHODS
Cloning, Expression, and Purification
An AR-LBD-(663–919) was obtained by PCR amplification from a full-length human AR expression construct (pCMVhAR; generously provided by Dr. Donald J. Tindall, Mayo Clinic and Mayo Foundation, Rochester, MN) with primers containing flanking restriction sites and inserted into the pGEX6P-1 plasmid (Amersham Biosciences). Mutations were created in the pGEX6P1-AR-(663–919) and the pCMVhAR via the Stratagene QuikChange mutagenesis kit according to the manufacturer's instructions. AR LBD expression and purification were performed essentially as previously described (5, 15, 16). The AR LBD was expressed as a glutathione S-transferase (GST) fusion protein in Escherichia coli BL21 DE3 at 15 °C for 16 h by induction with 30 μm isopropyl-1-thio-β-d-galactopyranoside. Cells were lysed in a buffer containing 150 mm NaCl, 50 mm Tris, pH 8.0, 5 mm EDTA, 10% glycerol, 1 mg/ml lysozyme, 10 units/ml DNase I, 10 mm MgCl2, 10 mm DTT, 0.5% CHAPS, 100 μm ligand, and 100 μm phenylmethylsulfonyl fluoride by three cycles of freeze-thaw. The supernatant from ultracentrifugation was incubated for 1 h at 4°C with glutathione-Sepharose resin (Amersham Biosciences) and washed with 150 mm NaCl, 50 mm Tris, pH 8.0, 5 mm EDTA, 10% glycerol, 10 mm ATP, 10 μm ligand (S-1, R-3, or HF), 0.1% n-octyl-β-glucoside, and 1 mm DTT. The GST-LBD fusion protein was cleaved in a buffer containing 150 mm NaCl, 50 mm Tris, pH 7.0, 10% glycerol, 10 μm ligand, 0.1% n-octyl-β-glucoside, 1 mm DTT, and 5 units/mg protein PreScission protease (Amersham Biosciences) at 4 °C overnight, releasing the AR LBD from the glutathione-Sepharose resin. The supernatant was then diluted 3-fold in 10 mm Hepes, pH 7.2, 10% glycerol, 10 μm ligand, 0.1% n-octyl-β-glucoside, and 1 mm DTT and loaded onto an HP SP cation exchange column (Amersham Biosciences). Protein was eluted with a gradient of 50–500 mm NaCl in the same dilution buffer. The buffer was exchanged in a Millipore 10-kDa cutoff concentrator to a buffer containing 150 mm Li2SO4, 50 mm Hepes, pH 7.2, 10% glycerol, 100 μm ligand, 0.1% n-octyl-β-glucoside, and 10 mm DTT, and protein was concentrated to above 4 mg/ml.
Crystallization, Data Collection, and Structure Determination
AR LBD crystals formed in 1–2 days using the hanging drop vapor diffusion method in 0.1 m Hepes, pH 7.5, and 0.5–0.8 m sodium citrate for the T877A-HF, T877A-S-1, and W741L-S-1 complexes. WT-S-1 crystals were obtained in 0.1 m Hepes, pH 7.5, and 0.6 m sodium citrate after 4 weeks. Nucleation sites from these WT-S-1 crystals were transferred with a cat whisker into freshly mixed protein and reservoir solution ranging from 0.3 to 0.6 m sodium citrate with 0.1 m Hepes, pH 7.5, to improve WT-S-1 crystal quality and size. Crystals formed in 1–2 days using this technique allowing for optimization. The WT-R-3 crystals were obtained by transferring microseeds with a cat whisker from T877A-HF crystals in 0.3-0.6 m sodium citrate with 0.1 m Hepes, pH 7.5. Prior to flash freezing in liquid nitrogen, AR LBD crystals were transferred to a solution consisting of 0.1 m Hepes, pH 7.5, 0.7 m sodium citrate, and 20% ethylene glycol. Diffraction data were collected using a Rigaku RU300 rotating anode generator and an R-axis IV++ image plate (Rigaku, The Woodlands, TX) and processed with Crystal Clear software (Molecular Structure Corporation, The Woodlands, TX). The W741L-R-bicalutamide structure (Protein Data Bank code 1Z95) was used as a starting structure for refinement using Crystallography and NMR System (CNS) (17). After an initial round of refinement, electron density maps allowed for accurate fitting of the ligand. Model building and water molecules were added using the program O (18), and further rounds of refinement were performed using rigid body, torsion angle simulated annealing, and individual temperature factor modules of CNS. Figures were prepared with PyMOL (The PyMOL Molecular Graphics System).
Cotransfection Assay
CV-1 cells (green monkey kidney cells) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 2 mm glutamine, and 1% streptomycin and penicillin to confluency in T175 tissue culture flasks. Cells were then transfected with 3 μg of the CMVhAR expression vector (WT or mutant), 30 μg of an androgen-dependent luciferase reporter construct (pMMTV-luc; generously provided by Dr. Ronald Evans at The Salk Institute, San Diego, CA), and 30 μg of a β-galactosidase expression construct (pSV-β-galactosidase; Promega, Madison, WI) via 30 μl of Plus reagent (Invitrogen) and 40 μl of Lipofectamine (Invitrogen) in serum-free Dulbecco's modified Eagle's medium. After 4 h, the medium was exchanged to Dulbecco's modified Eagle's medium supplemented with 0.2% fetal bovine serum and 2 mm glutamine. Cells were transferred 12 h later to 24-well tissue culture plates and after another 6 h were treated with 0.1–1000 nm, drug or no drug (control). After another 24 h, cells were washed twice with cold phosphate-buffered saline and harvested by incubation with 100 μl of passive lysis buffer (Promega) for 30 min. An aliquot (50 μl) of the lysate was then added to an opaque 96-well plate, and luciferase activity was monitored after automated injection of 50 μl of luciferase substrate (Promega) with a MicroLumatPlus LB96V luminometer (Berthold Technologies, Oak Ridge, TN) using the WinGlow software package. An aliquot (50 μl) of the lysate was also added to a clear 96-well plate along with 50 μl of β-galactosidase assay buffer. An absorbance measurement at 420 nm was taken following a 2-h incubation at 37 °C on a Dynex MRX plate reader. Luciferase activity was normalized with β-galactosidase activity to account for differences in cell number and/or loss in transfection efficiency. Transcriptional activation was interpreted as the -fold increase (relative luciferase units) over control (i.e. non-drug treated) wells.
RESULTS
All crystal structures of AR LBD solved to date have been in an orthorhombic P212121 space group (5, 15, 16, 19-21) with helix 12 in a similar conformation as other agonist-bound steroid receptor LBD structures (16, 22-24). Purification of AR LBD bound to agonist ligands is well documented and involves removal of contamination chaperones, dnak and groEL, through cation exchange chromatography (21). The AR LBD is not retained by cation exchange chromatography in the presence of antagonist compounds and co-elutes with groEL after anion exchange chromatography, suggesting that antagonist-bound AR LBD is tightly associated with groEL, possibly as a result of partial receptor unfolding (25-28). Thus, mutations to the AR that confer resistance to antiandrogens offer a viable method to obtain crystallographic evidence for antagonist binding conformations to the AR and provide insight as to how they would alter the conformation in the WT AR to stabilize association with heat shock proteins (3, 29). AR LBD bound to the partial agonists S-1 and R-3 demonstrated a fractional amount of cation exchange retention compared with DHT-bound LBD. The yields of purified S-1-associated AR LBD were, however, substantially improved in the T877A and W741L AR mutants. Additionally, crystal formation of the AR LBD-S-1 complex was also significantly facilitated in these mutants. WT-S-1 crystal formation took nearly one month as opposed to 1–2 days for the mutant complexes, likely because of slowed nucleation, and was circumvented by using microseeding. Furthermore, WT-R-3 crystals did not form in similar conditions or any of numerous crystal screens. Because WT-S-1 and T877A-HF complexes crystallized in the same space group with nearly the same unit cell dimensions and same overall protein fold (TABLE ONE), we predicted that the WT-R-3 crystals would be similar. Therefore, we obtained crystals of the WT-R-3 complex by transferring microseeds from T877A-HF crystals. Clear electron density for Thr-877 and the bromine atom insured that we had, in fact, obtained the WT-R-3 complex.
TABLE ONE. Crystallographic data and refinement statistics.
Values for data in the last resolution shell are shown in parenthesis. R.m.s.d., root mean square deviation.
| Complex | WT-R-3 | T877A-HF | WT-S-1 | T877A-S-1 | W741L-S-1 |
|---|---|---|---|---|---|
| Protein Data Bank code | 2AX9 | 2AX6 | 2AXA | 2AX7 | 2AX8 |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Unit cell | |||||
| A | 55.38 | 54.78 | 54.73 | 54.82 | 55.09 |
| B | 66.01 | 65.96 | 66.39 | 66.37 | 66.11 |
| C | 69.06 | 69.63 | 68.94 | 69.08 | 69.03 |
| Resolution range (Å) | 18.88–1.65 (1.71–1.65) | 18.03–1.50 (1.55–1.50) | 22.13–1.80 (1.86–1.80) | 30.64–1.90 (1.97–1.90) | 23.01–1.70 (1.76–1.70) |
| Number of unique reflections | 31,121 | 39,987 | 22,979 | 19,962 | 27,002 |
| Average redundancy | 8.27 (6.57) | 6.04 (4.88) | 12.55 (8.84) | 13.48 (9.71) | 11.95 (7.04) |
| % completeness | 100.0 (99.6) | 97.4 (85.2) | 96.0 (70.1) | 97.3 (80.8) | 95.0 (67.5) |
| Rmerge a | 0.095 (0.358) | 0.153 (0.480) | 0.092 (0.316) | 0.115 (0.53 | 0.079 (0.332) |
| I/σ | 12.6 (4.9) | 17.5 (3.5) | 16.9 (6.9) | 11.5 (3.6) | 20.5 (5.1) |
| Rfactor b | 0.224 (0.321) | 0.246 (0.409) | 0.210 (0.276) | 0.206 (0.244) | 0.212 (0.279) |
| Rfree | 0.248 (0.339) | 0.274 (0.455) | 0.250 (0.311) | 0.247 (0.284) | 0.251 (0.311) |
| R.m.s.d. bonds (Å) | 0.005 | 0.006 | 0.005 | 0.005 | 0.006 |
| R.m.s.d. angles | 1.1 | 1.1 | 1.1 | 1.1 | 1.1 |
| Mean B value (Å2) | 17.6 | 20.2 | 20.3 | 23.6 | 20.1 |
Rmerge = Σ|Ih − 〈I〉h|/ΣIh, where 〈I〉h is average intensity over symmetry equivalents.
Rfactor = Σ|Fobs − Fcalc|/ΣFobs. The free Rfactor is calculated from 10% of the reflections that are omitted from the refinement.
Structure of HF-like Nonsteroidal Agonist in the WT AR
R-3 is an analog of HF that differs only by the addition of a bromine atom to a methyl group, which creates a stereo-selective compound (8, 10) (Fig. 1). Addition of a bromine at this position results in a large increase in binding affinity to the AR (8). However, R-3 elicits agonist activity for the AR only at high concentrations (i.e. ≥100 nm) (Fig. 2a). The crystal structure of R-3 bound to the native AR LBD demonstrates a very similar bound conformation as R-bicalutamide in the W741L (5). Arg-752 and Gln-711 hydrogen bond with the nitro group of R-3 directly and by way of a water molecule (Fig. 3, a and b), resembling the hydrogen bond pattern seen with the cyano group of R-bicalutamide. However, Gln-711 is likely too distant (3.7 Å) from the cyano in the W741L-R-bicalutamide complex for direct hydrogen bond formation. The amide nitrogen of R-3 is within hydrogen bond distance to the backbone oxygen of Leu-704, and the chiral hydroxyl group of R-3 is within distance for hydrogen bonds with Asn-705 Oδ1 and the backbone oxygen of Leu-704, identical to the interactions observed with R-bicalutamide in the W741L mutant AR. Thr-877 is also oriented as in the W741L-R-bicalutamide complex, which is rotated 180° compared with steroidal bound AR structures. The Trp-741 indole ring, which is absent in the W741L-R-bicalutamide complex, borders the region occupied by bromine along with Met-742 and Met-895 residues as shown in Fig. 3b. Bulk provided by the bromine atom in this region therefore appears to greatly enhance binding affinity as compared with HF. Furthermore, the W741L and M895T mutations, which decrease bulk around the location of the bromine atom of R-3, demonstrate a decrease in transcriptional activation with R-3 relative to the WT (Fig. 2, c and d). A gain of AR-mediated transcription with R-3, however, is observed in the T877A AR (Fig. 2b).
FIGURE 1. Structures and binding affinities of AR ligands.
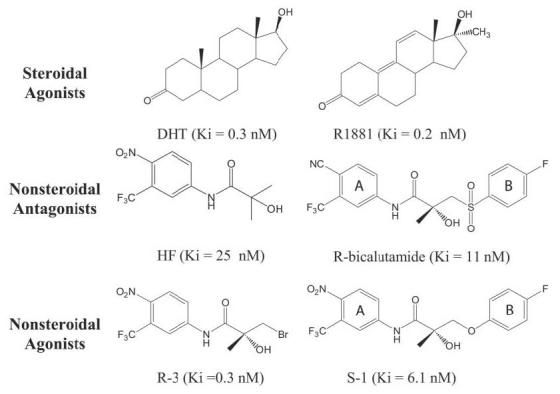
Binding affinity values were determined in our laboratory and previously reported (8, 9).
FIGURE 2. AR-mediated transcriptional activation by DHT, R-bicalutamide (R-bic), S-1, HF, and R-3 in the WT (a), T877A (b), W741L (c), and M895T (d) AR variants.
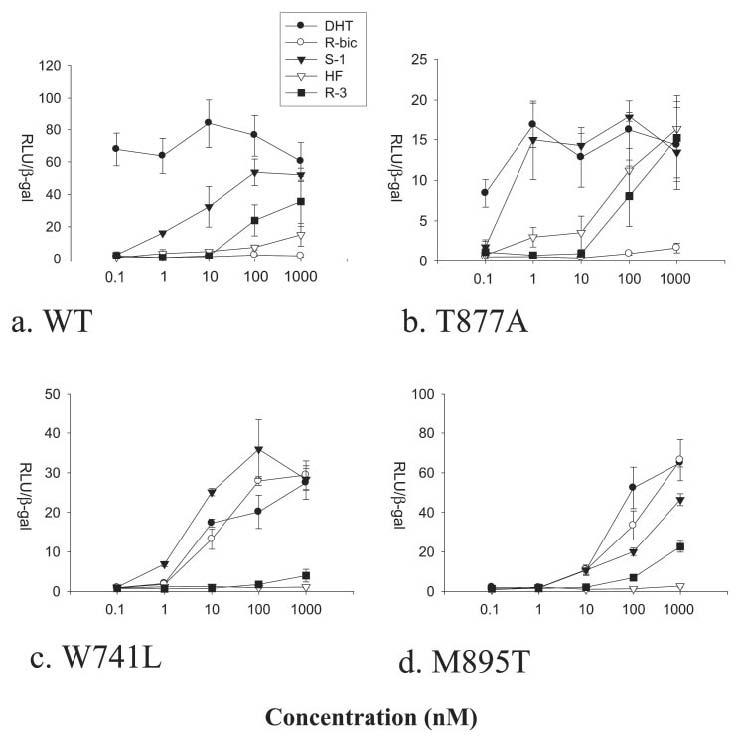
Relative luciferase units (RLU) normalized with β-galactosidase (β-gal) activity. Inter-experiment variation was observed with RLU/β-galactosidase values due to transfection efficiency, but relative drug-induced transcriptional activation was found consistent and therefore more accurately represents changes in functional activity observed in AR mutations.
FIGURE 3. Stereo representations of AR interactions with R-3 and S-1.
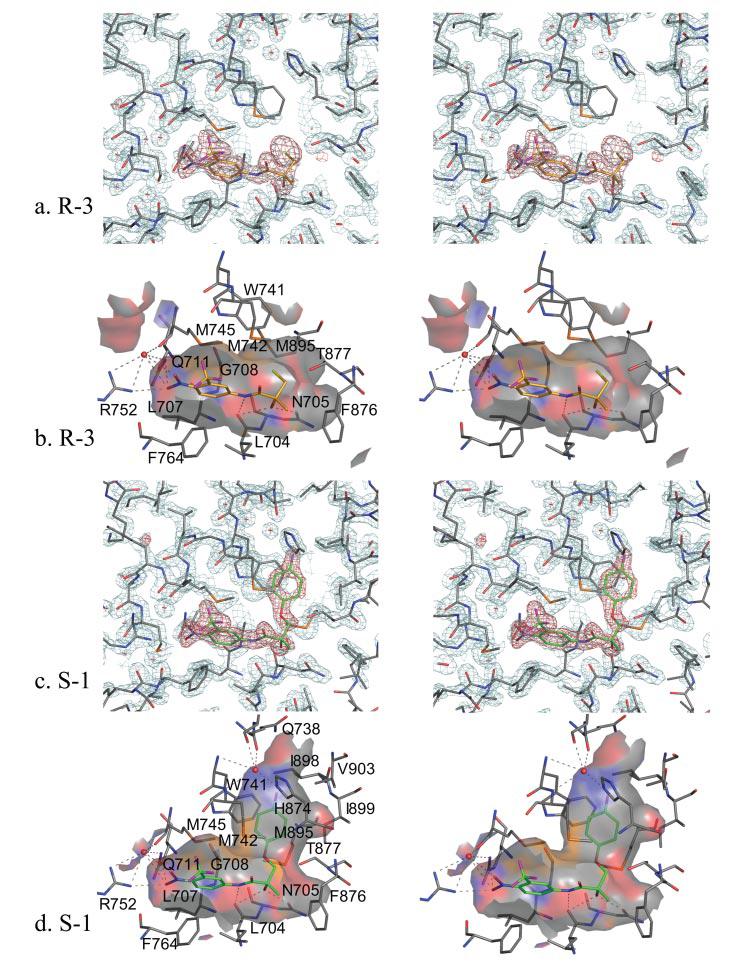
a, WT-R-3 complex fit into the 2Fo − Fc electron density maps contoured at the 2σ level (cyan) with R-3 shown in the Fo − Fc simulated annealing omit map contoured at the 4σ level (red). AR carbons, gray; R-3 carbons, gold; oxygen, red; nitrogen, blue; sulfur, orange; bromine, yellow. b, AR surface contacts with the R-3 binding pocket. Possible hydrogen bonds within 3.5 Å are represented by dashed lines. Notice that Gln-711 and Arg-752 hydrogen bond with the nitro group of R-3. Leu-704 and Asn-705 also form hydrogen bonds with R-3. Also notice the hydrophobic contacts from Trp-741, Met-742, and Met-895 with the R-3 bromine atom. Electron density for Trp-741 in the WT-R-3 structure cannot be visualized at the 2σ level, but electron density is more evident at lower σ levels. c, WT-S-1 complex fit into the 2Fo − Fc electron density maps contoured at the 2σ level (cyan) with S-1 (carbons in green) shown in the Fo − Fc simulated annealing omit map contoured at the 4σ level (red). d, AR surface contacts with the S-1 binding pocket. Similar hydrogen bonds to the AR are present in S-1 as with R-3. Notice the increased size of the S-1 binding pocket as compared with R-3 due to the S-1 B-ring. A water molecule hydrogen bonds to the His-874 side chain and backbone atoms of helices 4 and 5 within 3.0 Å of the B-ring fluorine atom, similar to that seen in the W741L-R-bicalutamide complex. Also notice the clear electron density for Trp-741 at the 2σ level as compared with the R-3-bound structure, likely because of decreased mobility of Trp-741 from binding of the S-1 B-ring.
Structure of Ether-linked Selective Androgen Receptor Modulator in WT AR
S-1 is an agonist for the AR that exhibits tissue-selective pharmacologic effects in vivo and therefore represents a novel class of AR ligands known as selective androgen receptor modulators (1, 30). S-1 resembles R-3 structurally but contains an ether-linked phenyl ring with a para-fluoro substituent in place of the bromine atom (Fig. 1). S-1 also closely resembles R-bicalutamide in structure, differing only at the linkage group and A-ring para position (Fig. 1). It is therefore surprising that S-1 elicits significantly more agonist activity than these structural analogs in the WT AR (Fig. 2a). The crystal structure of the WT-S-1 complex (Fig. 3, c and d) demonstrates that S-1 superimposes identically to the A-ring and amide portion of R-3. At the chiral hydroxyl group, S-1 bends and orients the B-ring nearly parallel to helix 12. Displacement of the Trp-741 indole ring by the B-ring of S-1 opens up an additional cavity in the AR binding pocket (Fig. 3, c and d). The B-ring is accommodated between the side chains of Trp-741 and Met-742, closely bordered by helix 12 residues Ile-898 and Ile-899. Interestingly, the B-ring of S-1 is positioned similar to that of R-bicalutamide in the W741L mutant AR (5). S-1, however, demonstrates an even more bent ligand conformation than R-bicalutamide, possibly because of intramolecular hydrogen bonding. The ether linkage group of S-1 is observed 3.3 Å from the amide nitrogen, similar to the close distance of 2.7 Å between the O15 sulfonyl oxygen and the amide nitrogen in R-bicalutamide in the W741L complex.
Ligand Interactions with Thr-877
An obvious difference between steroidal and nonsteroidal bound AR LBD crystal structures is the interaction with Thr-877. The 17β-OH group of R1881 and DHT forms a bifurcated hydrogen bond to Asn Oδ1 and Thr-877 Oγ (20). On the other hand, nonsteroidal AR ligands like S-1 and R-3 do not form a hydrogen bond with Thr-877, and the Thr-877 side chain is rotated 180° from its position in the steroidal structures (16, 20) (Fig. 4, a and b). As with HF, the T877A mutant increases the agonist activity of S-1 and R-3 (Fig. 2b). In fact, S-1 elicits nearly equal AR-mediated transcriptional activation as DHT at 1 nm in the T877A mutant AR. The slight loss in DHT activity is likely attributed to the loss of the hydrogen bond to this helix 11 residue, as seen in the T877A-DHT complex (20). S-1 maintains the same binding conformation in the T877A mutant as observed in the WT AR. Close contact distances between the Thr-877 side chain and carbon atoms attached to the chiral center of S-1 are increased by ∼0.4 Å to the alanine side chain in the T877A. Additionally, a water molecule in the T877A-S-1 complex was observed that would be precluded in the WT AR due to its proximity (2.0 Å) to the Thr-877 Oγ in the WT-S-1 complex. Interestingly, this water molecule bridges the ketone of S-1 to the backbone oxygen of Leu-873 by a hydrogen bond (Fig. 4c). HF binds to the T877A identically as the corresponding atoms on S-1. A water molecule is also present in the T877A-HF structure (Fig. 4, d and e), suggesting its conservation in T877A mutant AR bound to this nonsterodial pharmacophore.
FIGURE 4. Comparison of ligand interactions in the WT and the T877A mutant.
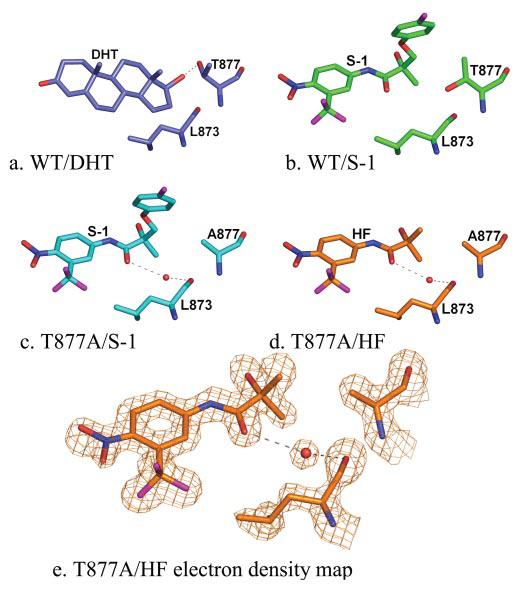
a, wild type-DHT complex, slate (Protein Data Bank code 1I37). The 17β-OH group of DHT forms a hydrogen bond with Thr-877. b, WT-S-1 complex, green. The side chain of Thr-877 is rotated 180° in the WT-S-1 complex, situating the hydroxyl group of Thr-877 in close range to the S-1 carbon atoms bound to the chiral center. c, T877A-S-1 complex, cyan. In the T877A-S-1 complex, a water molecule is present that hydrogen bonds to the backbone oxygen of Leu-873 and the ketone of S-1. d, T877A-HF complex, orange. The water molecule bridging the ketone to Leu-873 is also present in the T877A-HF structure. e, 2Fo − Fc electron density maps from the T877A-HF complex contoured at the 1.5σ level with density shown for HF, Leu-873, and Ala-877 by using the carve function in PyMol with a radius of 2.0 Å demonstrate the clear electron density for this water molecule.
Ligand-induced Changes to Trp-741
Flexibility of the Trp-741 indole ring is critical for accommodation of diverse AR ligands within the binding pocket. The presence of the 19-methyl group of DHT causes the side chain location of Met-745 and Trp-741 to be swapped as compared with R1881 (19) (Fig. 5a), which lacks the 19-methyl group. The Trp-741 side chain in the WT-R-3- and T877A-HF complexes is located in the same position as seen in the AR LBD crystal structures complexed to R1881 (19). S-1, however, displaces the indole ring even further from the steroidal binding pocket to allow the B-ring to fit between the Met-742 and Trp-741 side chains (Fig. 5, a and b).
FIGURE 5. Ligand-induced changes to Trp-741 and Met-895 in stereo overlay and individually as space-fill representations.
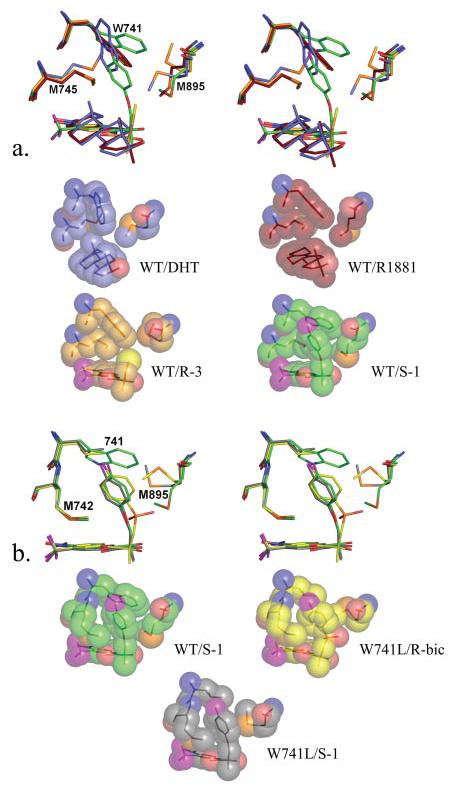
a, comparison of the changes induced by DHT (slate) (Protein Data Bank code 1I37), R1881 (ruby) (Protein Data Bank code 1XQ2), R-3 (gold), and S-1 (green) to the Trp-741, Met-745, Met-895 side chains. Notice the different location of Met-745 in the DHT-bound structure from displacement by the 19-methyl group, which causes the Trp-741 side chain to also move relative to its position in R1881. The Trp-741 indole ring is positioned similarly in R1881- and R-3-complexed structures; however, the bromine atom on R-3 displaces the Met-895 side chain. Also notice the change in position of the Trp-741 indole ring to allow accommodation of the S-1 B-ring. b, changes in the position of Met-895 in the W741L AR bound to S-1 (black) and R-bicalutamide (yellow). Compared with the WT-S-1 complex, Met-895 moves toward the Leu-741 side chain in the W741L-S-1 complex, compensating for the loss of bulk in this mutant. In the W741L-R-bicalutamide complex, the Met-895 side chain is wedged between the Leu-741 and the sulfonyl group of R-bicalutamide. Notice that the position of the Met-895 side chain in the WT-S-1 complex would be sterically precluded by the sulfonyl group of R-bicalutamide in the presence of Trp-741.
The W741L AR mutant, which is known to selectively confer agonism to bicalutamide and not HF, exhibits a decrease in AR-mediated transcription with DHT (Fig. 2c). This decrease is expected because of the large loss in binding affinity of DHT observed in the W741L variant (5). R-3 also loses activity in this mutation, suggesting that steric interactions between the bromine atom and the Trp-741 indole side chain do not account for the lesser ability of R-3 to stimulate AR-mediated transcription. Furthermore, the sustained agonist activity of S-1 in the W741L mutant demonstrates that the interaction with the Trp-741 indole ring does not play a significant role in S-1 binding. The Trp-741 indole ring therefore likely provides favorable contacts with compounds R-3 and DHT for binding and activity as opposed to bicalutamide and S-1, which contain significantly more bulk near this residue.
Ligand-induced Changes to Met-895
Resistance to AR antagonism with bicalutamide observed in Trp-741 mutations does not appear to be the direct result of increased pocket volume in this region or to be due to bulk from the bicalutamide B-ring. Further comparison of the W741L-R-bicalutamide and WT-S-1 structures demonstrates that the linkage group of these ligands represents the critical difference in receptor binding (Fig. 5b). The WT-S-1 structure shows that Met-895 forms close contacts with the ether linkage group of S-1 and is positioned in a similar location when the AR LBD is bound to steroidal agonists (16, 20). The sulfonyl linkage group of R-bicalutamide, however, results in displacement of Met-895 side chain in the W741L mutant into the position occupied by the Trp-741 indole ring in the WT-S-1 structure. Less bulk from the ether linkage in S-1 relative to R-bicalutamide therefore may play a role in accommodation of S-1 in the agonist AR conformation. S-1 exhibits little change in AR-mediated transcriptional activation in the W741L mutant as compared with the WT (Fig. 2c). However, the crystal structure does show that Met-895 is predominantly moved toward the Leu-741 side chain to replace the loss of bulk that occurs when the Trp-741 indole ring is absent (Fig. 5b).
The significantly bent binding conformation of S-1 observed in the WT is unchanged in the T877A and W741L mutations and is likely energetically favorable as a result of intramolecular hydrogen bonding between the oxygen of the ether linkage and the amide nitrogen, similar to the intramolecular hydrogen bonding between the sulfonyl oxygen and the amide nitrogen witnessed in the W741L-R-bicalutamide structure (5). This interaction could be responsible for pulling S-1 increasingly away from Met-895, permitting Met-895 to pack along the Trp-741 indole in the WT AR. The antagonist properties of R-bicalutamide therefore could be attributed to steric preclusion of Met-895 by the sulfonyl linkage group. To further explore this hypothesis, we tested the ability of R-bicalutamide to stimulate AR-mediated transcription in the M895T point mutation. This mutation to Met-895 was chosen because it appears in the AR mutation data base discovered in patients (31) and may also be clinically relevant to bicalutamide withdrawal syndrome similar to point mutations to Trp-741. As expected, M895T does confer agonism to R-bicalutamide at similar concentrations observed in the W741L mutation (Fig. 2d). Furthermore, failure of HF to stimulate AR-mediated transcription in the M895T supports the prediction that steric preclusion of Met-895 is specific for the antagonist effect of R-bicalutamide.
DISCUSSION
All AR LBD crystal structures solved to date demonstrate the same general AR conformation and resemble the conformation observed in other steroid receptor LBD crystal structures complexed to agonist ligands (16, 22, 24). The crystal structures of estrogen and glucocorticoid receptors have also been solved in an alternate conformation bound to antagonist compounds in which helix 12 is displaced over the coactivator binding pocket (22, 24). Whether helix 12 will undergo similar displacement when HF or R-bicalutamide is complexed to the WT AR is unknown. Compound R-3 is nearly identical to HF in structure, with the exception of a bromine atom, and displays only weak agonist activity for the AR despite its high binding affinity. Structural evidence that this ligand can invoke the same conformation as potent steroidal agonists suggests that HF could also potentially induce the same protein fold when associated with the WT AR LBD. However, our inability to obtain pure WT AR LBD-bound HF preparations due to failure to initiate chaperone dissociation prevented further attempts to crystallize the wild type-HF complex. Similarly, R-bicalutamide and S-1 are structurally very similar, yet WT AR LBD also could not be separated from groEL when complexed to R-bicalutamide. Evidence that AR LBD mutations, which confer agonist activity to these antagonist ligands, can be purified and crystallized with the same conformation as when complexed to agonists demonstrates that subtle structural differences recover AR activity. Induced dissociation from groEL is likely a result of the ability of AR ligands to incite a conformational change (3, 29), which invokes solubilization of the otherwise chaperone-stabilized protein. One explanation for this result is that AR agonists pull together the secondary structural elements through hydrogen bonds and favorable Van der Waals contacts, which buries hydrophobic surfaces that would otherwise be exposed in the unbound and antagonist-bound conformations.
Potent steroidal androgens, DHT and R1881, form hydrogen bonds to residues on helices 3, 5, and 11. Furthermore, the T877A mutation increases the agonist activity of the nonsteroidal AR ligands HF, R-3, and S-1. Evidence that this mutation results in a water-mediated hydrogen bond between S-1 or HF with helix 11, which is absent in the native AR, suggests that it may play a role in stimulating AR activation by allowing formation of a hydrogen bond between the carbonyl oxygen of the ligand, a water molecule, and the Leu-873 backbone. Maintenance of R-bicalutamide antagonism in the T877A AR, however, can be explained by its ability to sterically hinder receptor folding due to the bulkier structure of the compound. S-1 induces movement of the Trp-741 side chain to accommodate extra bulk compared with HF and steroidal ligands. A similar displacement of Trp-741 is therefore likely to occur upon R-bicalutamide binding to the WT AR. However, the sulfonyl group of R-bicalutamide would prevent the positioning of Met-895 observed in the WT AR complexed with S-1 (Fig. 5 b). Mutations to Trp-741 and Met-895, which alleviate this hindrance, therefore overcome the ability of R-bicalutamide to block receptor activation. This finding, however, does not explain why S-1 and R-bicalutamide, but not HF, activate the AR in the presence of Thr-877. One possibility is that the fluorine on the para position of S-1 and R-bicalutamide, which is within 3.0 Å of a water molecule that hydrogen bonds to His-874, provides a stabilizing interaction with helix 11. Interestingly, the H874Y mutation has been shown to increase activity of an array of compounds including HF (6, 32), indicating the involvement of this residue in AR regulation. The weak hydrogen-accepting capacity of the B-ring fluorine atom therefore may distinguish the ability of S-1 and R-bicalutamide to stimulate AR-mediated transcription in the presence of Thr-877 compared with HF. The water-mediated interaction with helix 11 similar to that seen in the T877A between the ketone of HF and Leu-873 offers an explanation for agonism of S-1 in the WT AR and R-bicalutamide in the M895T and W741L AR mutants.
AR mutations to residues that directly interact with the ligand and convert antagonists into agonists provide insight as to how antagonists block AR activation. The studies herein are the first to systematically explore the influence of ligands on AR structure. The location of Thr-877 and Met-895 on the end of helix 11 and beginning of helix 12, respectively, suggests that movement of helix 11 and 12 toward the ligand binding pocket is involved in initiating agonist activity in the AR (Fig. 6, a and b). Trp-741 is located at the core of the AR LBD on helix 5, which likely exhibits less mobility than helices 11 and 12. The ability of R-bicalutamide to invoke agonist activity in the W741L mutant appears to be the result of accommodation of Met-895, which would otherwise be hindered by the R-bicalutamide sulfonyl group (Fig. 6 c). Likewise, the ability of HF to stimulate AR-mediated transcription in T877A mutant AR appears to be the result of the ability of the ligand binding pocket to accommodate a water molecule that mediates hydrogen bonding interactions between nonsteroidal ligands and helix 11. Further studies are required, however, to determine the conformation of the unbound and antagonist-bound AR and the reason for its tight association with heat shock proteins.
FIGURE 6. Ligand interactions with helices 3, 5, 11, and 12.
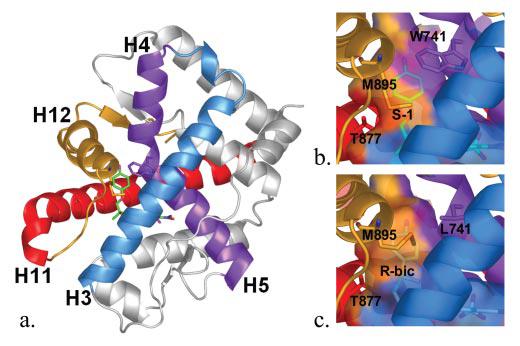
a, WT AR LBD complexed to S-1 (green) rotated 180° about the y-axis relative to Fig. 3, c and d. Helix 3, blue; helices 4/5, Trp741, purple; helix 11, Thr-877, red; helix 12, Met-895, gold. b, surface contacts of the secondary structural elements of the WT AR LBD with S-1. Notice the contacts from Trp-741, Thr-877, and Met-895 on helices 5, 11, and 12, respectively. c, surface contacts of the secondary structural elements of the W741L AR LBD with R-bicalutamide. Notice the loss of contacts of helix 5 from the loss of bulk in the W741L mutant and that helix 12 increases contacts to the ligand as a result of Met-895 repositioning. Also notice the location of the sulfonyl group of R-bicalutamide, which increases the binding pocket size relative to S-1.
Footnotes
This work was supported by National Institutes of Health Grants R01 DK59800 and R01 DK065227.
The abbreviations used are: AR, androgen receptor; DHT, dihydrotestosterone; HF, hydroxyflutamide;WT, wild type; LBD, ligand binding domain; DTT, dithiothreitol; CHAPS, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid.
REFERENCES
- 1.Negro-Vilar A. J. Clin. Endocrinol. Metab. 1999;84:3459–3462. doi: 10.1210/jcem.84.10.6122. [DOI] [PubMed] [Google Scholar]
- 2.Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M, Miyamoto M. Cancer Res. 2003;63:149–153. [PubMed] [Google Scholar]
- 3.Veldscholte J, Berrevoets CA, Ris-Stalpers C, Kuiper GG, Jenster G, Trapman J, Brinkmann AO, Mulder E. J. Steroid Biochem. Mol. Biol. 1992;41:665–669. doi: 10.1016/0960-0760(92)90401-4. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Prostate. 1996;29:153–158. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Proc. Natl. Acad. Sci. U. S. A. 2005;102:6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steketee K, Timmerman L, Ziel-van der Made AC, Doesburg P, Brinkmann AO, Trapman J. Int. J. Cancer. 2002;100:309–317. doi: 10.1002/ijc.10495. [DOI] [PubMed] [Google Scholar]
- 7.Poujol N, Wurtz JM, Tahiri B, Lumbroso S, Nicolas JC, Moras D, Sultan C. J. Biol. Chem. 2000;275:24022–24031. doi: 10.1074/jbc.M001999200. [DOI] [PubMed] [Google Scholar]
- 8.Dalton JT, Mukherjee A, Zhu Z, Kirkovsky L, Miller DD. Biochem. Biophys. Res. Commun. 1998;244:1–4. doi: 10.1006/bbrc.1998.8209. [DOI] [PubMed] [Google Scholar]
- 9.Marhefka CA, Gao W, Chung K, Kim J, He Y, Yin D, Bohl C, Dalton JT, Miller DD. J. Med. Chem. 2004;47:993–998. doi: 10.1021/jm030336u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin D, He Y, Perera MA, Hong SS, Marhefka C, Stourman N, Kirkovsky L, Miller DD, Dalton JT. Mol. Pharmacol. 2003;63:211–223. doi: 10.1124/mol.63.1.211. [DOI] [PubMed] [Google Scholar]
- 11.Marhefka CA, Moore BM, II, Bishop TC, Kirkovsky L, Mukherjee A, Dalton JT, Miller DD. J. Med. Chem. 2001;44:1729–1740. doi: 10.1021/jm0005353. [DOI] [PubMed] [Google Scholar]
- 12.Bohl CE, Chang C, Mohler ML, Chen J, Miller DD, Swaan PW, Dalton JT. J. Med. Chem. 2004;47:3765–3776. doi: 10.1021/JM0499007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soderholm AA, Lehtovuori PT, Nyronen TH. J. Med. Chem. 2005;48:917–925. doi: 10.1021/jm0495879. [DOI] [PubMed] [Google Scholar]
- 14.Balog A, Salvati ME, Shan W, Mathur A, Leith LW, Wei DD, Attar RM, Geng J, Rizzo CA, Wang C, Krystek SR, Tokarski JS, Hunt JT, Gottardis M, Weinmann R. Bioorg. Med. Chem. Lett. 2004;14:6107–6111. doi: 10.1016/j.bmcl.2004.09.049. [DOI] [PubMed] [Google Scholar]
- 15.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, Basler S, Schafer M, Egner U, Carrondo MA. J. Biol. Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 17.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998;54(Pt 5):905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 18.Jones TA, Zou JY, Cowan SW, Kjeldgaard Acta Crystallogr. Sect. A. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 19.He B, Gampe RT, Jr., Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM. Mol. Cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 20.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR, Jr., Weinmann R, Einspahr HM. Proc. Natl. Acad. Sci. U. S. A. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matias PM, Carrondo MA, Coelho R, Thomaz M, Zhao XY, Wegg A, Crusius K, Egner U, Donner P. J. Med. Chem. 2002;45:1439–1446. doi: 10.1021/jm011072j. [DOI] [PubMed] [Google Scholar]
- 22.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, Ohman L, Greene GL, Gustafsson JA, Carlquist M. Nature. 1997;389:753–758. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 23.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Proc. Natl. Acad. Sci. U. S. A. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, Alarcon M, Calles K, Engstrom O, Harlan J, Muchmore S, Ramqvist AK, Thorell S, Ohman L, Greer J, Gustafsson JA, Carlstedt-Duke J, Carlquist M. J. Biol. Chem. 2003;278:22748–22754. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 25.Ellis RJ, Hartl FU. FASEB J. 1996;10:20–26. doi: 10.1096/fasebj.10.1.8566542. [DOI] [PubMed] [Google Scholar]
- 26.Gragerov A, Nudler E, Komissarova N, Gaitanaris GA, Gottesman ME, Nikiforov V. Proc. Natl. Acad. Sci. U. S. A. 1992;89:10341–10344. doi: 10.1073/pnas.89.21.10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. Nature. 1992;356:683–689. doi: 10.1038/356683a0. [DOI] [PubMed] [Google Scholar]
- 28.Martin J, Langer T, Boteva R, Schramel A, Horwich AL, Hartl FU. Nature. 1991;352:36–42. doi: 10.1038/352036a0. [DOI] [PubMed] [Google Scholar]
- 29.Kallio PJ, Janne OA, Palvimo JJ. Endocrinology. 1994;134:998–1001. doi: 10.1210/endo.134.2.8299593. [DOI] [PubMed] [Google Scholar]
- 30.Gao W, Kearbey JD, Nair VA, Chung K, Parlow AF, Miller DD, Dalton JT. Endocrinology. 2004;145:5420–5428. doi: 10.1210/en.2004-0627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gottlieb B, Beitel LK, Wu JH, Trifiro M. Hum. Mutat. 2004;23:527–533. doi: 10.1002/humu.20044. [DOI] [PubMed] [Google Scholar]
- 32.Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, DeVere White RW, Pretlow TG, Harris SE, Wilson EM, Mohler JL, French FS. Mol. Endocrinol. 1997;11:450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]