

Abstract
Antibiotic-refractory Lyme arthritis, which may result from infection-induced autoimmunity, is associated with HLA-DR molecules that bind an epitope of Borrelia burgdorferi (Bb) outer-surface protein A (OspA165−173) and with T cell reactivity with this epitope. One potential mechanism to explain these associations is molecular mimicry between OspA165−173 and a self-peptide. Here, we searched the published human genome for peptides with sequence homology with OspA165−173. The two peptides identified with the greatest sequence homology with the OspA epitope were MAWD-BP276−288, which had identity at eight of the nine core amino acid residues, and T-span758−70, which had identity at six residues. MAWD-BP mRNA was expressed by synoviocytes, while T-span7 mRNA was not. However, neither peptide bound all of the HLA-DR molecules associated with antibiotic-refractory Lyme arthritis. Among 11 patients, nine had T cell reactivity with OspA161−170, six had responses to MAWD-BP276−288, and three had reactivity with T-span758−70, but reactivity with the self-peptides was lower than that induced by the spirochetal epitope. Thus, there remains an association between OspA165−173 and antibiotic-refractory Lyme arthritis, and infection-induced autoimmunity is an attractive hypothesis to explain this outcome. However, molecular mimicry due to sequence homology between OspA165−173 and a human peptide seems unlikely to be the critical mechanism.
Keywords: Borrelia burgdorferi, Lyme disease, T cells, molecular mimicry, MHC molecules
1. Introduction
Lyme arthritis, which is caused by the tick-borne spirochete, Borrelia burgdorferi (Bb), is characterized by intermittent or chronic arthritis in a few large joints, especially the knee (Steere, 2001; Steere et al., 1987). In most patients, the joint infection responds to either: a) a 1 or 2-month course of oral doxycycline therapy or b) a 2- to 4-week course of intravenous (IV) ceftriaxone (Dattwyler et al., 1988; Dattwyler et al., 2005; Steere et al., 1994). However, in a small percentage of patients, synovitis persists for months or even several years after having received ≥2 months of oral antibiotics or ≥1 month of IV antibiotics, or usually both (Steere and Angelis, 2006; Steere et al., 1994), which we have termed antibiotic-refractory Lyme arthritis. Although PCR results for Bb DNA in joint fluid are usually positive before antibiotic therapy, they are frequently negative by the end of 2−3 months of antibiotics (Nocton et al., 1994), and are almost always negative in synovial samples obtained months after treatment (Carlson et al., 1999; Steere and Angelis, 2006). Thus, in such patients, Lyme arthritis may continue after the near or total eradication of spirochetes, perhaps sustained by an infection-induced autoimmune response (Steere and Glickstein, 2004).
Many autoimmune diseases have a link with HLA genotypes. In initial studies, antibiotic-refractory Lyme arthritis was associated primarily with HLA-DR4 alleles (Steere et al., 1990) and with cellular and humoral immunity to Bb outer-surface protein A (OspA) (Akin et al., 1999; Kalish et al., 1993; Lengl-Janssen et al., 1994). The immunodominant epitope of OspA presented by the DRB1*0401 molecule was identified as OspA163−175 (Gross et al., 1998), and patients with antibiotic-refractory Lyme arthritis often had T cell recognition of this epitope (Chen et al., 1999). Recently, antibiotic-refractory Lyme arthritis was associated with HLA-DR molecules that showed moderate to strong binding of the OspA163−175 epitope, including the DRB1*0101, 0401, 0402, 0404, and 0405 molecules, and the DRB5 partner of the genetically linked DRB1*1501/DRB5*0101 molecules (Steere et al., 2006). In contrast, the DRB1*0301, 0801, 1101, and 1104 molecules, which showed negligible or no binding of the peptide, were more commonly found in patients with antibiotic-responsive arthritis. Only the DRB1*0102 and the genetically-linked DRB1*0701/DRB4*0101 molecules could not be categorized this way, since neither was clearly associated with a refractory or responsive outcome. Overall, 79% of the refractory arthritis patients had at least one of the seven known OspA peptide-binding HLA-DR molecules compared with 46% of those with antibiotic-responsive arthritis (odds ratio = 4.4; P<0.001).
It is not yet known how T cell reactivity to OspA163−175 might be involved in antibiotic-refractory Lyme arthritis. According to the molecular mimicry hypothesis of autoimmunity, T cells activated by a borrelial peptide during the infectious phase of the illness may continue to be stimulated by a homologous self-antigen during the post-infectious phase (Benoist and Mathis, 2001; Fujinami and Oldstone, 1985). In antibiotic-refractory Lyme arthritis, LFA-1αL332−340, a self-peptide with partial sequence homology with OspA165−173, was originally proposed as a candidate autoantigen (Gross et al., 1998). However, the LFA-1 peptide was later shown to act as only a weak, partial agonist for OspA165−173 –reactive DRB1*0401 T cells (Trollmo et al., 2001), and did not bind well to the refractory arthritis-associated DRB1*0101 molecule (Steere et al., 2003), making it unlikely to be a relevant autoantigen.
Other investigators attempted to identify OspA161−175 specific-TCR cross-reactivity with other peptides in an unbiased manner (Maier et al., 2000). In their study, the TCR recognition motifs of seven OspA164−175-specific HLA-DR4-restricted T cell hybridomas were determined using a peptide analog library in which individual residues of the OspA164−175 epitope were substituted for all 20 naturally occurring amino acids. Of 387 human or mouse peptides identified, 13 (3%) were recognized by at least one hybridoma cell line. However, none of the peptides were recognized by all seven hybridomas, and none of the hybridomas recognized all 13 peptides. A second search based on OspA164−175 sequence homology identified an additional 88 human or mouse peptides including LFA-1αL331−340. Peptides derived from glycerol kinase, complement receptor type I or complement receptor type II were each recognized by one hybridoma cell line, however the LFA-1α peptide was not recognized by any of the lines. These three cross-reactive peptides were identical at only four of the nine OspA165−173 amino acid residues and at only one of the major T cell contact sites. Though this study idenitified OspA165−173 cross-reactive self-epitopes, it was unable to address whether antibiotic-refractory Lyme arthritis patients have T cell responses to any of these self-antigens.
Since the human proteome is now much better characterized, the goal here was to search the proteome again for self-peptides with sequence homology with OspA165−173, and in an effort to determine pathogenicity, to test the best candidate autoantigens for mRNA expression in synoviocytes, in vitro HLA-DR-peptide binding, and patients' T cell proliferative responses.
2. Materials and methods
2.1 Computer database search for potential autoantigens
To identify potential autoantigens with sequence homology with OspA165−173, the Entrez protein databases (SwissProt, PIR, PRF, PDB, and translations from annotated coding regions in GenBank and RefSeq), which contain ∼ 34,000 unique human protein sequences, were searched for human peptides with sequence identity to OspA165−173 epitope using the blast program "search for short, nearly exact matches" (www.ncbi.nlm.nih.gov/BLAST/). The matrices selected were PAM30 and Blosum80.
2.2 PCR
To determine whether candidate autoantigen mRNA might be expressed in inflamed synovium or immune tissue, a human rheumatoid arthritis-positive synoviocyte lambda cDNA library (Stratagene) and a multiple tissue cDNA immune panel (Clontech, Mountain View, CA) were tested for the presence of the transcripts of interest. Each 50 ul reaction contained either 1 μl of the synoviocyte cDNA library or 4 μl from each tissue in the cDNA immune panel in addition to one unit of Qiagen Hot Star Taq polymerase, 1X Qiagen PCR Buffer, 1% Tween-20, 20 μM of each dNTP and 0.4 μM of each primer set. Primer sets, which were designed to amplify the coding region of each gene, were as follows: MAWD-BP: 5′-GTCCTGGATTTGGGAGTTGA-3′ and 5′-ATTCCCACGAAATGCTCTTG-3′; T-span7 5′-ACTGACACTCCCACCCAGTC-3′, and 5′-CGATGATGAGTTGCAGTTGG-3′; LFA-1αL: 5′-GTTCTTGGCCAAAGACCAAA-3′, and 5′-ATGCAGGAAGACAGGAAGGA-3′; G3PDH: 5′ - GAGTCAACGGATTTGGTCGT-3′, and 5′ - TTGATTTTGGAGGGATCTCG-3′. Amplifications were performed at 55°C for 40 cycles using a PTC-200 Peltier Thermal Cycler (MJ Research). MAWD-BP, T-span7, LFA-1αL and G3PDH generated 238, 190, 164 and 233 basepair fragments respectively.
2.3 HLA-DR molecules and MHC/peptide binding assays
Based on a previous analysis of DRB binding to OspA163−175 (Steere et al., 2006), an antibiotic-refractory arthritis-associated DRB binding profile was defined as one in which a peptide was bound by the HLA-DRB1*0101, 0401, 0402, 0404, and 0405 molecules and the DRB5*0101 or DRB1*1501 molecule, but not by the DRB1*0301, 1101 and 1104 molecules. Since antibiotic-refractory arthritis was not clearly associated with either the DRB1*0102 or the DRB1*0701/DRB4*0101 molecules, these molecules were not included in the definition. For human T cell epitopes with promising predicted MHC binding profiles, actual peptide binding was determined in vitro using recombinant forms of these 13 HLA-DRB molecules.
The DRB molecules were constructed and purified as previously described for the HLA-DRB1*0401 molecule (Novak et al., 1999). Briefly, the HLA-DR molecules were produced by subcloning the extracellular domain sequences of DRα and the various DRβ chains directly to the acidic and basic portion of the Jun/Fos leucine zipper sequences, respectively. The vectors were cotransfected into Schneider S-2 cells and soluble DR molecules were purified by affinity chromatography using the HLA-DR specific L243 antibody (American Type Culture Collection, Rockville, MD). All peptides were synthesized using an Applied Biosystems model 432 synthesizer and biotinylated as previously described (Kwok et al., 1995). Two aminohexanonic acid (AHA) spacers were placed between the biotin and the peptide to inhibit steric hindrance.
For binding assays, a 96-well plate (Costar 3590, Cambridge MA) was coated with 100 μl (12.5 μg/ml) of the anti-DR monoclonal antibody L243 diluted in 12.5 mM borate buffer (pH 8.1), and the plate was incubated overnight at 4°C. In a separate plate (Costar 3365), 1 μl (250 μM) of each biotinylated peptide diluted in DMSO (Sigma) was placed in duplicate wells. Afterwards, 50 μl of purified MHC molecules (4 ng/μl) diluted in 150 mM citrate phosphate buffer (pH 5.4) with 0.75% n-octyl-β-d-glucopyranoside (Sigma) and 1mM PefaBloc (Roche) was added to each well. This plate was incubated overnight at 37°C in a humidified chamber.
The following day, the antibody plate was washed five times with 0.05% Tween-20 in PBS (pH. 7.4) and blocked with 5% fetal calf serum in PBS for three hours at room temperature. After washing again, 50 μl of 50 mM Tris (pH 8.0) with 0.75% n-octyl-β-d-glucopyranoside was added to each well. The MHC/peptide complexes were transferred directly to the antibody plate and incubated over night at 4°C. The following day, the plate was washed again and 100 μl of europium-labeled streptavidin (0.1 mg/ml), diluted in Assay Buffer, (both from Wallac/PerkinElmer) was added to each well and incubated for 30 minutes at room temperature. The plate was washed a final time and 100 μl of Enhancement Buffer was added. Fluorescence was measured 10−15 minutes later with a Victor2 1420 multilabel counter (Wallac/PerkinElmer).
2.4 Proliferation Assays
To determine whether patients with antibiotic-refractory arthritis had T cell reactivity with candidate autoantigens, we tested cells from 11 patients with antibiotic-refractory arthritis who had at least one of the four HLA-DR molecules (HLA-DRB1*0101, 0401, 0404, or DRB5*0101) most commonly associated with antibiotic-refractory Lyme arthritis. All patients participated in a study, “Immunity in Lyme Arthritis”, which was approved by the Human Investigations Committees at Tufts-New England Medical Center and Massachusetts General Hospital.
T cell proliferation assays using bulk lymphocytes were performed as previously described (Chen et al., 1999). Briefly, PBMC or synovial fluid mononuclear cells (SFMC), collected using Ficoll-Hypaque density centrifugation, were stored in liquid nitrogen. On the day of testing, the cells were thawed quickly, washed with PBS (Invitrogen), and plated in round-bottom, 96-well plates (Costar, 3799) at 2 × 105 cells per well in 200 μl of complete RPMI 1640 containing 2 mM glutamine, penicillin (100 units/ml), streptomycin (100 μg/ml), 10 mM HEPES (all Invitrogen) and 10% human AB serum (Cellgro). Peptides were added at a final concentration of 1 μM in duplicate wells. After five days, 100 μl of culture media was removed from each well and replaced with an equal volume of fresh media containing 0.5 μCi of 3H-thymidine (PerkinElmer). Cells were harvested 16−18 hours later, and incorporated 3H-thymidine was measured using a TopCount scintillation counter (PerkinElmer).
2.5 MAWD-BP ELISA
Because six of the patients with antibiotic-refractory Lyme arthritis had moderate to weak T cell proliferative responses to MAWD-B276−288, we determined whether these patients and five others had antibody responses to the whole protein. The vector pCMV-Sport-MAWD-BP was obtained from the I.M.A.G.E. consortium (http://image.llnl.gov/image/) from which the MAWD-BP gene was subcloned into pGEX-4T-3 (GE Healthcare Life Sciences). Early attempts to purify GST-MAWD-BP from DH5α extracts using affinity chromatography were unsuccessful because the fusion protein would not elute from the glutathione beads. Consequently, the ELISA was performed using the GST 96-well detection module (GE Healthcare 27−4592−01) as directed by the manufacturer. Briefly, bacterial extracts expressing GST-MAWD-BP, GST (background control) or GST-Arp (positive control) at concentrations previously determined to be saturating, were added to wells coated with anti-GST antibodies. GST-Arp was chosen as a positive control because the majority of antibiotic-refractory Lyme arthritis patients were demonstrated to have a humoral response to this protein (Salazar et al., 2005). After a one-hour incubation, the plate was washed and either patient sera samples (diluted 1/100) or rabbit sera containing polyclonal anti-MAWD-BP antibodies (Alpha Diagnostics, San Antonio, TX), as a control, were added. The plate was again incubated for an hour and wash, after which anti-human-IgG-HRP or anti-rabbit-IgG-HRP secondary antibody (Biosource, Camarillo, CA) was added. Anti-MAWD-BP antibodies were detected using the substrate TMB (3,3′,5,5′-tetramethylbenzidine) and the reaction was stopped with 2 N H2SO4. The plate was read at 450 nm using a microplate reader (Bio-Rad model 550).
3. Results
3.1 Identification of candidate autoantigens with sequence homology with OspA165−173
In the initial search for candidate autoantigens in the late 1990s, the best match was human LFA-1αL332−340, which has five identical amino acids to B. burgdorferi OspA165−173, including the same P1 anchor residue and two identical amino acids in major T cell contact sites. However, this peptide acted as only a weak, partial agonist for OspA165−173-reactive T cells (Trollmo et al., 2001). A molecular characterization of the OspA epitope using a human OspA161−175 –specific HLA-DR4-restricted T cell line showed that the Val166 (P2) and Thr172 (P8) T cell contact sites were critical for T cell reactivity with this peptide (Drouin et al., 2004). Therefore, in the current study, we searched for self-peptides with equal or greater sequence homology to OspA165−173 than the LFA-1 peptide, particularly at the T cell contact sites. We identified 17 peptides with five or more amino acids identical to the OspA core epitope, an aromatic or hydrophobic amino acid at the proposed P1 MHC major anchor site (an important characteristic for MHC binding (Hill et al., 1991; Hill et al., 1994; Jardetzky et al., 1990; O'Sullivan et al., 1991)), and no more than one substitution at any of the three major T cell contact sites (P2, P5 and P8). Three of the peptides identified were added to the database since LFA-1 was identified as a candidate autoantigen in antibiotic-refractory Lyme arthritis in 1998 (Gross et al., 1998).
The current best match was MAWD-BP280−288, which was identical to OspA165−173 at eight of the nine core amino acid residues including all three major T cell contact sites (Table 1). The only substitution was Val for Tyr at the P1 MHC major anchor site, which would not be predicted to inhibit binding. However, the MAWD-BP280−288 epitope comprised the last 13 C-terminal amino acids of the protein, resulting in the absence of peptide flanking residues (PFR) on the carboxyl terminus, which might result in a less stable MHC/peptide complex (Nelson et al., 1993; Nelson et al., 1994; Sette et al., 1989). The second best match was T-span760−68, which shared identity with OspA165−173 at six residues, including the P1 anchor site and the three major T cell contact sites. The ubiquitin E4 factor466−482 epitope shared five amino acids in common with the OspA epitope, including the three major T cell contact sites and a compatible P1 anchor residue (Leu), but the glutamine in the P9 anchor site was likely to inhibit binding to DRB1*0101 and 0401 (Hammer et al., 1994; Hammer et al., 1997). The other peptides contained a substitution at one of the T cell contact sites that would make cross-reactivity less likely.
Table 1.
Human peptides with sequence homology to B. burgdorferi OspA165−173a
|
Major T Cell Contact Sites |
P2 |
P5 |
P8 |
Accession # |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| B. burgdorferi OspA165−173 | Y | V | L | E | G | T | L | T | A | NP_045688 |
| Human Homologues | ||||||||||
| Eight identical residues | ||||||||||
| MAWD-BP280−288b | V | " | " | " | " | " | " | " | " | NP_071412 |
| Six identical residues | ||||||||||
| T-span760−68c | " | " | " | I | " | " | G | " | T | NP_004606.2 |
| Ornithine Decarboxylase Inhibitor28−36 | " | " | Y | " | H | " | " | " | G | NP_680479 |
| Sorting Nexin 1621−629 | " | " | " | " | S | K | " | " | E | NP_722523.1 |
| Five identical residues | ||||||||||
| Ubiquitination Factor E4B341−349 | L | " | " | Q | " | S | " | " | Q | NP_006039 |
| Carnitine Transporter492−500d | " | I | " | M | " | S | " | " | I | NP_003051 |
| SOS-1444−452e | F | I | M | " | " | " | " | " | R | NP_005624.2 |
| Mitochondria P Carrier318−326f | I | I | M | I | " | " | " | " | " | NP_002626.1 |
| EMAP-1525−533g | F | " | " | Q | " | " | " | S | G | NP_004425 |
| Transmembrane NMB303−311h | " | " | " | N | " | " | F | S | L | NP_002501 |
| LFA-1332−340i | " | " | I | " | " | " | S | K | Q | NP_002200 |
| IL-1 Receptor accessory472−480 | " | " | " | Q | " | " | Q | A | L | NP_002173.1 |
| Pleckstrin homology domain324−332 | L | " | " | " | " | " | F | R | I | NP_001025055 |
| Erythroid Factor76−84 | " | " | " | " | " | L | K | F | V | AAC00001.1 |
| SHIP-2987−995j | " | " | " | " | " | V | P | H | Q | NP_001558.2 |
| LSP-1180−188k | L | " | " | " | " | " | I | E | Q | NP_002330 |
| ATP Binding Cassette530−538 | " | " | " | F | " | " | Y | I | I | NP_005680.1 |
Only peptides with a compatible P1 anchor residue (hydrophobic or aromatic) and no more than a single substitution at one of the major T cell contact sites are shown.
MAPK activator with WD-repeats binding protein
Tetraspanin 7
aka Solute carrier family 22 member 5
Son of Sevenless homolog 1
aka Solute carrier family 25 member 3 isoform b precursor
Echinoderm microtubule associated protein like 1 isoform b
glycoprotein nonmetastatic melanoma protein B
Integrin alpha L
Inositol polyphosphate phosphatase-like 1
lymphocyte-specific protein 1 isoform
Because availability of patients' T cells is limited and because these cells cannot be replenished, we focused on two peptides, MAWD-BP280−288 and T-span760−68, with the greatest degree of homology with OspA165−173, as well as the previously identified LFA-1αL332−340, in an effort to assess the potential pathogenicity of homologous self-peptides.
3.2 Tissue expression of candidate autoantigens
Though MAWD-BP and T-span7 were previously shown to be expressed in multiple tissues (Iriyama et al., 2001; Takagi et al., 1995), it was unknown whether either was expressed in joint tissue (e.g. synoviocytes), or whether MAWD-BP was expressed by any immune cells. Therefore, template cDNAs from immune cells or tissues, including synoviocytes from rheumatoid arthritis patients were tested for transcriptional expression of these two candidate autoantigens, and for the earlier identified candidate autoantigen LFA-αL. We detected MAWD-BP mRNA in all eight of the cells and tissue types, including PBL and synoviocytes (Fig. 1). T-span7 mRNA was also found in most immune cells or tissues, but not in PBL or synoviocytes and the well-characterized integrin LFA-1αL was expressed in all cell types tested except synoviocytes.
Fig. 1.
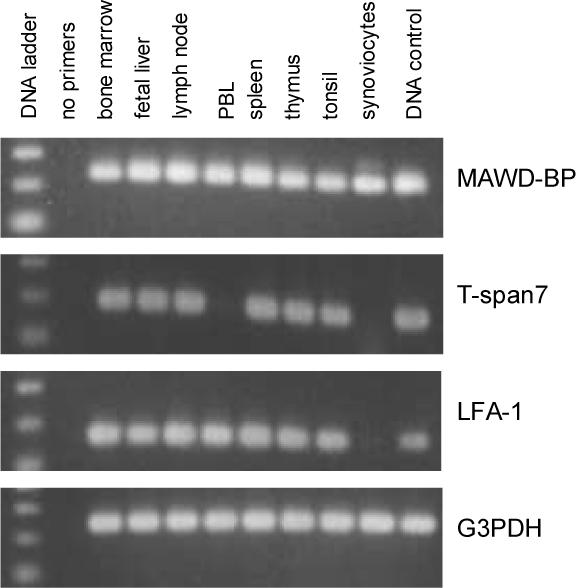
mRNA expression of candidate autoantigens in various types of immune cells including synoviocytes. First strand cDNA preparations from various human cells and tissues (Clontech, CA) and cDNA from a lambda rheumatoid arthritis synoviocyte library (Stratagene, CA) were used as templates for PCR with candidate autoantigen gene-specific primers. The cell types examined are indicated above each lane. The expression of glycerol-3-phosphate dehydrogenase (G3PDH) was examined as a control. The 100-bp ladder shown indicates bands of 100, 200 and 300 basepairs. MAWD-BP, T-span7, LFA-1αL and G3PDH generated 238, 190, 164 and 233 basepair fragments respectively.
3.3 In vitro MHC/peptide binding assays of candidate autoantigens
We next assessed whether 13 recombinant DRB molecules bound the MAWD-BP280−288, T-span 760−68, and LFA-1αL332−340 peptides, using in vitro MHC/peptide binding assays. As shown previously (Steere et al., 2006), all six antibiotic-refractory associated DR molecules (DRB1*0101, 0401, 0402, 0404, 0405 and DRB5*0101) bound OspA163−175, whereas the antibiotic-responsive associated DR molecules (DRB1*0301, 1101 and 1104 molecules) did not. In contrast, MAWD-BP276−288 was bound well by only three of the refractory arthritis associated DR molecules (Fig. 2), a difference that may result from the peptide's absence of peptide flanking regions (PFRs) (Nelson et al., 1993; Nelson et al., 1994; Sette et al., 1989). In support of this idea, when the MAWD-BP peptide was tested with a human OspA161−175 –specific HLA-DR4-restricted T cell line, proliferation was less than half of that induced by the OspA peptide. However, when a modified MAWD-BP peptide containing two additional C-terminal amino acids was tested, proliferation nearly doubled relative to the parent peptide, almost reaching the level induced by OspA163−175 (unpublished data). As for the other two self-peptides, T-span758−70 was bound by only four of the antibiotic-refractory associated DR molecules, while LFA-1αL332−340 was bound by six. However, LFA-1αL332−340 binding to the DRB1*0101 molecule, which is highly associated with antibiotic-refractory arthritis, was weak. Furthermore, using a competition assay (Steere et al., 2003), we previously found that the LFA-1 peptide was not able to compete well with a known positive control peptide for binding to the 0101 molecule.
Fig. 2.

In vitro binding profiles of OspA163−175 and self-peptides to 13 recombinant HLA-DRB molecules. MHC molecules are aligned on the X-axis according to their association with antibiotic-refractory Lyme arthritis (DRB1*1501/B5*0101 and DRB1*0701/B4*0101 are in linkage disequilibrium) . MHC molecules were incubated with labeled peptides and MHC/labeled-peptide complexes were detected by fluorescence. The bars represent mean relative fluorescence of duplicate wells and the error bars denote the standard deviation. The amino acid sequence for each peptide is shown with core amino acids capitalized. Amino acids that are identical to OspA165−173 sequence are in bold, while identical T cell contact site residues are underlined. All positive control peptides gave markedly positive results. The sequences of the positive control peptides were 0102-PC, PKYVKLNALKLAT for the DRB1*01 molecules; influenza NSI32−45, FLDRLRRDQRSLRG for 0301; GAD65557I, NFIRMVMSNPAAT for the 04 molecules, 1501 and B4*0101; 0701-PC, TSLYVRTSSFVIVSI for 0701; 1104-PC, PKYVKLNKLKSAT for 1101 and 1104; and HA307−319, PKYVKQNTLKLAT for B5*0101. OspA165A, KGAVLEGTLTAEK was used as a negative control peptide with all MHC molecules except the 04 molecules in which 04-NC, VSAATRGSLQATV was used.
3.4 Immune recognition of candidate autoantigens
PBMC or SFMC (synovial fluid mononuclear cells) from 11 patients with antibiotic-refractory Lyme arthritis who had the DRB1* 0101, 0401, 0404 or B5*0101 alleles were stimulated with the OspA epitope and the three human peptides in proliferation assays (Fig. 3). Of the 11 patients, nine had reactivity with OspA161−170, and six had reactivity with MAWD-BP276−288, though their responses to the MAWD-BP peptide were lower than those induced by the OspA peptide. Patients expressing one or two of the four MHC molecules tested responded to both peptides, including those with the DRB1*0101 or DRB5*0101 molecules. This was surprising since binding of the MAWD-BP peptide by these two DRB molecules was below background in our in vitro MHC/peptide binding assays. However, these T cell proliferation results imply that these two MHC molecules on antigen presenting cells can bind and present the MAWD-BP276−288 peptide. As for the other peptides, T-span758−70 stimulated T cells weakly in only three patients, and LFA-1αL332−340 weakly activated T cells from only two patients.
Fig. 3.

Proliferation assays testing the recognition of candidate autoantigenic peptides by antibiotic-refractory Lyme arthritis patients' T cells. Bulk PBMC or SFMC (synovial fluid mononuclear cells) from 11 patients with antibiotic-refractory Lyme arthritis who expressed the antibiotic-refractory arthritis-associated DRB1* 0101, 0401, 0404 or B5*0101 molecules were stimulated with three self-peptides and the borrelial peptide OspA161−175 (all 1 μM). Two patients expressed two refractory arthritis-associated HLA-DR alleles, patient 1 (0402/0404) and patient 2 (0101/0401). There were not enough cells from patients two or four to determine their proliferative responses to LFA-1αL332−340. The average CPM of duplicate wells is indicated. The HLA-DR alleles of the control subjects were unknown.
Since six patients had a T cell response to MAWD-BP276−288, we determined whether patients with antibiotic-refractory Lyme arthritis had linked B cell responses to this protein. Serum samples from these six patients and five others were tested for MAWD-BP specific antibodies by ELISA (Fig. 4). While 10 of 11 patients had a significant antibody response to the recombinant B. burgdorferi protein (GST-Arp), in agreement with previous data [24], none had detectable autoantibodies to recombinant GST-MAWD-BP over background. Moreover, serial serum samples from five patients obtained over a 6- to 24-month period did not reveal any MAWD-BP-specific autoantibodies (data not shown).
Fig. 4.

Determining if antibiotic-refractory Lyme arthritis patients produce MAWD-BP specific autoantibodies. Sera from eleven antibiotic-refractory Lyme arthritis patients, including all six with a T cell response to MAWD-BP276−280 (Fig 3) were tested for anti-MAWD-BP specific antibodies by ELISA. Because the fusion protein, GST-MAWD-BP, could not be purified from bacterial lysates, ELISA were performed by incubating bacterial lysates on plates precoated with anti-GST antibodies. Also tested were extracts from cells expressing GST (background control) and recombinant B. burgdorferi GST-Arp (positive control), a protein against which most antibiotic-refractory Lyme arthritis patients have an antibody response (Salazar et al., 2005). After binding of the GST proteins, patient sera (diluted 1/100) or rabbit sera containing polyclonal anti-MAWD-BP antibodies were added. Anti-MAWD-BP antibodies were detected with anti-human-IgG-HRP or anti-rabbit-IgG-HRP and the substrate TMB (3,3′,5,5′-tetramethylbenzidine). MAWD-BP autoantibodies were not detected in patient sera, however the polyclonal anti-MAWD-BP sera clearly detected the recombinant GST-MAWD-BP.
4. Discussion
In this study, we searched the nearly complete human genome for peptides with sequence homology with B. burgdorferi OspA165−173. The most promising self-peptide appeared to be MAWD-BP280−288, which shares eight of the nine core amino acid residues with the spirochetal peptide. Little is known about this human protein (Iriyama et al., 2001). It was initially identified based on its ability to bind MAWD (MAPK activator with WD-repeats), a protein that may be involved in cell signaling pathways (Matsuda et al., 2000). Consistent with previous reports (Iriyama et al., 2001), we detected mRNA for MAWD-BP in all tissues tested, including synoviocytes, suggesting that this protein is ubiquitously expressed. However, it was not bound in vitro by all six antibiotic-refractory associated DRB molecules. In addition, although six of the 11 antibiotic-refractory patients had mild-to-moderate T cell proliferative responses to MAWD-BP276−288, reactivity was always lower than that induced by OspA163−175. Finally, refractory patients lacked an antibody response to MAWD-BP protein, though we recognize that a pathogenic T cell response would not necessarily have to induce a linked B cell response. Taken together, it seemed unlikely that MAWD-BP280−288 was a relevant autoantigen in antibiotic-refractory Lyme arthritis.
The other newly identified human peptide tested was T-span760−68, which shares identity at six OspA165−173 core amino acid residues including the P1 anchor site and all the T cell contact sites. T-span7, a cell surface glycoprotein that mediates signal transduction events (Berditchevski, 2001), was not expressed in synoviocytes; the T-span7 peptide was not bound in vitro by all antibiotic-refractory associated DRB molecules, and it induced weak T cell proliferation in only a few patients. Similarly, LFA-1αL332−340, which has five amino acids in common with OspA165−173, including two at T cell contact sites, was not expressed by synoviocytes, and it induced weak T cell proliferative responses in only a few patients. In a previous analysis (Gross et al., 1998), synovial fluid T cells from 6 of 11 patients with antibiotic-refractory Lyme arthritis produced IFN-γ when stimulated with OspA164−175 or LFA-1αL326−345, as determined by an ELISA spot assay. However, when OspA165−184-reactive T cells were cloned, the OspA peptide induced T cell proliferation and IFN-γ and IL-13 secretion in almost all clones tested, whereas the LFA-1 peptide stimulated only ∼10% of these clones to proliferate, and they secreted only IL-13 (Trollmo et al., 2001). Moreover, we previously found that patients with Lyme arthritis lacked an antibody response to LFA-1 (unpublished data). Thus, neither T-span760−68 nor LFA-1αL332−340 appeared to be a relevant autoantigen.
Our analysis does not exclude the possibility that a structural mimic of OspA165−173, rather than one with simple sequence homology, might have a pathogenic role in antibiotic-refractory Lyme arthritis (Kamradt and Volkmer-Engert, 2004). It is known that individual substitutions in a peptide sequence may inhibit TCR recognition, whereas a series of substitutions may produce an activating peptide (Ausubel et al., 1996). However, the rules that govern structural recognition of a peptide are not yet clear. In an effort to get around this problem, Ghosh et al. generated recombinant antibody probes from synovial lesions of two patients with antibiotic-refractory Lyme arthritis in an attempt to reproduce disease-relevant antigen-binding specifities at the site of inflammation (Ghosh et al., 2006). These probes identified human cytokeratin 10 as a candidate autoantigen, and several of the antibody probes cross-reacted with OspA. However, only 3 of 15 patients with antibiotic-refractory arthritis had weak antibody responses to cytokeratin 10 that were slightly above the responses in patients with antibiotic-responsive arthritis, rheumatoid arthritis, or in healthy control subjects.
Previous analyses of patients with antibiotic-refractory Lyme arthritis have not examined whether T cell activation by the OspA165−173 epitope is sustained in the post-infectious period. Therefore, using tetramer reagents, we recently determined the frequencies of OspA165−173–reactive T cells in peripheral blood or synovial fluid samples of patients throughout the course of Lyme arthritis. Approximately 60% of HLA-DRB1*0401-positive patients with either antibiotic-responsive or antibiotic-refractory arthritis initially had increased frequencies of OspA165−173–reactive T cells (Kannian et al., 2006). These responses tended to be higher in antibiotic-refractory patients than in antibiotic-responsive patients. However, the frequencies of OspA165−173–reactive T cells declined to baseline levels in both refractory and responsive patients within weeks after the initiation of antibiotic therapy, months before the resolution of arthritis in the refractory group. Thus, since some antibiotic-refractory patients did not develop a response to OspA and since this response declined after antibiotic therapy, these results argue against molecular mimicry with OspA165−173 as a critical mechanism in antibiotic-refractory Lyme arthritis.
Insulin-dependent diabetes mellitus (IDDM) is another illness in which infection-induced autoimmunity may play a pathogenic role. It is known that Coxsackie B4 viral infection is strongly associated with the development of IDDM. Based on sequence homology between the viral protein P2-C and the islet autoantigen glutamic acid decarboxylase (GAD65), molecular mimicry was proposed as the mechanism of autoimmunity. However, when NOD mice with susceptible MHC alleles were virally infected, they developed diabetes at the same rate as uninfected control mice. In comparison, when mice expressing a T cell receptor transgene specific for a different islet cell autoantigen (BDC2.5) were infected, they developed diabetes rapidly, while uninfected controls never did (Horwitz et al., 1998). These results suggest that tissue damage and inflammation resulted in the release of sequestered antigen(s), which triggered autoimmunity (bystander T cell activation), rather than molecular mimicry between viral and self-epitopes.
Recent evidence is consistent with this alternative hypothesis of autoimmunity in antibiotic-refractory Lyme arthritis. In this study, antibiotic-refractory patients had significantly higher levels of TH1 chemoattractants and cytokines in joint fluid, particularly CXCL9 and IFN-γ, than antibiotic-responsive patients. Moreover, in refractory patients, these inflammatory responses remained high or even increased in the post-antibiotic period, though reactivity with borrelial proteins declined (Shin et al., 2007). The idea that an autoantigen may continue to drive this highly inflammatory immune response after spirochetal killing remains an attractive hypothesis. Thus, similar to the situation with IDDM and coxsackie virus, our current model is that OspA165−173 (or other currently unidentified borrelial epitopes) may cause exceptionally high inflammatory responses in genetically susceptible individuals, and this response might serve as a bridge to activate autoreactive T cells that recognize a non-OspA related self-epitope. With techniques now available, it may be possible to isolate pathogenic autoantigenic peptides directly from the MHC/peptide complexes in synovial tissue, identify them by mass spectrometry, and test candidate autoantigens using patients' T cells.
In conclusion, there remains an association among particular HLA-DR molecules, OspA165−173 and antibiotic-refractory Lyme arthritis (Steere et al., 2006) and infection-induced autoimmunity is an attractive hypothesis to explain this outcome. However, molecular mimicry due to sequence homology between OspA165−173 and a human peptide seems unlikely to be the critical mechanism.
Acknowledgements
We would like to thank Dr. Lee Ann Baxter-Lowe for HLA typing of patients. This work was supported by grants AR-20358 from the National Institutes of Health (NIH), the Mathers Foundation, the English, Bonter, Mitchell Foundation, the Lyme/Arthritis Research Fund, and the Esche Fund. Dr. Drouin was supported by a scholarship for Lyme disease studies by the Lillian B. Davey Foundation.
Glossary
Abbreviations:
- OspA
outer-surface protein A
- Bb
Borrelia burgorferi
- SFMC
synovial fluid mononuclear cells
- MAWD-BP
MAPK activator with WD-repeats binding protein
- T-span7
tetraspanin 7
- LFA-1α
lymphocyte function-associated antigen 1α
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akin E, McHugh GL, Flavell RA, Fikrig E, Steere AC. The immunoglobulin (IgG) antibody response to OspA and OspB correlates with severe and prolonged Lyme arthritis and the IgG response to P35 correlates with mild and brief arthritis. Infect Immun. 1999;67:173–81. doi: 10.1128/iai.67.1.173-181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ausubel LJ, Kwan CK, Sette A, Kuchroo V, Hafler DA. Complementary mutations in an antigenic peptide allow for crossreactivity of autoreactive T-cell clones. Proc Natl Acad Sci U S A. 1996;93:15317–22. doi: 10.1073/pnas.93.26.15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist C, Mathis D. Autoimmunity provoked by infection: how good is the case for T cell epitope mimicry? Nat Immunol. 2001;2:797–801. doi: 10.1038/ni0901-797. [DOI] [PubMed] [Google Scholar]
- Berditchevski F. Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci. 2001;114:4143–51. doi: 10.1242/jcs.114.23.4143. [DOI] [PubMed] [Google Scholar]
- Carlson D, Hernandez J, Bloom BJ, Coburn J, Aversa JM, Steere AC. Lack of Borrelia burgdorferi DNA in synovial samples from patients with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 1999;42:2705–9. doi: 10.1002/1529-0131(199912)42:12<2705::AID-ANR29>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Chen J, Field JA, Glickstein L, Molloy PJ, Huber BT, Steere AC. Association of antibiotic treatment-resistant Lyme arthritis with T cell responses to dominant epitopes of outer surface protein A of Borrelia burgdorferi. Arthritis Rheum. 1999;42:1813–22. doi: 10.1002/1529-0131(199909)42:9<1813::AID-ANR4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Dattwyler RJ, Halperin JJ, Volkman DJ, Luft BJ. Treatment of late Lyme borreliosis--randomised comparison of ceftriaxone and penicillin. Lancet. 1988;1:1191–4. doi: 10.1016/s0140-6736(88)92011-9. [DOI] [PubMed] [Google Scholar]
- Dattwyler RJ, Wormser GP, Rush TJ, Finkel MF, Schoen RT, Grunwaldt E, Franklin M, Hilton E, Bryant GL, Agger WA, Maladorno D. A comparison of two treatment regimens of ceftriaxone in late Lyme disease. Wien Klin Wochenschr. 2005;117:393–7. doi: 10.1007/s00508-005-0361-8. [DOI] [PubMed] [Google Scholar]
- Drouin EE, Glickstein LJ, Steere AC. Molecular characterization of the OspA161−175 T cell epitope associated with treatment-resistant Lyme arthritis: differences among the three pathogenic species of Borrelia burgdorferi sensu lato. J Autoimmun. 2004;23:281–92. doi: 10.1016/j.jaut.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Fujinami RS, Oldstone MB. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science. 1985;230:1043–5. doi: 10.1126/science.2414848. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Seward R, Costello CE, Stollar BD, Huber BT. Autoantibodies from synovial lesions in chronic, antibiotic treatment-resistant Lyme arthritis bind cytokeratin-10. J Immunol. 2006;177:2486–94. doi: 10.4049/jimmunol.177.4.2486. [DOI] [PubMed] [Google Scholar]
- Gross DM, Forsthuber T, Tary-Lehmann M, Etling C, Ito K, Nagy ZA, Field JA, Steere AC, Huber BT. Identification of LFA-1 as a candidate autoantigen in treatment-resistant Lyme arthritis. Science. 1998;281:703–6. doi: 10.1126/science.281.5377.703. [DOI] [PubMed] [Google Scholar]
- Hammer J, Bono E, Gallazzi F, Belunis C, Nagy Z, Sinigaglia F. Precise prediction of major histocompatibility complex class II-peptide interaction based on peptide side chain scanning. J Exp Med. 1994;180:2353–8. doi: 10.1084/jem.180.6.2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer J, Sturniolo T, Sinigaglia F. HLA class II peptide binding specificity and autoimmunity. Adv Immunol. 1997;66:67–100. doi: 10.1016/s0065-2776(08)60596-9. [DOI] [PubMed] [Google Scholar]
- Hill CM, Hayball JD, Allison AA, Rothbard JB. Conformational and structural characteristics of peptides binding to HLA-DR molecules. J Immunol. 1991;147:189–97. [PubMed] [Google Scholar]
- Hill CM, Liu A, Marshall KW, Mayer J, Jorgensen B, Yuan B, Cubbon RM, Nichols EA, Wicker LS, Rothbard JB. Exploration of requirements for peptide binding to HLA DRB1*0101 and DRB1*0401. J Immunol. 1994;152:2890–8. [PubMed] [Google Scholar]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N. Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med. 1998;4:781–5. doi: 10.1038/nm0798-781. [DOI] [PubMed] [Google Scholar]
- Iriyama C, Matsuda S, Katsumata R, Hamaguchi M. Cloning and sequencing of a novel human gene which encodes a putative hydroxylase. J Hum Genet. 2001;46:289–92. doi: 10.1007/s100380170081. [DOI] [PubMed] [Google Scholar]
- Jardetzky TS, Gorga JC, Busch R, Rothbard J, Strominger JL, Wiley DC. Peptide binding to HLA-DR1: a peptide with most residues substituted to alanine retains MHC binding. Embo J. 1990;9:1797–803. doi: 10.1002/j.1460-2075.1990.tb08304.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalish RA, Leong JM, Steere AC. Association of treatment-resistant chronic Lyme arthritis with HLA-DR4 and antibody reactivity to OspA and OspB of Borrelia burgdorferi. Infect Immun. 1993;61:2774–9. doi: 10.1128/iai.61.7.2774-2779.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamradt T, Volkmer-Engert R. Cross-reactivity of T lymphocytes in infection and autoimmunity. Mol Divers. 2004;8:271–80. doi: 10.1023/b:modi.0000036236.11774.1b. [DOI] [PubMed] [Google Scholar]
- Kannian P, Glickstein LJ, Drouin EE, Kwok WW, Nepom GT, Steere AC. Frequencies of Borrelia burgdorferi OspA161−175-specific T cells in HLA-DRB1*0401-positive patients with Lyme disease. Arthritis Rheum. 2006;54:S546. doi: 10.4049/jimmunol.179.9.6336. [DOI] [PubMed] [Google Scholar]
- Kwok WW, Nepom GT, Raymond FC. HLA-DQ polymorphisms are highly selective for peptide binding interactions. J Immunol. 1995;155:2468–76. [PubMed] [Google Scholar]
- Lengl-Janssen B, Strauss AF, Steere AC, Kamradt T. The T helper cell response in Lyme arthritis: differential recognition of Borrelia burgdorferi outer surface protein A in patients with treatment-resistant or treatment-responsive Lyme arthritis. J Exp Med. 1994;180:2069–78. doi: 10.1084/jem.180.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier B, Molinger M, Cope AP, Fugger L, Schneider-Mergener J, Sonderstrup G, Kamradt T, Kramer A. Multiple cross-reactive self-ligands for Borrelia burgdorferi-specific HLA-DR4-restricted T cells. Eur J Immunol. 2000;30:448–57. doi: 10.1002/1521-4141(200002)30:2<448::AID-IMMU448>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Matsuda S, Katsumata R, Okuda T, Yamamoto T, Miyazaki K, Senga T, Machida K, Thant AA, Nakatsugawa S, Hamaguchi M. Molecular cloning and characterization of human MAWD, a novel protein containing WD-40 repeats frequently overexpressed in breast cancer. Cancer Res. 2000;60:13–7. [PubMed] [Google Scholar]
- Nelson CA, Petzold SJ, Unanue ER. Identification of two distinct properties of class II major histocompatibility complex-associated peptides. Proc Natl Acad Sci U S A. 1993;90:1227–31. doi: 10.1073/pnas.90.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CA, Petzold SJ, Unanue ER. Peptides determine the lifespan of MHC class II molecules in the antigen-presenting cell. Nature. 1994;371:250–2. doi: 10.1038/371250a0. [DOI] [PubMed] [Google Scholar]
- Nocton JJ, Dressler F, Rutledge BJ, Rys PN, Persing DH, Steere AC. Detection of Borrelia burgdorferi DNA by polymerase chain reaction in synovial fluid from patients with Lyme arthritis. N Engl J Med. 1994;330:229–34. doi: 10.1056/NEJM199401273300401. [DOI] [PubMed] [Google Scholar]
- Novak EJ, Liu AW, Nepom GT, Kwok WW. MHC class II tetramers identify peptide-specific human CD4(+) T cells proliferating in response to influenza A antigen. J Clin Invest. 1999;104:R63–7. doi: 10.1172/JCI8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan D, Arrhenius T, Sidney J, Del Guercio MF, Albertson M, Wall M, Oseroff C, Southwood S, Colon SM, Gaeta FC, et al. On the interaction of promiscuous antigenic peptides with different DR alleles. Identification of common structural motifs. J Immunol. 1991;147:2663–9. [PubMed] [Google Scholar]
- Salazar CA, Rothemich M, Drouin EE, Glickstein L, Steere AC. Human Lyme arthritis and the immunoglobulin G antibody response to the 37-kilodalton arthritis-related protein of Borrelia burgdorferi. Infect Immun. 2005;73:2951–7. doi: 10.1128/IAI.73.5.2951-2957.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sette A, Lamont A, Buus S, Colon SM, Miles C, Grey HM. Effect of conformational propensity of peptide antigens in their interaction with MHC class II molecules. Failure to document the importance of regular secondary structures. J Immunol. 1989;143:1268–73. [PubMed] [Google Scholar]
- Shin JJ, Glickstein L, Steere AC. High levels of inflammatory chemokines and cytokines in joint fluid and synovial tissue throughout the course of antibiotic-refractory Lyme arthritis. Arthritis Rheum. 2007 doi: 10.1002/art.22441. In press. [DOI] [PubMed] [Google Scholar]
- Steere AC. Lyme disease. N Engl J Med. 2001;345:115–25. doi: 10.1056/NEJM200107123450207. [DOI] [PubMed] [Google Scholar]
- Steere AC, Angelis SM. Therapy for Lyme arthritis: strategies for the treatment of antibiotic-refractory arthritis. Arthritis Rheum. 2006;54:3079–86. doi: 10.1002/art.22131. [DOI] [PubMed] [Google Scholar]
- Steere AC, Dwyer E, Winchester R. Association of chronic Lyme arthritis with HLA-DR4 and HLA-DR2 alleles. N Engl J Med. 1990;323:219–23. doi: 10.1056/NEJM199007263230402. [DOI] [PubMed] [Google Scholar]
- Steere AC, Falk B, Drouin EE, Baxter-Lowe LA, Hammer J, Nepom GT. Binding of outer surface protein A and human lymphocyte function- associated antigen 1 peptides to HLA-DR molecules associated with antibiotic treatment-resistant Lyme arthritis. Arthritis Rheum. 2003;48:534–40. doi: 10.1002/art.10772. [DOI] [PubMed] [Google Scholar]
- Steere AC, Glickstein L. Elucidation of Lyme arthritis. Nat Rev Immunol. 2004;4:143–52. doi: 10.1038/nri1267. [DOI] [PubMed] [Google Scholar]
- Steere AC, Klitz W, Drouin EE, Falk BA, Kwok WW, Nepom GT, Baxter-Lowe LA. Antibiotic-refractory Lyme arthritis is associated with HLA-DR molecules that bind a Borrelia burgdorferi peptide. J Exp Med. 2006;203:961–71. doi: 10.1084/jem.20052471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steere AC, Levin RE, Molloy PJ, Kalish RA, Abraham JH,, 3rd, Liu NY, Schmid CH. Treatment of Lyme arthritis. Arthritis Rheum. 1994;37:878–88. doi: 10.1002/art.1780370616. [DOI] [PubMed] [Google Scholar]
- Steere AC, Schoen RT, Taylor E. The clinical evolution of Lyme arthritis. Ann Intern Med. 1987;107:725–31. doi: 10.7326/0003-4819-107-5-725. [DOI] [PubMed] [Google Scholar]
- Takagi S, Fujikawa K, Imai T, Fukuhara N, Fukudome K, Minegishi M, Tsuchiya S, Konno T, Hinuma Y, Yoshie O. Identification of a highly specific surface marker of T-cell acute lymphoblastic leukemia and neuroblastoma as a new member of the transmembrane 4 superfamily. Int J Cancer. 1995;61:706–15. doi: 10.1002/ijc.2910610519. [DOI] [PubMed] [Google Scholar]
- Trollmo C, Meyer AL, Steere AC, Hafler DA, Huber BT. Molecular mimicry in Lyme arthritis demonstrated at the single cell level: LFA-1 alpha L is a partial agonist for outer surface protein A-reactive T cells. J Immunol. 2001;166:5286–91. doi: 10.4049/jimmunol.166.8.5286. [DOI] [PubMed] [Google Scholar]